

Abstract
The adaptive response to hypoxia, low oxygen tension, involves inhibition of energy-intensive cellular processes including protein translation. This effect is mediated in part through a decrease in the kinase activity of mammalian target of rapamycin complex 1 (mTORC1), a master regulator of protein translation. The principle mechanism for hypoxia-induced mTORC1 inhibition however was not elucidated until recently. Our work has demonstrated that the stress-induced protein REDD1 is essential for hypoxia regulation of mTORC1 activity and has further defined the molecular mechanism whereby REDD1 represses mTORC1 activity under hypoxic stress. Using our studies with REDD1 as an example, we describe in detail biochemical approaches to assess mTORC1 activity in the hypoxic response. Here we provide methodologies to monitor signaling components both downstream and upstream of the hypoxia-induced mTORC1 inhibitory pathway. These methodologies will serve as valuable tools for researchers seeking to understand mTORC1 dysregulation in the context of hypoxic stress.
Keywords: mTOR, hypoxia, phospho-4E-BP1, phospho-p70S6K, phospho-S6, REDD1, TSC2, 14-3-3, protein translation
1. Introduction
Under conditions of hypoxia, defined here as low to moderate oxygen availability, mammalian cells exhibit a reduced capacity for oxidative metabolism and as a result initiate ATP conservation by limiting energy-consuming processes, including protein synthesis (1-3). The effect of hypoxia on protein translation is mediated in part through inhibition of mammalian target of rapamycin complex 1 (mTORC1) kinase (4,5), a central regulator of cellular growth, proliferation, and protein translation (6,7). Regulation of cap-dependent translation initiation by mTORC1 involves the direct phosphorylation of its downstream substrates eukaryotic initiation factor 4E binding protein 1 (4E-BP1) and ribosomal protein kinase S6 (p70S6K) (8-10). Several studies have indeed shown that cells exposed to hypoxic stress display a pronounced dephosphorylation of 4E-BP1 and p70S6K (4,11). Therefore, the examination of the phosphorylation status of 4E-BP1 and p70S6K, as well as that of the p70S6K substrate ribosomal protein S6, by immunoblot analysis using commercially available phospho-site specific antibodies can serve as readouts for mTORC1 activity under hypoxic conditions.
We along with others have demonstrated that the hypoxia-inducible protein REDD1 (for regulated in development and DNA damage, also known as RTP801, DDIT4, Dig2) is necessary for hypoxia-induced inhibition of mTORC1 activity (12-14) (Figure 1). Work in both Drosophila and in mammalian cells showed that REDD1 functions upstream of the tuberous sclerosis tumor suppressor complex proteins TSC1 and TSC2 to inhibit mTORC1 activity. Parallel studies showed that under normoxia, inhibitory 14-3-3 protein binds to TSC2 to suppress the function of the TSC1/2 complex, a key inhibitor of mTORC1 activity (15). We found that in response to hypoxia, REDD1 gene expression is induced, leading to REDD1-dependent dissociation of 14-3-3 and TSC2 (16) (Figure 2). This dissociation, which appears to depend on direct, competitive binding of REDD1 to 14-3-3 within a membrane compartment, activates the TSC1/2 complex to down-regulate mTORC1 activity. Thus, the assessment of REDD1/14-3-3 association and TSC2/14-3-3 dissociation by co-immunoprecipitation studies followed by immunoblot analysis provide insight into mTORC1 regulation in response to hypoxia.
Figure 1. REDD1 is required for inhibition of mTORC1 activity under hypoxia.

Hypoxia inhibits mTORC1 activity in wild-type but not REDD1-/- MEFs, as evidenced by dephosphorylation of S6K (T389) and 4E-BP1 (T70). MEFs of each genotype growing in 10% serum were exposed to hypoxia (1% O2) for the indicated times. The same blot was stripped and reprobed for the respective total proteins. Note the prominence of hypophosphorylated 4E-BP1 (lower band) upon hypoxic exposure of wild-type cells. Beta tubulin serves as a loading control. Adapted from Genes Dev. 22:239.
Figure 2. REDD1 is required for hypoxia-induced TSC2/14-3-3 dissociation.
