

Abstract
In a search for novel agents that boost the anti-neoplastic effects of polo-like kinase 1 (PLK1) inhibitor volasertib, we found that a sepantronium and volasertib combination at the nano mole concentration potently inhibited growth of various non-small cell lung cancer (NSCLC) cell lines than either drug alone in vitro. Combination use of volasertib with sepantronium inhibited adaptation of cells to polo arrest. Addition of sepantronium to volasertib prevented accumulation of survivin and cyclin B protein at a concentration causing no appreciable survivin down regulation. Sepantronium induced cell cycle arrest from G1 or G2/M phase. Further studies demonstrated DNA damage of cancer cells when they are treated with sepantronium, which is evidenced by induction of phospho-γH2AX. In line with induction of a DNA damage response in cancer cells, known DNA damage response sensors and transducers ATM, ATR, CHK1, CHK2, p53 are phosphorylated following drug treatment. Meanwhile, expression of CDKN1A, BAX and XRCC5 are induced at the mRNA level as determined by quantitative real time PCR. A single cell electrophoresis assay (Comet assay) of cells treated with sepantronium revealed severe DNA strand breaks. M-phase arrest does not increase the lethality of DNA damage by sepantronium as compared to G1 phase arrest. Knock down of survivin did not cause DNA damage. Hence, sepantronium is a DNA damaging agent that synergizes with volasertib and down-regulation of survivin is likely the consequence of DNA damage induced by sepantronium.
Keywords: Sepantronium, volasertib, DNA damage, lung cancer, synergy
Introduction
Deregulation in multiple signaling pathways exists in cancer cells [1]. Collectively, they contribute to the expression of phenotypes characteristic of cancers known as the hallmarks of cancer [2]. Although specific agents that target only one pathway usually have lower toxicity, such agents have lower response rate due to existence in cancer cells of redundant pathway(s) that provide compensation for the loss. Even the cancer cells respond initially to such agents, they usually fail quickly due to development of secondary resistance because cancer cells are genetically unstable [3]. To overcome resistance, drugs with distinct molecular mechanisms of action were used in combination in lung cancer therapy successfully. As a result, platinum based doublet became the first line therapy in the metastatic settings [4-7]. Unfortunately, the overall response rate to platinum based chemo is low, and patients do not benefit from the same beyond six cycles (18 weeks) even in responders [8]. Combination chemotherapies are known to have serious side effects such as renal failure, peripheral neuropathy etc. This is in the large part due to the fact that chemo agents cannot distinguish between cancer and normal cells. Side effects show up as part of the collateral damage. Many patients could not receive the standard of care because too many lung cancer patients are elderly (median age 70) and sick [9]. Therefore new drug combinations that target multiple hallmarks of cancer with distinct mechanism of action are needed to overcome resistance and minimize treatment toxicity in lung cancer therapy. Combination therapies were successfully used to overcome cross-resistance in both leukemia and solid tumor chemotherapy, as well as to treat HIV infection disease [10].
PLK1 is an essential cell cycle kinase [11]. Inactivation of PLK1 cause G2/M arrest and blocks mitotic reentry/adaptation after DNA damage checkpoint arrest [12]. Previous studies have shown that PLK1 is overexpressed in cancer cells [13], and suppressing the kinase activity of PLK1 with PLK1 inhibitor was as effective as down regulation of PLK1 mRNA or protein expression by shRNA. Cells exposed to PLK1 inhibitor enter into a “polo arrest” [14] with elevated phospho histone H3 levels and aberrant spindle formation before going into apoptosis. As the earlier version of small molecule dihydroperidinone derivative PLK1 inhibitor targeting the Polo Box Domain (PDB), BI2536 was highly selective and potent (IC50 0.83 nm/L). This enabled targeting only some but not all the Plks. In a panel of 64 kinases tested, BI2536 exhibited 10,000 fold more selectivity towards PLK1 compared to 63 other kinases [15]. In animal models, it inhibited mouse xenograft tumor of human lung cancer (A549 and NCI-H460). Because the microtubules are not the target, it does not cause peripheral neuropathy like a typical taxane or vincristine. So it is well tolerated in phase 1 studies and the dose limiting toxicities (DLT) is hematological and all reversible [16]. Single agent was tested in three phase 2 trials in patients with NSCLC, head and neck cancer and pancreatic cancer [17-19]. These were either modestly effective or not effective at all. However, BI 2536, used in combination with pemetrexed resulted in 2 partial responses and 54% stable disease in 41 patients in a phase 1 clinical trial [20]. These patients were all previously treated and had advanced or metastatic lung cancer, thus rising hope that combination treatment with other agents might be the way to go. Volasertib, also known as BI6727 [21] represents the new generation of PLK1 inhibitor. It has a large volume distribution and a long half-life in mouse compared to the previous generation thus can be given intravenously or orally. In a phase 1 clinical dose-escalation trial [22], volasertib was found to be safe and had signs of efficacy. The dose limiting toxicities are again hematological which were all reversible.
First reported in 2007 [23], sepantronium, also known as YM155 was discovered by screening a drug library for molecules that suppressed luciferase activity under a 1 kb survivin promoter. Preclinical studies showed that sepantronium suppressed survivin expression in a dose and time dependent manner, causing apoptosis in a broad range of cancer cell lines including lung cancer, induced tumor regression in animal models and rapid distribution into the tissue with a 20 fold increase in tumors. Despite subsequent favorable report in two phase 1 clinical trials showing well tolerance [23,24], the performance of single agent in four phase 2 trials in patients with NSCLC [25], melanoma [26], prostate cancer [27] as well as diffused large B cell lymphoma [28] in patients failing one or more lines of chemotherapy were disappointing. Combination use with other drugs is considered highly desirable.
For agents that are considered for use in a combination, one of the biggest concerns is added toxicity. A defined mechanism of action for each drug is essential in choosing the partners for synergy and lower toxicities. The mechanism of action of sepantronium up until now however is still elusive [29,30], hampering efforts in the discovery of a rational partner. In a search for drug combination(s) that might boost the efficacy of volasertib, overcome resistance without adding significant toxicity, we found that sepantronium synergizes with volasertib in NSCLC cell killing. Detailed studies have characterized sepantronium as a DNA damaging agent that causes DNA strand breaks in contrast to the well accepted notion that sepantronium is a survivin inhibitor. Thus, sepantronium is a DNA damaging agent that synergizes with volasertib in NSCLC. DNA damage caused by sepantronium is not dependent on survivin down-regulation.
