

CONSPECTUS
The use of Au(I) complexes for the catalytic activation of C-C π-bonds has been the subject of intense investigation in the last decade or so. The facile formation of carbon-carbon and carbon-heteroatom bonds facilitated by gold naturally led to efforts to render these transformations enantioselective. Early examples of enantioselective gold-catalyzed transformations have focused on bis(phosphinegold) complexes derived from axially chiral scaffolds. Although these complexes were highly successful in some reactions like cyclopropanation, careful choice of the weakly coordinating ligand (or counterion) was needed to obtain high levels of enantioselectivity for the case of allene hydroamination. These counterion effects led us to use the anion itself as a source of chirality, which was successful in the case of allene hydroalkoxylation. In order to expand the scope of reactions amenable to enantioselective gold catalysis to cycloadditions and other carbocyclization processes, a new class of mononuclear phosphite and phosphoramidite ligands was developed to supplement the previously widely utilized phosphines. Finally carbene ligands, in particular, the acyclic diaminocarbenes, have also been successfully applied to enantioselective transformations.

Introduction
The development of homogeneous gold catalysis in the last decade has been remarkable in its rapidity. As strong yet air- and moisture-tolerant carbophilic Lewis acids, gold complexes have been shown to catalyze transformations involving C-C multiple bonds with unparalleled mildness and selectivity. Although Hg(II) and Tl(III) salts have long been used in the activation of C-C π-bonds, the ability for cationic Au(I) complexes to effect these transformations was poorly recognized. A handful of early reports document the use of Au(III) catalysts for π-activation.1-3 In 1998, Teles and coworkers reported the exceptionally high activity of cationic Au(I)-phosphine complexes for alkyne hydroalkoxylation.4 The cationic Au(I) complexes were generated by protonolysis of the corresponding Au(I)-methyl (eq 1a), although subsequently, it was found that halide abstraction using a silver salt of a non-coordinating anion was a more general approach (eq 1b). Since Teles's initial report, gold catalyzed alkyne hydrofunctionalization has been extended to a variety of nucleophiles, as well as to alkene and allene substrates. Mechanistically, these reactions are believed to proceed by anti attack on a gold-activated C-C multiple bond (a π-complex), followed by protodemetalation of the resultant σ-complex (Scheme 1).
| (1a) |
| (1b) |
Scheme 1.

General mechanistic scheme for gold(I)-catalyzed hydrofunctionalization of C-C multiple bonds.
A second class of reactions, gold-catalyzed skeletal rearrangements, which includes cycloisomerizations, cycloadditions and other carbocyclization reactions, was reported soon after the initial reports of gold-catalyzed hydrofunctionalization. Earlier, Murai and coworkers observed that π-acidic Pt(II/IV) complexes catalyze rearrangements of enyne substrates.5 In 2004, Echavarren and coworkers reported the cationic Au(I)-catalyzed cycloisomerization of 1,6-enynes.6 Conceptually, these carbocyclization reactions are related to the hydrofunctionalization reactions in that initial activation of an alkyne or allene takes place. Attack by a pendant unsaturated C-C bond follows, leading to a diverse array of subsequent rearrangement steps. Later studies have revealed a multitude of pathways for C-C bond formation and the rearrangement steps that follow, with reaction outcome often dependent on properties of the ancillary ligand on gold.7
The rapid rise in interest in gold catalysis has been accompanied by efforts to develop enantioselective variants of gold-catalyzed reactions to further increase the synthetic utility of these transformations.8The gold-catalyzed Aldol-type reaction between aldehydes and isocyanoacetate esters reported by Hayashi, Ito, and Sawamura in 1986 represents the first example of a gold-catalyzed enantioselective reaction.9 Although this reaction has been well studied, it stands apart from more recent developments in the timeline of its development as well as in its activation mode.10 Shortly after their discovery that cationic gold complexes were highly efficient catalysts for 1,6-enyne alkoxycyclization, the Echavarren group made the first steps in rendering this reaction enantioselective through the synthesis of chiral bis(phosphine)digold(I) dichlorides as precatalysts. For one substrate, an enantioselectivity of 94% ee was achieved (eq 2), although all others gave under 60% ee.11
In this Account, we survey the development of catalysts and ligands for enantioselective gold catalysis by the Toste research group as well as related developments by others. We focus on some of the strategies that our group has developed to address the challenges of enantioselective gold(I) catalysis. In many cases, the use of the bisphosphinedigold(I) complexes proved sufficient in achieving high enantioselectivities. In other cases, other strategies needed to be employed, including the tuning the weakly coordinating X-type ligand (or counterion) on gold, the use of chiral phosphate counterions, and the development of other ligand classes besides the bisphosphines. Generally, these strategies serve to augment steric influence beyond the 2-coordinate ligand environment around the reactive gold center. Binuclear complexes allow the second gold center and its associated ligand (or counterion) environment to exert a steric influence. Similarly, the use of a chiral anion (in place or in addition to a chiral ligand) can potentially move chiral information to closer proximity to the reactive center. Finally, the use of mononuclear complexes is possible, but careful design of steric environment is often required to create “walls” that enclose the gold center.
 |
(2) |
Enantioselective reactions catalyzed by bis(phosphine)digold(I) complexes involving gold(I)-carbene intermediates
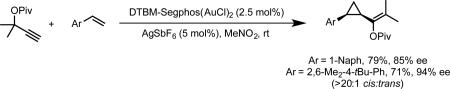
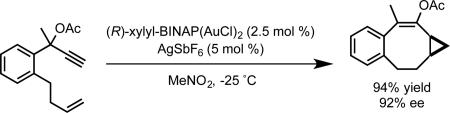
Our initial investigations into enantioselective gold catalysis focused on the development of an enantioselective variant of the gold-catalyzed Conia-ene reaction.12 The lack of success in rendering the gold-catalyzed reaction enantioselective, led us to (a) development the enantioselective palladium-catalyzed variant of this reaction,13 and (b) consider other modes of activation for asymmetric gold catalysis that did not involve addition of a prochiral nucleophile to a gold-activated alkyne. To this end, we were inspired by observations made during the course of our investigation of gold-catalyzed alkene cyclopropanation using propargyl esters as carbene precursors.14 During these studies we noted that the cis-trans diastereomeric ratio of the cyclopropanation product depended on the steric properties of the phosphine ligand on gold. The observed selectivity for the cis isomer was rationalized by a concerted cyclopropanation model, involving the intermediacy of a cationic gold(I)-carbene intermediate, whose bonding and reactivity have subsequently been investigated by our research group and others (Scheme 2).15-18 Given the important steric influence exerted on the diastereoselectivity by the ancillary ligand in this model, we reasoned that it might be possible to use the same steric effect to render the reaction enantioselective. After surveying chiral bisphosphine ligands for gold, we found that DTBM-Segphos afforded product with 81% ee, and with >20:1 cis diasteroselectivity. Several styrene substrates were evaluated giving enantioselectivities ranging from 76-94% ee, with the highest enantioselectivities observed for sterically encumbered substrates (eq 3). The enantioselective cyclopropanation could be extended in an intramolecular sense to generate cyclopropanated medium-sized rings by using xylyl-BINAP as the bisphosphine ligand (eq 4).19
Scheme 2.

