

Abstract
Background
Endogenous prostanoids have been suggested to modulate sensitization during experimental allergic asthma, but the specific role of prostaglandin E2 (PGE2) or of specific E prostanoid (EP) receptors is not known.
Objective
Here we tested the role of EP2 signaling in allergic asthma.
Methods
Wild type (WT) and EP2−/− mice were subjected to ovalbumin sensitization and acute airway challenge. The PGE2 analog misoprostol was administered during sensitization in both genotypes. In vitro culture of splenocytes and of flow-sorted dendritic cells and T cells defined the mechanism by which EP2 exerted its protective effect. Adoptive transfer of WT and EP2−/− CD4 T cells was used to validate the importance of EP2 expression on T cells.
Results
As compared to WT mice, EP2−/− mice had exaggerated airway inflammation in this model. Splenocytes and lung lymph node cells from sensitized EP2−/− mice produced more IL-13 than did WT cells, suggesting increased sensitization. In WT but not EP2−/− mice, subcutaneous administration of a stable PGE2 analog during sensitization inhibited allergic inflammation. PGE2 decreased cytokine production and inhibited STAT6 phosphorylation by CD3/CD28-stimulated CD4pos T cells. Co-culture of flow cytometry-sorted splenic CD4pos T cells and CD11cpos dendritic cells from WT or EP2−/− mice suggested that the increased IL-13 production in EP2−/− mice was due to the lack of EP2 specifically on T cells. Adoptive transfer of CD4pos EP2−/− T cells caused greater cytokine production in the lungs of WT mice than did transfer of WT CD4pos T cells.
Conclusion
We conclude that the PGE2-EP2 axis is an important endogenous brake on allergic airway inflammation, primarily targets T cells, and its agonism represents a potential novel therapeutic approach to asthma.
Keywords: asthma, allergic sensitization, prostaglandin E2, CD4 T cells
INTRODUCTION
A link between prostanoids – lipid mediators derived from the cyclooxygenase (COX) metabolism of arachidonic acid – and asthma has long been appreciated. Approximately 10% of asthmatics develop acute bronchoconstriction after taking aspirin or other nonsteroidal anti-inflammatory drugs (NSAIDs) that inhibit COX 1. Furthermore, COX inhibition by NSAIDs and deletion of genes encoding COX isoenzymes have both been found to increase allergic inflammation in mouse models of asthma 2–5. While these data strongly suggest the presence of an endogenous suppressive COX metabolite whose removal unmasks allergic inflammation, its precise identity remains uncertain. COX metabolites include five major bioactive prostanoids – prostaglandin E2 (PGE2), prostacyclin, PGD2, PGF2α, and thromboxane A2 – and all of these have been implicated in various aspects of asthma pathogenesis 6–10.
PGE2 is the most abundant prostanoid of most tissues and it can act on virtually all cell types. However, its effects on inflammatory and immune events are widely pleiotropic, in part reflecting its ability to ligate any of four G protein-coupled receptors, termed E prostanoid (EP) receptors 1–4, which activate distinct intracellular signaling programs. EP2 and EP4 signal by stimulating adenyl cyclase to generate the second messenger cAMP, which is typically suppressive of inflammatory cell functions. By contrast, EP3, by inhibiting adenyl cyclase, and EP1, by increasing intracellular Ca2+, typically activate leukocyte functions 11. EP2 ligation by PGE2 is a strong candidate mechanism to account for the suppressive effects of an endogenous prostanoid on allergic inflammation. First, EP2 has been shown to mediate many of the inhibitory actions of PGE2 on various functions of immune cells 12–14, including T cells 15,16. Second, a polymorphism in the EP2 gene was reported to be associated with aspirin-induced asthma 17.
Investigating the process of sensitization to allergen provides a window onto the generation of an adaptive immune response, a process which is integral to allergic asthma, but which is clinically silent. PGE2 has been implicated in the generation of Th2 immune responses 18. On the other hand, its ability to inhibit T cell proliferation 19 and MHC class II expression 20,21 also endows PGE2 with the potential to interfere with adaptive immunity. In this study, we sought to specifically interrogate the role of the PGE2-EP2 axis in controlling sensitization and Th2 polarization in a murine model of allergic asthma. Our data support an important suppressive role for endogenous PGE2-EP2 signaling in allergic asthma, and further identify EP2 on CD4pos T cells as the critical target mediating PGE2-induced suppression of sensitization.
METHODS
Animals
Mice harboring a targeted deletion of both alleles of the ptger2 gene encoding the EP2 receptor were originally generated by Dr. Richard Breyer (Vanderbilt University) 22 and bred in the University of Michigan Unit for Laboratory Animal Medicine. Eight-to-twelve week old male mice were used for all experiments. Animals were treated according to National Institutes of Health guidelines for the use of experimental animals, with the approval of the University of Michigan Committee for the Use and Care of Animals.
Reagents
Albumin from chicken egg (ovalbumin, OVA) was obtained from Sigma-Aldrich (St. Louis, MO). EP2 agonist (butaprost free acid), PGE2 and PGE2 stable analog (misoprostol) were obtained from Cayman Chemicals (Ann Arbor, MI). EP4 agonist (Ono-AE1-329) and EP3 agonist (Ono-AE-248) were kind gifts from Ono Pharmaceuticals (Osaka, Japan). DMSO served as vehicle control for PGE2 and EP agonists. The PKA-specific cAMP analog, 6-Bnz-cAMP (N6-benzoyladenosine-3′,5′-cyclic monophosphate), and the Epac-1-specific cAMP analog, 8-pCPT-2′-O-Me-cAMP (8–4-chlorophenylthio-2′-O-methyladenosine-3′,5′-cyclicmonophosphate), were obtained from Biolog Life Science Institute (Howard, CA).