Materials and methods
Cell culture, proliferation assays
All cell lines (H1299, H661 and H2228) were purchased from ATCC and cultured in RPMI 1640 (Invitrogen) with 5% FBS (Hyclone), at 37°C in 10% CO2. For drug treatments, 5,000-8,000 cells/well, dependent on the cell lines, were seeded in 96-well plates and incubated overnight. Cells were then treated with either DMSO (control), sepantronium, volasertib or sepantronium and volasertib combination at various concentrations. Cell viability was determined using the Cell Titer 96 Aqueous One Solution Cell Proliferation Assay (Promega Inc). Cytotoxic single drug and combination effects of sepantronium and volasertib were assessed with 6 different concentrations (1, 3, 10, 30, 100 and 300, and 1/8 to 8 x IC50Sep/IC50Vol). Each concentration was tested in 6 duplicate wells and repeated for total of three times. Combination Indices (CIs) were calculated using CalcuSyn software (Biosoft, Cambridge, UK) based on the Chou-Talalay method. This method defines synergy as CI<1, additive effects as CI=1 and antagonism as CI>1 [31].
Cell apoptosis assay and cell cycle analysis
For analysis of apoptosis, unsynchronized cells or cells synchronized with 200 μM of mimosine for 24 hours or 50 nM of nocodazole for 16 hour were treated with either DMSO or different drugs/drug combinations for up to 72 hours. The cells were then washed with PBS, stained with annexin V-APC and 7-AAD (BD Biosciences) following the manufacturer’s instruction. Appearance of Annexin V and 7-AAD in flow cytometric analysis indicated onset of apoptosis. Cell cycle analysis was performed using standard flow cytometry procedures following propidium iodide staining as described previously [32]. Cells were analyzed using a LSRII flow cytometer. All flow cytometry data were analyzed using FlowJo software (Tree Star, Ashland, OR).
Western blot analyses and antibodies
Proteins were isolated using standard procedures. Whole-cell lysates containing 50 μg of proteins, were separated by SDS-PAGE and immunoblotted with the following specific antibodies. Antibodies for survivin, p53 or phosphorylated p53, H2AX, ATM, CHK1, ATR, CHK2 were purchased from Cell Signaling Technology and beta-actin from Sigma. Antibody for p21cip1, cyclinD1, cyclin D3, p27kip1, p18 were included in the Cell Cycle Sampler Kit (Cell Signaling). Antibodies were used at 1:1,000 as recommended by the manufacturer. Immunoblot was performed using standard protocols as described previously [33].
Immunofluorescence staining
Immunocytochemistry analysis was performed as described previously [32]. Briefly, the cells were grown on sterile glass coated coverslip, and treated with DMSO, various concentration of drugs or drug concentration for indicated length of time. Cells were fixed and permeabilized. Cells were first blocked with 3% ovalbumin for 30 min at 25°C. They were then stained with primary antibody (e.g. H2AX or anti-α-tubulin) at 4°C overnight. The cells were then washed with PBS and incubated further with the Cy2-conjugated goat anti-rabbit IgG or Cy3-conjugated donkey anti-mouse IgG (Jackson ImmunoResearch) secondary antibody (1:200) for 30 min at 37°C. After two more washings, the slides were counterstained with Hoechst 33342 DNA dye before mounting. Confocal images of Cy3 fluorescence were acquired using Plan-Apochromat × 63/1.4 oil objective in a Zeiss LSM 510 confocal microscope using Zen2009 software.
Molecular analyses
Total RNA was isolated using TRIzol (Invitrogen) reagent, and digested with RNAse free DNase to eliminate genomic DNA contamination. Reverse-transcription was achieved using a SuperScript first-strand synthesis system (Invitrogen), and amplified by PCR or quantitated by real-time RT-PCR using conditions described previously [34]. The PCR primers used for amplifying CDKN1A are 5’cctgcccaagctctaccttc3’ for the sense strand and 5’ttccaggactgcaggcttcc3’ for the antisense strand respectively. For amplification of BAX, the primers used for the sense strand is 5’ctcaggatgcgtccaccaag3’, and 5’tggtgggaccagaacctagg3’ for the antisense strand. For XRCC5, the sense and antisense strand primers are 5’ggcagctgttgtgctgtgta3’ and 5’ttcagtcggacaactcttgg3’ respectively. All primers were synthesized from IDT DNA Technology.
Clonogenicity assay
Five-hundred to 1,000 cells/well were seeded in six-well plates, allowed to attach overnight to the plastic substrate before treatment with sepantronium, volasertib, sepantronium and volasertib or vehicle (DMSO only). After 48 hr treatment, the media were replaced with drug-free media and cells grown for the desired length of time. As the colonies became visible (usually 2-3 weeks), cells were fixed with methanol, stained with Giemsa (1:10 in distilled water), and counted.
shRNA knock-down of survivin
TRIPZ Inducible Lentiviral shRNAs for survivin were purchased from Thermo Scientific (clone ID # for the last four digits are 3705, 3706, 0788, 0789, 2484, and 2796 targeting different exons of survivin). pTRIPZ non-silencing vector was used as a control. Retroviral supernatants (≥5 × 106 CFU/μl) were generated and introduced into cells as described previously, puromycin was used for selection of clones where needed. Doxycycline at 0.2 μg/ml was used for induction of protein expression.
Single cell gel electrophoresis (comet assay)
A single-cell gel electrophoresis system (Comet assay kit; Trevigen) was used to analyze and quantitate DNA damage caused by sepantronium following manufacture’s instruction under either alkaline or neutral condition [35]. Briefly, cells were harvested by trypsinization and mixed with 1% low-melting-point agarose. A total of 100 μl (5,000 to 10,000 cells) of the cell suspension was spread on a precoated glass slide and placed at 4°C for 30 min. Cells were lysed by submerging the slides in ice cold lysis solution (10 mM Tris, 100 mM EDTA, 2.5 M NaCl, with 1% Triton X-100 and 10% dimethyl sulfoxide added just prior to use) at 4°C for at least 1 h. After lysis, the slides were placed in freshly made alkaline buffer (300 mM NaOH and 1 mM EDTA, pH>13.0) for 45 min at room temperature. The samples were subjected to electrophoresis in 0.5 × Tris-borate-EDTA buffer at 1.0 V/cm for 30 min. After electrophoresis, the slides were rinsed gently with 0.4 M Tris-HCl (pH 7.5), fixed with ethanol and the DNA was stained with Sybr Gold (Molecular Probes). Fluorescently stained nucleoids were scored visually using an epifluorescence microscope (Zeiss Axioplan 3). Images were captured at a magnification of × 20. The tail moment, and % of DNA in the tail a measure of the amount of DNA damage, was calculated using CometScore from TriTek Corporation (a minimum of 50 cells was used for each condition).