Observed diastereoselectivity and mechanistic rationale for gold(I)-catalyzed cyclopropanation.
Given our success with enantioselective cyclopropanation, intermediates, we considered the reaction of related gold(I)-carbenoid intermediates with nucleophiles other than olefins. To this end, we investigated the reactivity of propargylic ester tethered to allylic ethers. As expected, the allylic ether participated in nucleophilic attack of the highly electrophilic gold(I)-carbene intermediate to generate an alkylgold-oxonium species, which undergoes O-to-C allyl transfer to generate a benzopyran bearing an all carbon quaternary stereocenter with excellent enantiocontrol (Scheme 3).18
 |
(3) |
 |
(4) |
Scheme 3.

Proposed mechanism of enantioselective allylic ether rearrangement to generate benzopyrans.
Enantioselective hydrofunctionalizations catalyzed by bis(phosphine)digold(I) complexes
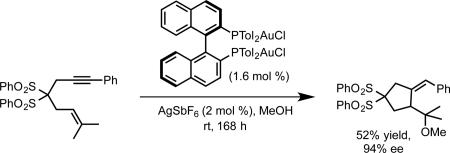
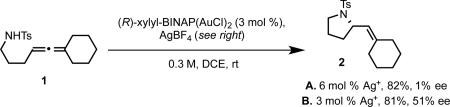
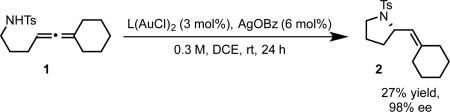
Our success in the development of the enantioselective cyclopropanation led us to consider employing similar bisphosphine gold complexes for asymmetric allene functionalization reactions such as hydroamination.21 The gold catalyzed allene hydroamination had previously been reported by the Krause, Yamamoto, and Widenhoefer groups.22-24 Using a model allene substrate 1, we initially surveyed conditions analogous to those developed for cyclopropanation (2:1 silver salt activator/bisphosphine gold precatalyst, eq 5, A). Under those conditions, hydroamination to give 2 was observed, but with essentially no enantioselectivity. However, using a 1:1 ratio of silver salt to gold precatalyst (eq 5, B) resulted in the formation of product in moderate enantioselectivity.
We rationalized these observations by postulating that the monocationic complex in which one of the gold center remains ligated to chloride catalyzes the hydroamination with moderate enantioselectivity, while the dicationic complex exhibits poor enantioinduction (Scheme 4A-B). We further suspected that the steric properties of this remaining ligand could have a strong impact on enantioselectivity. We investigated bis(phosphine)digold(I) dicarboxylate complexes as potential catalysts, hypothesizing that a catalytically active monocationic species would exist in equilibrium with the inactive coordinatively saturated form, and, at the same time provide a more sterically demanding chiral environment (Scheme 4C). In accord with our hypothesis, the in situ generated benzoate complex (R)-xylyl-BINAP(AuOBz)2 catalyzed the hydroamination with a remarkable increase in enantioselectivity, albeit with poor yield even over extended reaction times (eq 6), likely reflecting the low equilibrium concentration of the active monocationic species. Wishing to reduce the basicity (and ligating ability) of the counterion/ligand, we prepared (R)-xylyl-BINAP(AuOPNB)2 as an isolable complex. As we had hoped, this complex exhibited better catalyst activity while maintaining selectivity, providing 2 in excellent yield and enantioselectivity (eq 7).
 |
(5) |
 |
(6) |
 |
(7) |
Scheme 4.

The effect of counterion/X-type ligand on enantioselectivity of hydroamination.
In work conducted independently and reported simultaneously, Widenhoefer and coworkers developed the hydroalkoxylation of allenes.25 In their report, the counterion on gold was also important, with p-toluenesulfonate (OTs) found to be optimal, and less coordinating anions giving poor enantioselectivities. Although high enantioselectivities were achieved, bulky gemdiphenyl groups on the alkyl chain appeared to be required.
 |
(8) |
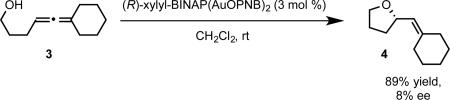
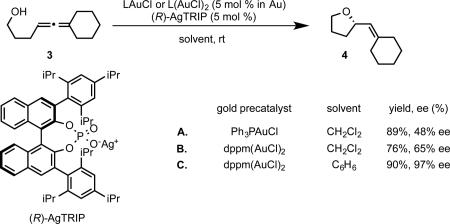
The insights gained from the development of asymmetric allene amination led us to initiate our own studies into hydroalkoxylation. Efforts to apply our earlier reaction conditions to the sterically unconstrained allenol 3 resulted in formation of hydroalkoxylation product with poor enantioselectivity (eq. 8). We conjectured that we could overcome the limitations on enantioinduction posed by the linear geometry of gold(I) complexes by moving the source of chirality to an outersphere counterion. We selected the BINOL-derived chiral phosphate anions to test this idea. Encouragingly, the use of [Ph3PAu+][TRIP−] generated in situ from halide abstraction of Ph3PAuCl by the silver salt of TRIP phosphoric acid (AgTRIP) resulted in the formation of hydroalkoxylation product in moderate ee (eq. 9A). Using the dinuclear achiral catalyst dppm(AuCl)2 (dppm=bis(diphenylphosphino)methane) and switching to a solvent of low polarity (benzene) to favor the formation of a tighter ion pair ultimately led to conditions that afforded hydroalkoxylation production with exceptional enantiocontrol (eq 9B-C).26
 |
(9) |
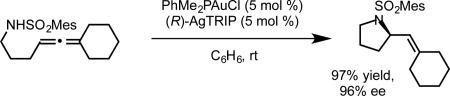
The use of PhMe2PAuCl as precatalyst instead of dppm(AuCl)2 allowed the chiral counterion strategy to be extended to hydroamination (eq 10). Attempts to effect the corresponding hydrocarboxylation of 5 using either chiral bisphosphine ligands or chiral counterions resulted in only low levels of enantioinduction (eq 11, A-B). However, the use of a chiral gold complex and chiral phosphate counterion together allowed the chiral information imparted by each to act in concert. A combination of (S)-BINAP(AuCl)2 and (R)-AgTRIP gave hydrocarboxylation product 6 with good enantioselectivity (82% ee, eq 11, D). A strong matched-mismatched pairing effect between ligand and counterion was observed, and the mismatched combination (R)-BINAP(AuCl)2/(R)-AgTRIP provided nearly racemic product (eq 11, C). The strategies developed for the hydroamination, hydroalkoxylation, and hydrocarboxylation allowed us achieve the enantioselective cyclizations of allenes using other nucleophiles, including hydrazines and both N- and O-terminated hydroxylamines bearing a variety of protecting groups and substitution patterns, to generate enantioenriched pyrazolidines, isoxazolidines, and tetrahydrooxazines.27
 |
(10) |
 |
(11) |
More recently, we sought to extend the synthetic versatility of allene cyclization by achieving catalytic turnover through the means of electrophilic halodemetalation, rather than protodemetalation, to generate haloalkene-substituted heterocyclic products.28 Though superficially similar to hydrofunctionalization, finding a reagent of suitable reactivity proved challenging. Using allene 5 as a model substrate, and the previously developed (R)-xylyl-BINAP(AuOPNB)2 as catalyst, more active reagents like N-iodo- or N-bromosuccinimide exhibited strong background reactivity, and consequently, provided nearly racemic haloamination products (6a, 6b, resp., eq 12 A,B). On the other hand, reaction with N-chlorosuccinimide, which gave the desired chloroamination product 6c in moderate enantioselectivity (76% ee), suffered from competitive protodemetalation to give hydroamination product 6d as a side product (eq 12 C). Reasoning that the succimidate anion formed upon halogen transfer was insufficiently basic to efficiently sequester the proton equivalent generated, we turned to N-haloamides as possible alternatives. Thus, switching to N-bromopyrrolidinone (NBL-5) as the halogenating reagent, excellent enantioselectivity was observed for the bromocyclization of 5, with competitive hydroamination completely suppressed (eq 12 D). While application of standard conditions to carboxylic acid 5 delivered racemic lactone 9, the combination of a chiral ligand and chiral counterion developed earlier afforded halocyclization product with good enantioselectivity and moderate yield (eq 13 A). By switching to N-bromoazepanone (NBL-7) as the brominating reagent, both yield and enantioselectivity were improved substantially (eq 13 B), further illustrating the importance of reagent choice in this transformation.