Purification and culture of mouse splenic CD4pos T cells and CD11cpos DCs
For co-culture of splenic T cells and DCs a single cell suspension of splenocytes from OVA sensitized mice was washed with PBS/2 mM EDTA/0.5% fetal calf serum (FCS) and Fc receptor-mediated and nonspecific antibody binding was blocked by addition of FcR Blocking Reagent (Miltenyi Biotec, Auburn, CA). Cells were incubated with magnetic bead-conjugated anti-CD11c antibodies (Miltenyi Biotec) followed by magnetic separation according to the manufacturer’s instructions. Subsequently, the cell population enriched in CD11cpos cells was stained with CD11c antibody and the cell population negative for CD11c cells was stained with CD4 antibody, and both populations were flow-sorted to high purity (>96%). >93% of the CD4pos cell population that we isolated from spleens of WT or EP2−/− mice were CD3pos T cells. After sorting, purified CD11cpos DCs and CD4pos T cells were co-cultured with 100 μg/ml of OVA at a ratio of 1:10 in U-bottom 96 well plate for 3 days and supernatant was collected for cytokine analysis by ELISA.
In vitro expansion and activation of CD4pos T cells
96 well plates were coated with 0.5 μg/ml of CD3 (Biolegend) and 0.5 μg/ml CD28 (BD Bioscience) antibodies for 4 h in 37°C and washed with PBS to removed unbound soluble antibodies. Splenic CD4pos T cells were plated at a concentration of 2×105 per well and cultured for 3 days.
OVA-induced asthma protocol and sample harvest
Asthma was induced as previously described by intraperitoneal injection with 20 μg of OVA (Sigma) mixed with 2 mg of alum (Thermo Scientific) 23 followed seven days later by two airway challenges with 1 % OVA, unless indicated otherwise. This well-established protocol is known to result in eosinophilic inflammation and induction of Th2 cytokines in BALF. Samples were collected 24 h after the last airway challenge. Total cells were counted followed by differential counting of Wright-Giemsa prepared cytospins. To assess cytokines, lavage fluid recovered from the first 0.6 ml aliquot injected into the lung was analyzed by ELISA. For IgE measurements blood was collected from the inferior vena cava and serum was isolated by centrifugation; after 1:1000 dilution serum was analyzed for IgE using ELISA.
In vivo misoprostol treatment
Misoprostol was given in vivo according to a previously established protocol 14. To determine the effects of a stable PGE2 analog on asthma, mice were injected subcutaneously with 200 μl saline containing 50 μg of misoprostol in 0.5% DMSO two hours before and ten hours after intraperitoneal sensitization or airway challenge with OVA; control mice received 200 μl saline containing 0.5% DMSO alone.
CD4pos T cell transfer
CD4pos T cells were isolated from spleens of wild type or EP2−/− OVA-sensitized mice after airway challenge using magnetic beads for CD4 positive selection (Miltenyi Biotech). Isolation was performed according to manufacturer’s instructions and purity of CD4pos T cells was over 90%. Five million CD4pos T cells were injected intravenously and 24 h later mice were challenged with 3% OVA for three consecutive days; 24 h after the last airway challenge mice were sacrificed and samples were collected.
Data analysis
All data are displayed as mean values ± SEM from 3 to 6 independent experiments, each employing a different mouse, unless indicated otherwise. Statistical differences among treatment groups were estimated by ANOVA with Tukey’s post hoc test for multiple comparisons, or by Student’s t-test, as appropriate, using GraphPad Prism 5.0 (GraphPad Software, San Diego, CA). A p value < 0.05 was considered statistically significant.
RESULTS
Enhanced allergic inflammation in EP2-deficient animals
PGE2-EP2 signaling has been shown to be immunosuppressive in many different cell types and in vivo models 12,15. Here we utilized EP2-deficient and -sufficient (WT) mice to assess the importance of endogenous PGE2-EP2 signaling in a model of ovalbumin (OVA)-induced allergic asthma 24. Following sensitization and subsequent airway challenge using the protocol outlined in Fig. 1A, differential cell counts from BALF cytospins revealed higher numbers of both total leukocytes and eosinophils in the airways of EP2−/− mice (Fig. 1B–D). Additionally, mice deficient in EP2 exhibited increased BALF levels of the Th2 cytokines IL-4, IL-5 and IL-13, as well as IL-17 without any difference in levels of the Th1 cytokine IFN-γ (Fig. 1E). Except for IL-5, which was present at very low levels (~20 pg/ml) and did not differ between the two genotypes, none of these other cytokines were detectable in serum. Lymphocytes and granulocytes each comprised < 5% of total cells found in BALF and there were no differences in their relative numbers between the two genotypes (data not shown). Consistent with an increased systemic Th2 immune response, mice lacking EP2 also manifested higher serum IgE levels (Fig. 1H).
Figure 1. Enhanced allergic inflammation in EP2-deficient animals.

(A) Schematic representation of experimental protocol. (B–D) EP2 −/− mice have increased numbers of eosinophils and total cells in BALF after airway challenge. (E) EP2 −/− mice have increased cytokines in BALF after airway challenge. (n=4).* p<0.05, ** p<0.01. (H) EP2 −/− mice have increased serum IgE levels. Samples were collected 24 h after the second airway challenge (n=3) ** p<0.01.
Augmented Th2 responses in EP2-deficient mice are present during sensitization
Lung lymph node cells and splenocytes were isolated from mice 24 h after the second airway challenge and cultured for 3 days with varying concentrations of OVA. Cells of both types from EP2−/− mice exhibited dose-dependent IL-13 production which was greater than that from WT cells (Fig. 2A and 2B). IFN–γ production was likewise up-regulated in splenocytes from EP2−/− (data not shown), suggesting that enhanced immune responses in the absence of PGE2-EP2 signaling are not Th2-specific. IL-4 was not detected in our in vitro system (data not shown), consistent with similar findings reported in another study of allergic asthma 25. These heightened cellular responses (Fig. 2A and B) and allergic lung inflammatory responses (Fig. 1) in EP2−/− mice do not discriminate between enhanced allergen sensitization vs. enhanced airway challenge phases of this model. In order to specifically focus on the former, we isolated splenocytes from mice that underwent sensitization but not airway challenge, and cultured them with OVA as depicted in Fig. 2A and B. Splenocytes isolated at this point in the protocol from EP2−/− mice likewise exhibited greater IL-13 (Fig. 2C) and IFN-γ (data not shown) production than did those from WT mice (Fig. 2C), suggesting the presence of an increased immune response during sensitization to antigen in EP2−/− mice.
Figure 2. Augmented Th2 responses in EP2-deficient mice are present during the sensitization phase.