Statistical analysis
Data from the cell proliferation assay and clonogenicity assay are presented as means ± SD. Differences between groups were analyzed using the Student’s t-test for independent samples. The level of significance was set at p<0.05.
Results
Sepantronium synergizes with volasertib in killing of NSCLC
In a screening for drug combination(s) that significantly increase inhibition of growth of NSCLC cells by volasertib, we found that sepantronium did so at nanomole concentration (2-50 nM) in a MTT assay. This synergistic effect existed across cell lines (H1299, H661 and H2228). The growth inhibition is further confirmed in a colony formation assay (CFA) where 5 nM of volasertib and sepantronium drastically suppressed the formation of colonies by volasertib and sepantronium than either alone (Figure 1A) in the H1299 and H661 cell lines. Cell death often follows Polo arrest. We thus investigated whether the increase in growth inhibition by sepantronium is associated with an increase in the number of cells undergoing programmed cell death. As expected, the drug combination indeed resulted in a remarkable increase of apoptosis 28.7% versus ~8% in either drug alone after 72 h treatment (Figure 1B). To study the degree of synergy in detail, the cell lines were assayed for IC50 first. They were then treated with a drug combination at a fixed ratio based on their IC50s. The combination effects were calculated using the CalcuSyn software. At effect levels of ED90, ED95 and ED97, CIs for different cell lines ranged from 0.205 to 0.708, indicating synergy to strong synergy (Table 1) [31]. Thus, sepantronium was chosen for further mechanistic study. Sepantronium was chosen due to its potency, very favorable side effects profile, and non-overlapping toxicity shown in prior clinical trials.
Figure 1.
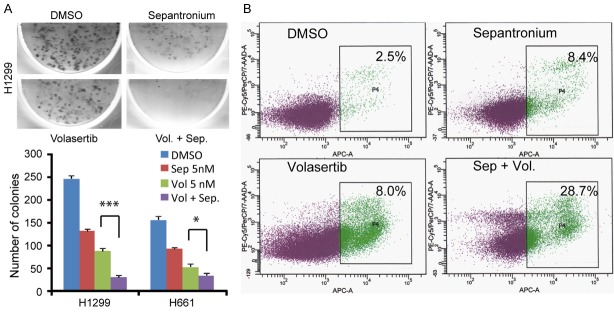
Sepantronium synergizes with volasertib in killing of different Non-small cell lung cancer cells (H1299, H661). A: Clonogenic assay showing the colony-forming capacity of H1299 and H661 cells treated briefly with DMSO, sepantronium (5 nM), volasertib (5 nM), sepantronium and volasertib (5 nM each). B: Assessment for apoptosis in cells (H1299) treated for 72 hours with DMSO, sepantronium (20 nM), volasertib (40 nM), sepantronium and volasertib. Percentage of apoptotic cells are indicated in the box. *P<0.05, **P<t0.01, ***P<0.001.
Table 1.
Synergism between YM155 and BI6727
| Effect level (Fa) | H1299 | H661 | H2228 | |
|---|---|---|---|---|
| 0.90 | YM155 | 0.181 | 0.008 | 0.361 |
| BI6727 | 0.638 | 0.028 | 0.708 | |
| CI | 0.718 | 0.390 | 0.560 | |
| 0.95 | YM155 | 0.319 | 0.018 | 0.503 |
| BI6727 | 1.128 | 0.038 | 0.098 | |
| CI | 0.465 | 0.270 | 0.311 | |
| 0.97 | YM155 | 0.479 | 0.146 | 0.636 |
| BI6727 | 1.690 | 0.467 | 0.124 | |
| CI | 0.348 | 0.210 | 0.205 |
CI: Combination index. Concentration in μM. CI<1, synergism, CI 0.1-0.3 strong synergy, 0.3-0.7 synergy and 0.7-0.85 moderate synergy.
Sepantronium blocks cell adaptation to polo arrest
Volasertib induces polo arrest. To determine whether adding sepantronium to volasertib have any effects on the polo arrest, we have performed cell cycle analysis of H1299 cells when they are treated with DMSO, sepantronium and volasertib alone or in combination. As shown in Figure 2A, while sepantronium (10 nM) alone did not affect the cell cycle distribution in unsynchronized cells, volasertib (40 nM) blocked the majority of H1299 cells in G2/M phase as expected. Volasertib together with sepantronium did not show any combination effect on cell cycle distribution after 24 h of treatment. Cells in the volasertib and combination treatment groups were largely arrested with a 4N DNA content in G2/M phase of the cell cycle 24 hours into the treatment indicating that cells were able to replicate their DNA but were not able to finish cell division. Interestingly, when these cells were followed for cell cycle progression for 72 hours, we found that the majority of cells that underwent polo arrest before progressed into G1 phase in volasertib treated cells. However, more than half of cells in the drug combination group failed to progress towards G1 indicating poor adaption to polo arrest and/or a tighter G2/M arrest after exposure to sepantronium (Figure 2B). Under the microscope, cells treated with volasertib for 24 hours were arrested in the metaphase with abnormal spindles (Figure 2C). While sepantronium treatment alone did not change the cell cycle distribution, cells in the sepantronium and volasertib treatment group had significantly fewer cells in the metaphase suggesting the progression into metaphase is delayed (Figure 2C). The delay of cell cycle progression is supported by failure of cyclin B1 accumulation in sepantronium treated cells compared to cells arrested by volasertib (Figure 2D). Interestingly, in this case low concentration of sepantronium has not caused significant down regulation of survivin expression in the protein level yet. These data point out that sepantronium might be causing delay in cell cycle progression independent of survivin suppression.
Figure 2.