 |
(12) |
 |
(13) |
Upon the successful implementation of enantioselective heteroatom additions to allenes, we investigated the expansion of this reactivity to carbon nucleophiles. We anticipated that a latent carbon nucleophile in the form of a neighboring cyclopropanol would undergo ring expansion Meerwein-Wagner shift onto an allene to deliver an enantioenriched vinylcyclobutanone. Indeed, under optimized conditions, a variety of allenylcyclopropanols underwent ring expansion to generate diverse vinylcyclobutanones bearing all carbon quaternary stereocenters with excellent enantioselectivity (eq 14).29 The use of NaBARF (sodium tetrakis(3,5-trifluoromethylphenyl)borate) to abstract the chloride ligand from the precatalyst led to higher levels of enantioselectivity. Use of more typical halide abstracting reagents such as AgNTf2 resulted in trace acid formation, and thus, lowered enantioselectivities due to a competitive acid-catalyzed racemic pathway to product formation.
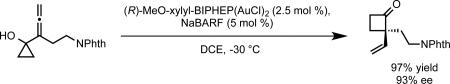 |
(14) |
We further speculated that nucleophilic additions onto C-C multiple bonds might be extended to alkene nucleophiles, provided another nucleophile is present to trap the incipient carbocation. In this way, we envisioned a polycyclization process in which gold would selectively initiate cyclization at the alkyne functionality to the exclusion of olefins also present in the substrate. While it was well known that enynes undergo cycloisomerization reactions in the presence of gold, we posited that polycylization would be favored over competing reactions due to the availability of a low energy Stork-Eschenmoser-type transition state that has been proposed for other cationic polycyclizations.28 Fürstner had previously articulated this possibility while addressing the importance of charge stabilization in gold-catalyzed reactions.15a Indeed, the carboxylic acid terminated 1,6-enyne cyclized in the presence of chiral gold catalyst to afford the 6-exo-dig dicyclization product as a single diastereomer (eq 15a). Under similar conditions, the reactivity could be extended to a tricyclization to yield the tetracyclic product in excellent enantioselectivity as a single diastereomer (eq 15b).31 Additionally, we found that 5-endo-dig dicyclization process could be rendered enantioselectve using related (bisphosphine)gold catalysts.32
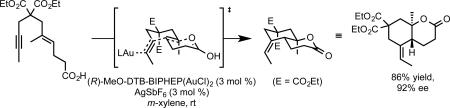 |
(15a) |
 |
(15b) |
Development of chiral phophoramidite and phosphite ligands for gold-catalyzed cycloadditions
In our earlier studies of the reactivity of allene-dienes in the presence of cationic gold complexes, we observed a product distribution strongly dependent on the ancillary ligand on gold. Subjection of allene-diene 10 to Ph3PAuCl/AgSbF6 resulted in the formation of a mixture of formal [4+2] and [4+3] cycloadddition products, 11 and 12.
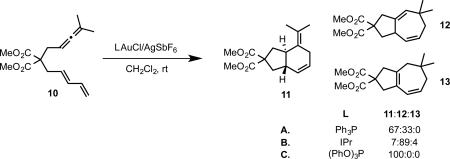 |
(16) |
In contrast, the use of an NHC ligand, IPr (1,3-bis(2,6-diisopropropylphenyl)imidazol-2-ylidene) strongly favored the formation of [4+3] product 12, while a phosphite ligand produced the [4+2] product 11 exclusively (eq 16).33 A subsequent density functional study of this system in collaboration with Goddard and coworkers provided insight into the origin of this strong ligand effect.17 The reaction was predicted to proceed primarily through an initial [4+3] cycloaddition pathway. The resultant gold-carbene intermediate could undergo 1,2-hydrogen shift (Scheme 5, path a) or 1,2-alkyl shift (path b) to provide the [4+3] and [4+2] products, respectively. While the 1,2-alkyl shift was predicted to be relatively insensitive to the nature of the ligand, the computed barrier to 1,2-hydrogen shift was found to be strongly disfavored by phosphite ligands on gold, in agreement with the observed selectivity for the [4+2] product.
Scheme 5.

Proposed mechanistic pathways of allene-diene cyclizations, as supported by DFT calculations.
Given the excellent regioselectivity provided by phosphite ligands, we wished to explore chiral variants of these and electronically similar phosphoramidite ligands to effect the [4+2] cycloaddition enantioselectively.34 Although we13a and others11 had previously reported BINOL-derived mononuclear phosphoramiditegold(I) complexes, reactions catalyzed by these complexes had not exhibited significant enantioselectivity. Due to the 2-coordinate linear geometry of gold, we anticipated substantial difficulty in achieving good enantioselectivity with these mononuclear complexes. Indeed, in our initial studies, the previously reported phosphoramidite complex 14 catalyzed the cycloaddition of allene-dienes 15 and 16 with good [4+2] selectivity, but poor enantioselectivity(eq 17).
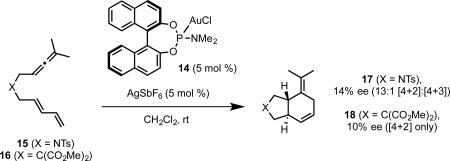 |
(17) |
We turned to the C3-symmetric phosphite ligands first reported by Reetz and coworkers for Rhcatalyzed hydrogenation as an alternative, reasoning that the outward projecting ester moiety would provide an effective chiral environment for linear Au(I).35 Indeed, X-ray crystallographic analysis of phosphitegold complex 19 showed the adamantyl ester groups enclosing the gold center (Figure 1).
