

Abstract
Multidrug resistance (MDR) mediated by ATP-binding cassette (ABC) transporters through efflux of antineoplastic agents from cancer cells is a major obstacle to successful cancer chemotherapy. The inhibition of these ABC transporters is thus a logical approach to circumvent MDR. There has been intensive research effort to design and develop novel inhibitors for the ABC transporters to achieve this goal. In the present study, we evaluated the ability of UMMS-4 to modulate P-glycoprotein (P-gp/ABCB1)-, breast cancer resistance protein (BCRP/ABCG2)- and multidrug resistance protein (MRP1/ABCC1)-mediated MDR in cancer cells. Our findings showed that UMMS-4, at non-cytotoxic concentrations, apparently circumvents resistance to ABCB1 substrate anticancer drugs in ABCB1-overexpressing cells. When used at a concentration of 20 μmol/L, UMMS-4 produced a 17.53-fold reversal of MDR, but showed no effect on the sensitivity of drug-sensitive parental cells. UMMS-4, however, did not significantly alter the sensitivity of non-ABCB1 substrates in all cells and was unable to reverse ABCG2- and ABCC1-mediated MDR. Additionally, UMMS-4 profoundly inhibited the transport of rhodamine 123 (Rho 123) and doxorubicin (Dox) by the ABCB1 transporter. Furthermore, UMMS-4 did not alter the expression of ABCB1 at the mRNA and protein levels. In addition, the results of ATPase assays showed that UMMS-4 stimulated the ATPase activity of ABCB1. Taken together, we conclude that UMMS-4 antagonizes ABCB1-mediated MDR in cancer cells through direct inhibition of the drug efflux function of ABCB1. These findings may be useful for the development of safer and more effective MDR modulator.
Keywords: UMMS-4, multidrug resistance, ATP binding cassette transporters, ABCB1, chemotherapeutic drugs
Introduction
Multidrug resistance (MDR) is the major cause of cancer chemotherapy failure. It is usually caused by the overexpression of ABC transporters on cell surface of cancer cells, which use the energy derived from ATP hydrolysis to efflux numerous structurally and functionally unrelated anticancer drugs out of the cells [1-3]. In the human genome, there are 48 different ABC transporter family genes, which are divided into seven subfamilies (A-G) based on amino acid sequence similarities and phylogeny [4]. Among them, the ABCB1, ABCG2 and ABCC1 are the major ABC transporters reported to confer MDR in cancer cells [5,6].
ABCB1, encoded by human mdr1 gene, is located on chromosome 7 of humans and is a plasma membrane protein 1280 amino acids long that consists of two homologous halves [7]. Each half contains six hydrophobic transmembrane domains and two ATP binding and utilization sites [8,9]. ABCB1 recognizes and transports a broad range of hydrophobic substrates, either un-charged or slightly positively charged, including most natural product anti-cancer drugs such as colchicine, doxorubicin (daunorubicin, anthracyclines), paclitaxel (taxanes), vinblastine (vinca alkaloids), epipodophyllotoxins (etoposide, teniposide) and actinomycin D [10-12].
ABCB1 is not only expressed in a wide variety of cancer cells, including solid tumors and hematological malignancies, but also in some normal tissues, such as liver, kidney, pancreas and intestine [13,14]. Moreover, cancer cells can become progressively refractory to antitumor compounds during chemotherapy or at relapse after treatment because of ABCB1 upregulation [15]. Many compounds that inhibit ABCB1-mediated transport have been studied to circumvent MDR [16,17]. To date, a few ABCB1 inhibitors have been developed. They exhibit potent inhibition of ABCB1 function to increase the intracellular accumulation and anticancer effects of conventional anticancer drugs on MDR cancer cells in vitro and/or in vivo [18,19]. The first-generation of ABCB1 inhibitors, including verapamil, quinine and cyclosporin A, were substrates of ABCB1 and significantly inhibited the function of ABCB1 [15]. However, they failed to show an improvement in therapeutic outcome and adverse effects were common in clinical trials. The second-generation of ABCB1 modulators, PSC-388, displayed a superior pharmacologic profile compared to the first-generation compounds [20]. However, they were found to inhibit the activity of CYP3A4, which led to a decrease in the metabolism of antineoplastic agents, thereby producing unacceptable toxicity. Therefore, a lower concentration of the second-generation ABCB1 modulators has to be used for MDR reversal, thus limiting their clinical usefulness. The third-generation ABCB1 inhibitors, including Tariquidar (XR9576) [21], Zosuquidar (LY335979) [22], Laniquidar (R102933) [23], ONT-093 (OC144-193) [24], GF120918 [25] and Biricodar (VX-710) [26], potently reversed ABCB1-mediated MDR in vitro and in vivo, and did not affect CYP3A4 and the pharmacokinetic profile of conventional chemotherapeutic agents at relevant concentration. The third-generation inhibitors are currently being studied for their clinical efficacy. However, no ABCB1 inhibitor has yet been approved for clinical use, which spurred on efforts to search for new and more effective ABCB1 modulators.