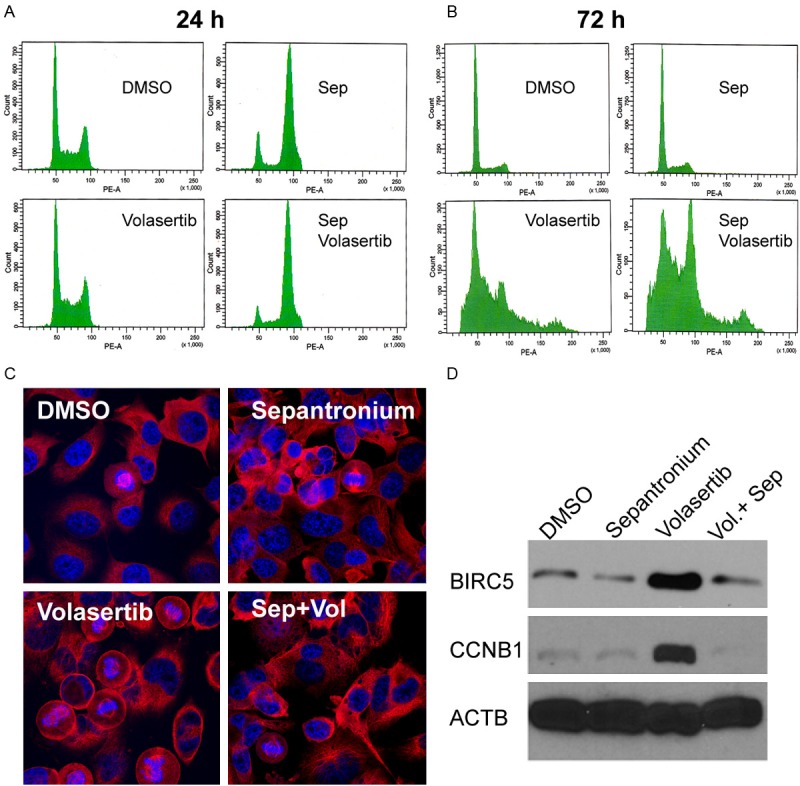
H1299 cells were treated with DMSO, sepantronium (20 nM), volasertib (40 nM), sepantronium and volasertib. Cell cycle distribution was analyzed at 24 h (A), 72 h (B). Confocal laser microscopy images were taken to show details of chromosomes and mitotic spindles (C). In (D), Western blot of cyclin B and survivin expression in H1299 cells treated for 24 hours at the same concentrations.
Sepantronium treatment induces cell cycle arrest irrespective of cell cycle phase
To determine whether sepantronium is indeed causing cell cycle perturbation, we decided to approach using synchronized cells. In our preliminary study, we were able to arrest H1299 cells at G1 phase by treating them for 24 hours with mimosine at 200 μM and M phase by treating them for 16 hours with nocodazole at 50 ng/ml (data not shown). To determine the effects of sepantronium on cell cycle progression, cells were either arrested in G1 phase by mimosine or M phase by nocodazole. Drugs were then washed out and the cells were reseeded into media containing either DMSO or 40 nM of sepantronium. As expected, cells arrested in G1 phase and then released into DMSO progressed synchronously through S phase 8 hours later and by 24 hours, 62% of cells cycled back to G1 again. In comparison, cells that were released into media containing sepantronium arrested themselves tightly in G1 with no progression even after 24 hours (Figure 3, left panel). Similarly, in M phase arrested cells by nocodazole, 43% of cells in the DMSO group progressed to G1 compared to only 2% in the sepantronium group 4 hours after release. Even after 24 hours, still 52% cells were not able to progress beyond M phase after release (Figure 3, right panel). All these data suggest that sepantronium delays cell cycle progression regardless whether they are in G1 or M phase of the cell cycle.
Figure 3.
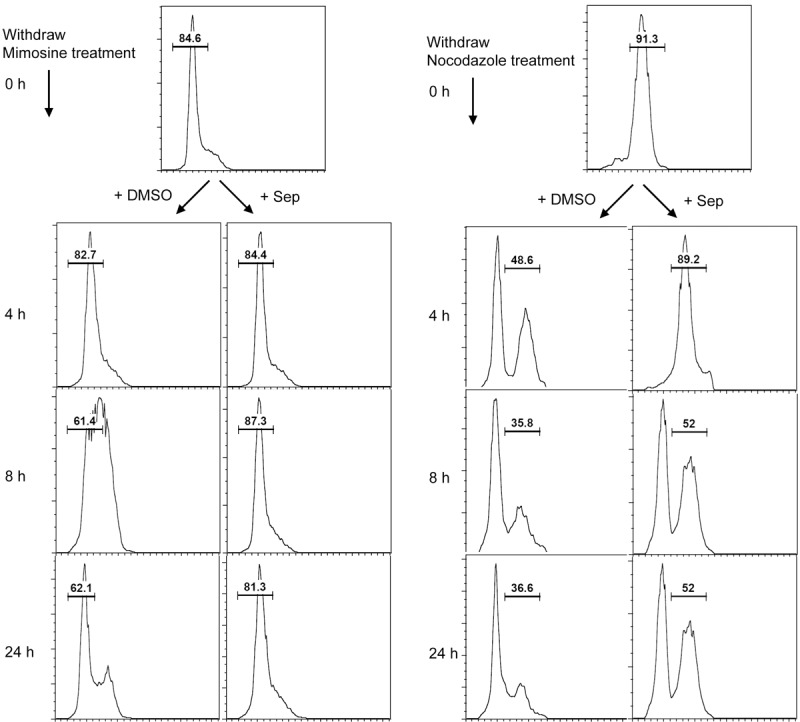
Sepantronium induces cell cycle arrest. H1299 cells were synchronized to G1 phase with mimosine for 24 hours (left) and to M phase with nocodazole for 16 hours (right). Cell cycle distribution was followed after cells were released into DMSO or sepantronium. Sepantronium caused cell cycle delay in both cases.
Sepantronium damages DNA and triggers DNA damage response and checkpoint signaling
The cell cycle perturbation and prior data of sepantronium triggered γH2AX accumulation [30] prompted us to hypothesize that cell cycle arrest is a result of DNA damage caused by sepantronium. To determine whether sepantronium induces DNA damage, we treated H1299 and H661 cells with DMSO, sepantronium, etoposide, a podophyllotoxin derivative known to cause double strand breaks (DSB). Cells were then stained for γH2AX that normally appears when DNA damage occurs. Similar to etoposide, sepantronium treatments also led to a remarkable increase in the number of γH2AX positive cells compared with control (Figure 4A). This result suggest that sepantronium induced cell-cycle arrest may be secondary to DNA damage. To further confirm this hypothesis, CDKN1A, BAX1 and XRCC5 genes induction at the transcriptional level was determined by quantitative RT-PCR. The mRNA levels of BAX and XRCC5 significantly increased in H1299 cells treated with sepantronium (Figure 4B). Their expression levels returned back to normal after 4 hours. In contrast, the mRNA level of CDKN1A was not increased until sepantronium treatment (Figure 4B). To characterize the DNA damage induced by sepantronium further, single cell electrophoresis assay (comet assay) was performed in cells treated with either DMSO or sepantronium. Etoposide was used as a control. As shown in Figure 4C, sepantronium treatment both at 80 nM and 320 nM dosages resulted in remarkable DNA damage that the nucleoid DNA stained like light bulbs. Quantitative analysis of the percentage of DNA in the comet tails showed 9 fold more in H1299 and H661 cells treated with 320 nM of sepantronium. Cells treated with high concentration of etoposide. (Figure 4D) showed 4 fold as much DNA in the tail compared to DMSO alone. To determine whether DNA checkpoint signaling is triggered, we conducted a survey of phosphorylation of multiple known checkpoint sensors and transducers in H661 cells. γH2AX levels increase when cells are treated. p53 stabilizes while increased phosphorylation was seen across the board in ATM/CHK2 and ATR/CHK1 (Figure 4E).