Figure 1.
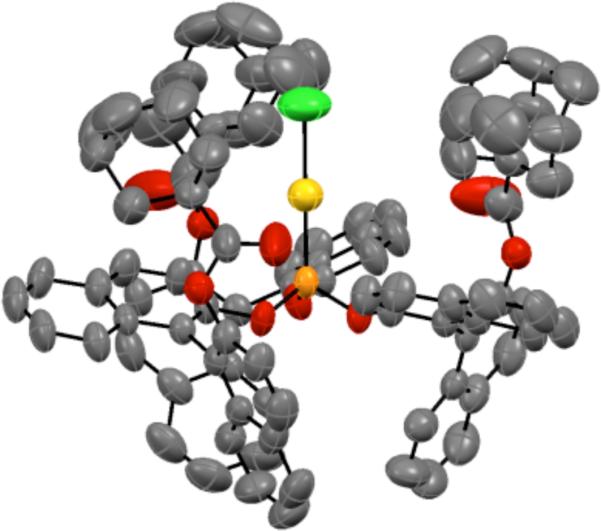
X-ray structure of 19.
In the event, complex 19 catalyzed the [4+2]-cycloaddition of malonate-tethered substrate 16 to give [4+2] cycloadduct 18 with good enantioselectivity (eq 18A). Further optimization using the H8-BINOL derived complex 20 furnished 18 with high regio- and enantioselectivity (eq 18C). Unfortunately, these results could not be extended to the more conformationally unconstrained sulfonamide-tethered substrate 15 using phosphite ligands (eq 18D).
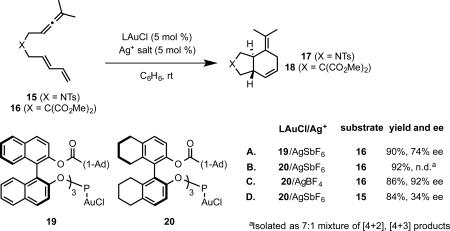 |
(18) |
To address this limitation, we returned to the phosphoramidite ligand system. Examination of the X-ray structure of phosphoramidite complex 14 suggested that we might more effectively enclose the gold center through a combination of substituents on 3, 3’ positions of the binaphthyl and the nitrogen moiety of the phosphoramidite. Furthermore, we anticipated that the modularity of this ligand class would be advantageous toward catalyst optimization. We were encouraged to observe modestly increased enantioselectivity (21% ee vs. 14% ee) by switching from 14 to complex 21 prepared from (S,S)-bis(1-phenylethyl)amine and (S)-BINOL (eq 19B). In contrast, complex 21’ with mismatching amine and binaphthyl chiralities ((S,S)-amine and (R)-BINOL) furnished racemic product (eq 19C). Installing the bulky 1-pyrenyl substituents on the binaphthyl framework, complex 22 catalyzed the [4+2] cycloaddition of sulfonamide 15 with an exceedingly high level of enantiocontrol (eq 19D). The X-ray structure of 22 supported our design principle of enclosing the gold center (Figure 2).
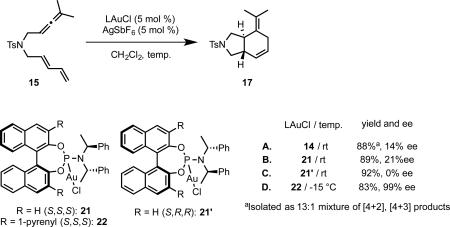 |
(19) |
Figure 2.

X-ray structure of 22.
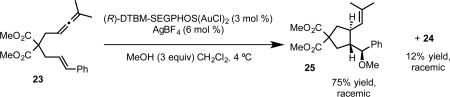
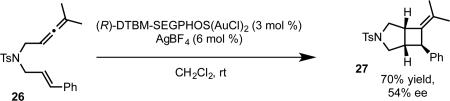
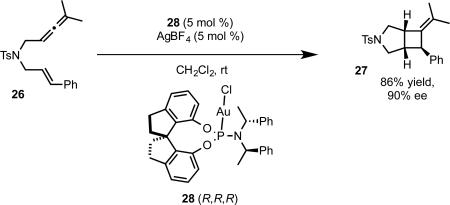
The use of phosphoramidite complexes could be extended to the allene-enes, which afford cis [2+2] cycloaddition product or trans cyclization/addition product in the absence and presence of external nucleophile, respectively.36 While we were able to achieve enantioselectivity with the [2+2] cycloaddition using the bisphosphinedigold complexes, high enantioselectivity was limited to malonate tethered allene-enes. Moreover, the cyclization/addition reaction in the presence of nucleophile gave only racemic product under these conditions (eq 20b).37 A thorough investigation of phosphoramidite structure revealed that a SPINOL (1,1’-spirobiindane-7,7’-diol) derived complex 28 catalyzed the [2+2] cycloaddition of sulfonamide tethered allene-ene 26 with a high level of enantioselectivity (eq 21b).
 |
(20a) |
 |
(20b) |
 |
(21a) |
 |
(21b) |
We next turned to the trans cyclization/addition reaction of 26 in the presence of methanol. Although the previously optimal complex 28 produced methoxycyclization product 29 with low enantioselectivity, 3,3’-bis(1-pyrenyl) substituted 22 developed from the previous study of the [4+3] allene-diene cycloaddition provided product in moderate enantioselectivity. Further improvements in enantioselectivity were achieve by switching to the H8-BINOL scaffold and increasing the bulk on the 3,3’-binaphthyl substituent. Computational investigation in collaboration with Goddard and coworkers indicated the importance of the larger biaryl dihedral angle in the H8-BINOL substituted catalysts. Thus, after successive rounds of catalyst structure optimization, including the use of an electron deficient pyrenyl substituent for better reactivity, we found thatadduct 29 could be obtained in 94% ee using complex 32. Although the scope of this transformation was limited to nitrogen-tethered allene-enes, the generation of pyrrolidines with three contiguous stereocenters imparts substantial synthetic utility to this reaction. As an illustration, we completed the formal total synthesis of (-)-isocynometrine in 86% ee and complete diastereocontrol using a hydroxycyclization as a key transformation (Scheme 6).
Scheme 6.

Optimization of cyclization/alkoxylation reaction and application to the synthesis of (-)-isocynometrine.
Concurrent with our studies of the phosphite and phosphoramidite ligand systems for gold, the Mascareñas group conducted studies of allene-diene cycloaddition on very similar ligand systems.38 High enantioselectivity was achieved using a 9-anthracenyl substituted BINOL phosphoramidite ligand. In addition, computational studies performed by the Mascareñas group reached similar conclusions to ours regarding the origin of the strong preference for [4+2] cycloaddition using π-acidic ligands.