(A) Thoracic lymph node cells and (B) splenocytes were isolated after the second airway challenge and cultured for 3 days in the presence of OVA. Controls were cultured without OVA. (n=4).* p<0.05, ** p<0.01. (C) Splenocytes isolated from mice after sensitization and without airway challenge were cultured for 3 days. Culture supernatants were analyzed for IL-13 by ELISA. (n=4).* p<0.05.
Exogenous PGE2 signaling strongly decreases IL-13 production in vitro
Data presented in Fig. 1 and 2 reveal a suppressive effect of endogenous PGE2-EP2 signaling on antigen sensitization and subsequent airway responses. We next utilized pharmacologic tools applied in vitro to further characterize the mechanisms by which PGE2 inhibits splenocytes isolated from sensitized wild type and EP2 −/− mice. While PGE2 itself was able to decrease OVA-induced production of IL-13 by ~85% (Fig. 3) and IFN-γ by ~100% (data not shown) from wild type mice, it failed to reduce production of both cytokines in splenocytes from EP2−/− mice. Exogenous administration of PGE2 analogs selective for either EP2 or EP4 also reduced IL-13 production, while an EP3 agonist had no such effect. cAMP can act through either of two effector proteins, protein kinase A (PKA) or guanine nucleotide exchange protein directly activated by cAMP (Epac) 26. cAMP analogs selective for activation of either PKA or Epac both strongly reduced IL-13 levels, indicating that both effectors may participate in the process of inhibition of antigen sensitization by PGE2.
Figure 3. Exogenous PGE2 suppresses splenocyte IL-13 production via cAMP signaling.
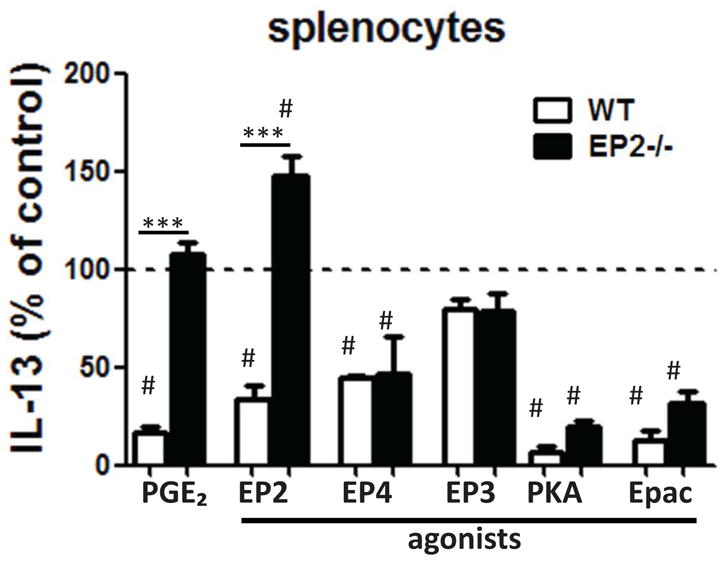
Splenocytes from sensitized wild type and EP2−/− mice were cultured with 10 μg/ml OVA in the presence of PGE2, EP2 agonist EP3 EP4 agonist (all at 1 μM), or the PKA-specific cAMP analog 6-Bnz-cAMP or the Epac-specific cAMP analog 8-pCPT-2′-O-Me-cAMP (both at 500 μM); All treatments were statistically different compared to control (#) (n=4) *** p<0.001.
In vivo administration of a PGE2 analog during sensitization inhibits allergic inflammation
To assess the effects of PGE2 on in vivo allergic sensitization, misoprostol – a stable but receptor-nonselective PGE2 analog – was administered subcutaneously using a regimen reported previously 14. Specifically, misoprostol was administered 2 h before and 10 h after intraperitoneal OVA/alum injection, and seven days later mice underwent two consecutive daily airway challenges with OVA; BALF and serum samples were collected 24 h after the second airway challenge (Fig. 4A). The potential receptor dependence of misoprostol was evaluated by determining its actions in EP2−/−, as compared to WT, mice. Misoprostol-treated WT mice showed reduced eosinophil numbers (Fig. 4B) and IL-5 and IL-13 levels (Fig. 4D) in BALF and reduced IgE levels in the serum (Fig. 4E). By contrast, misoprostol effects in EP2−/− mice were less pronounced and not statistically significant (Fig. 4B–E). Mononuclear cell numbers were unaffected by misoprostol (Fig. 4C). Lymphocytes and granulocytes each accounted for < 5% of total BALF cells and their numbers did not differ between genotypes (data not shown). IL-13 production by cultured splenocytes isolated from mice treated with misoprostol was reduced in WT but not in EP2−/− mice (Fig. 4F). These experiments suggest that in vivo PGE2, mainly acting through EP2, inhibits the immunological events occurring during sensitization and this action is sufficient to attenuate allergic inflammation following subsequent airway challenge.
Figure 4. Subcutaneous administration of a PGE2 analog during the sensitization phase inhibits allergic inflammation following airway challenge.

(A) Schematic representation of misoprostol treatment protocol (50 μg misoprostol or DMSO vehicle administered 2 h before and 10 h after sensitization) (B, C) Misoprostol reduces eosinophilia in wild type but not EP2 −/− mice. (D) Misoprostol reduces BALF IL-5 and IL-13 levels in wild type but not EP2 −/− mice. (E) Misoprostol reduces serum IgE in wild type but not EP2 −/− mice. (F) Splenocytes isolated from wild type but not EP2−/− mice treated with misoprostol have decreased IL-13 production in vitro. (n=4)* p<0.05, ** p<0.01.