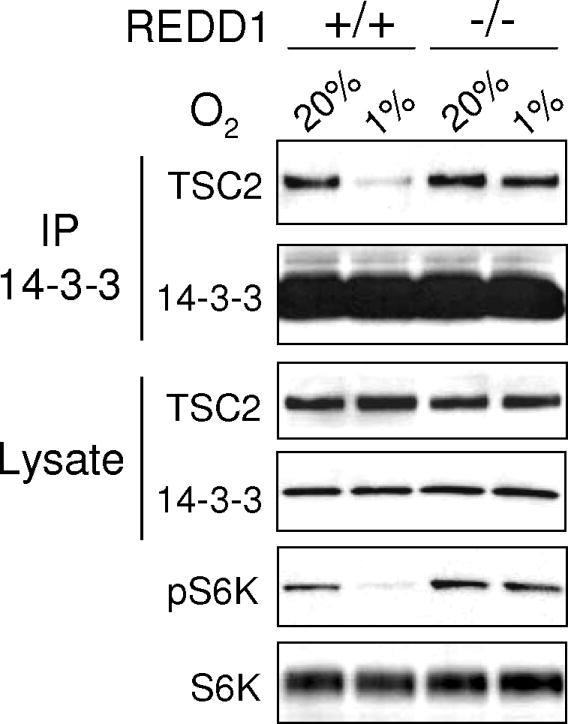
MEFs of the indicated genotype were treated with hypoxia (3 hours) followed by western analysis or IP for endogenous 14-3-3. Hypoxia-induced TSC2/14-3-3 dissociation and S6K1 (T389) dephosphorylation are both absent in REDD1-/- MEFs. Adapted from Genes Dev. 22:239.
Accumulating evidence suggest that the inappropriate control of mTORC1 activity in hypoxic cells confers a growth advantage and likely contributes to tumorigenesis and tumor maintenance (11, 16-18). However, the mechanism(s) by which mTORC1 activity is maintained in tumor cells under hypoxic stress remains to be fully elucidated, and further studies are warranted to clarify the interplay between aberrant mTORC1 activity, hypoxia, and tumorigenesis. The use of methodologies that provide accurate assessment of mTORC1 regulation and activity will be critical to this research effort.
2. Materials
2.1 Cell culture
Primary mouse embryonic fibroblasts (MEFs) derived from 12.5-14.5 postcoitum embryos are maintained in Dulbecco's Modified Eagle's Medium containing 4.5 g/L glucose and L-glutamine, supplemented with 10% (v/v) fetal bovine serum and 1% (v/v) penicillin/streptomycin (see Note 1). Alternatively, other cell model systems of interest and their corresponding culture media can be used in lieu of primary MEFs.
Phosphate buffered saline (PBS), sterilized (see Note 2).
Trypsin solution 0.25% in 1mM EDTA.
Hypoxia cell culture incubator Heracell 150 (see Note 3)
2.2 Preparation of cell lysates
PS6 lysis buffer for phospho-4E-BP1, phospho-p70S6K, phospho-S6 and corresponding total proteins (19) contains 0.5% Nonidet P-40, 150mM NaCl, protease and phosphatase inhibitor cocktails. Store the buffer at 4°C.
Denaturing lysis buffer for co-immunoprecipitation of 14-3-3 complexes (16) contains 0.75% Nonidet P-40, 1mM dithiothreitol (DTT) in PBS, along with protease and phosphatase inhibitor cocktails (see Note 4). Store the buffer at 4°C.
2.3 SDS-Polyacrylamide Gel Electrophoresis (PAGE) and membrane transfer
Bio-rad protein assay dye reagent concentrate.
12% Tris-Glycine polyacrylamide pre-cast gels (see Note 5).
SDS-PAGE running buffer (10×) contains 250mM Tris, 1.92M glycine, and 1% (w/v) sodium dodecyl sulfate (SDS) (see Note 6). Prepare 1× working solution with a 1:10 dilution of deionized distilled water. Store the 10× stock solution and the 1× working solution at room temperature.
5× Laemmli sample buffer is prepared with 62.5mM Tris-HCl pH 6.8, 20% (v/v) glycerol, 2% (w/v) SDS, 5% (v/v) 2-mercaptoethanol, and 1% (w/v) bromophenol blue (see Note 7). Store sample buffer in small aliquots at −20°C.
Pre-stained standard protein molecular weight marker.
Polyvinylidene fluoride (PVDF) Immobilon-FL transfer membrane cut to the dimensions of 7×8.4 cm.
Extra thick blot paper (mini-blot size).
Thick chromatography paper cut to the dimensions of 7×8.4 cm.