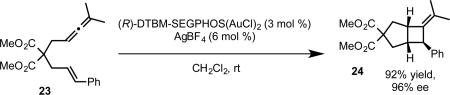
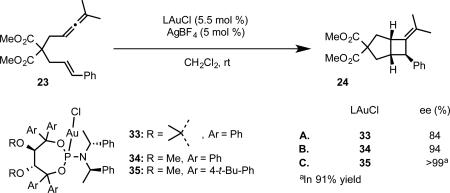
The Fürstner group has also developed a series of chiral phosphoramidite gold complexes based on a TADDOL-related backbone.39 In initial studies, Fürstner and coworkers observed good enantioselectivity in the [2+2] cycloaddition of allenene 23 employing complex 33 as the catalyst. However, by switching from the archetypal isopropylidene acetal group on the TADDOL backbone to acyclic dimethyl ethers, significantly higher enantioselectivities were observed (complexes 34 and 35, eq 22). The improvement in enantioinduction was attributed to the relief of ring-strain on the seven-membered ring, which allows the two axial aryl groups on the TADDOL backbone and axial aryl group on the chiral amine to form a pseudo-three-fold symmetric steric environment around the gold center. Notably, the enantioselective synthesis of clinical candidate GSK 1360707 by gold-catalyzed cycloisomerization was achieved with this catalyst system.40
 |
(22) |
Development of chiral gold-carbene complexes
Despite substantial efforts to design chiral carbene ligands for gold, high levels of enantioselectivity have not been observed for NHC-Au(I) complexes. The difficulty of constructing an enclosing steric environment around the gold center is a major challenge, as the NHC substituents tend to project outward rather than around the gold center. The continued development of chiral carbene ligands for gold would be an important goal, given the strong σ-donating capability of these ligands, to which the different reactivity and selectivity of NHCAu(I) complexes is ascribed. In our attempts to optimize a gold-catalyzed dynamic kinetic transformation of a phenol substituted propargyl ester, only moderate reactivity and enantioselectivity could be achieved with bisphosphinedigold catalysts.41 Since the transformation proceeded through an allene intermediate, and computational evidence by Cavallo and coworkers suggested that NHC ligands may stabilize Au(I)-allene complexes relative to other potential intermediates in propargyl ester rearrangements,42 we explored the use of chiral carbene ligands for this transformation.
Although the chiral Au(I)-NHC reported by Tomioka and coworkers43 was competent in this transformation, the observed enantioselectivity was low. We reasoned that, in analogy to bisphosphinedigold(I) complexes, dinuclearity could be used to construct a more enclosed steric environment. We were drawn to the modularly constructed complexes reported by Espinet and coworkers featuring an intramolecular hydrogen-bond that locks the conformation of the 2-pyridyl substituent of the carbene framework.44 We anticipated that, as in the case of the phosphoramidite complexes, substitution at the 3,3’ position could provide another “wall” around the gold center. This strategy proved effective, with complex 36 bearing a 4-trifluoromethylphenyl substituent allowing for the synthesis of chromenyl pivalates in high enantioselectivity (eq 23). The X-ray structure of 36 was in line with our original suppositions (Figure 3).
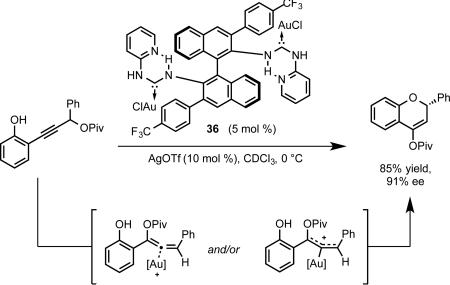 |
(23) |
Figure 3.

X-ray structure of 36.
Subsequently, Slaughter and coworkers reported a mononuclear carbene gold complex which catalyzed the enantioselective alkoxycyclization of alkynylbenzaldehydes. Interestingly, the substituent on the biaryl played a major role in controlling the conformation of the gold complex, with computational and crystallographic studies indicating a gold-arene interaction that is accentuated in the case of electron poor aryl groups.45 More recently, the Mascareñas group has recently reported axially chiral triazoloisoquinolinylidene ligands for gold, which were highly active and selective catalysts for the formal [4+2] cycloadditions of allenamides and dienes.46
Development of bifunctional monophosphine ligands for three-component coupling reaction
Our group was interested in the development of reactions in which the gold catalyst operates in multiple distinct modes of activation. After significant efforts in reaction developments, a three-component coupling was realized in which a terminal alkyne, an aldimine and a tosylisocyanate are combined to form cyclic carbamimidates in the presence of cationic phosphinegold complexes. The transformation is hypothesized to proceed through gold acetylide formation, subsequent addition into iminium, acetylation of the formed amine, and 5-exo-dig cyclization of the carbamimidate onto the gold-activated internal alkyne (Scheme 7).
Scheme 7.

Two activation modes required for three-component coupling.
Previous strategies for the development of an enantioselective variant (use of bisphosphine ligands, monodentate phosphoramidite ligands, or chiral counterions) proved ineffective, with bulky ligands leading to lower product yields and regioselectivity. As an alternative, the use of small chiral bifunctional ligands that may induce asymmetry based on attractive interactions47 rather than steric repulsion was explored. We postulated that a phosphine ligand containing a hydrogen-bond accepting moiety could help order the transition state by bringing the active iminium intermediate into proximity with the phosphine-bound gold acetylide. Extensive screening led to the identification of the tosylurea-containing complex 37 as a competent catalyst for the enantioselective addition of terminal alkynes into aldimines (eq 24). A slight modification of the protocol proved suitable for the three component coupling (eq 25).48
 |
(24) |
 |
(25) |
Summary and Outlook
Our studies in enantioselective catalysis using cationic gold complexes have been intimately related to our development of the reactivity of gold complexes in general. Our observation of ligand effects on the diastereoselectivity and regioselectivity of gold-catalyzed reactions led to insights on the mechanism of these reactions, which provided guidance for finding suitable conditions for obtaining high enantioselectivities. While bisphosphinedigold(I) complexes have proven to be relatively general and versatile for many classes of gold catalyzed reactions, understanding the role of the counterion or weakly coordinating ligand was required for the successful development of these reactions. The use of weakly coordinating chiral counterions as sole or auxiliary source of chirality developed logically from these insights.
More generally, we recognized that additional enantiocontrol elements were often required to mitigate the linear geometry of gold complexes which could place the center of reactivity far away from the chirality source. The use of dinuclearity along with carboxylate ligands or chiral anions constituted one approach. In the case of phosphite, phosphoramidite, and carbene ligands, the idea of building a “wall” around gold proved to be a more general strategy. Although great strides have been made, many challenges await for the development of enantioselective gold catalysis. Notably, carbocyclization reactions continue to pose challenges with respect to cyclization mode as well as enantioselectivity. In many cases, enantioselective versions remain limited in scope. Intermolecular reactivity has seen slower development in gold catalysis, and enantioselective variants are especially rare. Finally, effective enantioselective catalysis that relies on the higher oxidations of gold (Au(II) or Au(III)) have yet to be realized.