PGE2 prevents IL-13 production primarily by targeting EP2 on T cells and inhibits STAT6 phosphorylation
To dissect the relative roles of antigen-presenting cells versus T cells in the inhibitory actions of PGE2, CD4pos T cells and CD11cpos dendritic cells (DCs) from OVA-sensitized spleens of WT and EP2−/− mice were sorted by flow cytometry to high purity and co-cultured in a “mix and match” design (Fig. 5A). Marked elevation of IL-13 production was only observed in co-cultures containing EP2−/− T cells, whereas cytokine generation was not influenced by the absence of EP2 on DCs. These data strongly suggest that PGE2 inhibition of sensitization is mediated by suppressive actions targeting EP2-bearing CD4pos T cells, rather than DCs. To further explore the inhibitory effect of PGE2 directed at T cells, splenic CD4pos T cells were purified using magnetic beads from sensitized wild type and EP2−/− mice and cultured for 3 days on plates pre-coated with CD3/CD28 antibodies. CD4pos T cells from EP2−/− mice exhibited significantly higher IL-13 production than did cells from wild type mice (Fig. 5B). PGE2 and the EP2 agonist inhibited IL-13 production by ~75% in CD4pos T cells from wild type, but not EP2−/− animals; the EP3 agonist had no effect and the effect of the EP4 agonist here was significant, but less than that of the EP2 agonist (Fig. 5C). Additionally, PKA and Epac agonists both significantly inhibited IL-13 production in T cells from both genotypes (Fig. 5C). Exogenous PGE2 also inhibited CD4pos T cell production of other Th2 cytokines (IL-4 and IL-5) as well as of IFN-γ, but did not affect IL-17 production (data not shown). Generation of Th2 cytokines depends on phosphorylation at residue Y641 and subsequent activation of the key transcription factor, STAT6 27,28. Interestingly, PGE2 treatment reduced both constitutive (Fig. 6A) and IL-4-stimulated (Fig. 6B) STAT6 phosphorylation in splenic T cells isolated from sensitized mice. Together, these data suggest that CD4pos T cells are important targets for the inhibitory actions of PGE2 on allergen sensitization and subsequent lung inflammation.
Figure 5. PGE2 inhibits IL-13 production primarily by inhibiting T cells through EP2.

(A) and (A) EP2 deficiency on T cells primarily contributes to enhanced Th2 responses in EP2−/− mice. Purified DCs and T cells were co-cultured at a ratio of 1:10 for 3 days in the presence of 100 μg/ml OVA. Data are from one experiment representative of 3 independent experiments. (B) CD4pos T cells from EP2−/− mice exhibited significantly higher IL-13 production than did cells from wild type mice (C) PGE2 suppresses Th2 polarization. Purified CD4pos T cells were cultured in the presence of 1 μM PGE2, EP2, EP3, or EP4 agonist or 500 μM PKA-specific cAMP analog 6-Bnz-cAMP or the Epac-specific cAMP analog 8-pCPT-2′-O-Me-cAMP. Treatments statistically different compared to control are indicated with (#). (n=3) ** p<0.01, *** p<0.001.
Figure 6. PGE2 inhibits STAT6 phosphorylation in T cells.

(A) CD4 T cells cultured for 3 days in the presence of 1 μM PGE2 exhibit decreased STAT6 phosphorylation on residue Y641. (B) CD4 T cells pretreated for 30 min with 1 μM PGE2 and subsequently treated for 5 min with recombinant IL-4 (25 ng/ml) have decreased STAT6 phosphorylation. n=4 * p<0.05
Adoptive transfer of CD4pos T cells from EP2−/− mice induces a greater cytokine response in the lung
To validate the role and importance of EP2 expression on CD4pos T cells as a brake on allergic inflammation, we employed a CD4pos T cell transfer model in unsensitized mice subsequently subjected to airway challenges using a protocol outlined in Fig. 7A. Transfer of EP2−/− CD4pos T cells from mice that underwent sensitization and airway challenge resulted in greater cytokine levels in BALF of unsensitized recipient wild type mice than did that of wild type T cells. Although the increases reached statistical significance only for the Th2 cytokines IL-4 and IL-5, a trend towards increased levels in mice receiving EP2−/− CD4pos T cells was also observed for IL-13, IFN-γ, and IL-17 (Fig. 7B). These data validate the important role of EP2-bearing T cells as a suppressive influence on allergic inflammation, but also suggest that the inhibitory effects of PGE2-EP2 signaling in CD4pos T cells may extend to polarization of T cells in general.
Figure 7. Adoptive transfer of CD4pos T cells from EP2−/− mice induces increased cytokine production in wild type mice.
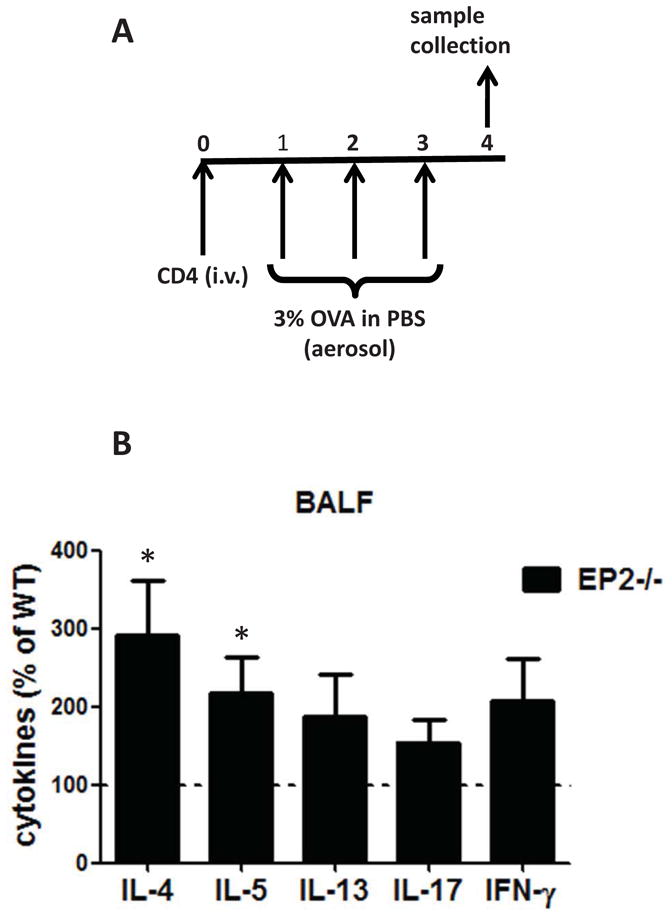
(A) Schematic representation of experimental protocol. (B) Naïve wild type mice were injected intravenously with 5 × 106 purified CD4pos T cells from asthmatic wild type or EP2−/− mice. 24 h later mice were challenged with OVA and BALF was collected 24 thereafter for cytokine determination by ELISA. Wild type values were set at 100%. (n=4).* p<0.05
DISCUSSION
Exogenous PGE2 has long been known to be a direct bronchodilator 29–32 and to protect against airway inflammatory responses to challenges such as aspirin 33,34 and allergen 35–37. However, this abundant prostanoid is also well known to exert pleiotropic effects on immune responses, and its influence on the initiation of the allergic response and the subsequent consequences thereof in a model of allergic asthma remain uncertain. Precedent exists for EP2 mediating suppressive actions of PGE2 on both maturation of DCs 15 as well as T cell proliferation 16. Here we have definitively shown for the first time that PGE2-EP2 signaling acts as an endogenous brake on the development of adaptive Th2-polarized immunity, and this is sufficient to blunt local inflammatory responses upon subsequent airway challenge. Although modest effects on DCs were observed, T cells appear to be the main target of PGE2 action. Moreover, we have identified that PGE2 inhibits phosphorylation of STAT6, which is a transcription factor involved in Th2 differentiation. Such a decrease in STAT6 activation represents a possible mechanism by which PGE2-EP2 attenuates the generation of Th2 cytokines. Th2 cells are widely regarded as the main producers of Th2 cytokines and are sufficient for the induction of inflammation and the physiologic and pathologic changes associated with asthma 38. Although we have not established in our study that CD4 T cells are entirely responsible for the increase in Th2 cytokines in vivo, it is highly likely that they are major contributors to this phenomenon. As effects of PGE2 extend also to other polarization states, our findings may extend beyond asthma to other conditions where generation of polarized T cells is important.
