

Abstract
Dendritic cells (DC) are potent antigen-presenting cells essential for initiating adaptive immunity. Following pathogen exposure, trafficking of DC to lymph nodes (LN) through afferent lymphatic vessels constitutes a crucial step in the execution of their functions. The mechanisms regulating this process, however, are poorly understood, although the involvement of certain chemokines in this process has recently been reported. Herein, we demonstrate that genetically altering the fine structure (N-sulfation) of heparan sulfate specifically in mouse lymphatic endothelium significantly reduces DC trafficking to regional lymph nodes in vivo. Moreover, this alteration had the unique functional consequence of reducing CD8+ T cell proliferative responses in draining lymph nodes in an ovalbumin immunization model. Mechanistic studies suggested that lymphatic endothelial heparan sulfate regulates multiple steps during DC trafficking, including optimal presentation of chemokines on the surface of DC, thus acting as a co-receptor that may function “in trans” to mediate chemokine-receptor binding. This study not only identifies novel glycan-mediated mechanisms that regulate lymphatic DC trafficking, but also validates the fine structure of lymphatic-vascular specific heparan sulfate as a novel molecular target for strategies aiming to modulate DC behavior and/or alter pathologic T cell responses in lymph nodes.
INTRODUCTION
Dendritic cells (DC) are the most potent antigen-presenting cells, and their usual roles center on initiating both protective immunity as well as immunological tolerance. Upon exposure to antigens, DC exhibit superior capacity to take up and process antigens, undergo maturation, migrate to draining lymph nodes (LN), and present antigens to naïve T cells. The trafficking of DC from the periphery to the LN via afferent lymphatic vessels represents a critical element for the execution of DC functions. The overall process has multiple steps including local chemotaxis through extracellular matrix (ECM), adhesion to lymphatic endothelium, trans-endothelial migration, intra-lumen transit following lymph flow, and adhesion to as well as extravasation from lymphatic vessels in LN. Trafficking of DC to the LN depends on many chemokine-receptor interactions, among which the CCR7-CCL21/CCL19 axis appears to be the most potent and best characterized. The LN-homing chemokine receptor CCR7 is up-regulated upon DC maturation. Through interactions with its cognate ligands CCL19 and CCL21, which are produced by LN stromal cells and lymphatic endothelial cells (LEC), CCR7 facilitates directional migration of antigen-presenting DC toward regional LN(1, 2). Besides CCR7-CCL21/CCL19 crosstalk, other receptor-chemokine complexes, such as CXCR4-CXCL12, are also implicated in regulating DC migration to LN(3). Despite the biological significance of chemokines, the mechanistic importance of macromolecules that simultaneously control the actions of multiple chemokines along the intervening lymphatic conduit remains poorly understood. However, complex carbohydrates (glycans) bound on cell-surface as well as matrix proteoglycans are thought to be important regulators.
Heparan sulfate (HS) is a negatively charged, linear polysaccharide that regulates many cellular processes including proliferation, adhesion, migration, endocytosis and signal transduction. The glycan achieves this regulation through interactions with a variety of protein ligands, including growth factors such as basic fibroblast growth factor, various species of vascular endothelial growth factors, chemokines such as interleukin 8 (IL-8), CCL21, and CXCL12, and possibly others (4). Recent studies in our laboratory demonstrate that lymphatic endothelial HS modulates the optimal presentation of chemokine CCL21 both in cis (on CCR7+ lymphatic endothelium) and in trans (on CCR7+ migrating cells), thereby cooperating with the formation of chemokine gradients, to ultimately facilitate chemokine receptor-mediated migration signaling(5). As a functional consequence of this regulation, disrupting the biosynthesis of lymphatic endothelial HS, by targeting either HS chain initiation or chain sulfation, significantly reduces chemokine-dependent trafficking of tumor cells in the lymphatic system(5). So far, however, our understanding of how lymphatic traffic by DC is modulated in the lymphatic microenvironment, including mechanisms that control multiple chemokines, remains very limited. For CCL21 in particular, work in our laboratory, as well as a study examining the effects of a global-vascular disruption of HS chain elongation on multiple forms of cell traffic to the LN, showed that altering HS chains on the lymphatic endothelial cell surface impaired adhesion between CCL21 and lymphatic endothelium(5, 6). In addition, gradients of CCL21 in the peri-lymphatic space have been shown to be altered following chemical disruption of heparan sulfate in the extracellular matrix(7, 8). These observations raise questions as to what a lymphatic-vascular specific genetic disruption in the fine structure of heparan sulfate might do not only to DC traffic, but also to the ultimate T cell responses in the lymph node upstream from an antigenic stimulus.
In this study, we hypothesize that disruption of lymphatic HS will inhibit the trafficking of antigen-loaded DC to regional LN as well as antigen-dependent T cell activation. We also hypothesize that specific fine structural modifications of lymphatic HS (which can be genetically targeted) may uniquely affect DC trafficking. To test our hypothesis, we examined the significance of genetically altering the sulfation of lymphatic endothelial HS on the trafficking of DC to regional LN, and explored underlying mechanisms. We also assessed the subsequent effects on T-cell immunity. Our results reveal novel glycan-specific modifications and mechanisms that regulate lymphatic DC traffic, and validate the fine structure of lymphatic HS as a potential molecular target for therapeutic approaches to modulate DC behavior and/or correct pathologic immune responses.
MATERIALS AND METHODS
Cell culture and treatments
Primary human lung lymphatic endothelial cells (hLEC; Lonza, Basle, Switzerland) were cultured in EBM2 endothelial basal medium supplemented with EGM2 bullet kit (Lonza). Bone-marrow-derived dendritic cells (BMDC) were isolated from the femurs and tibias of C57Bl/6 mice (8–12 weeks of age) and differentiated with granulocyte macrophage colony-stimulating factor (GM-CSF; 20 ng/mL; Peprotech, Rocky Hill, NJ) as previously described(9). Unless otherwise stated, BMDC at day 9 of differentiation were used for experiments. All siRNA duplexes were from IDT (Coralville, IA) and transfected into cells according to the manufacturer’s instructions. For heparinase treatment, cells were incubated with heparinase (heparin lyases I, II and III; 2.5 mU/mL; generously provided by Dr. Jeffrey D. Esko, UCSD) in serum-free EBM2 medium at 37 °C, 5% CO2 for 1 h. To block specific chemokine or chemokine receptor signaling, cells were incubated with antibodies against CCL21 (1:100), CXCL12 (1:50), CCL5 (1:100), CXCR4 (1:100) or CCR7 (1:100, R&D, Minneapolis, MN) at 37°C, 5% CO2 for one hour (for adhesion and transmigration assays under shear flow), six hours (for transwell migration assays) or overnight (for transwell invasion assays).
Animals
All animal experiments were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of UCSD. Mice between four to eight weeks of age were used in this study. Lymphatic-specific Ndst1 mutants (designated Ndst1f/fProx1+/CreERT2) and their wild-type littermates (Ndst1f/fProx1−/CreERT2) were generated by breeding tamoxifen-inducible Prox1+/CreERT2 transgenic mice (generously provided by Dr G. Oliver at St. Jude Children’s Research Hospital, Memphis, TN) extensively backcrossed onto the C57Bl/6 background with Ndst1f/f conditional mutant mice. To induce the activity of Cre recombinase, tamoxifen (Sigma) dissolved in corn oil was intraperitoneally (i.p.) injected into the mice at 0.12 mg/g body weight daily for five consecutive days. In separate pilot studies, mice were found to tolerate this schedule well, remained with stable body weight, and behaved normally following tamoxifen injections. All mice were maintained in a pathogen-free facility on a 12 h:12 h light-dark cycle with food and water provided ad libitum.