CONSPECTUS.
During the past decade, the use of Au(I) complexes for the catalytic activation of C-C π-bonds has been investigated intensely. Over this time period, the development of homogeneous gold catalysis has been extraordinarily rapid and has yielded a host of mild and selective methods for the formation of carbon-carbon and carbon-heteroatom bonds. The facile formation of new bonds facilitated by gold naturally led to efforts toward rendering these transformations enantioselective.
In this Account, we survey the development of catalysts and ligands for enantioselective gold catalysis both by our research group as well as related work by others. We also discuss some of our strategies to address the challenges of enantioselective gold(I) catalysis. Early on, our work with enantioselective gold-catalyzed transformations focused on bis(phosphinegold) complexes derived from axially chiral scaffolds. Although these complexes were highly successful in some reactions like cyclopropanation, the careful choice of the weakly coordinating ligand (or counterion) was necessary to obtain high levels of enantioselectivity for the case of allene hydroamination. These counterion effects led us to use the anion itself as a source of chirality, which was successful in the case of allene hydroalkoxylation. In general, these tactics enhance the steric influence around the reactive gold center beyond the 2-coordinate ligand environment. The use of binuclear complexes allowed us to use the second gold center and its associated ligand (or counterion) to exert a further steric influence. In a similar vein, we employed a chiral anion (in place or in addition to a chiral ligand) to move the chiral information closer to the reactive center.
In order to expand the scope of reactions amenable to enantioselective gold catalysis to cycloadditions and other carbocyclization processes, we also developed a new class of mononuclear phosphite and phosphoramidite ligands to supplement the previously widely utilized phosphines. However, we needed to judiciously design the steric environment to create “walls” that enclose the gold center. We also successfully applied these same considerations were also to the development of binuclear carbene ligands for gold. Finally, we describe the design of bifunctional urea-monophosphine ligands used in a gold-catalyzed three component coupling.
ACKNOWLEDGMENT
We are gratefully acknowledge the NIHGMS (RO1 GM073932) for funding this work. We thank Takasago International Corporation (Segphos ligands), Solvias (Biphep ligands) and Johnson Matthey (AuCl3) for their generation donations. We are greatly indebted to our coworkers; without their creativity, insights and efforts, the work highlighted in this account would not have been possible.
Biography
Yi-Ming Wang was born in Shanghai, China and grew up in Boulder, Colorado. He graduated with an A.B./A.M. degree in chemistry/physics and mathematics from Harvard University in 2008 after conducting research in the group of Prof. Andrew Myers. He did his graduate studies under the supervision of Prof. F. Dean Toste at the University of California, Berkeley and obtained his Ph.D. in 2013. He is currently pursuing postdoctoral studies in the laboratory of Prof. Stephen Buchwald at the Massachusetts Institute of Technology.
Aaron D. Lackner received a B.A. degree in chemistry from Carleton College in Northfield, Minnesota in 2006. After two years working in Merck Research Laboratories in Rahway, New Jersey, he carried out doctoral research under the supervision of Prof. F. Dean Toste at the University of California, Berkeley. He obtained his Ph.D. in 2013. He is currently a postdoctoral fellow in the laboratory of Prof. Alois Fürstner at the Max-Planck-Institut für Kohlenforschung in Mülheim, Germany.
F. Dean Toste was born in Terceira, Azores, Portugal but soon moved to Toronto, Canada. He obtained a BSc and MSc from the University of Toronto and his Ph.D. from Stanford University in 2000 from Professor Barry M. Trost. Following postdoctoral research at the California Institute of Technology in the laboratory of Professor Robert H. Grubbs, he joined the faculty at the University of California, Berkeley in 2002. His research interests are in catalysis, in particular homogenous catalysis, and its application to chemical synthesis.
Footnotes
The authors declare no competing financial interest
REFERENCES
- 1.Norman ROC, Parr WJE, Thomas CB. Gold(III) as an Electrophilic Oxidant of Alkenes. J. Chem. Soc., Perkin Trans. 1976;1:811–817. [Google Scholar]
- 2.Norman ROC, Parr WJE, Thomas CB. The Reactions of Alkynes, Cyclopropanes, and Benzene Derivatives with Gold(III). J. Chem. Soc., Perkin Trans. 1976;1:1983–1987. [Google Scholar]
- 3.Fukuda Y, Utimoto K. Effective Transformation of Unactivated Alkynes into Ketones or Acetals with a Gold(III) Catalyst. J. Org. Chem. 1991;56:3729–3731. [Google Scholar]
- 4.Teles JH, Brode S, Chabanas M. Cationic Gold(I) Complexes: Highly Efficient Catalysts for the Additions of Alcohols to Alkynes. Angew. Chem. Int. Ed. 1998;37:1415–1418. doi: 10.1002/(SICI)1521-3773(19980605)37:10<1415::AID-ANIE1415>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 5.Chatani N, Furukawa N, Sakurai H, Murai S. PtCl2-Catalyzed Conversion of 1,6-and 1,7-Enynes to 1-Vinylcycloalkenes. Anomalous Bond Connection in Skeletal Reorganization of Enynes. Organometallics. 1996;15:901–903. [Google Scholar]
- 6.Nieto-Oberhuber C, Muñoz MP, Buñuel E, Nevado C, Cárdenas DJ, Echavarren AM. Cationic Gold(I) Complexes: Highly Alkynophilic Catalysts for the exo- and endo-Cyclization of Enynes. Angew. Chem. Int. Ed. 2004;43:2402–2406. doi: 10.1002/anie.200353207. [DOI] [PubMed] [Google Scholar]
- 7.Jiménez-Núñez E, Echavarren AM. Molecular Diversity Through Gold Catalysis with Alkynes. Chem. Commun. 2007:333–346. doi: 10.1039/b612008c. [DOI] [PubMed] [Google Scholar]
- 8.Reviews on enantioselective gold catalysis [Google Scholar]; a Pradal A, Toullec PY, Michelet V. Recent Developments in Asymmetric Catalysis in the Presence of Chiral gold complexes. Synthesis. 2011;10:1501–1514. [Google Scholar]; b Sengupta S, Shi X. Recent Advances in Asymmetric Gold Catalysis. ChemCatChem. 2010;2:609–619. [Google Scholar]
- 9.Ito Y, Sawamura M, Hayashi T. Catalytic Asymmetric Aldol Reaction: Reaction of Aldehydes with Isocyanoacetate Catalyzed by a Chiral Ferrocenylphosphine-Gold(I) Complex. J. Am. Chem. Soc. 1986;108:6405–6406. [Google Scholar]
- 11.For related use of gold(I) complexes as σ-acid (rather than π-acid) catalysts, see Melhado AD, Amarante GW, Wang ZJ, Luparia M, Toste FD. Gold(I)-Catalyzed Diastereo- and Enantioselective 1,3-Dipolar Cycloaddition and Mannich Reactions of Azlactones. J. Am. Chem. Soc. 2011;133:3517–3527. doi: 10.1021/ja1095045. for use of gold(I) complexes for Lewis acid activation of Brønsted acids: see Cheon CH, Kanno O, Toste FD. Chiral Brønsted Acid from a Cationic Gold(I) Complex: Catalytic Enantioselective Protonation of Silyl Enol Ethers of Ketones. J. Am. Chem. Soc. 2011;133:13248–13252. doi: 10.1021/ja204331w.Muñoz MP, Adrio J, Carretero JC, Echavarren AM. Ligand Effects in Gold- and Platinum-Catalyzed Cyclization of Enynes: Chiral Gold Complexes for Enantioselective Alkoxycyclization. Organometallics. 2005;24:1293–1300.