The following assumes the use of a semi-dry membrane transfer system. We use the Trans-blot semi-dry transfer cell from Bio-rad.
Prepare transfer solutions. Anode I buffer consists of 0.3M Tris and 10% (v/v) methanol, pH 10.4. Anode II buffer contains 25mM Tris and 10% (v/v) methanol, pH 10.4. Cathode buffer consists of 25mM Tris, 192mM glycine, and 10% (v/v) methanol, pH 9.4. Store buffers at room temperature.
2.4 Immunoblotting for phosphorylated downstream targets of mTORC1
Prepare solution of PBS containing 0.1% (v/v) Tween-20 (PBST). Store the solution at room temperature.
Blocking solution is prepared either with 5% (w/v) non-fat dry milk or 5% (w/v) bovine serum albumin (BSA) fraction V in PBST (see Note 8).
Pertinent information regarding primary and secondary antibodies for immunoblotting, including supplier, product code, and recommended use are detailed in Table 1. It should be emphasized that these primary antibodies can be used to detect both mouse and human species. Thus, these reagents would also be useful for investigators wishing to use human cell model systems.
Western lightening plus-enhanced chemiluminescence (ECL) substrate reagents.
BioMax XAR film.
Autoradiography cassette.
Table 1. Antibody reagents and suppliers.
| Antibody | Supplier | Product no. | Recommended use |
|---|---|---|---|
| Phospho-4E-BP1 (Thr70) | Cell Signalling Technologies | #9455 | (WB) 1:1,000 1% BSA-PBST O/N |
| Phospho-p70S6K (Thr389) | Cell Signalling Technologies | #9205 | (WB) 1:1,000 1% BSA-PBST O/N |
| Phospho-S6 (Ser235/Ser236) | Cell Signalling Technologies | #2211 | (WB) 1:1,000 1% BSA-PBST O/N |
| 4E-BP1 | Santa Cruz Biotechnology | #6936 | (WB) 1:1,000 5% milk-PBST 1h |
| p70S6K | Cell Signalling Technologies | #9202 | (WB) 1:1,000 5% milk-PBST 1h |
| S6 | Cell Signalling Technologies | #2217 | (WB) 1:1,000 5% milk-PBST 1h |
| β-Tubulin | Millipore | #MAB 3408 | (WB) 1:10,000 5% milk-PBST 1h |
| Pan-14-3-3 | Thermo Scientific | #MS-1504P | (IP) 1ug/mg protein, (WB) 1:1,000 5% milk-PBST O/N |
| TSC2 | Santa Cruz Biotechnology | #893 | (WB) 1:1,000 5% milk-PBST O/N |
| Goat α-mouse IgG-HRP | Biorad | #170-6516 | (WB) 1:5000 either 1% BSA or 5% milk-PBST 1h |
| Goat α-rabbit IgG-HRP | Biorad | #170-6515 | (WB) 1:5000 either 1% BSA or 5% milk-PBST 1h |
2.5 Stripping and re-probing membrane for total protein
Restore western blot stripping buffer.
2.6 Co-immunoprecipitation of 14-3-3 complexes
Protein G Sepharose beads.
Relevant information regarding antibodies for co-immunoprecipitation studies, including supplier, product code, and recommended use, are provided in Table 1.
3. Methods
The methods described below outline our established protocols for (1) treatment of cells under hypoxic conditions; (2) preparation of cell samples; (3) detection of mTORC1 downstream targets by immunoblot analysis; and (4) detection of 14-3-3 immunocomplexes by co-immunoprecipitation and immunoblot analysis. We have also highlighted some important considerations for using these techniques.
3.1 Culturing cells under normoxic and hypoxic conditions
Seed cells for the described experiments when passaged cells reach 80-90% confluency. Detach cells from tissue culture plates by washing with sterile PBS, adding trypsin-EDTA, and then placing plates back into a 37°C humidified incubator for 2-5 minutes.
Verify detachment of cells using an inverted phase contrast microscope. Once the majority of the cells have rounded up, inactivate the trypsin with the addition of pre-warmed media containing serum (trypsin inhibitors are present in serum). Resuspend the cells using a serological pipette and pipette-aid (see Note 9).
Seed primary MEFs at a cell density of 5×105 cells per 10-cm plate (see Note 10).