Skin painting
Fluorescein isothiocyanate (FITC; 6.6 mg/mL; Sigma) and oxazolone (30 mg/mL, Sigma) were dissolved in 95% ethanol and painted onto the shaved abdomen of mice (200 μL/mouse). After 40 h, inguinal and axillary lymph nodes were isolated, and digested with 0.2% type I collagenase at 37 °C for 1 h. Digested samples were filtered through a 40-μm cell strainer and then stained with PE-labeled anti-mouse CD11c antibody (eBioscience), and PE/FITC double-positive cells were analyzed by FACSCalibur (BD).
Whole-mount immunofluorescence staining of mouse ear
Unstimulated tamoxifen-induced mutant versus wild-type mice were sacrificed, and their ears dissected and separated into dorsal and ventral sheets. The sheets were fixed in 1% paraformaldehyde, permeabilized with PBS containing 0.3% Triton X-100 (PT buffer), blocked with PT buffer supplemented with 3% goat serum, and then incubated with rabbit anti-Lyve1 antibody (1:800, Abcam, Cambridge, MA) overnight at 4°C. After three washes in 1% BSA/PT, Cy3-conjugated anti-rabbit secondary antibody (Jackson ImmunoResearch, West Grove, PA) was added and incubated at room temperature for 2 h. At least five random images were acquired from each sample using the fluorescence microscope, and the Lyve1+ lymphatic-vascular area per image was quantified using NIH Image J software.
Quantification of skin DC
Skin tissues were mechanically chopped into small pieces, and digested in DMEM medium containing 0.8% trypsin and 20 μg/mL DNase I (Sigma) at 37 °C for 1 h on a rocking plate. Digested cells were filtered through a 40-μm cell strainer (BD), stained with FITC-labeled anti-mouse CD11c antibody as well as PE-labeled anti-mouse MHCII antibody (eBioscience), and FITC/PE double-positive cells were analyzed by FACSCalibur.
Trafficking of in situ implanted BMDC
BMDC at day 9 of differentiation in culture were labeled with calcein AM and injected into the left foot dorsum of Ndst1f/fProx1+/CreERT2 mutants and their Ndst1f/fProx1−/CreERT2 wild-type littermates (2 × 106 cells/mouse). After 40 h, the left popliteal LN was isolated, imaged under the fluorescence microscope, and digested with 0.2% type I collagenase into a single-cell suspension. Cells from each LN digest were resuspended into 100 μL of PBS with 1.5 μL spotted onto the well of a Terasaki microtiter plate, and imaged under the fluorescence microscope. The number of calcein+ cells was quantified using NIH Image J software.
In vivo T cell proliferation in draining LN
Ovalbumin (OVA, Sigma) was dissolved into PBS at 5 mg/mL, and mixed thoroughly with Incomplete Freund’s Adjuvant (IFA, Sigma, v:v=1:1) until a uniform oil-in-water emulsion was formed. The emulsion was then injected intraperitoneally into the mice (200 μL/mouse). Seven days later, 40 μL of freshly prepared OVA: IFA emulsion was injected intradermally into the footpad. Five days later, the mice were sacrificed, with popliteal and inguinal LN from the injected side isolated, digested into single-cell suspension, stained with Alexa Fluor 488-labeled anti-CD3 together with APC-labeled anti-CD4 or APC-labeled anti-CD8 antibodies (eBioscience), and analyzed by FACSCalibur.
Adoptive transfer of CFSE-labeled T cells and their proliferation in vivo
CD8 T cells were purified from spleens of OT-I mice and CD4 T cells from spleens of OT-II mice (both OT-I and OT-II mice were kindly provided by Dr. Stephen Hedrick, UCSD Division of Biological Sciences) using corresponding Dynabeads Untouched Cell Isolation kits (Invitrogen, Carlsbad, CA), respectively. The purified T cells were labeled with carboxyfluorescein diacetate succinimidyl ester (CFSE) following an established protocol(10) and a mixture of equal numbers of CFSE-labeled CD4 and CD8 cells (3×106 each/mouse) were adoptively transferred into the mice via retro-orbital injection as previously described(11). After 24 h, the mice were injected with a mixture of the OT-I and OT-II respective peptides, OVA257-264 and OVA323-339 (1 mg/mL in normal saline and mixed thoroughly with IFA at v:v=1:1) into the footpad (40 μL/footpad). After 72 h, mice were sacrificed, and the popliteal lymph node on the injected side was isolated and digested into a single-cell suspension using 0.2% type I collagenase at 37 °C for 1 h followed by filtration through a 40-μm cell strainer. Cells were then stained with APC-labeled anti-CD4 or APC-labeled anti-CD8 antibodies (eBioscience), and analyzed by FACSCalibur.
BMDC interactions with hLEC under flow
The hLEC were seeded onto microfluidic channels of BioFlux plates (48-well, 0–20 dyn/cm2; Fluxion Biosciences, San Francisco, CA) pre-coated with matrigel (1:50 in PBS; BD, San Jose, CA). At approximately 90% confluence, hLEC were either treated with heparinase, blocking antibodies, or transfected with siRNA. For adhesion assays, BMDC pre-labeled with calcein AM (eBioscience, San Diego, CA) following the manufacturer’s instruction were perfused into the channel at a concentration of 2 × 106 cells/mL in EBM2 medium under an initial shear stress of 2 dyn/cm2 for 2 min followed by a constant flow at 0.14 dyn/cm2 for 15 min. After removing the residual BMDC from the inlet well, the channel was washed with EBM2 medium at 1 dyn/cm2 for 5 min to remove any unattached BMDC. Adherent BMDC across the whole channel were imaged using Perkin Elmer UltraVIEW Vox Spinning Disk Confocal microscope (100×, UCSD Light Microscopy Facility) and quantified with Metamorph software (Molecular Devices, Sunnyvale, CA). Following the capture of adhesion images at time 0 (T0), transmigration of adherent BMDC (defined by disappearance of adherent cells visibly from the surface of the hLEC monolayer to the space beneath the monolayer, as opposed to detachment and carriage out of the viewing field under flow) was observed under a constant shear of 0.14 dyn/cm2 for another 30 min using the same microscope, with the end-point image taken at that time (T30). The % transmigration was calculated as [(No. of adherent BMDC at T0)−(No. of adherent BMDC at T30)/No. of adherent BMDC at T0] × 100%.
Transwell invasion and chemotaxis migration assays
To assess the mobility of BMDC toward hLEC in vitro, a modified transwell invasion and chemotaxis assay was performed as previously described(5). For collagen matrix-based invasion assays, 1 × 105 hLEC were embedded into 100 μL of type I collagen gel containing 3 mg/mL PureCol (Advanced Biomatrix, San Diego, CA) in DMEM (pH 7.3), applied to cover the lower side of transwell insert (3.0 μm in pore size, Corning) and allowed to gel in a 37°C, 5% CO2 incubator for six hours. The insert was then inverted and placed into a 24-well plate containing pre-warmed serum-free EBM2 (500 μL/well). For chemotaxis assays, 5 × 104/well hLEC were seeded directly into the bottom of a 24-well plate and treated. For both assays, 2–5 × 105 BMDC pre-labeled with calcein AM were resuspended in 100 μL of EBM2 and loaded on top of the insert. For the invasion assay, the plates were placed in a 37 °C, 5% CO2 incubator overnight, and for six hours in the case of the chemotaxis assays. At the end of the invasion/chemotaxis period, transwells were transferred to a new 24-well plate and treated with either 0.2% type I collagenase (Sigma, St. Louis, MO) for 1 h (for collagen matrix-based invasion assay) or trypsin (Invitrogen, Grand Island, NY) for 5 min (for chemotaxis assay) with gentle rocking at 37 °C. Bottom-well solutions were collected, transferred to clean eppendorf tubes and centrifuged at 5,000 × g for 5 min. Cell pellets were resuspended in 20 μL PBS with 1.5 μL aliquots loaded onto a 96-well Terasaki plate (Robbins Scientific, Sunnyvale, CA), and images were taken at 40 × magnification with a Nikon Eclipse 80i fluorescence microscope and analyzed with NIH Image J software. All experiments were set up in triplicate with at least three independent experiments performed.