- 12.Kennedy-Smith JJ, Staben ST, Toste FD. Gold(I)-Catalyzed Conia-Ene Reaction of β-Ketoesters with Alkynes. J. Am. Chem. Soc. 2004;126:4526–4527. doi: 10.1021/ja049487s. [DOI] [PubMed] [Google Scholar]
- 13.a Corkey BK, Toste FD. Catalytic Enantioselective Conia-Ene Reaction. J. Am. Chem. Soc. 2005;127:17168–17169. doi: 10.1021/ja055059q. [DOI] [PubMed] [Google Scholar]; b Corkey BK, Toste FD. Palladium-Catalyzed Enantioselective Cyclization of Silyloxy-1,6-Enynes. J. Am. Chem. Soc. 2006;129:2764–2765. doi: 10.1021/ja068723r. [DOI] [PubMed] [Google Scholar]; c Corkey BK, Heller ST, Wang Y-M, Toste FD. Tetrahedron. 2013;69:5640–5646. doi: 10.1016/j.tet.2013.02.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johansson MJ, Gorin DJ, Staben ST, Toste FD. Gold(I)-Catalyzed Stereoselective Olefin Cyclopropanation. J. Am. Chem. Soc. 2005;127:17616–17617. doi: 10.1021/ja0552500. [DOI] [PubMed] [Google Scholar]
- 15.a Fürstner A, Morency L. On the Nature of the Reactive Intermediates in Gold-Catalyzed Cycloisomerization Reactions. Angew. Chem. Int. Ed. 2008;47:5030–5033. doi: 10.1002/anie.200800934. [DOI] [PubMed] [Google Scholar]; b Seidel G, Mynott R, Fürstner A. Elementary Steps of Gold Catalysis: NMR Spectroscopy Reveals the Highly Cationic Character of a “Gold Carbenoid”. Angew Chem. Int. Ed. 2009;48:2510–2513. doi: 10.1002/anie.200806059. [DOI] [PubMed] [Google Scholar]
- 16.Benitez D, Shapiro ND, Tkatchouk E, Wang Y, Goddard WA, Toste FD. A Bonding Model for Gold(I) Carbene Complexes. Nature Chem. 2009;1:482–486. doi: 10.1038/nchem.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Benitez D, Tkatchouk E, Gonzalez AZ, Goddard WA, Toste FD. On the Impact of Steric and Electronic Properties of Ligands on Gold(I)-Catalyzed Cycloaddition Reaction. Org. Lett. 2009;11:4798–4801. doi: 10.1021/ol9018002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.López-Carrillo V, Huguet N, Mosquera Á, Echavarren AM. Nature of the Intermediates in Gold(I)-Catalyzed Cyclizations of 1,6-Enynes. Chem. Eur. J. 2011;17:10972–10978. doi: 10.1002/chem.201101749. [DOI] [PubMed] [Google Scholar]
- 19.Watson IDG, Ritter S, Toste FD. Asymmetric Synthesis of Medium-Sized Rings by Intramolecular Au(I)-Catalyzed Cyclopropanation. J. Am. Chem. Soc. 2009;131:2056–2057. doi: 10.1021/ja8085005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Uemura M, Watson IDG, Katsukawa M. Gold(I)-Catalyzed Enantioselective Synthesis of Benzopyrans via Rearrangement of Allylic Oxonium Intermediates. J. Am. Chem. Soc. 2009;131:3464–3465. doi: 10.1021/ja900155x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lalonde RL, Sherry BD, Kang EJ, Toste FD. Gold(I)-Catalyzed Enantioselective Intramolecular Hydroamination of Allenes. J. Am. Chem. Soc. 2007;129:2452–2453. doi: 10.1021/ja068819l. [DOI] [PubMed] [Google Scholar]
- 22.Morita N, Krause N. Gold Catalysis in Organic Synthesis: Efficient Cycloisomerization of α-Aminoallenes to 3-Pyrrolines. Org. Lett. 2004;6:4121–4123. doi: 10.1021/ol0481838. [DOI] [PubMed] [Google Scholar]
- 23.Nishina N, Yamamoto Y. Gold-Catalyzed Intermolecular Hydroamination of Allenes with Arylamines and Resulting High Chirality Transfer. Angew. Chem. Int. Ed. 2006;45:3314–3317. doi: 10.1002/anie.200600331. [DOI] [PubMed] [Google Scholar]
- 24.Zhang Z, Liu C, Kinder RE, Han X, Qian H, Widenhoefer RA. Highly Active Au(I) Catalyst for the Intramolecular exo-Hydrofunctionalization of Allenes with Carbon, Nitrogen, and Oxygen Nucleophiles. J. Am. Chem. Soc. 2006;128:9066–9073. doi: 10.1021/ja062045r. [DOI] [PubMed] [Google Scholar]
- 25.Zhang Z, Widenhoefer RA. Gold(I)-Catalyzed Intramolecular Enantioselective Hydroalkoxylation of Allenes. Angew. Chem. Int. Ed. 2007;46:283–285. doi: 10.1002/anie.200603260. [DOI] [PubMed] [Google Scholar]
- 26.Hamilton GL, Kang EJ, Mba M, Toste FD. A Powerful Chiral Counterion Strategy for Asymmetric Transition Metal Catalysis. Science. 2007;317:496–499. doi: 10.1126/science.1145229. [DOI] [PubMed] [Google Scholar]
- 27.Lalonde R, Wang ZJ, Mba M, Lackner AD, Toste FD. Gold(I)-Catalyzed Enantioselective Synthesis of Pyrazolidines, Isoxazolidines, and Tetrahydrooxazines. Angew. Chem. Int. Ed. 2010;49:598–601. doi: 10.1002/anie.200905000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miles DH, Veguillas M, Toste FD. Gold(I)-Catalyzed Enantioselective Bromocyclization Reactions of Allenes. Chem. Sci. 2013;4:3427–3431. [Google Scholar]
- 29.Kleinbeck F, Toste FD. Gold(I)-Catalyzed Enantioselective Ring Expansion of Allenylcyclopropanols. J. Am. Chem. Soc. 2009;131:9178–9179. doi: 10.1021/ja904055z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.a Stork G, Burgstahler AW. The Stereochemistry of Polyene Cyclization. J. Am. Chem. Soc. 1955;77:5068–5077. [Google Scholar]; b Eschenmoser A, Ruzicka L, Jeger O, Arigoni D. Zur Kenntnis der Triterpene. 190. Mitteilung. Eine stereochemische Interpretation der biogenetischen Isoprenregel bei den Triterpenen. Helv. Chim. Acta. 1955;38:1890–1904. [Google Scholar]