Place plates in a 37°C humidified incubator at 5% CO2 and atmospheric oxygen level or normoxia (20% O2) for a minimum of 3-5 hours to allow adherence of cells to the plates. We generally culture cells for 12-24 hours post-seeding.
Transfer plates to a 37°C hypoxia cell culture incubator and expose cells to 1% oxygen for various time points (see Note 11). Keep control plates in the normoxia incubator. Note that in our study with primary MEFs we evaluated mTORC1 activity in response to hypoxia at time points ranging from 15 minutes to 6 hours (see Figure 1). We observed dephosphorylation of phospho-p70S6K (Thr389) and phospho-4E-BP1 (Thr70) within 3 hours in wildtype MEFs. In contrast, REDD1-null MEFs exhibited a defect in dephosphorylation of mTORC1 substrates at all time points examined.
3.2 Preparation of cell lysates following hypoxia treatment
Remove plates from the hypoxia and normoxia cell culture incubators (see Note 11).
Quickly place the plates on ice and wash them with ice-cold, non-sterile PBS. Wash twice (see Note 12).
Add 400μL of the appropriate cell lysis buffer, either PS6 or denaturing cell lysis buffer, directly to the plates.
Collect cells from the plates using a cell scraper then transfer cells into microcentrifuge tubes.
Allow the cells to lyse for 20 minutes on ice, inverting the microcentrifuge tubes every few minutes. Alternatively, place the microcentrifuge tubes on a 4°C rocker.
Centrifuge tubes at 13,000 rpm for 20 minutes to pellet nuclei. Use a 4°C refrigerated tabletop centrifuge.
Carefully transfer supernatants to new microcentrifuge tubes and place tubes on ice (see Note 13).
3.3 SDS-PAGE and protein transfer
Determine protein concentrations of lysates using the Bio-rad protein assay dye reagent concentrate. Both the PS6 and denaturing cell lysis buffers are compatible with this Bradford protein assay. Briefly, dilute 200μL of the protein dye concentrate with 800μL of deionized distilled water, pipette 1μL of lysate into the solution, wait 5 minutes, and then measure the absorbance at 595nm.
Samples containing 25-40μg of lysate are diluted 4:1 (v/v) with 5× Laemmli sample buffer, boiled at 95°C in a heat block for 5 minutes, briefly vortexed and centrifuged, and then placed on ice until use.
Cut open a pouch containing a 12% Tris-Glycine polyacrylamide pre-cast gel and remove the tape at the bottom of the cassette.
Assemble the pre-cast gel cassette into the electrophoresis chamber. We use the Bio-rad mini-protean II apparatus.
Carefully lift the comb straight up to remove it from the cassette.
Fill the inner buffer chamber with 1× SDS-PAGE running buffer and allow the running buffer to overflow into the outer buffer chamber. This will wash and displace any air bubbles in the wells of the gel.
Load pre-stained standard protein molecular weight marker (∼10μL) and samples slowly into the wells using a P20 micropipettor (see Note 14).
Connect the unit to a power supply. Then run the gel at a constant voltage of 125 volts until the bromophenol blue dye front has migrated to the bottom of the gel (see Note 15). This will take approximately 90 minutes.
Once electrophoresis is completed, disconnect the device from the power supply and take out the gel cassette. Open the cassette, and using a razor blade cut the upper and lower most portions of the gel and discard them. This will leave behind only the separating gel. Mark the orientation of the separating gel by cutting the top left corner. Then equilibrate the gel in Cathode buffer for 5 minutes.
The following steps detail the electrophoretic transfer of proteins from the gel to PVDF membrane using the Trans-blot semi-dry transfer system from Bio-rad.
Activate the PVDF membrane for protein transfer by wetting the membrane with 100% methanol for 1-2 minutes (the membrane should change from opaque to semi-translucent). Then immerse the membrane in Anode II buffer for 5 minutes (see Note 16).
-
Assemble the semi-dry transfer stack in the following manner:
Wet one extra thick blot paper in Anode I buffer and place it on the anode plate of the transfer apparatus.
Wet one piece of thick chromatography paper in Anode II buffer and place it on top of the extra thick blot paper.
Place the activated PVDF membrane on top of the stack.
Place the gel on top of the membrane, and using a serological pipette remove air bubbles between the gel and membrane by rolling it gently over the gel.
Wet one extra thick blot paper in Cathode buffer and place it on top of the gel, and remove air bubbles as before.
Place the cathode plate on top of the transfer stack.