Reverse-transcription followed by quantitative real-time PCR (RT-qPCR)
Total RNA was extracted from cells using RNAqueous 4-PCR kit (Ambion) and reverse transcribed into cDNA with SuperScript III kit (Invitrogen) according to the manufacturers’ instructions. Real-time PCR was performed with iQ Sybr Green Supermix Kit (BioRad). The primer sequences (5′ to 3′) used for real-time PCR were as follows: human Ndst1 forward, GGACATCTGGTCTAAG, and reverse, GATGCCTTTGTGATAG; human XylT2 forward ACGTTCAACCGCAAACTACC, and reverse, ATTGCTCAGTTCCCCATCTG; human CCL21 forward, GCCTTGCCACACTCTTTCTC, and reverse, CAAGGAAGAGGTGGGGTGTA; human CCR7 forward, TTCTTCACTGTCCTCCAAGC, and reverse, ACATTTCCCTTGTCCTCTCC. The PCR program was as follows: 95°C for 3 min followed by 40 cycles of 95°C for 30 sec, 59°C for 30 sec and 72°C for 30 sec. Relative expression of target gene against β-actin was calculated using 2−ΔΔCt method(12).
Preparation of conditioned media from hLEC
To harvest conditioned media (CM), hLEC were transfected with different siRNA molecules, washed once with PBS, and incubated in EBM2/5% horse serum (post-treatment over a DEAE column to clear endogenous sulfated glycosaminoglycans from the serum) for 24 h. The supernatant was then collected and briefly centrifuged to remove the cell debris.
Duolink assay
The specific interactions between chemokines from CM and chemokine receptors on cytospined BMDC, were determined with the Duolink proximity ligation assay (PLA, Olink Bioscience, Uppsala, Sweden) following the manufacturer’s instruction using the following primary antibodies: anti-CCL21 (R&D, MAB3661, 1:100), anti-CCR7 (Novus, St. Charles, MO; 1:250), anti-CXCL12 (R&D, AF-310-NA, 1:100), and anti-CXCR4 (R&D, MAB172, 1:200).
Purification of HS from hLEC
Purification of HS was performed as previously described(13). Briefly, human primary LECs (hLEC) in 10-cm tissue culture plates were transfected with different siRNA molecules, washed with PBS once and digested in 1x Pronase buffer containing 167 μg/mL pronase (Sigma), 40 mM sodium acetate and 320 mM sodium chloride in PBS (10 mL buffer/10-cm plate) at 37 °C for 24 h. The cellular digest was then filtered through 0.2-μm PES syringe filters (GE Healthcare, Piscataway, NJ) and run through a DEAE-Sephacel (GE Healthcare) column by gravity. After wash with DEAE equilibration buffer (0.2 M sodium chloride, 20 mM sodium acetate, pH 6.0), the glycan components were eluted from the column with DEAE elution buffer containing 2 M sodium chloride and 20 mM sodium acetate, pH 6.0. The salt in the sample was then removed by running the eluent through the PD-10 desalting column (GE Healthcare) following the manufacturer’s instruction. The eluted glycans (containing both HS and chondroitin sulfate) were lyophilized, redissolved in condroitinase buffer (50 mM Tris-HCl pH 8.0, 50 mM sodium acetate), and digested with chondroitinase A, B and C (a kind gift from Dr. Jeffrey Esko, UCSD Dept. of Cellular & Molecular Medicine) at 37 °C for at least 2 h. After repeated passage through DEAE-Sephacel and PD-10 desalting columns as described above, the purified and lyophilized HS from was resupended in PBS (500 μL/10-cm plate).
Oligomerization analysis of CCL21 and CXCL12
0.1 μL and 0.3 μL of purified HS were incubated separately with recombinant human CCL21 or CXCL12 (20 ng/reaction, Peprotech) in a total reaction volume of 30 μL at room temperature for 1 h followed by crosslinking using BS3 (Thermo Scientific, Rockford, IL). After quenching the crosslinking reaction with 50 mM Tris, non-reducing protein loading buffer (5 × concentration) (Thermo Scientific) was added to the samples, which were then boiled for 5 min and separated on 4%–20% gradient gels (BioRad, Hercules, CA). Samples were then electrotransferred from the gel onto a nitrocellulose membrane, and probed with anti-CCL21 or anti-CXCL12 antibody (R&D, 1:1000) followed by IRDye 800 secondary antibody (LI-COR, Lincoln, NE). The signal was detected using the Odyssey Infrared Imaging System (LI-COR).
Statistical analysis
Quantitative data were presented as mean ± SD for three replicates or three independent experiments where indicated. Significance between groups was calculated using two-tailed Student’s t test. Differences in LN colonization of skin DC following FITC+oxazalone painting and that from in situ implanted BMDC between mutant and littermate-control mice were examined using the rank order test. A value of P < 0.05 was considered statistically significant.
RESULTS
Tissue-specific inhibition in the sulfation of lymphatic HS impairs in vivo DC trafficking
To test the degree to which the sulfation of HS on lymphatic endothelium plays a role in regulating DC trafficking in vivo, we inducibly and selectively knocked down the gene N-deacetylase/N-sulfotransferase-1 (Ndst1) in lymphatic endothelial cells. The Ndst family of enzymes is involved in N-sulfation of glucosamine residues during the biosynthesis of nascent HS chains on proteoglycan core proteins(14). The LEC-specific knockdown of Ndst1 was achieved by crossing mice bearing both loxP-floxed alleles of Ndst1 (Ndst1f/f mice)(13) with mice harboring a tamoxifen-inducible Cre allele driven by the lymphatic endothelial specific Prox1 promoter (Prox1+/CreERT2 transgenic mice)(5). Previous work has shown that in Ndst1f/fProx1+/CreERT2 mutant mice, five consecutive intraperitoneal injections of tamoxifen potently and selectively reduced Ndst1 expression in lymphatic endothelial cells, as compared to that of Ndst1f/fProx1−/CreERT2 wild-type control mice(5). After topical application of a solution containing skin allergen oxazolone together with the FITC, FITC+ DC from the skin to the draining LN were quantified by flow cytometric staining of FITC+CD11c+ cells. As shown in Figure 1A, targeting the sulfation of lymphatic endothelial HS in Cre+ mutant mice significantly reduced the number of FITC+CD11c+ cells that colonized the draining LN, as compared to that in the Cre- controls. To examine whether a transient reduction in lymphatic endothelial Ndst1 expression might lead to any alterations in skin lymphatic vascular density or basal numbers of DC within skin, we determined the density of Lyve1+ lymphatic vessels in the ear by whole-mount staining, and quantified CD11c+MHCII+ cells in skin by flow cytometry. As shown in Figure 1B and 1C, at baseline, there were no significant differences in either lymphatic vascular density or total DC quantity within the skin, suggesting that transient reduction in the biosynthesis of lymphatic endothelial HS in pre-existing unstimulated lymphatic vasculature does not alter the conduit by which DC travel or the basal peripheral pool of DC prior to antigen uptake, but rather the trafficking process itself.
Figure 1. Lymphatic endothelial-specific mutation impairing the sulfation of heparan sulfate inhibits in vivo trafficking of DC.