Figure 4.

Sepantronium damages DNA. Immunofluorescence staining of γH2AX (green) in H1299 and H661 cells treated with DMSO, sepantronium and etoposide. Red color is tubulin counterstaining (A). Quantitative real time PCR results showing induction of BAX and XRCC5 RNA at 2 hours and CDKN1A at 4 hours (B). GAPDH was used as control. (C) Comet assay images of H1299 treated with DMSO, sepantronium and etoposide. (D) Quantitation of tail length, tail area, %DNA in tails and tail moment performed with software. (E) Analysis of checkpoint phospho protein by Western blot. *P<0.05, **P<0.01, ***P<0.001.
Effects of sepantronium on the vitality of cells arrested at G1 or M phase
To compare the effects of DNA damage on cell vitality in cells transiently arrested in G1 or M phase, we first arrested H1299 cells with mimosine or nocodazole as described above. DMSO or sepantronium were then added to the culture. Apoptosis was monitored for 72 hours after addition of sepantronium. The number of apoptotic cells kept increasing in both groups (Figure 5A). Overall more cells died when sepantronium is added in cells arrested in G1 phase compared to cells arrested in M phase suggesting that sepantronium caused DNA damage is at least as lethal if not more in G1-arrested as M-arrested cells. Over the whole period, cells were arrested tightly in G1 or M phase (Figure 5B). Follow up for the extent of DNA damage revealed that cells arrested in G1 phase had significant bigger increase in γH2AX levels from the baseline than cells arrested in M phase (Figure 5C).
Figure 5.

DNA damage by sepantronium is cell cycle non-specific. H1299 cells synchronized with nocodazole or mimosine and treated with sepantronium were analyzed for apoptosis by flow cytometry for 72 hours (A). The cell cycle distribution was followed at the same time point when apoptosis was analyzed (B). Cells were checked for γH2AX and BIRC5 expression at the time point indicated (C).
Knockdown survivin does not cause DNA damage
To determine the effects of survivin knock down on DNA damage, we have created several shRNA knockdown clones of survivin in H1299 cells. In these clones, survivin shRNAs are expressed under the control of a tet-on inducible promoter. Addition of doxycycline to the media effectively induced knockdown of survivin level by 90% after 72 h of induction (Figure 6A). Western blot analyses with specific antibodies for DNA damage checkpoint protein show that there were no differences in the level of phosphorylation regardless of whether survivin level is reduced (Figure 6A). These data suggest that knockdown of survivin does not trigger DNA damage checkpoint signaling. To test whether knock down of survivin is sufficient to increase the amount of DNA strand breaks, the same shRNA knockdown clones were subjected to comet assay 72 hours after survivin shRNAs are induced. There was no increase in the amount of DNA damage seen in the tails (Figure 6B). Together, these results indicate that sepantronium induced DNA damage is not mediated by inhibition of survivin.
Figure 6.
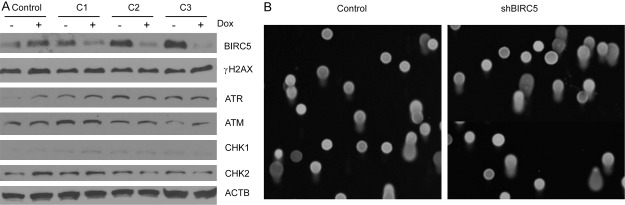
Survivin knockdown does not contribute to DNA damage. Three knock down clones of survivin were induced with doxycycline for 72 hours before they were processed for analysis of BIRC5 knock down levels and checkpoint signaling (A). Comet assays were performed to assess DNA damage. Shown are representative pictures from the comet assay (B).
Discussion
In a search for drug(s) that boost the killing of NSCLC cells by volasertib, we screened more than a dozen small molecule compounds known to be antiproliferative in lung cancer cells. One compound named sepantronium stood out. At nanomolar level, sepantronium potently augmented the growth inhibitory effects of volasertib, triggering programed cell death. This effect existed in all three cell lines tested (H1299, undifferentiated, H661, large cell, and H2228 adenocarcinoma). The combination effects are clearly synergistic since the CIs ranged from 0.205 to 0.708 [31]. This provides a potentially a start point for thinking of combination chemotherapy using sepantronium and volasertib in NSCLC.
Volasertib is known to ablate PLK1 kinase potently and selectively, causing a polo arrest, a cell cycle arrest at M phase with abnormally formed spindles [36]. Cell cycle progression was significantly delayed 72 h after treatment by the sepantronium and volasertib drug combination. Adaptation of cells to volasertib induced polo arrest and further progression into the G1 phase was significantly delayed. Immunofluorescence staining of the mitotic spindle and the chromosomes revealed cell cycle arrest before metaphase also consistent with delayed progression through the S and G2/M phase when sepantronium is added to volasertib. This concept of cell cycle delay is also supported by the fact that survivin and cyclin B1 level did not peak in the drug combination group compared with the volasertib alone 24 hours into the treatment.
Using cells synchronized at G1 phase by mimosine and M phase by nocodazole, we were able to further strengthen the notion that sepantronium is causing cell cycle arrest and cell cycle perturbation. Therefore, sepantronium induces cell cycle arrest irrespective the phase cells reside. Sepantronium is best known as a survivin suppressant [37-39]. The dependency of sepantronium effects on survivin suppression has never been demonstrated. It is interesting to note here that, at low concentration (<100 nM) and short treatment (<24 hours), sepantronium has not had a big impact on survivin expression at the protein level suggesting that the cell cycle delay may not be dependent on survivin suppression. Also in our hands and others, 1) survivin level did not correlate with sepantronium sensitivity, 2) sepantronium did not cause a sharp increase percentage of cells going into apoptosis instead it was cytostatic when Merkel cell carcinoma were treated [29]. 3) sepantronium and NSC80467 share structural similarity with doxorubicin, mitomycin C known to damage DNA [30]. All these prompted us to think that sepantronium is a DNA damaging agent. This would be consistent also with the fact that sepantronium causes cell cycle arrest.