Connect the transfer apparatus to a power supply and apply a constant current of 0.25mA for 35 minutes.
After the transfer is completed, disconnect the apparatus from the power supply and remove the membrane. Mark the orientation of the membrane, as with the gel, by cutting the top left corner of the membrane.
3.4 Immunoblotting for phosphorylated downstream targets of mTORC1
Block the membrane with 10mL of 5% BSA-PBST. Incubate the membrane for 30 minutes at room temperature on a shaker.
Discard the blocking solution. Refer to Table 1 for the appropriate primary antibody against phospho-4E-BP1 (Thr70), phospho-p70S6K (Thr389), and phospho-S6 (Ser235/Ser236) (see Note 17). Incubate the membrane with the recommended primary antibody diluted 1:1000 in 1% BSA-PBST overnight at 4°C on a rocker. Care should be taken to add sufficient volume of the diluted primary in order to cover the entire membrane.
Remove the primary antibody and rinse the membrane twice with 0.1% PBST. Next, wash the membrane twice with 10mL 0.1% PBST for 10 minutes at room temperature on a shaker (see Note 18).
Incubate the membrane with the appropriate horseradish peroxidase-conjugated secondary antibody (diluted 1:5000 in 1% BSA-PBST) for 40-60 minutes at room temperature on a shaker.
Discard secondary antibody and repeat washes as before.
Immunoreactivity signal is visualized using enhanced chemiluminescence (ECL) solutions (mixture of 1:1). The membrane is blotted on a paper towel, placed on top of a piece of plastic wrap, overlaid with ECL solution for 1 minute, and after which time blotted again on a paper towel.
Wrap the membrane in plastic wrap, making sure to remove air bubbles. Then place the membrane inside an autoradiography cassette and expose the membrane to x-ray film for 30 seconds to 5 minutes.
3.5 Stripping and re-probing membrane for total protein
Once the phosphoprotein blot has been obtained, the membrane can then be stripped of primary and secondary antibodies so that it can be reprobed with an antibody that recognizes the corresponding total protein. This will allow the assessment of total protein loading. To proceed with stripping the membrane, quickly rinse the membrane of residual ECL with 0.1% PBST.
Incubate the membrane with 10mL of Restore western blot stripping buffer for 15-20 minutes at room temperature on a shaker (see Note 19).
Discard the stripping buffer. Wash twice with 0.1% PBST for 10 minutes at room temperature on a shaker.
Block the membrane with 10mL of 5% milk for 30 at room temperature on a shaker.
Incubate the membrane with the appropriate primary antibody against the corresponding total protein. Refer to Table 1. Since we have found that these primary antibodies work well and provide strong detection signal, we generally incubate the membrane with the diluted primary antibody (1:1000 in 5% milk-PBST) for 1 hour at room temperature on a shaker.
Refer to steps 3-7 in the methods section 3.4 for immunoblotting (see Note 20).
3.6 Co-immunoprecipitation of 14-3-3 complexes
Culture cells under normoxia and hypoxia as described in section 3.1.
Prepare cell lysates in denaturing cell lysis buffer as described in section 3.2.
Determine protein concentration as described in section 3.3.
Save an aliquot of 50μL of lysate samples to be used as an input control for immunoblot analysis. These samples can be stored at −80°C until use.
Use equivalent amounts of cell lysates (400μg-1mg protein) for co-immunoprecipitation. Transfer cell lysates into new microcentrifuge tubes and bring the volume of the lysate up to 800μL with the denaturing cell lysis buffer.
Pre-clear lysates with the addition of 40μL of Protein G Sepharose beads. Rotate microcentrifuge tubes for 20 min at 4°C (see Note 21). The pre-clearing step will help to reduce non-specific binding of proteins to the beads and improve the signal-to-noise ratio.
Use a refrigerated centrifuge to pulse-spin the tubes at 8000 rpm. Then transfer the supernatant to new microcentrifuge tubes.
Add 1μg of the 14-3-3 antibody to the lysates (refer to Table 1) and rotate the microcentrifuge tubes for 90 minutes (see Note 22).
Incubate lysates with 40μL of pre-washed Protein G Sepharose beads. Continue to rotate the microcentrifuge tubes for an additional 30 minutes at 4°C.
Pulse-spin microcentrifuge tubes at 8000 rpm. Remove the supernatant with a P1000 micropipettor and discard the supernatant.