A. Ndst1f/fProx1+/CreERT2 mutant mice (N=11) and their Ndst1f/fProx1−/CreERT2 littermates (N=12) were painted with FITC plus oxazolone on the abdomen. After 40 h, the draining inguinal and axillary lymph nodes (LN) were isolated and digested into a single-cell suspension. Percentage of FITC+CD11c+ cells within LN were analyzed by flow cytometry, and averaged following normalization to the mean value of the Ndst1f/fProx1+/CreERT2 group. Data represents the mean of three independent experiments. A representative example of the flow cytometric data is shown in the inset. B. Lymphatic vascular density was determined in unstimulated Ndst1f/fProx1+/CreERT2 mutant mice (N=4) and in Ndst1f/fProx1−/CreERT2 littermates (N=5) by immunofluorescence staining of Lyve1 (green signal) on ear whole-mount. Representative images are shown in upper section (scale bar: 100 μm), and the averages of Lyve1+ lymphatic area per field are shown in lower graph. C. Percentage of CD11c+MHCII+ DC in the skin of Ndst1f/fProx1+/CreERT2 mutants (N=7) and Ndst1f/fProx1−/CreERT2 littermate controls was determined by flow cytometry. D. BMDC at day 9 of differentiation in culture were labeled with calcein AM and injected into the left foot dorsum of Ndst1f/fProx1+/CreERT2 mutants (N=6) and Ndst1f/fProx1−/CreERT2 littermate controls (N=9) (2 × 106 cells/mouse). After 40 h, the left popliteal LN was isolated, imaged under the fluorescence microscope (upper images, scale bar: 200 μm), and digested into a single-cell suspension. Cells from each LN digest were resuspended into 100 μL of PBS with 1.5 μL spotted onto the well of a Terasaki microtiter plate, and imaged under the fluorescence microscope. The number of calcein+ cells was quantified using NIH Image J software, plotted, and averaged for each genotype (horizontal bars in D). *P < 0.05, #P < 0.01, as compared to the Ndst1f/fProx1−/CreERT2 group.
Considering that painted FITC may not only be taken up by skin-resident DC, but also might freely diffuse from the skin, reach the draining LN via afferent lymphatic vessels, and be taken up by LN-resident DC, we also examined in vivo DC trafficking by implanting mutant versus wild-type mice with equal numbers of fluorescence-labeled BMDC into the foot dorsum, and quantified DC that trafficked to the draining LN by fluorescence microscopy. To achieve this, we followed a well-established protocol for DC differentiation(15), and obtained approximately 80% marrow-derived CD11c+ DC after 9-day culture in the presence of GM-CSF (Supplemental Fig. 1A). Further characterization showed that close to 99% of CD11c+ BMDC were also positive for CD11b, while negative for B220 (Supplemental Fig. 1B), consistent with the myeloid immunophenotype of these BMDC. Figure 1D shows that by 40 h after in situ DC injection, a significantly lower number BMDC travel to the popliteal LN in Cre+ mutant mice as compared to that in Cre-negative littermate controls.
Genetic targeting of the N-sulfation of lymphatic endothelial specific HS inhibits in vivo DC-dependent CD8+ T cell proliferation upon OVA challenge
To test the biological significance of targeting lymphatic endothelial HS on DC-trafficking-dependent T cell responses, we sensitized the mice with an initial dose of the classical antigen OVA. Following a re-application of OVA into the footpad, we examined the populations of distinct T cell subtypes within the draining LN (Fig. 2A). Using initially a model characterized by a pan-endothelial mutation in Ndst1, we found that CD8+ T cell populations were significantly lower in Ndst1f/fTekCre+ mutant mice than in Ndst1f/fTekCre− control littermates (Fig. 2B). Upon restricting the mutation to solely the lymphatic endothelium in vivo (using an inducible Prox1Cre transgenic model to drive transient lymphatic-specific disruption in Ndst1 expression), a similar reduction in CD8+ T cell proliferation in response to immunization was also observed in Ndst1f/fProx1+/CreERT2 mutants as compared to that in Ndst1f/fProx1−/CreERT2 littermate controls (Fig. 2C). In this model, when compared to vehicle (IFA)-injected mice, we observed a significant increase in the total T cell (CD3+) population upon OVA challenge in both Ndst1f/fProx1+/CreERT2 mutants and Ndst1f/fProx1−/CreERT2 littermates (with only a slight but insignificant reduction in the response of mutants), indicating that T cell proliferation occurred in both groups of animals. Importantly, however, while a significant CD8+ T cell proliferation response occurred in Cre-negative wild-type littermates, the CD8+ T cell response was strikingly absent in Ndst1f/fProx1+/CreERT2 mutants. On the other hand, a relatively lesser degree of CD4+ T cell proliferation occurred in both groups (Fig. 2C, middle panel), although significance relative to the response in IFA-only controls was only achieved in the mutant group. Thus, the most dramatic effect in this model involving a highly lymphatic-specific deficiency in the N-sulfation of HS was the contrast between the CD8+ T cell proliferative response in wild-type mice and the complete lack of such a response in the Ndst1 mutants (Fig. 2C, right panel).
Figure 2. Lymphatic endothelial specific reduction in the N-sulfation of heparan sulfate results in the inhibition of CD8+ T cell proliferation upon OVA challenge.

A. Scheme of the experiment. All animals were pre-treated with daily tamoxifen for 5 consecutive days to activate Cre recombinase. On day 12, a sensitizing dose of OVA (500 μg/mouse) was administered intraperitoneally. Seven days later (day 19), 10 μg of OVA was delivered to the left footpad by intradermal injection. On day 24, the left popliteal LNs were isolated, with distinct populations of T cells examined by flow cytometry. The experiment was first carried out in animals bearing a pan-vascular mutation in Ndst1. B. Number of CD3+, CD3+CD4+, and CD3+CD8+ T cells in the draining LN was compared between Ndst1f/fTekCre+ (N=5) and Ndst1f/fTekCre− control littermates (N=5). *P < 0.05, as compared to Ndst1f/fTekCre− control group. C. Number of CD3+, CD3+CD4+, and CD3+CD8+ T cells in draining LN was compared between Ndst1f/fProx1+/CreERT2 mutants (N=8) and Ndst1f/fProx1−/CreERT2 littermates (N=9) after either vehicle control (IFA only) or OVA challenge (OVA+IFA). *P < 0.05, #P < 0.01.
The OVA model employed in Fig. 2 involves two-step stimulation with antigen. To more directly examine how a single immunization on the mutant background might, as a result of altered DC trafficking, affect lymph node T cell proliferation, we adoptively transferred CFSE-labeled OTI CD8 and OTII CD4 T cells (expressing transgenic T cell receptors for peptides OVA257-264 and OVA323-339, respectively) into Ndst1f/fProx1+/CreERT2 versus Ndst1f/fProx1−/CreERT2 mice. At 72 h following footpad injection of a mixture of two specific OVA-derived antigens, namely OVA257-264 and OVA323-339 peptides, we examined CD8 and CD4 proliferation within the draining popliteal lymph nodes by flow cytometry. As shown in Supplemental Figure 2, we observed a significant reduction in the ratio of CFSEintermediate (CFSEint)CD8+ to CFSEhigh (CFSEhi)CD8+ in Ndst1f/fProx1+/CreERT2 mutant mice as compared to that in Ndst1f/fProx1−/CreERT2 controls. Considering that CFSEintCD8+ cells represented adoptively transferred and actively proliferating CD8 T cells (as compared with non-proliferating CDhiCD8+ cells), the ratio of these two populations reflects the proliferative activity of the adopitively transferred CD8 T cells in vivo. On the other hand, no dramatic difference was noted in proliferating CFSE+ CD4 T cells between the two genotypes.
In addition to a phenotype that appears to depend upon the trafficking of DC in distinct lymphatic beds in the two-step model in Fig. 2, secondary effects by cell types other than DC in the model nevertheless have the potential to influence T cell responses. Accordingly, we characterized the baseline level of distinct cellular components within the peripheral blood (by complete blood count, CBC) and the skin-draining lymph nodes (by flow cytometry) from Ndst1f/fProx1+/CreERT2 versus Ndst1f/fProx1−/CreERT2 mice following tamoxifen injection. The two genotypes at baseline showed no significant differences in the CBC profile (Supplemental Table 1A), and there were no significant differences in baseline levels of T/B cells, NK cells, and monocytes within the skin-draining lymph nodes (Supplemental Table 1B). We also compared distinct DC subpopulations, including CD8α+ resident DC, PDCA1+ plasmacytoid DC, and migratory DC in the skin-draining lymph nodes of Ndst1f/fProx1+/CreERT2 versus Ndst1f/fProx1−/CreERT2 mice at baseline, and identified no dramatic differences (Supplemental Table 1C).