DNA damage was first revealed here by the observation that increased γH2AX level happened 4-6 hours after low concentration (80 nM) of sepantronium. So this is an event that happens much quicker, under very low concentration of sepantronium, and when survivin level is not suppressed. Importantly, we also detected events associated with DNA damage when cells are treated with sepantronium. These include transcriptional induction of negative cell cycle regulator CIP1 which is important for cell cycle arrest [40], apoptosis (BAX1) [41] and DNA repair (XRCC5, also known as KARP1, Ku86 Autoantigen Related Protein-1) [42]. XRCC5 plays an important role in DNA double strand break repair as a regulator of the DNA dependent kinase complex, known to be induced up to 100 fold after DNA damage. This concept is further backed by the Comet assay results that showed unambiguous evidence of DNA strand breaks under both alkaline and neutral condition (data not shown). The percentage of DNA in the comet tail and the tail moment in sepantronium treated cells are even more prominent than etoposide treated cells, providing the strongest evidence that sepantronium is a DNA damaging agent. Consistent with this, survey of DNA damage checkpoint signaling revealed changes consistent with active signaling inside the pathway with increased phosphorylation of p53, ATM, ATR, CHK1, CHK2. Cell cycle regulators also displayed changes consistent with occurrence of DNA damage.
One explanation for the sepantronium and volasertib synergy might be that DNA damage incurred in M phase is more detrimental to cell vitality than cells arrested in G1 because DNA damage checkpoint signaling mechanism may not be active until cells finishes mitosis [43]. Our results do not support such hypothesis. Compared to cells transiently arrested in M phase, cells transiently arrested in G1 phase were at least as susceptible if not more to sepantronium treatment. This notion is supported by our Western results showing a bigger increase in the amount of γH2AX when G1 arrested cells are treated with sepantronium compared with M arrested cells. Consistently, There seems to be more DNA damage when G1 arrested cells are treated with sepantronium.
Previous work has shown association of survivin suppression with sepantronium toxicity [23,44]. We also observed the suppression of survivin expression by sepantronium, in addition to DNA damage. The question is whether survivin downregulation mediates DNA damage. Our survivin knockdown experiments do not seem to support such possibility. This is because knockdown of survivin neither increased level of phosphorylation in known DNA damage checkpoint sensors or transducers, nor there was a difference in the percentage of DNA resides in the comet tails when survivin shRNA were induced. Therefore, sepantronium damages DNA and its effects does not seem to be mediated by survivin down-regulation. Instead, survivin down-regulation might be the consequence of DNA damage. The question for future study is how sepantronium damages DNA. Due to its positive charge, sepantronium could be a good DNA binding agent. Binding to DNA by way of major/minor groove binding or intercalating may cause DNA damage. High concentration of sepantronium could compete out transcription factor such as sp1 poised to bind the survivin promoter [45]. Currently we are investigating whether sepantronium is a DNA binding agent.
Acknowledgements
We thank Sam Chang and Lesleyann Hawthorn of the core facility for help with the transcriptome analysis and Ms Haiyan Qin for providing excellent technical assistance. This work is supported in part by the institution start-up fund to Z Hao.
Disclosure of conflict of interest
No conflict of interests disclosed by the authors.
References
- 1.Sarkar FH, Li Y. Harnessing the fruits of nature for the development of multi-targeted cancer therapeutics. Cancer Treat Rev. 2009;35:597–607. doi: 10.1016/j.ctrv.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 3.Lengauer C, Kinzler KW, Vogelstein B. Genetic instabilities in human cancers. Nature. 1998;396:643–649. doi: 10.1038/25292. [DOI] [PubMed] [Google Scholar]
- 4.Schiller JH, Harrington D, Belani CP, Langer C, Sandler A, Krook J, Zhu J, Johnson DH. Comparison of four chemotherapy regimens for advanced non-small-cell lung cancer. N Engl J Med. 2002;346:92–98. doi: 10.1056/NEJMoa011954. [DOI] [PubMed] [Google Scholar]
- 5.Scagliotti GV, De Marinis F, Rinaldi M, Crino L, Gridelli C, Ricci S, Matano E, Boni C, Marangolo M, Failla G, Altavilla G, Adamo V, Ceribelli A, Clerici M, Di Costanzo F, Frontini L, Tonato M. Phase III randomized trial comparing three platinum-based doublets in advanced non-small-cell lung cancer. J. Clin. Oncol. 2002;20:4285–4291. doi: 10.1200/JCO.2002.02.068. [DOI] [PubMed] [Google Scholar]
- 6.Smit EF, van Meerbeeck JP, Lianes P, Debruyne C, Legrand C, Schramel F, Smit H, Gaafar R, Biesma B, Manegold C, Neymark N, Giaccone G. Three-arm randomized study of two cisplatin-based regimens and paclitaxel plus gemcitabine in advanced non-small-cell lung cancer: a phase III trial of the European Organization for Research and Treatment of Cancer Lung Cancer Group--EORTC 08975. J. Clin. Oncol. 2003;21:3909–3917. doi: 10.1200/JCO.2003.03.195. [DOI] [PubMed] [Google Scholar]
- 7.Souquet PJ, Tan EH, Rodrigues Pereira J, Van Klaveren R, Price A, Gatzemeier U, Jaworski M, Burillon JP, Aubert D. GLOB-1: a prospective randomised clinical phase III trial comparing vinorelbine-cisplatin with vinorelbine-ifosfamide-cisplatin in metastatic non-small-cell lung cancer patients. Ann Oncol. 2002;13:1853–1861. doi: 10.1093/annonc/mdf316. [DOI] [PubMed] [Google Scholar]