An inhibitory effect of PGE2 on T cell proliferation has been very well documented 39. However, its effect on T cell cytokine secretion is much more controversial. PGE2 has been reported to inhibit synthesis of Th1 cytokines such as IFN-γ 40. By contrast, a number of studies over the years have reported that PGE2 promotes Th2 responses 18,41,42, and this conclusion has largely been taken as axiomatic. Inhibition of Th2 cytokines by an endogenous prostanoid was strongly suggested by studies showing their upregulation by pharmacologic inhibition or genetic deletion of COX isoenzymes in mouse models of allergic asthma 3–5,43–45. These experimental approaches were not designed to implicate a particular prostanoid in such suppressive actions; however, in view of its known immunosuppressive actions exerted on T cells and other cell types pertinent to asthma 15,16, PGE2 is an attractive candidate to mediate this COX-dependent suppression of allergen sensitization. One approach to evaluating this possibility involved the use of mice deficient in the key PGE synthase, microsomal PGE synthase-1. However, conflicting results were observed, as allergic inflammation in mpges−/− mice was reported to be reduced in an OVA model 42 but increased in a house dust mite model 9. These conflicting findings may underscore the divergent signaling from, and biologic actions attributable to, specific EP receptors. One report used a genetic approach to address the role of specific EP receptors in allergic asthma. In that study, mice deficient in each individual EP subtype were examined in response to OVA-induced asthma 46. Only EP3 receptor-deficient mice exhibited increased allergic inflammation, and in vivo administration of an EP3 agonist was found to be protective 46. These findings were unexpected, since EP3 signaling generally reduces cAMP and typically results in stimulatory effects that oppose those elicited by EP2. We were unable to identify any such suppressive actions of this same EP3 agonist on splenocytes or purified T cells. Here we focused on PGE2-EP2 signaling in an OVA-induced asthma model and identified enhanced allergic inflammation in EP2-deficient mice. The protective role of PGE2 in our model was achieved by a reduction in Th2 cytokines. This finding is consistent with the effects of PGE2 on IL-13 production in dispersed nasal polyp cells from patients with chronic rhinosinusitis 47, its inhibition of Th2 (as well as Th1) responses in human T cells 48, and its suppression of IL-4 production by T cells leading to reduced IgE biosynthesis 49. Moreover, our study is supported by results showing that both EP2 and EP4 agonists suppress OVA-induced methacholine reactivity 50 – a parameter that typically correlates with allergic inflammation.
Although our findings point to the importance of EP2 on CD4 T cells, we acknowledge that the absence of EP2 receptors on other cell types could contribute to the exaggerated allergic inflammatory phenotype observed in these mice. Addressing this possibility further would require mice deficient in EP2 receptors selectively on T cells. Indeed, it is well documented that PGE2 can influence asthma pathogenesis by modulating the functions of cell types other than T cells. One example of this is its ability to directly inhibit eosinophil trafficking in the lung 6,51,52. We accordingly tested the ability of misoprostol to reduce eosinophilic lung inflammation in OVA-sensitized mice when administered exclusively around the time of airway challenge (Fig. S1). Indeed, misoprostol used in such a protocol significantly reduced Th2 cytokine levels as well as eosinophil numbers in BALF, but not serum IgE levels (Fig. S1). This ability of PGE2 to attenuate lung inflammation by targeting the events driving both sensitization and challenge responses is particularly attractive from a therapeutic perspective.
PGE2 levels are well recognized to increase in a variety of disease states such as inflammation and cancer and to decrease upon treatment with drugs including NSAIDs and glucocorticoids. Furthermore, EP2 expression is also subject to dynamic regulation 53. Our data suggest that states characterized by deficient PGE2-EP2 signaling may facilitate antigen sensitization. This idea is supported by epidemiological studies indicating that the use of COX inhibitors, which include acetaminophen 54, is associated with increased risk of developing allergic diseases including asthma 55–57 and that EP2 polymorphism is associated with aspirin-induced asthma 17 – a condition also typically characterized by increased sensitization to allergens. Overall, we have demonstrated that endogenous and exogenous PGE2 suppresses allergen sensitization predominantly by ligating EP2 on T cells, thereby diminishing Th2 polarization. Effects of exogenous PGE2 administered exclusively during the sensitization phase were sufficient to decrease airway inflammation during the airway challenge phase one week later, and persisted during in vitro culture. These data provide new insights into the regulation of immune responses and identify EP2-mediated suppression as an attractive new therapeutic strategy.
Supplementary Material
Key message.
PGE2-EP2 signaling targets CD4 T cells to suppress the generation of adaptive immune responses
This is sufficient to inhibit allergic lung inflammation upon antigen challenge.