Wash beads with 900μL denaturing cell lysis buffer four times, and pulse-spin as described in step 10.
Remove the entire volume of lysis buffer after the last wash using a P200 micropipettor.
To the beads, add 30μL of 2× Laemmli sample buffer (diluted in deionized distilled water). Then tap microcentrifuge tubes to resuspend the beads.
Boil the microcentrifuge tubes at 95°C in a heat block for 5 minutes. Pulse-spin tubes and transfer equal volume of supernatant to new microcentrifuge tubes (see Note 23).
Load immunoprecipitated complexes as well as input lysate samples (25-40μg) onto a SDS-PAGE gel then perform membrane transfer as described in section 3.3. Subsequently, probe the membrane for detection of TSC2 in the 14-3-3 immunoprecipitated samples using the method described in section 3.4 (see Note 24). In our studies, we observed that hypoxia (1% O2) induced the dissociation of TSC2 and 14-3-3 in wildtype primary MEFs but not in REDD1-null MEFs (see Figure 2).
4. Notes
Primary MEFs replicate for a limited number of population doubling, typically exhibiting rapid growth at passage 1-5, slow growth at passage 5-10, and then no growth or senescence at passage 10-25. At higher passage, a subset of primary MEFs will escape senescence through spontaneous immortalization. Accordingly, we recommend the use of primary MEFs at an early passage of less than 6 to preclude confounding effects associated with senescence or spontaneous immortalization.
Unless noted otherwise, all solutions are prepared in deionized distilled water with a specific resistance of 18.2MΩ-cm.
A hypoxia cell culture incubator such as the Heracell 150 has an oxygen sensor probe that allows the precise control of oxygen content. This instrument utilizes nitrogen gas to displace or flush oxygen, thereby reducing the oxygen content inside the chamber. We prefer the use of a hypoxia cell culture incubator over hypoxia mimetics such as cobalt chloride (CoCl2) and desferrioxamine (DFO). There may be differences in the response of cells elicited by chemical-induced hypoxia versus ambient hypoxia as different intracellular signaling pathways might be activated or inactivated by hypoxia mimetic drugs.
For both the PS6 lysis buffer and denaturing lysis buffer, protease and phosphatase inhibitors are added at the time of use to maximize their efficacy. Similarly, we add DTT (1M stock solution stored at −20°C) to the denaturing lysis buffer before use as well.
We have found that the use of polyacrylamide pre-cast gels ensure a high degree of consistency and minimize the handling of toxic chemicals that are associated with preparation of self-cast gels. Note the expiration date of pre-cast gels prior to use.
Caution should be used when preparing SDS-PAGE running buffer as SDS powder is hazardous. Prepare the solution in a ventilated chemical fume hood and wear a face mask when weighing SDS powder. We suggest the use of 3M Particulate Respirator N95.
When dispensing 2-mercaptoethanol for preparation of 5× Laemmli sample buffer, work in a ventilated chemical fume hood as it is toxic and has an unpleasant odor.
The choice between the use of BSA or milk as blocking agents as well as for diluting primary and secondary antibodies is dependent on whether the protein of interest is a phosphoprotein. Milk contains phosphatases which may result in the dephosphorylation of phosphoproteins. Moreover, the phosphoprotein casein is also present in milk. Primary antibodies against phosphorylated proteins may bind to casein, resulting in high background. Consequently, we commonly use BSA when probing for phosphorylated proteins.
It is crucial to break up any cell clumps in order to achieve at least a 95% single cell suspension for accurate cell counting with a hemacytometer and for uniform plating of cells. We suggest two methods to facilitate obtaining a single cell suspension. One effective method to mechanically dissociate cell clumps is to place the tip of a serological pipette against the bottom of the plate while dispensing the cell suspension. As vigorous pipetting may damage the cell membrane and reduce viability of some cell types, an alternative method is to briefly rinse cells with trypsin, aspirate the solution, and then add additional trypsin to the cells followed by placing the plate back into a 37°C incubator for 2-5 minutes.
It is rarely appreciated that cells cultured in a monolayer to a high density are under some degree of hypoxic stress. This is mainly due to the high rate of oxygen consumption and limited diffusion of oxygen in a confluent cell layer. Thus, we recommend seeding cells at a moderate density such that at the time of experimental use and exposure to hypoxia, cells do not exceed 80% confluency.