Altering lymphatic endothelial HS affects DC adhesion but not transmigration under low shear flow
To understand which biological processes during DC trafficking are affected by targeting lymphatic endothelial HS, we first set up an in vitro flow system whereby DC derived from bone marrow progenitor cells were introduced under low shear flow into a chamber lined with a confluent layer of hLEC. To target hLEC HS in vitro, two general approaches were applied: (1) enzymatic pre-treatment of hLEC with heparinase (destroys HS chains(16)), and (2) altering HS biosynthesis (including specific sulfate modifications) in the hLEC through efficient siRNA targeting of specific HS biosynthetic enzymes (Supplemental Figure 3A). After 15 min under shear flow, a moderate number of DC were adherent to hLEC, while this was dramatically reduced if the hLEC were pre-treated with heparinase or blocking antibody to CCR7 (Fig. 3A, upper panels; and 3B, upper graph). Significant reduction in DC adhesion was also observed for hLEC pre-treated with CCL21-neutralizing antibody (Fig. 3B, upper graph). When hLEC were transfected with siRNA targeting Ndst1 or XylT2 (the latter is required for initiating the biosynthesis of glycosaminoglycan chains on core proteins), adhesion was also significantly reduced (Fig. 3B, lower graph). However, pre-treatment with CCL5-neutralizing antibody, or transfection with siRNA targeting Hs3st1 (enzyme responsible for glucoronyl 3-O-sulfation of HS) did not alter adhesion. Following DC adhesion under flow, we monitored DC transmigration across the hLEC monolayer. In addition to the effect on adhesion, antibodies for CCR7 or CCL21 markedly inhibited DC transmigration (Fig. 3A, lower panels; Fig. 3C, upper graph). In contrast, neither heparinase nor siRNA targeting the different HS biosynthetic enzymes significantly affected DC transmigration (Fig. 3C). These findings imply that CCL21-CCR7 binding and signaling plays an essential role in mediating both DC adhesion to and transmigration across hLEC layers. On the other hand, it appears that HS-mediated adhesion, at least in this model, is not required for transmigration. (It should be noted that the force of gravity in such studies may also potentially contribue to engagement interactions that may be necessary for diapedesis.)
Figure 3. Lymphatic endothelial heparan sulfate critically regulates chemokine-dependent adhesion of BMDC under low-level physiologic shear flow.

Human lymphatic endothelial cells (hLEC) were seeded into a Bioflux flow chamber (edges marked by dashed lines) and pre-treated with neutralizing antibodies (against CCL21, CCR7 or CCL5), or siRNA targeting the indicated HS biosynthetic enzymes (siDS indicates control siRNA). Day 9 BMDC were labeled with calcein AM and introduced into the chamber at a constant shear stress of 0.14 dyne/cm2 at 37°C. After 15 min, non-adherent cells were washed off and the adherent BMDC were imaged using fluorescence microscopy (A upper panels, fluorescence in white/grey over black flow-channel background), quantified with Metamorph software, and normalized to hLEC control group (B, upper graph) and to the siDS control group for siRNA targeting experiments (B, lower graph). The transmigration of adherent BMDC was recorded following an additional 30-min cell-free flow period (from T0 to T30), imaged under the fluorescence microscope (A lower panels), and quantified using Metamorph software, with the % transmigration calculated as [(No. of adherent BMDC at T0)−(No. of adherent BMDC at T30)/No. of adherent BMDC at T0] × 100%. All transmigration data were normalized to hLEC control group (C, upper graph) and to the siDS control group for siRNA experiments (C, lower graph). *P < 0.05, #P < 0.01, as compared to hLEC control group in B and C upper graphs, and to siDS control group in B and C lower graphs.
Lymphatic endothelial HS promotes the directional invasion and chemotaxis of DC
We also assessed the role of lymphatic endothelial HS in modulating DC invasion/chemotaxis transwell-based systems in vitro. To mimic the in vivo physiological step by which DC invade into extracellular matrix surrounding LEC, we designed a modified in vitro collagen matrix-based transwell assay(5) where hLEC were embedded in type I collagen on the underside of transwell filters, and fluorescence-labeled BMDC were loaded on top of the insert. Invading BMDC into the collagen gel were quantified under various conditions that alter or inhibit HS produced by hLEC (Fig. 4A and 4B). The presence of normal hLEC in the collagen was able to drive invasion by DC more than 200-fold over basal invasion into hLEC-free collagen. If invasion proceeded in the presence of blocking antibody to either CCR7 or its cognate ligands CCL19 and CCL21, invasion was significantly reduced, suggesting that CCR7 mediated signaling (in addition to the presence of the cognate CCR7 chemokine ligands) plays an important role in the invasion of DC toward hLEC across collagen. The production of HS by hLEC was required for DC invasion, as initially evidenced by marked inhibition in the setting of heparinase-treated hLEC. Targeting the HS chain-initiating enzyme XylT2 or the sulfating enzyme Ndst1 also led to significant reductions in DC invasion. In contrast, antibody blockade of CCL5 or treatment with siRNA targeting Hs3st1 did not lead to significant reduction in DC invasion. These findings suggest distinct and specific requirements with respect to chemokines as well as the fine structure of lymphatic HS in the system.
Figure 4. Invasion and migration of BMDC depends on lymphatic heparan sulfate.

A. and B. Invasion of BMDC at day 9 of differentiation into a collagen gel containing either no cells (“NC”) or hLEC treated as indicated was quantified and normalized to NC. C. to F. Transwell migration of BMDC into wells containing no cells (“NC”) or hLEC monolayer treated as indicated was quantified and normalized to NC. αCCR7 or αR7 (in 4F), αCCL19, αCCL21 or αL21 (in 4F), αCXCR4, αCXCL12 or αCCL5, blocking antibodies to CCR7, CCL19, CCL21, CXCR4, CXCL12 or CCL5, respectively; H’ase, hLEC pre-treated with heparinase; siDS, hLEC transfected with control siRNA; siNdst1, siXylT2 or siHs3st1, hLEC transfected with siRNA targeting corresponding HS biosynthetic enzymes. *P < 0.05, #P < 0.01, as compared to hLEC control group in A, C, D, F, and to siDS control group in B and E.
Since LEC-derived HS may either be associated with core proteins (eg. syndencans or glypicans) on the lymphatic cell surface or secreted into the extracellular space (eg. perlecans), we further investigated whether targeting HS secreted by LEC might alter the ability of DC to migrate, with the idea that LEC-secreted HS might play a trans-acting role in DC migration. Specifically, fluorescence-labeled BMDC on transwell filters were separated from the HS-targeted hLEC monolayer in lower wells by liquid medium, and DC migration into lower wells was quantified(5). The presence of hLEC in the bottom well was sufficient to drive DC migration (Fig. 4C and 4D), and destroying HS with heparinase, blocking CCR7/CCL21 (Fig. 4C), CXCR4/CXCL12 (Fig. 4D), or interfering with the biosynthesis of hLEC HS (Fig. 4E, using siRNA targeting Ndst1 and XylT2) significantly reduced DC migration. In contrast, CCL5 blocking antibody, CCL19 blocking antibody or treatment of hLEC with Hs3st1 siRNA was not sufficient to alter migration (Fig. 4C to 4E). This suggests that the presence as well as specific sulfation properties (i.e., N-sulfation but not 3-O-sulfation) of HS produced into the hLEC conditioned medium is critical for mediating CCL21- and CXCL12-dependent migration of DC cells toward the hLEC. Furthermore, when we combined targeting approaches, that is, heparinase treatment of the hLEC together with blocking antibodies to CCR7 or CCL21, we detected no further reduction in BMDC chemotaxis as compared to that noted for each individual treatment alone (Fig. 4F), suggesting that the CCL21/CCR7-mediated effects under these conditions appear to fully depend upon lymphatic endothelial HS.