Acknowledgments
Funding: This work was supported by the Deutsche Forschungsgemeinschaft (German Research Foundation) (ZZ), American Lung Association Senior Research Fellowships (ZZ, KO, EB), T32 AI007413 and American Heart Association (RD-G) and NIH grants R01 HL94311 and R01 HL58897 (MP-G), AI065543 (BBM), AI032302 (NWL), HD057176 (DMA).
We thank Thomas Moore for help with flow cytometry, Teresa Murphy, Sally Przybranowski and Carol Wilke for technical assistance, as well as the members of the Peters-Golden laboratory for helpful input.
Abbreviations
- PG
prostaglandin
- WT
wild type
- COX
cyclooxygenase
- NSAIDs
nonsteroidal anti-inflammatory drugs
- OVA
ovalbumin
- DC
dendritic cells
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Szczeklik A. Prostaglandin E2 and aspirin-induced asthma. Lancet. 1995;345(8956):1056. doi: 10.1016/s0140-6736(95)90799-8. [DOI] [PubMed] [Google Scholar]
- 2.Peebles RS, Jr, Hashimoto K, Morrow JD, et al. Selective cyclooxygenase-1 and -2 inhibitors each increase allergic inflammation and airway hyperresponsiveness in mice. Am J Respir Crit Care Med. 2002;165(8):1154–1160. doi: 10.1164/ajrccm.165.8.2106025. [DOI] [PubMed] [Google Scholar]
- 3.Gavett SH, Madison SL, Chulada PC, et al. Allergic lung responses are increased in prostaglandin H synthase-deficient mice. J Clin Invest. 1999;104(6):721–732. doi: 10.1172/JCI6890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nakata J, Kondo M, Tamaoki J, et al. Augmentation of allergic inflammation in the airways of cyclooxygenase-2-deficient mice. Respirology. 2005;10(2):149–156. doi: 10.1111/j.1440-1843.2005.00687.x. [DOI] [PubMed] [Google Scholar]
- 5.Peebles RS, Jr, Dworski R, Collins RD, et al. Cyclooxygenase inhibition increases interleukin 5 and interleukin 13 production and airway hyperresponsiveness in allergic mice. Am J Respir Crit Care Med. 2000;162(2 Pt 1):676–681. doi: 10.1164/ajrccm.162.2.9911063. [DOI] [PubMed] [Google Scholar]
- 6.Sturm EM, Schratl P, Schuligoi R, et al. Prostaglandin E2 inhibits eosinophil trafficking through E-prostanoid 2 receptors. J Immunol. 2008;181(10):7273–7283. doi: 10.4049/jimmunol.181.10.7273. [DOI] [PubMed] [Google Scholar]
- 7.Jaffar Z, Ferrini ME, Buford MC, Fitzgerald GA, Roberts K. Prostaglandin I2-IP signaling blocks allergic pulmonary inflammation by preventing recruitment of CD4+ Th2 cells into the airways in a mouse model of asthma. J Immunol. 2007;179(9):6193–6203. doi: 10.4049/jimmunol.179.9.6193. [DOI] [PubMed] [Google Scholar]
- 8.Kawikova I, Barnes PJ, Takahashi T, Tadjkarimi S, Yacoub MH, Belvisi MG. 8-Epi-PGF2 alpha, a novel noncyclooxygenase-derived prostaglandin, constricts airways in vitro. Am J Respir Crit Care Med. 1996;153(2):590–596. doi: 10.1164/ajrccm.153.2.8564103. [DOI] [PubMed] [Google Scholar]
- 9.Liu T, Laidlaw TM, Feng C, et al. Prostaglandin E2 deficiency uncovers a dominant role for thromboxane A2 in house dust mite-induced allergic pulmonary inflammation. Proc Natl Acad Sci U S A. 2012;109(31):12692–12697. doi: 10.1073/pnas.1207816109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oguma T, Asano K, Ishizaka A. Role of prostaglandin D(2) and its receptors in the pathophysiology of asthma. Allergol Int. 2008;57(4):307–312. doi: 10.2332/allergolint.08-RAI-0033. [DOI] [PubMed] [Google Scholar]
- 11.Regan JW. EP2 and EP4 prostanoid receptor signaling. Life Sci. 2003;74(2–3):143–153. doi: 10.1016/j.lfs.2003.09.031. [DOI] [PubMed] [Google Scholar]
- 12.Aronoff DM, Canetti C, Peters-Golden M. Prostaglandin E2 inhibits alveolar macrophage phagocytosis through an E-prostanoid 2 receptor-mediated increase in intracellular cyclic AMP. J Immunol. 2004;173(1):559–565. doi: 10.4049/jimmunol.173.1.559. [DOI] [PubMed] [Google Scholar]
- 13.Medeiros AI, Serezani CH, Lee SP, Peters-Golden M. Efferocytosis impairs pulmonary macrophage and lung antibacterial function via PGE2/EP2 signaling. J Exp Med. 2009;206(1):61–68. doi: 10.1084/jem.20082058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zaslona Z, Serezani CH, Okunishi K, Aronoff DM, Peters-Golden M. Prostaglandin E2 restrains macrophage maturation via E prostanoid receptor 2/protein kinase A signaling. Blood. 2012;119(10):2358–2367. doi: 10.1182/blood-2011-08-374207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang L, Yamagata N, Yadav R, et al. Cancer-associated immunodeficiency and dendritic cell abnormalities mediated by the prostaglandin EP2 receptor. J Clin Invest. 2003;111(5):727–735. doi: 10.1172/JCI16492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nataraj C, Thomas DW, Tilley SL, et al. Receptors for prostaglandin E(2) that regulate cellular immune responses in the mouse. J Clin Invest. 2001;108(8):1229–1235. doi: 10.1172/JCI13640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jinnai N, Sakagami T, Sekigawa T, et al. Polymorphisms in the prostaglandin E2 receptor subtype 2 gene confer susceptibility to aspirin-intolerant asthma: a candidate gene approach. Hum Mol Genet. 2004;13(24):3203–3217. doi: 10.1093/hmg/ddh332. [DOI] [PubMed] [Google Scholar]