Reoxygenation of cells is a major issue that is often encountered with the use of a hypoxia cell culture incubator. There are two precautions that we use to minimize reoxygenation of cells. Foremost, move quickly. This includes promptly closing incubator doors after placing or removing plates from the incubator. Importantly, process all samples as soon as possible once plates are removed from the hypoxia incubator. Another precaution is to increase the volume of cell culture media in the tissue culture plates.
For a 10-cm plate, wash with approximately 10mL of PBS. We wash twice, and aspirate any residual PBS to ensure complete removal of serum-containing media as it can interfere with accurate determination of protein concentration.
At this point, lysates prepared in PS6 lysis buffer can be stored frozen at −80°C until later use. Samples prepared in denaturing lysis buffer however should be processed immediately to maximize detection of 14-3-3 immunocomplexes. However, if co-immunoprecipitation of 14-3-3 complexes cannot be performed immediately then add 10% glycerol (v/v) to the lysates and store the samples at −80°C. This will help to preserve protein complexes upon freezing and thawing.
For best results, load empty wells with 1× Laemmli sample buffer diluted in lysis buffer to ensure that adjacent sample lanes do not spread.
Ensure proper running of the gel by making certain that the electrodes are connected correctly.
Do not allow the PVDF membrane to dry as this will result in poor and blotchy transfer of proteins. Should drying of the membrane occur re-wet it with methanol and rinse with Anode II buffer.
It should be noted that phosphorylation at Thr70 on 4E-BP1 as well as phosphorylation at Thr389 on p70S6K is rapamycin-sensitive. Furthermore several studies have confirmed that both Thr70 on 4E-BP1 and Thr389 on p70S6K are phosphorylated directly by mTORC1. Thus, these particular phospho-site specific antibodies can be used to monitor mTORC1 activity.
Retain these primary antibodies as they can be re-used for 10-15 subsequent experiments over several months. Exposure time may increase following extensive re-use of the diluted primary antibody.
It is best to use a closed container to minimize the unpleasant odor of the stripping buffer.
After detection of total protein by immunoblot analysis, membranes can be stored for later re-probing of other desired proteins. Rinse the membrane of ECL and allow it to dry at room temperature. The dried membrane can be stored at 4°C and is stable for up to 6 months. To re-probe the dried membrane, simply immerse it in 100% methanol for 30 seconds to 1 minute.
Use a cut micropipette tip to draw up the beads in order to prevent them from being shattered or damaged. Additionally, for the pre-clearing step use beads that have been pre-washed in the denaturing cell lysis buffer twice. Pulse-spin the microcentrifuge tube containing the beads at 8000 rpm to pellet the beads. Remove the lysis buffer with a P1000 micropipette. Then resuspend the beads in the denaturing cell lysis buffer.
One technical issue with the use of a pan 14-3-3 antibody is that such reagents may not allow determination of the exact 14-3-3 isoform that binds to TSC2.
Following the elution of captured proteins from the beads with 2× Laemmli sample buffer, the samples can be stored at −80°C until later use.
Refer to Table 1 for the TSC2 antibody and recommended dilutions and conditions.
Acknowledgments
The authors would like to thank Nicole Forster and Zachary M. Nash for critical reading of the manuscript and for discussion. This work was funded by RO1 CA122589 to LWE and by NRSA postdoctoral fellowship award F32 CA150633 to DDV.
Footnotes
The authors declare no competing conflicts of interest.