HS secreted from LEC is required for optimal binding of chemokines to cognate receptors on DC
The finding that the migration of BMDC is sensitive to altering HS produced by hLEC across liquid medium prompted us to explore the molecular mechanisms by which HS secreted by hLEC modulate DC migration. Given that CCR7/CCL21 and CCR4/CXCL12 signaling is essential for DC migration (Fig. 4C and 4D), and that HS is known to interact with basic amino-acid motifs on several chemokines, including CCL21 and CXCL12(8), we focused on the chemokine-receptor binding that might be affected by altering HS secreted by hLEC. For this purpose, conditioned medium (CM) was harvested from either control siRNA (siDS)-transfected hLEC or an equal number of hLEC transfected with siNdst1 or siXylT2, and applied to BMDC. The interaction between CCL21 in the CM and CCR7 expressed on BMDC was measured using the proximity ligation assay (PLA), wherein a fluorescent signal will be generated when and only when the two target proteins are in close proximity (i.e., when the chemokine binds to its cognate receptor). The engagement of numerous cell-surface CCL21:CCR7 complexes was noted upon exposure of BMDC to CM from control hLEC (Fig. 5A; CM-siDS). Significant reduction in CCL21:CCR7 complexes was noted upon the application of CM from siNdst1-targeted hLEC (CM-siNdst1), and further reduction occurred upon exposing BMDC to siXylT2-targeted CM (CM-siXylT2; Fig. 5A and 5B). Similar (albeit somewhat less dramatic effects) were also observed for the interaction between CXCL12 and CXCR4 on BMDC (Fig. 5C and 5D). Vehicle medium, the basal medium used to collect CM from hLEC, produced minimal CCL21:CCR7 or CXCL12:CXCR4 complexes (Fig. 5A and 5C), implying that PLA signal depends on the presence of these chemokines in the CM. Given that transient transfection of hLEC with different siRNA did not dramatically change the expression of CCL21 (Supplemental Figure 3B) or its secretion into CM(5), the PLA data suggests that the optimal binding of chemokine to receptor on the BMDC surface depends upon the presence of intact lymphatic HS produced into the CM (where it essentially acts “in trans” as a co-receptor).
Figure 5. Presence of appropriately sulfated heparan sulfate in lymphatic endothelial conditioned medium is required for binding of CCL21 to bone marrow-derived DC.

Conditioned medium (CM) was collected from hLEC transfected with either control siRNA (siDS) or siRNA targeting the HS biosynthetic enzymes Ndst1 or XylT2. Cytospin samples of day 9 BMDC were incubated with the different CM. Binding of CCL21 in the CM to CCR7 on BMDC was detected by proximity ligation assay (PLA). Representative merged images showing PLA signal (red) and nuclear DAPI stain (blue) were taken by fluorescence microscopy (400 ×). Signal for BMDC incubated in the vehicle control medium alone (EBM2 containing 5% horse serum; which served as the basal medium for all hLEC CM) is shown on the upper left panel. PLA signal from each field was quantified and indexed to total nuclear area within the same field. The average of at least 5 random fields from each group was included for analysis, with mean data normalized to control signal (CM-siDS) (see graph below). *P < 0.05, #P < 0.01, as compared to CM-siDS group.
Lymphatic endothelial HS facilitates oligomerization of lymphatic chemokines
The diversity of chemokine oligomerization has been shown to initiate distinct signaling responses, and is known to be stabilized by HS glycosaminoglycans(17). To understand whether targeting lymphatic endothelial HS leads to alterations in chemokine oligomerization, we purified HS from confluent monolayers of hLEC transfected with either control siRNA (siDS), siXylT2, siNdst1 or siHs3st1, and incubated two different doses of the various purified siRNA-targeted HS species with recombinant human CCL21 (molecular weight: 12.2 kDa). We then examined the pattern of CCL21 oligomerization following crosslinking and gel-electrophoretic separation of multimeric products. As shown in Fig. 6A, at the lower dose tested, HS from siDS- or siHs3st1-transfected hLEC led to robust CCL21 oligomerization, with distinct bands separated by approximately the size of a CCL21 monomer and the highest detectable oligomer being what appears to be an octamer (and a smear-like extension toward possibly larger products). In contrast, the same dose of HS from siXylT2- or siNdst1-transfected hLEC could only facilitate the formation of CCL21 trimers and tetramers, respectively. In the latter case, even at the higher HS dose, the purified siNdst1-altered HS could not support the formation of CCL21 oligomers greater than tetramer-size (Fig. 6A), suggesting that targeting lymphatic endothelial HS biosynthesis at the level of initial chain formation or N-sulfation, but not 3O-sulfation, dramatically reduces its capability to support the complexing of larger chemokine oligomers in this assay. A similar HS effect on CXCL12 (molecular weight: 8 kDa) oligomerization was also observed (Fig. 6B), with HS isolated from mock-transfected (siDS) control cells, but not from siXylT2 or siNdst1-transfected cells, strongly supporting CXCL12 multimerization.
Figure 6. Lymphatic endothelial heparan sulfate is essential for optimal oligomerization of CCL21 and CXCL12.
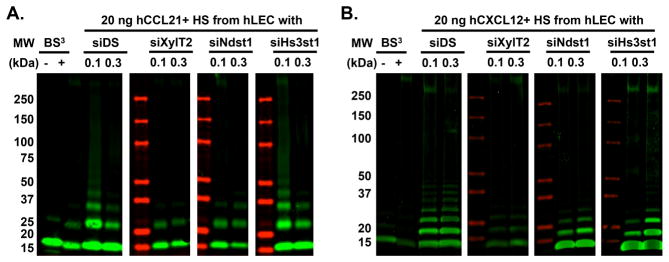
HS was purified from cultured hLEC monolayers (grown in 10-cm plates) that were transfected with either control siRNA (siDS) or siRNA targeting the indicated HS biosynthetic enzymes, and resuspended in 500 μL of PBS. Recombinant human CCL21 (hCCL21, 20 ng/reaction, molecular weight 12.2 kDa, A) or hCXCL12 (20 ng/reaction, molecular weight: 8 kDa, B) was incubated with 0.1 μL and 0.3 μL of each HS prep, followed by BS3 mediated crosslinking, and separation on SDS-PAGE gels with detection by Western immunoblotting. Green signal: hCCL21 (A) or hCXCL12 (B) as detected by Western immunoblot; red signal: protein size marker. − and +: pure hCCL21 or hCXCL12 in the absence or presence of BS3 crosslinker, respectively.
DISCUSSION
In this study, we provide genetic evidence that targeting the N-sulfation of lymphatic endothelial heparan sulfate significantly reduces in vivo DC trafficking from the periphery to the draining LN, with the functional consequence of inhibiting CD8+ T cell proliferation in the draining LN in response to OVA immunization. Mechanistically, lymphatic endothelial HS may mediate DC trafficking at multiple steps, including adhesion to LEC under flow as well as chemokine-dependent migration toward lymphatic endothelium. Moreover, appropriately sulfated lymphatic HS appears to be required for chemokine oligomerization and optimal presentation of certain chemokines such as CCL21 and CXCL12 on the DC cell-surface.
The development of effective approaches to manipulate DC traffic has been limited by a lack of understanding of the molecular controls for this process. A few studies, including our own, have demonstrated that altering vascular HS biosynthesis, either through an alteration in N-sulfation of nascent HS chains (Ndst1 mutation) or chain polymerization (Ext1 mutation), results in altered chemokine-dependent interactions of endothelial cells with trafficking neutrophils, DCs, and tumor cells as they home to sites of inflammation or colonize lymphoid organs via blood-vascular or lymphatic vascular routes (5, 6, 18). For example, a pan-endothelial mutation in Ext1 inhibits CCL21/CCL19 binding to LEC and impairs DC migration to the draining LN(6). While this suggests what may occur when HS chains are absent or enzymatically ablated in the entire circulation, the degree to which lymphatic endothelial-specific alteration in HS affects DC traffic and specific T cell responses to antigen has not been reported. We stringently interrogated the role of lymphatic-vascular specific HS, separate from any role that blood-endothelial HS might play in affecting T cell traffic through the blood vasculature (such as LN high endothelial venules). Moreover, HS is endowed with functional specificity for distinct ligands such as chemokines that may variably bind as a result of specific sulfate modifications. Targeting of the latter has also not been explored in the lymphatic system.