- 31.Sethofer SG, Mayer T, Toste FD. Gold(I)-Catalyzed Enantioselective Polycyclization Reactions. J. Am. Chem. Soc. 2010;132:8276–8277. doi: 10.1021/ja103544p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brazeau JF, Zhang S, Colomer I, Corkey BK, Toste FD. Enantioselective Cyclization of Silyloxyenynes Catalyzed by Cationic Metal Phosphine Complexes. J. Am. Chem. Soc. 2012;134:2742–2749. doi: 10.1021/ja210388g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mauleón P, Zeldin RM, González AZ, Toste FD. Ligand-Controlled Access to [4+2] and [4+3] Cycloadditions in Gold-Catalyzed Reactions of Allene-Dienes. J. Am. Chem. Soc. 2009;131:6348–6349. doi: 10.1021/ja901649s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.González AZ, Toste FD. Gold(I)-Catalyzed Enantioselective [4+2]-Cycloaddition of Allene-Dienes. Org. Lett. 2010;12:200–203. doi: 10.1021/ol902622b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reetz MT, Guo H, Ma J-A, Goddard R, Mynott RJ. Helical Triskelion Monophosphites as Ligands in Asymmetric Catalysis. J. Am. Chem. Soc. 2009;131:4136–4142. doi: 10.1021/ja809297a. [DOI] [PubMed] [Google Scholar]
- 36.González AZ, Benitez D, Tkatchouk E, Goddard WA, Toste FD. Phosphoramidite Gold(I)-Catalyzed Diastereo- and Enantioselective Synthesis of 3,4-Substituted Pyrrolidines. J. Am. Chem. Soc. 2011;133:5500–5507. doi: 10.1021/ja200084a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luzung MR, Mauleón P, Toste FD. Gold(I)-Catalyzed [2+2]-Cycloaddition of Allenenes. J. Am. Chem. Soc. 2007;129:12402–12403. doi: 10.1021/ja075412n. [DOI] [PubMed] [Google Scholar]
- 38.Alonso I, Trillo B, López F, Montserrat S, Ujacque G, Castedo L, Lledós A, Mascareñas JL. Gold-Catalyzed [4C+2C] Cycloadditions of Allenedienes, including an Enantioselective Version with New Phosphoramidite-Based Catalysts: Mechanistic Aspects of the Divergence between [4C+3C] and [4C+2C] Pathways. J. Am. Chem. Soc. 2009;131:13020–13030. doi: 10.1021/ja905415r. [DOI] [PubMed] [Google Scholar]
- 39.Teller H, Corbet M, Mantilli L, Gopakumar G, Goddard R, Thiel W, Fürstner A. One-Point Binding Ligands for Asymmetric Gold Catalysis: Phosphoramidites with a TADDOL-Related but Acyclic Backbone. J. Am. Chem. Soc. 2012;134:15331–15342. doi: 10.1021/ja303641p. [DOI] [PubMed] [Google Scholar]
- 40.Teller H, Fürstner A. Concise Synthesis of the Antidepressive Drug Candidate GSK1360707 by a Highly Enantioselective Gold-Catalyzed Enyne Cycloisomerization Reaction. Chem. Eur. J. 2011;17:7764–7767. doi: 10.1002/chem.201101346. [DOI] [PubMed] [Google Scholar]
- 41.Wang Y-M, Kuzniewski CN, Rauniyar V, Hoong C, Toste FD. Chiral (Acyclic Diaminocarbene)Gold(I)-Catalyzed Dynamic Kinetic Asymmetric Transformation of Propargyl Esters. J. Am. Chem. Soc. 2011;133:12972–12975. doi: 10.1021/ja205068j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Correa A, Marion N, Fensterbank L, Malacria M, Nolan SP, Cavallo L. Golden Carousel in Catalysis: The Cationic Gold/Propargyl Ester Cycle. Angew. Chem. Int. Ed. 2008;47:718–721. doi: 10.1002/anie.200703769. [DOI] [PubMed] [Google Scholar]
- 43.Matsumoto Y, Selim KB, Nakanishi H, Yamada K, Yamamoto Y, Tomioka K. Chiral Carbene Approach to Gold-Catalyzed Asymmetric Cyclization of 1,6-Enynes. Tetrahedron Lett. 2010;51:404–406. [Google Scholar]
- 44.Bartolomé C, García-Cuadrado D, Ramiro Z, Espinet P. Synthesis and Catalytic Activity of Gold Chiral Nitrogen Acyclic Carbenes and Gold Hydrogen Bonded Heterocyclic Carbenes in Cyclopropanation of Vinyl Arenes and in Intramolecular Hydroalkoxylation of Allenes. Inorg. Chem. 2010;49:9758–9764. doi: 10.1021/ic101059c. [DOI] [PubMed] [Google Scholar]
- 45.Handa S, Slaughter LM. Enantioselective Alkynylbenzaldehyde Cyclizations Catalyzed by Gold(I) Acyclic Diaminocarbene Complexes Containing Weak Au-Arene Interactions. Angew. Chem. Int. Ed. 2012;51:2912–2915. doi: 10.1002/anie.201107789. [DOI] [PubMed] [Google Scholar]
- 46.Francos J, Grande-Carmona F, Faustino H, Iglesia-Sigüenza J, Díez E, Alonso I;, Fernández R, Lassaletta JM, López F, Mascareñas JL. Axially Chiral Triazoloquinolin-3-ylidene Ligands in Gold(I)-Catalyzed Asymmetric Intermolecular (4+2) Cycloadditions of Allenamides and Dienes. J. Am. Chem. Soc. 2012;134:14322–14325. doi: 10.1021/ja3065446. [DOI] [PubMed] [Google Scholar]
- 47.Knowles RR, Jacobsen EN. Attractive Noncovalent Interactions in Asymmetric Catalysis: Links between Enzymes and Small Molecule Catalysts. Proc. Natl. Acad. Sci. USA. 2010;107:20678–20875. doi: 10.1073/pnas.1006402107. for a recent example of multi-point binding in enantioselective gold catalysis: see Bandini M, Bottoni A, Chiarucci M, Cera G, Miscione GP. J. Am. Chem. Soc. 2012;134:20690–20700. doi: 10.1021/ja3086774. and references therein.
- 48.Cambpell MJ, Toste FD. Enantioselective Synthesis of Cyclic Carbamimidates via a Three-component Reaction of Imines, Terminal Alkynes, and p-Toluenesulfonylisocyanate Using a Monophosphine Gold(I) Catalyst. Chem. Sci. 2011;2:1369–1378. doi: 10.1039/C1SC00160D. [DOI] [PMC free article] [PubMed] [Google Scholar]