References
- 1.Liu L, Cash TP, Jones RG, Keith B, Thompson CB, Simon MC. Hypoxia-induced energy stress regulates mRNA translation and cell growth. Mol Cell. 2006;21:521–531. doi: 10.1016/j.molcel.2006.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wouters BG, van den Beucken T, Magagnin MG, Koritzinsky M, Fels D, Koumenis C. Control of the hypoxic response through regulation of mRNA translation. Semin Cell Dev Biol. 2005;16:487–501. doi: 10.1016/j.semcdb.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 3.Hochachka PW, Buck LT, Doll CJ, Land SC. Unifying theory of hypoxia tolerance: molecular/metabolic defense and rescue mechanisms for surviving oxygen lack. Proc Natl Acad Sci. 1996;93:9493–9498. doi: 10.1073/pnas.93.18.9493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arsham AM, Howell JJ, Simon MC. A novel hypoxia-inducible factor-independent hypoxic response regulating mammalian target of rapamycin and its targets. J Biol Chem. 2003;278:29655–29660. doi: 10.1074/jbc.M212770200. [DOI] [PubMed] [Google Scholar]
- 5.Koritzinsky M, Magagnin MG, van den Beucken T, Seigneuric R, Savelkouls K, Dostie J, Pyronnet S, Kaufman RJ, Weppler SA, Voncken JW, Lambin P, Koumenis C, Sonenberg N, Wouters BG. Gene expression during acute and prolonged hypoxia is regulated by distinct mechanisms of translational control. EMBO J. 2006;25:1114–1125. doi: 10.1038/sj.emboj.7600998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Foster KG, Fingar DC. Mammalian target of rapamycin (mTOR): conducting the cellular signaling symphony. J Biol Chem. 2010;285:14071–14077. doi: 10.1074/jbc.R109.094003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ma XM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol. 2009;10:307–318. doi: 10.1038/nrm2672. [DOI] [PubMed] [Google Scholar]
- 8.Mamane Y, Petroulakis E, LeBacquer O, Sonenberg N. mTOR, translation initiation and cancer. Oncogene. 2006;25:6416–6422. doi: 10.1038/sj.onc.1209888. [DOI] [PubMed] [Google Scholar]
- 9.Burnett PE, Barrow RK, Cohen NA, Snyder SH, Sabatini DM. RAFT1 phosphorylation of the translational regulators p70 S6 kinase and 4E-BP1. Proc Natl Acad Sci. 1998;95:1432–1437. doi: 10.1073/pnas.95.4.1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gingras AC, Raught B, Gygi SP, Niedzwiecka A, Miron M, Burley SK, Polakiewicz RD, Wyslouch-Cieszynska A, Aebersold R, Sonenberg N. Hierarchical phosphorylation of the translation inhibitor 4E-BP1. Genes Dev. 2001;15:2852–2864. doi: 10.1101/gad.912401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Connolly E, Braunstein S, Formenti S, Schneider RJ. Hypoxia inhibits protein synthesis through a 4E-BP1 and elongation factor 2 kinase pathway controlled by mTOR and uncoupled in breast cancer cells. Mol Cell Biol. 2006;26:3955–3965. doi: 10.1128/MCB.26.10.3955-3965.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shoshani T, Faerman A, Mett I, Zelin E, Tenne T, Gorodin S, Moshel Y, Elbaz S, Budanova A, Chajut A, Kalinski H, Kamer I, Rozen A, Mor O, Keshet E, Leshkowitz D, Einat P, Skaliter R, Feinstein E. Identification of a novel hypoxia-inducible factor 1-responsive gene, RTP801, involved in apoptosis. Mol Cell Biol. 2002;22:2283–2293. doi: 10.1128/MCB.22.7.2283-2293.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brugarolas J, Lei K, Hurley RL, Manning BD, Reiling JH, Hafen E, Witters LA, Ellisen LW, Kaelin WG., Jr Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004;18:2893–2904. doi: 10.1101/gad.1256804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reiling JH, Hafen E. The hypoxia-induced paralogs Scylla and Charybdis inhibit growth by down-regulating S6K activity upstream of TSC in Drosophila. Genes Dev. 2004;18:2879–2892. doi: 10.1101/gad.322704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cai SL, Tee AR, Short JD, Bergeron JM, Kim J, Shen J, Guo R, Johnson CL, Kiguchi K, Walker CL. Activity of TSC2 is inhibited by AKT-mediated phosphorylation and membrane partitioning. J Cell Biol. 2006;173:279–289. doi: 10.1083/jcb.200507119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.DeYoung MP, Horak P, Sofer A, Sgroi D, Ellisen LW. Hypoxia regulates TSC1/2-mTOR signaling and tumor suppression through REDD1-mediated 14-3-3 shuttling. Genes Dev. 2008;22:239–251. doi: 10.1101/gad.1617608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12:9–22. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 18.Schneider A, Younis RH, Gutkind JS. Hypoxia-induced energy stress inhibits the mTOR pathway by activating an AMPK/REDD1 signaling axis in head and neck squamous cell carcinoma. Neoplasia. 2008;10:1295–1302. doi: 10.1593/neo.08586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sofer A, Lei K, Johannessen CM, Ellisen LW. Regulation of mTOR and cell growth in response to energy stress by REDD1. Mol Cell Biol. 2005;25:5834–5845. doi: 10.1128/MCB.25.14.5834-5845.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]