We inducibly knocked down Ndst1 in lymphatic vasculature, and demonstrate impairments in DC traffic in models that allow examination of movement of endogenous DC (utilizing FITC+oxazolone skin painting) as well as the traffic of exogenously loaded bone-marrow-derived DC to regional LN. Since pathologically stimulated lymphatic endothelium may potentially undergo impaired growth/remodeling responses in the setting of HS mutations(19), a control experiment confirmed that transient induction of lymphatic endothelial Ndst1 knockdown in the skin under basal/non-stimulated conditions does not alter lymphatic vascular density(Fig. 1B). Moreover, no significant changes in the total DC number within the skin were noted upon Ndst1 knockdown at baseline, suggesting that reduced DC colonization within the draining LN (Fig. 1A and 1D) is caused by the effect of lymphatic-specific HS mutation on the DC trafficking process itself. To what extent the mutation might result in a defect in slowing/arresting of DC as they enter the interfollicular sinuses from afferent lymphatics entering the LN is unknown, although a sophisticated analysis of this may be worthwhile in future studies.
To test downstream DC-dependent immunologic events, we asked how targeting the N-sulfation of lymphatic HS would affect T cell responses in the draining LN upon antigen stimulation. In a sensitization-elicitation allergic reaction model employing OVA antigen, we observed significant reduction in the total CD8+ T cell population within the draining LN (Fig. 2). Although this is a complex immune response that involves in vivo lymphatic DC trafficking at both sensitization and elicitation steps, the lack of a LN CD8+ T cell response in lymphatic-HS mutants highlights the functional importance of appropriately sulfated lymphatic HS in modulating the magnitude of the DC-dependent LN immune response. Moreover, OVA contains multiple epitopes capable of activating distinct subtypes of T cells, including CD8+ cytotoxic and CD4+ helper T cells(20), potentially mediated by DC of different subsets or at distinct maturation or activation stages. With this in mind, the specific reduction of a CD8+ T cell proliferative response but not that of the CD4+ T-cells reveals the unique effect of silencing lymphatic HS on the LN balance of T cell responses. As a final control, we “dissected” the importance of lymphatic-specific HS from that of blood-vascular HS in this immunization process by repeating the experiment in Tie2-Cre+Ndst1f/f mutant mice (with a pan-endothelial deficiency in HS sulfation). A nearly identical result in terms of the CD8+ T cell phenotype in the LN was observed, highlighting the importance of lymphatic-specific HS in the immune phenotype.
The molecular diversity of heparan sulfate is highlighted by the ability of its unique sulfate-modified structural motifs to facilitate and/or mediate protein interactions that are essential for a variety of biological processes(14). However, knowledge is lacking with respect to how specific HS modifications contribute to mechanistic steps involved in functional immune responses. In cell-based assays, we found that by targeting the integrity of HS with heparinase or by blocking LEC biosynthesis of HS via siRNA, DC adhesion under flow and LEC-driven collagen invasion and matrix-independent migration were all significantly reduced. This was associated with chemokine specificity as well as HS fine-structure specificity in the system: blockade was achieved using antibodies to CCR7/CCL21 or CXCR4/CXCL12 but not CCL5; and the effects of silencing N-sulfation of LEC HS as opposed to 3-O sulfation (siHs3st1) are consistent with data regarding the importance of N-sulfation and 2-O-sulfation (both of which result from Ndst1 silencing(5, 18)) as opposed to 3-O-sulfation for binding of CCL21 or CXCL12 to immobilized HS on glycan arrays(8). The trends in specificity suggest that chemokines that drive this system may work cooperatively with key sulfate motifs on HS produced by the LEC.
Other soluble regulators that might affect DC trafficking to the LN include selectin adhesion molecules, integrin family members, metalloproteinases, and possibly other chemokines(21–23). Importantly, we found that upon disruption of lymphatic HS biosynthesis in a low shear-rate flow model of DC over primary LECs, DC-LEC adhesion events were markedly inhibited. On the other hand, HS-mediated adhesion did not appear to be required for transmigration in this model, while antibody-mediated blocking of CCR7/CCL21 had a significant impact on both DC-LEC adhesion as well as DC transmigration across hLEC under flow. The lack of HS effect on transmigration in this in vitro assay, as opposed to our in vivo findings supporting the role HS in orchestrating DC entry into lymphatic vessels by modulating CCL21 haptotaxis, may also be attributed to the fact that the majority of adhesion events required for subsequent transmigration in some in vitro models may be less dependent on HS-mediated chemokine sequestration since the adherent DC are in direct contact with the LEC monolayer under the force of gravity alone, which might be sufficient to engage the necessary “machinery” for diapedesis. In contrast, in vivo DC may require greater assistance from sequestered chemokines to navigate efficiently through tissues and make such contacts.
We followed phenotypic characterizations with mechanistic studies suggesting that LEC HS may essentially regulate chemokine-dependent signaling by DC in multiple ways depending on the context in which HS is presented. Recent work demonstrates that peri-lymphatic CCL21 gradients likely depends upon HS in the extracellular matrix(5–7). However, the reduced in vitro DC migration observed upon disrupting HS prompted us to look specifically into the importance of HS secreted by LEC in the presentation of chemokine to the migrating DC. Expression of CCL21 by LEC in these systems did not appear to be reduced by treating the LEC with heparinase or transient transfection of LEC with siNdst1 or siXylT2 (Supplemental Fig. 3B). Nevertheless, we noted that the ability of conditioned medium from siXylT2 or siNdst1 targeted LECs to support partnerships between CCL21 and CCR7 (or CXCL12 and CXCR4) on the DC surface was markedly reduced in comparison to that of conditioned medium from siRNA-control LECs (Fig. 5). These observations suggest that LEC HS plays a critical role in supporting “in-trans” presentation of these lymphatic chemokines to their receptors. A mechanistic explanation and extension of these findings highlights the possibility that LEC HS (whether bound to secreted HS proteoglycans or released as free chains) may serve as a soluble co-receptor for lymphatic chemokines to optimally interact with their receptors on the surface of trafficking DC. This is supported by the fact that genetic alterations in LEC HS biosynthesis resulted in marked alterations in the ability of CCL21 to oligomerize on purified soluble HS from mutant LEC as opposed to HS from wild-type/control LEC (Fig. 6).
The findings of this study contribute to our understanding of basic mechanisms that regulate DC migration and immunity. More generally, since DC traffic may become dysregulated in disorders such as autoimmunity, transplant rejection, or cancer, these findings suggest strategies for possibly re-programming immunity through rational therapeutic interventions that modulate HS fine-structure.
Supplementary Material
Acknowledgments
We acknowledge assistance from Dr Guillermo Oliver (St. Jude Children’s Research Hospital, Memphis TN) for provision of inducible Prox1Cre transgenic mutant mice as well as Dr. Jeffrey Esko (University of California, San Diego) for provision of heparin lyases. We acknowledge Steven Kendall and Fluxion Biosciences (San Francisco, CA) for assistance with BioFlux plates and microfluidics system as well as Dr Jennifer Meerloo of the UCSD Microscopy Shared Facility (University of California, San Diego). We thank Dr Klaus Ley of the La Jolla Institute for Allergy and Immunology for assistance with analysis of DC subsets in the lymph node and Dr Stephen Hedrick for provision of OT-I and OT-II transgenic mice. We also thank Dr Phil Gordts for initial assistance with adoptive T cell transfers, and Faye Nourollahi for additional assistance with mouse studies.