- 8.Soon YY, Stockler MR, Askie LM, Boyer MJ. Duration of chemotherapy for advanced non-small-cell lung cancer: a systematic review and meta-analysis of randomized trials. J. Clin. Oncol. 2009;27:3277–3283. doi: 10.1200/JCO.2008.19.4522. [DOI] [PubMed] [Google Scholar]
- 9.Earle CC, Venditti LN, Neumann PJ, Gelber RD, Weinstein MC, Potosky AL, Weeks JC. Who gets chemotherapy for metastatic lung cancer? Chest. 2000;117:1239–1246. doi: 10.1378/chest.117.5.1239. [DOI] [PubMed] [Google Scholar]
- 10.Hull MW, Lima VD, Hogg RS, Harrigan PR, Montaner JS. Epidemiology of treatment failure: a focus on recent trends. Curr Opin HIV AIDS. 2009;4:467–473. doi: 10.1097/COH.0b013e328331d353. [DOI] [PubMed] [Google Scholar]
- 11.Petronczki M, Lenart P, Peters JM. Polo on the Rise-from Mitotic Entry to Cytokinesis with Plk1. Dev Cell. 2008;14:646–659. doi: 10.1016/j.devcel.2008.04.014. [DOI] [PubMed] [Google Scholar]
- 12.van Vugt MA, Bras A, Medema RH. Polo-like kinase-1 controls recovery from a G2 DNA damage-induced arrest in mammalian cells. Mol Cell. 2004;15:799–811. doi: 10.1016/j.molcel.2004.07.015. [DOI] [PubMed] [Google Scholar]
- 13.Takai N, Hamanaka R, Yoshimatsu J, Miyakawa I. Polo-like kinases (Plks) and cancer. Oncogene. 2005;24:287–291. doi: 10.1038/sj.onc.1208272. [DOI] [PubMed] [Google Scholar]
- 14.Lenart P, Petronczki M, Steegmaier M, Di Fiore B, Lipp JJ, Hoffmann M, Rettig WJ, Kraut N, Peters JM. The small-molecule inhibitor BI 2536 reveals novel insights into mitotic roles of polo-like kinase 1. Curr Biol. 2007;17:304–315. doi: 10.1016/j.cub.2006.12.046. [DOI] [PubMed] [Google Scholar]
- 15.Steegmaier M, Hoffmann M, Baum A, Lenart P, Petronczki M, Krssak M, Gurtler U, Garin-Chesa P, Lieb S, Quant J, Grauert M, Adolf GR, Kraut N, Peters JM, Rettig WJ. BI 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo. Curr Biol. 2007;17:316–322. doi: 10.1016/j.cub.2006.12.037. [DOI] [PubMed] [Google Scholar]
- 16.Mross K, Frost A, Steinbild S, Hedbom S, Rentschler J, Kaiser R, Rouyrre N, Trommeshauser D, Hoesl CE, Munzert G. Phase I dose escalation and pharmacokinetic study of BI 2536, a novel Polo-like kinase 1 inhibitor, in patients with advanced solid tumors. J. Clin. Oncol. 2008;26:5511–5517. doi: 10.1200/JCO.2008.16.1547. [DOI] [PubMed] [Google Scholar]
- 17.Sebastian M, Reck M, Waller CF, Kortsik C, Frickhofen N, Schuler M, Fritsch H, Gaschler-Markefski B, Hanft G, Munzert G, von Pawel J. The efficacy and safety of BI 2536, a novel Plk-1 inhibitor, in patients with stage IIIB/IV non-small cell lung cancer who had relapsed after, or failed, chemotherapy: results from an open-label, randomized phase II clinical trial. J Thorac Oncol. 2010;5:1060–1067. doi: 10.1097/JTO.0b013e3181d95dd4. [DOI] [PubMed] [Google Scholar]
- 18.Schoffski P, Blay JY, De Greve J, Brain E, Machiels JP, Soria JC, Sleijfer S, Wolter P, Ray-Coquard I, Fontaine C, Munzert G, Fritsch H, Hanft G, Aerts C, Rapion J, Allgeier A, Bogaerts J, Lacombe D. Multicentric parallel phase II trial of the polo-like kinase 1 inhibitor BI 2536 in patients with advanced head and neck cancer, breast cancer, ovarian cancer, soft tissue sarcoma and melanoma. The first protocol of the European Organization for Research and Treatment of Cancer (EORTC) Network Of Core Institutes (NOCI) Eur J Cancer. 2010;46:2206–2215. doi: 10.1016/j.ejca.2010.03.039. [DOI] [PubMed] [Google Scholar]
- 19.Mross K, Dittrich C, Aulitzky WE, Strumberg D, Schutte J, Schmid RM, Hollerbach S, Merger M, Munzert G, Fleischer F, Scheulen ME. A randomised phase II trial of the Polo-like kinase inhibitor BI 2536 in chemo-naive patients with unresectable exocrine adenocarcinoma of the pancreas - a study within the Central European Society Anticancer Drug Research (CESAR) collaborative network. Br J Cancer. 2012;107:280–286. doi: 10.1038/bjc.2012.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ellis PM, Chu QS, Leighl N, Laurie SA, Fritsch H, Gaschler-Markefski B, Gyorffy S, Munzert G. A Phase I Open-Label Dose-Escalation Study of Intravenous BI 2536 Together With Pemetrexed in Previously Treated Patients With Non-Small-Cell Lung Cancer. Clin Lung Cancer. 2013;14:19–27. doi: 10.1016/j.cllc.2012.04.003. [DOI] [PubMed] [Google Scholar]
- 21.Rudolph D, Steegmaier M, Hoffmann M, Grauert M, Baum A, Quant J, Haslinger C, Garin-Chesa P, Adolf GR. BI 6727, a Polo-like kinase inhibitor with improved pharmacokinetic profile and broad antitumor activity. Clin Cancer Res. 2009;15:3094–3102. doi: 10.1158/1078-0432.CCR-08-2445. [DOI] [PubMed] [Google Scholar]
- 22.Schoffski P, Awada A, Dumez H, Gil T, Bartholomeus S, Wolter P, Taton M, Fritsch H, Glomb P, Munzert G. A phase I, dose-escalation study of the novel Polo-like kinase inhibitor volasertib (BI 6727) in patients with advanced solid tumours. Eur J Cancer. 2012;48:179–186. doi: 10.1016/j.ejca.2011.11.001. [DOI] [PubMed] [Google Scholar]
- 23.Nakahara T, Kita A, Yamanaka K, Mori M, Amino N, Takeuchi M, Tominaga F, Hatakeyama S, Kinoyama I, Matsuhisa A, Kudoh M, Sasamata M. YM155, a novel small-molecule survivin suppressant, induces regression of established human hormone-refractory prostate tumor xenografts. Cancer Res. 2007;67:8014–8021. doi: 10.1158/0008-5472.CAN-07-1343. [DOI] [PubMed] [Google Scholar]
- 24.Satoh T, Okamoto I, Miyazaki M, Morinaga R, Tsuya A, Hasegawa Y, Terashima M, Ueda S, Fukuoka M, Ariyoshi Y, Saito T, Masuda N, Watanabe H, Taguchi T, Kakihara T, Aoyama Y, Hashimoto Y, Nakagawa K. Phase I study of YM155, a novel survivin suppressant, in patients with advanced solid tumors. Clin Cancer Res. 2009;15:3872–3880. doi: 10.1158/1078-0432.CCR-08-1946. [DOI] [PubMed] [Google Scholar]
- 25.Giaccone G, Zatloukal P, Roubec J, Floor K, Musil J, Kuta M, van Klaveren RJ, Chaudhary S, Gunther A, Shamsili S. Multicenter phase II trial of YM155, a small-molecule suppressor of survivin, in patients with advanced, refractory, non-small-cell lung cancer. J. Clin. Oncol. 2009;27:4481–4486. doi: 10.1200/JCO.2008.21.1862. [DOI] [PubMed] [Google Scholar]