- 18.Kuroda E, Ishii KJ, Uematsu S, et al. Silica crystals and aluminum salts regulate the production of prostaglandin in macrophages via NALP3 inflammasome-independent mechanisms. Immunity. 2011;34(4):514–526. doi: 10.1016/j.immuni.2011.03.019. [DOI] [PubMed] [Google Scholar]
- 19.Anastassiou ED, Paliogianni F, Balow JP, Yamada H, Boumpas DT. Prostaglandin E2 and other cyclic AMP-elevating agents modulate IL-2 and IL-2R alpha gene expression at multiple levels. J Immunol. 1992;148(9):2845–2852. [PubMed] [Google Scholar]
- 20.Figueiredo F, Uhing RJ, Okonogi K, et al. Activation of the cAMP cascade inhibits an early event involved in murine macrophage Ia expression. J Biol Chem. 1990;265(21):12317–12323. [PubMed] [Google Scholar]
- 21.Li G, Harton JA, Zhu X, Ting JP. Downregulation of CIITA function by protein kinase a (PKA)-mediated phosphorylation: mechanism of prostaglandin E, cyclic AMP, and PKA inhibition of class II major histocompatibility complex expression in monocytic lines. Mol Cell Biol. 2001;21(14):4626–4635. doi: 10.1128/MCB.21.14.4626-4635.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kennedy CR, Zhang Y, Brandon S, et al. Salt-sensitive hypertension and reduced fertility in mice lacking the prostaglandin EP2 receptor. Nat Med. 1999;5(2):217–220. doi: 10.1038/5583. [DOI] [PubMed] [Google Scholar]
- 23.Stumm CL, Wettlaufer SH, Jancar S, Peters-Golden M. Airway remodeling in murine asthma correlates with a defect in PGE2 synthesis by lung fibroblasts. Am J Physiol Lung Cell Mol Physiol. 2011;301(5):L636–644. doi: 10.1152/ajplung.00158.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nials AT, Uddin S. Mouse models of allergic asthma: acute and chronic allergen challenge. Dis Model Mech. 2008;1(4–5):213–220. doi: 10.1242/dmm.000323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lundequist A, Nallamshetty SN, Xing W, et al. Prostaglandin E(2) exerts homeostatic regulation of pulmonary vascular remodeling in allergic airway inflammation. J Immunol. 2010;184(1):433–441. doi: 10.4049/jimmunol.0902835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aronoff DM, Canetti C, Serezani CH, Luo M, Peters-Golden M. Cutting edge: macrophage inhibition by cyclic AMP (cAMP): differential roles of protein kinase A and exchange protein directly activated by cAMP-1. J Immunol. 2005;174(2):595–599. doi: 10.4049/jimmunol.174.2.595. [DOI] [PubMed] [Google Scholar]
- 27.Kaplan MH, Schindler U, Smiley ST, Grusby MJ. Stat6 is required for mediating responses to IL-4 and for development of Th2 cells. Immunity. 1996;4(3):313–319. doi: 10.1016/s1074-7613(00)80439-2. [DOI] [PubMed] [Google Scholar]
- 28.Zheng W, Flavell RA. The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell. 1997;89(4):587–596. doi: 10.1016/s0092-8674(00)80240-8. [DOI] [PubMed] [Google Scholar]
- 29.Melillo E, Woolley KL, Manning PJ, Watson RM, O’Byrne PM. Effect of inhaled PGE2 on exercise-induced bronchoconstriction in asthmatic subjects. Am J Respir Crit Care Med. 1994;149(5):1138–1141. doi: 10.1164/ajrccm.149.5.8173753. [DOI] [PubMed] [Google Scholar]
- 30.Pavord ID, Tattersfield AE. Bronchoprotective role for endogenous prostaglandin E2. Lancet. 1995;345(8947):436–438. doi: 10.1016/s0140-6736(95)90409-3. [DOI] [PubMed] [Google Scholar]
- 31.Sastre B, del Pozo V. Role of PGE2 in asthma and nonasthmatic eosinophilic bronchitis. Mediators Inflamm. 2012;2012:645383. doi: 10.1155/2012/645383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smith WG, Thompson JM, Kowalski DL, McKearn JP. Inhaled misoprostol blocks guinea pig antigen-induced bronchoconstriction and airway inflammation. Am J Respir Crit Care Med. 1996;154(2 Pt 1):295–299. doi: 10.1164/ajrccm.154.2.8756797. [DOI] [PubMed] [Google Scholar]
- 33.Szczeklik A, Mastalerz L, Nizankowska E, Cmiel A. Protective and bronchodilator effects of prostaglandin E and salbutamol in aspirin-induced asthma. Am J Respir Crit Care Med. 1996;153(2):567–571. doi: 10.1164/ajrccm.153.2.8564099. [DOI] [PubMed] [Google Scholar]
- 34.Sestini P, Armetti L, Gambaro G, et al. Inhaled PGE2 prevents aspirin-induced bronchoconstriction and urinary LTE4 excretion in aspirin-sensitive asthma. Am J Respir Crit Care Med. 1996;153(2):572–575. doi: 10.1164/ajrccm.153.2.8564100. [DOI] [PubMed] [Google Scholar]
- 35.Pavord ID, Wong CS, Williams J, Tattersfield AE. Effect of inhaled prostaglandin E2 on allergen-induced asthma. Am Rev Respir Dis. 1993;148(1):87–90. doi: 10.1164/ajrccm/148.1.87. [DOI] [PubMed] [Google Scholar]
- 36.Gauvreau GM, Watson RM, O’Byrne PM. Protective effects of inhaled PGE2 on allergen-induced airway responses and airway inflammation. Am J Respir Crit Care Med. 1999;159(1):31–36. doi: 10.1164/ajrccm.159.1.9804030. [DOI] [PubMed] [Google Scholar]
- 37.Hartert TV, Dworski RT, Mellen BG, Oates JA, Murray JJ, Sheller JR. Prostaglandin E(2) decreases allergen-stimulated release of prostaglandin D(2) in airways of subjects with asthma. Am J Respir Crit Care Med. 2000;162(2 Pt 1):637–640. doi: 10.1164/ajrccm.162.2.9904038. [DOI] [PubMed] [Google Scholar]
- 38.Cohn L, Elias JA, Chupp GL. Asthma: mechanisms of disease persistence and progression. Annu Rev Immunol. 2004;22:789–815. doi: 10.1146/annurev.immunol.22.012703.104716. [DOI] [PubMed] [Google Scholar]