FUNDING SUPPORT: We acknowledge funding from NIH/NHLBI (R01-HL107652-01A1 to MMF; with additional support from P01 HL57345-14), NIH/NIAID (R01-AI37113 to TMH), NIGMS Ruth L. Kirschstein MARC Predoctoral Fellowship funding (to CLS), and training grant funding from NIH/NHLBI (T32HL098062 to XY). We acknowledge NIH/NIAID (R01-081923 and R21-AI102247 to EIZ). We also acknowledge the VA Merit Review Program (VA-Merit I01BX000987-01 to MMF) and the American Cancer Society (RSG-08-153-01-CSM to MMF).We also acknowledge assistance and support from the Veterans Medical Research Foundation.
NONSTANDARD ABBREVIATIONS
- BMDC
Bone marrow dendritic cells
- CM
Conditioned medium
- DC
Dendritic cells
- hLEC
Human lymphatic endothelial cells
- HS
Heparan sulfate
- LN
Lymph node
References
- 1.Forster R, Davalos-Misslitz AC, Rot A. CCR7 and its ligands: balancing immunity and tolerance. Nat Rev Immunol. 2008;8:362–371. doi: 10.1038/nri2297. [DOI] [PubMed] [Google Scholar]
- 2.Randolph GJ, Angeli V, Swartz MA. Dendritic-cell trafficking to lymph nodes through lymphatic vessels. Nat Rev Immunol. 2005;5:617–628. doi: 10.1038/nri1670. [DOI] [PubMed] [Google Scholar]
- 3.Delgado-Martin C, Escribano C, Pablos JL, Riol-Blanco L, Rodriguez-Fernandez JL. Chemokine CXCL12 uses CXCR4 and a signaling core formed by bifunctional Akt, extracellular signal-regulated kinase (ERK)1/2, and mammalian target of rapamycin complex 1 (mTORC1) proteins to control chemotaxis and survival simultaneously in mature dendritic cells. J Biol Chem. 2011;286:37222–37236. doi: 10.1074/jbc.M111.294116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Capila I, Linhardt RJ. Heparin-protein interactions. Angew Chem Int Ed Engl. 2002;41:391–412. doi: 10.1002/1521-3773(20020201)41:3<390::aid-anie390>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 5.Yin X, Truty J, Lawrence R, Johns SC, Srinivasan RS, Handel TM, Fuster MM. A critical role for lymphatic endothelial heparan sulfate in lymph node metastasis. Mol Cancer. 2010;9:316:1–20. doi: 10.1186/1476-4598-9-316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bao X, Moseman EA, Saito H, Petryniak B, Thiriot A, Hatakeyama S, Ito Y, Kawashima H, Yamaguchi Y, Lowe JB, von Andrian UH, Fukuda M. Endothelial heparan sulfate controls chemokine presentation in recruitment of lymphocytes and dendritic cells to lymph nodes. Immunity. 2010;33:817–829. doi: 10.1016/j.immuni.2010.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weber M, Hauschild R, Schwarz J, Moussion C, de Vries I, Legler DF, Luther SA, Bollenbach T, Sixt M. Interstitial dendritic cell guidance by haptotactic chemokine gradients. Science. 2013;339:328–332. doi: 10.1126/science.1228456. [DOI] [PubMed] [Google Scholar]
- 8.de Paz JL, Moseman EA, Noti C, Polito L, von Andrian UH, Seeberger PH. Profiling heparin-chemokine interactions using synthetic tools. ACS Chem Biol. 2007;2:735–744. doi: 10.1021/cb700159m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Inaba K, Inaba M, Romani N, Aya H, Deguchi M, Ikehara S, Muramatsu S, Steinman RM. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med. 1992;176:1693–1702. doi: 10.1084/jem.176.6.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Quah BJ, Warren HS, Parish CR. Monitoring lymphocyte proliferation in vitro and in vivo with the intracellular fluorescent dye carboxyfluorescein diacetate succinimidyl ester. Nat Protoc. 2007;2:2049–2056. doi: 10.1038/nprot.2007.296. [DOI] [PubMed] [Google Scholar]
- 11.Yardeni T, Eckhaus M, Morris HD, Huizing M, Hoogstraten-Miller S. Retro-orbital injections in mice. Lab Anim (NY) 2011;40:155–160. doi: 10.1038/laban0511-155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 13.Grobe K, Inatani M, Pallerla SR, Castagnola J, Yamaguchi Y, Esko JD. Cerebral hypoplasia and craniofacial defects in mice lacking heparan sulfate Ndst1 gene function. Development. 2005;132:3777–3786. doi: 10.1242/dev.01935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bishop JR, Schuksz M, Esko JD. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature. 2007;446:1030–1037. doi: 10.1038/nature05817. [DOI] [PubMed] [Google Scholar]
- 15.Lutz MB, Kukutsch N, Ogilvie AL, Rossner S, Koch F, Romani N, Schuler G. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J Immunol Methods. 1999;223:77–92. doi: 10.1016/s0022-1759(98)00204-x. [DOI] [PubMed] [Google Scholar]
- 16.Desai UR, Wang HM, Linhardt RJ. Substrate specificity of the heparin lyases from Flavobacterium heparinum. Arch Biochem Biophys. 1993;306:461–468. doi: 10.1006/abbi.1993.1538. [DOI] [PubMed] [Google Scholar]
- 17.Salanga CL, Handel TM. Chemokine oligomerization and interactions with receptors and glycosaminoglycans: the role of structural dynamics in function. Exp Cell Res. 2011;317:590–601. doi: 10.1016/j.yexcr.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ando T, Jordan P, Joh T, Wang Y, Jennings MH, Houghton J, Alexander JS. Isolation and characterization of a novel mouse lymphatic endothelial cell line: SV-LEC. Lymphat Res Biol. 2005;3:105–115. doi: 10.1089/lrb.2005.3.105. [DOI] [PubMed] [Google Scholar]
- 19.Yin X, Johns SC, Lawrence R, Xu D, Reddi K, Bishop JR, Varner JA, Fuster MM. Lymphatic endothelial heparan sulfate deficiency results in altered growth responses to VEGF-C. J Biol Chem. 2011 doi: 10.1074/jbc.M110.206664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beacock-Sharp H, Donachie AM, Robson NC, Mowat AM. A role for dendritic cells in the priming of antigen-specific CD4+ and CD8+ T lymphocytes by immune-stimulating complexes in vivo. Int Immunol. 2003;15:711–720. doi: 10.1093/intimm/dxg067. [DOI] [PubMed] [Google Scholar]
- 21.Forster R, Braun A, Worbs T. Lymph node homing of T cells and dendritic cells via afferent lymphatics. Trends Immunol. 2012;33:271–280. doi: 10.1016/j.it.2012.02.007. [DOI] [PubMed] [Google Scholar]
- 22.Johnson LA, Jackson DG. Inflammation-induced secretion of CCL21 in lymphatic endothelium is a key regulator of integrin-mediated dendritic cell transmigration. Int Immunol. 2010;22:839–849. doi: 10.1093/intimm/dxq435. [DOI] [PubMed] [Google Scholar]
- 23.Rouzaut A, Garasa S, Teijeira A, Gonzalez I, Martinez-Forero I, Suarez N, Larrea E, Alfaro C, Palazon A, Dubrot J, Hervas-Stubbs S, Melero I. Dendritic cells adhere to and transmigrate across lymphatic endothelium in response to IFN-alpha. Eur J Immunol. 2010;40:3054–3063. doi: 10.1002/eji.201040523. [DOI] [PubMed] [Google Scholar]
- 24.Li P, Zhao ZJ, Liu FY, Sun LY, Ding X, Zhang WZ, Shang DH, Sun CF. The chemokine receptor 7 regulates cell adhesion and migration via beta1 integrin in metastatic squamous cell carcinoma of the head and neck. Oncol Rep. 2010;24:989–995. doi: 10.3892/or.2010.989. [DOI] [PubMed] [Google Scholar]
- 25.Seth S, Oberdorfer L, Hyde R, Hoff K, Thies V, Worbs T, Schmitz S, Forster R. CCR7 essentially contributes to the homing of plasmacytoid dendritic cells to lymph nodes under steady-state as well as inflammatory conditions. J Immunol. 2011;186:3364–3372. doi: 10.4049/jimmunol.1002598. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.