- 26.Lewis KD, Samlowski W, Ward J, Catlett J, Cranmer L, Kirkwood J, Lawson D, Whitman E, Gonzalez R. A multi-center phase II evaluation of the small molecule survivin suppressor YM155 in patients with unresectable stage III or IV melanoma. Invest New Drugs. 2011;29:161–166. doi: 10.1007/s10637-009-9333-6. [DOI] [PubMed] [Google Scholar]
- 27.Tolcher AW, Quinn DI, Ferrari A, Ahmann F, Giaccone G, Drake T, Keating A, de Bono JS. A phase II study of YM155, a novel small-molecule suppressor of survivin, in castration-resistant taxane-pretreated prostate cancer. Ann Oncol. 2012;23:968–973. doi: 10.1093/annonc/mdr353. [DOI] [PubMed] [Google Scholar]
- 28.Cheson BD, Bartlett NL, Vose JM, Lopez-Hernandez A, Seiz AL, Keating AT, Shamsili S, Papadopoulos KP. A phase II study of the survivin suppressant YM155 in patients with refractory diffuse large B-cell lymphoma. Cancer. 2012;118:3128–3134. doi: 10.1002/cncr.26510. [DOI] [PubMed] [Google Scholar]
- 29.Arora R, Shuda M, Guastafierro A, Feng H, Toptan T, Tolstov Y, Normolle D, Vollmer LL, Vogt A, Domling A, Brodsky JL, Chang Y, Moore PS. Survivin is a therapeutic target in Merkel cell carcinoma. Sci Transl Med. 2012;4:133ra156. doi: 10.1126/scitranslmed.3003713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Glaros TG, Stockwin LH, Mullendore ME, Smith B, Morrison BL, Newton DL. The “survivin suppressants” NSC 80467 and YM155 induce a DNA damage response. Cancer Chemother Pharmacol. 2012;70:207–212. doi: 10.1007/s00280-012-1868-0. [DOI] [PubMed] [Google Scholar]
- 31.Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006;58:621–681. doi: 10.1124/pr.58.3.10. [DOI] [PubMed] [Google Scholar]
- 32.Ren M, Hong M, Liu G, Wang H, Patel V, Biddinger P, Silva J, Cowell J, Hao Z. Novel FGFR inhibitor ponatinib suppresses the growth of non-small cell lung cancer cells overexpressing FGFR1. Oncol Rep. 2013;29:2181–2190. doi: 10.3892/or.2013.2386. [DOI] [PubMed] [Google Scholar]
- 33.Ren M, Qin H, Ren R, Tidwell J, Cowell JK. Src activation plays an important key role in lymphomagenesis induced by FGFR1 fusion kinases. Cancer Res. 2011;71:7312–7322. doi: 10.1158/0008-5472.CAN-11-1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ren M, Li X, Cowell JK. Genetic fingerprinting of the development and progression of T-cell lymphoma in a murine model of atypical myeloproliferative disorder initiated by the ZNF198-fibroblast growth factor receptor-1 chimeric tyrosine kinase. Blood. 2009;114:1576–1584. doi: 10.1182/blood-2009-03-212704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Palii SS, Van Emburgh BO, Sankpal UT, Brown KD, Robertson KD. DNA methylation inhibitor 5-Aza-2’-deoxycytidine induces reversible genome-wide DNA damage that is distinctly influenced by DNA methyltransferases 1 and 3B. Mol Cell Biol. 2008;28:752–771. doi: 10.1128/MCB.01799-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Medema RH, Lin CC, Yang JC. Polo-like kinase 1 inhibitors and their potential role in anticancer therapy, with a focus on NSCLC. Clin Cancer Res. 2011;17:6459–6466. doi: 10.1158/1078-0432.CCR-11-0541. [DOI] [PubMed] [Google Scholar]
- 37.Kaneko N, Yamanaka K, Kita A, Tabata K, Akabane T, Mori M. Synergistic antitumor activities of sepantronium bromide (YM155), a survivin suppressant, in combination with microtubule-targeting agents in triple-negative breast cancer cells. Biol Pharm Bull. 2013;36:1921–1927. doi: 10.1248/bpb.b13-00515. [DOI] [PubMed] [Google Scholar]
- 38.Kaneko N, Kita A, Yamanaka K, Mori M. Combination of YM155, a survivin suppressant with a STAT3 inhibitor: a new strategy to treat diffuse large B-cell lymphoma. Leuk Res. 2013;37:1156–1161. doi: 10.1016/j.leukres.2013.05.010. [DOI] [PubMed] [Google Scholar]
- 39.Chen J, Pise-Masison CA, Shih JH, Morris JC, Janik JE, Conlon KC, Keating A, Waldmann TA. Markedly additive antitumor activity with the combination of a selective survivin suppressant YM155 and alemtuzumab in adult T-cell leukemia. Blood. 2013;121:2029–2037. doi: 10.1182/blood-2012-05-427773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reinhardt HC, Hasskamp P, Schmedding I, Morandell S, van Vugt MA, Wang X, Linding R, Ong SE, Weaver D, Carr SA, Yaffe MB. DNA damage activates a spatially distinct late cytoplasmic cell-cycle checkpoint network controlled by MK2-mediated RNA stabilization. Mol Cell. 2010;40:34–49. doi: 10.1016/j.molcel.2010.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yamada Y, Coffman CR. DNA damage-induced programmed cell death: potential roles in germ cell development. Ann N Y Acad Sci. 2005;1049:9–16. doi: 10.1196/annals.1334.002. [DOI] [PubMed] [Google Scholar]
- 42.Myung K, Braastad C, He DM, Hendrickson EA. KARP-1 is induced by DNA damage in a p53- and ataxia telangiectasia mutated-dependent fashion. Proc Natl Acad Sci U S A. 1998;95:7664–7669. doi: 10.1073/pnas.95.13.7664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Heijink AM, Krajewska M, van Vugt MA. The DNA damage response during mitosis. Mutat Res. 2013;750:45–55. doi: 10.1016/j.mrfmmm.2013.07.003. [DOI] [PubMed] [Google Scholar]
- 44.Iwasa T, Okamoto I, Suzuki M, Nakahara T, Yamanaka K, Hatashita E, Yamada Y, Fukuoka M, Ono K, Nakagawa K. Radiosensitizing effect of YM155, a novel small-molecule survivin suppressant, in non-small cell lung cancer cell lines. Clin Cancer Res. 2008;14:6496–6504. doi: 10.1158/1078-0432.CCR-08-0468. [DOI] [PubMed] [Google Scholar]
- 45.Cheng Q, Ling X, Haller A, Nakahara T, Yamanaka K, Kita A, Koutoku H, Takeuchi M, Brattain MG, Li F. Suppression of survivin promoter activity by YM155 involves disruption of Sp1-DNA interaction in the survivin core promoter. Int J Biochem Mol Biol. 2012;3:179–197. [PMC free article] [PubMed] [Google Scholar]