- 39.Sreeramkumar V, Fresno M, Cuesta N. Prostaglandin E2 and T cells: friends or foes? Immunol Cell Biol. 2012;90(6):579–586. doi: 10.1038/icb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Betz M, Fox BS. Prostaglandin E2 inhibits production of Th1 lymphokines but not of Th2 lymphokines. J Immunol. 1991;146(1):108–113. [PubMed] [Google Scholar]
- 41.Woolard MD, Wilson JE, Hensley LL, et al. Francisella tularensis-infected macrophages release prostaglandin E2 that blocks T cell proliferation and promotes a Th2-like response. J Immunol. 2007;178(4):2065–2074. doi: 10.4049/jimmunol.178.4.2065. [DOI] [PubMed] [Google Scholar]
- 42.Church RJ, Jania LA, Koller BH. Prostaglandin E(2) produced by the lung augments the effector phase of allergic inflammation. J Immunol. 2012;188(8):4093–4102. doi: 10.4049/jimmunol.1101873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhou W, Newcomb DC, Moore ML, Goleniewska K, O’Neal JF, Peebles RS., Jr Cyclooxygenase inhibition during allergic sensitization increases STAT6-independent primary and memory Th2 responses. J Immunol. 2008;181(8):5360–5367. doi: 10.4049/jimmunol.181.8.5360. [DOI] [PubMed] [Google Scholar]
- 44.Hashimoto K, Sheller JR, Morrow JD, et al. Cyclooxygenase inhibition augments allergic inflammation through CD4-dependent, STAT6-independent mechanisms. J Immunol. 2005;174(1):525–532. doi: 10.4049/jimmunol.174.1.525. [DOI] [PubMed] [Google Scholar]
- 45.Peebles RS, Jr, Hashimoto K, Sheller JR, et al. Allergen-induced airway hyperresponsiveness mediated by cyclooxygenase inhibition is not dependent on 5-lipoxygenase or IL-5, but is IL-13 dependent. J Immunol. 2005;175(12):8253–8259. doi: 10.4049/jimmunol.175.12.8253. [DOI] [PubMed] [Google Scholar]
- 46.Kunikata T, Yamane H, Segi E, et al. Suppression of allergic inflammation by the prostaglandin E receptor subtype EP3. Nat Immunol. 2005;6(5):524–531. doi: 10.1038/ni1188. [DOI] [PubMed] [Google Scholar]
- 47.Okano M, Fujiwara T, Haruna T, et al. Prostaglandin E(2) suppresses staphylococcal enterotoxin-induced eosinophilia-associated cellular responses dominantly through an E-prostanoid 2-mediated pathway in nasal polyps. J Allergy Clin Immunol. 2009;123(4):868–874. e813. doi: 10.1016/j.jaci.2009.01.047. [DOI] [PubMed] [Google Scholar]
- 48.Okano M, Sugata Y, Fujiwara T, et al. E prostanoid 2 (EP2)/EP4-mediated suppression of antigen-specific human T-cell responses by prostaglandin E2. Immunology. 2006;118(3):343–352. doi: 10.1111/j.1365-2567.2006.02376.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Parker CW, Huber MG, Godt SM. Suppression of IL-4 Production in Murine Lymphocytes by Orally Effective Prostaglandin E(1) Analogs. Am J Ther. 1995;2(10):772–776. doi: 10.1097/00045391-199510000-00007. [DOI] [PubMed] [Google Scholar]
- 50.Tanaka H, Kanako S, Abe S. Prostaglandin E2 receptor selective agonists E-prostanoid 2 and E-prostanoid 4 may have therapeutic effects on ovalbumin-induced bronchoconstriction. Chest. 2005;128(5):3717–3723. doi: 10.1378/chest.128.5.3717. [DOI] [PubMed] [Google Scholar]
- 51.Herrerias A, Torres R, Serra M, et al. Activity of the cyclooxygenase 2-prostaglandin-E prostanoid receptor pathway in mice exposed to house dust mite aeroallergens, and impact of exogenous prostaglandin E2. J Inflamm (Lond) 2009;6:30. doi: 10.1186/1476-9255-6-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Herrerias A, Torres R, Serra M, et al. Subcutaneous prostaglandin E(2) restrains airway mast cell activity in vivo and reduces lung eosinophilia and Th(2) cytokine overproduction in house dust mite-sensitive mice. Int Arch Allergy Immunol. 2009;149(4):323–332. doi: 10.1159/000205578. [DOI] [PubMed] [Google Scholar]
- 53.Katsuyama M, Ikegami R, Karahashi H, Amano F, Sugimoto Y, Ichikawa A. Characterization of the LPS-stimulated expression of EP2 and EP4 prostaglandin E receptors in mouse macrophage-like cell line, J774.1. Biochem Biophys Res Commun. 1998;251(3):727–731. doi: 10.1006/bbrc.1998.9540. [DOI] [PubMed] [Google Scholar]
- 54.Boutaud O, Aronoff DM, Richardson JH, Marnett LJ, Oates JA. Determinants of the cellular specificity of acetaminophen as an inhibitor of prostaglandin H(2) synthases. Proc Natl Acad Sci U S A. 2002;99(10):7130–7135. doi: 10.1073/pnas.102588199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Allmers H. Frequent acetaminophen use and allergic diseases: is the association clear? J Allergy Clin Immunol. 2005;116(4):859–862. doi: 10.1016/j.jaci.2005.07.019. [DOI] [PubMed] [Google Scholar]
- 56.Amberbir A, Medhin G, Hanlon C, Britton J, Venn A, Davey G. Frequent use of paracetamol and risk of allergic disease among women in an Ethiopian population. PLoS One. 2011;6(7):e22551. doi: 10.1371/journal.pone.0022551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Thomsen SF, Kyvik KO, Skadhauge LR, Steffensen I, Backer V. Regular use of non-steroidal anti-inflammatory drugs increases the risk of adult-onset asthma: a population-based follow-up study. Clin Respir J. 2009;3(2):82–84. doi: 10.1111/j.1752-699X.2008.00113.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.