In a compound library of imidazole-based ABCB1 modulators, our structure-activity relationship analysis revealed that potent ABCB1 inhibitors are all hydrophobic compounds with multiple amine groups. We have subsequently selected one of the newly synthesized compound, UMMS-4, N’-(4-(Dimethylamino)benzylidene)-2-(4-methyl-5,5-dioxido-3-Phenyl-benzo[e]pyrazolo[4,3-c][1,2]thiazin-1(4H)-yl)acetohydrazide, and investigated its potential ABCB1 inhibition and circumvention of MDR.
Materials and methods
Chemicals
UMMS-4, a novel synthetic compound [27], with a molecular structure as shown in Figure 1. Rhodamine 123 (Rho 123), doxorubicin (Dox), vincristine (VCR), paclitaxel, topotecan, cisplatin, fumitremorgin C (FTC), 1-(4,5-dimethylthiazol-2-yl)-3,5-diphenylformazan (MTT) and other chemicals were purchased from Sigma Chemical Co (St. Louis, MO, USA). DMEM and RPMI 1640 were products of Life Technologies, Inc. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody was purchased from Kangchen Co. (Shanghai, China). Monoclonal antibody against ABCB1 was from Santa Cruz Biotechnology. Other routine laboratory reagents were of analytical grade and obtained from commercial sources.
Figure 1.
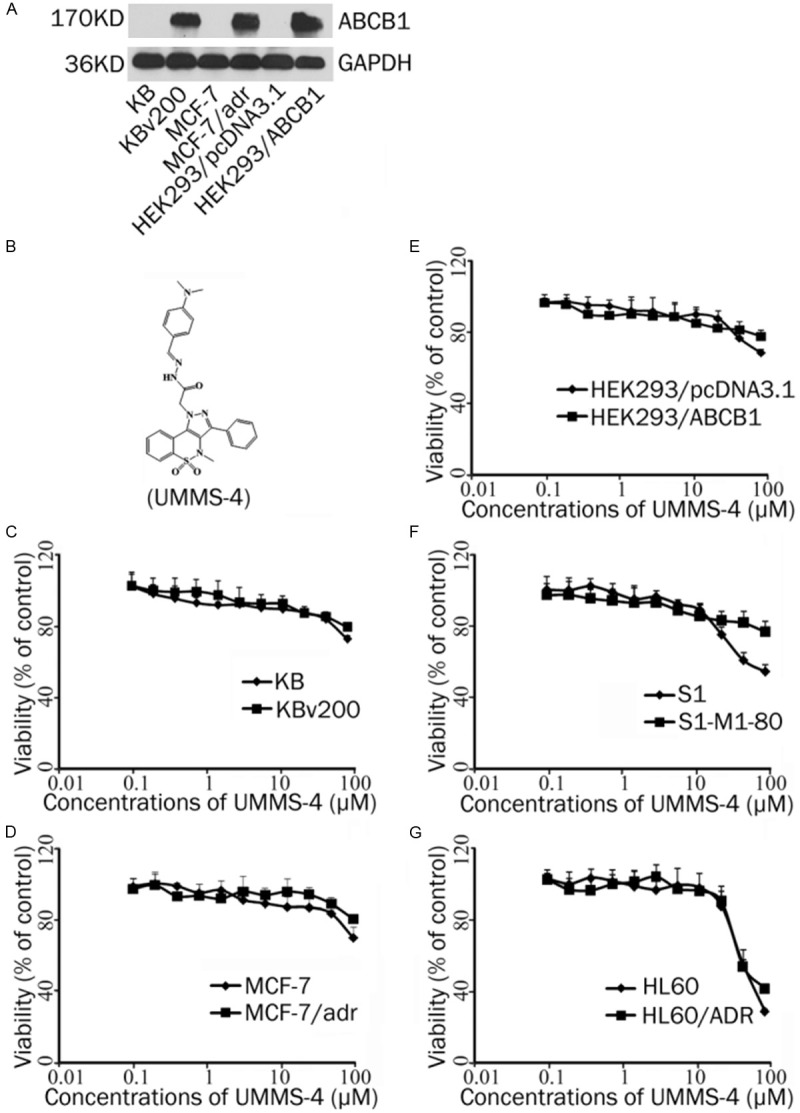
Cytotoxicity of UMMS-4 alone in the drug-resistant and parental sensitive cancer cells. A: The protein expression of ABCB1 in KB, KBv200, MCF-7, MCF-7/adr, HEK293/pcDNA3.1 and HEK293/ABCB1. B: The structure of UMMS-4. C-G: MTT cytotoxicity assay was used to examine the cytotoxicity effect of UMMS-4 alone in KB and KBv200, MCF-7 and MCF-7/adr, HEK293/pcDNA3.1 and HEK293/ABCB1, S1 and S1-M1-80, HL60 and HL60/ADR after drug treatment for 72 h. Each point represents the mean±standard deviations for three independent determinations. Each experiment was performed in four replicate wells.
Cell lines and cell culture
The following cell lines were cultured in DMEM or RPMI 1640 supplemented with 10% FBS at 37°C in a humidified atmosphere of 5% CO2: the human oral epidermoid carcinoma cell lines KB and its vincristine-selected derivative ABCB1-overexpressing KBv200 [28]; the human breast carcinoma cell lines MCF-7 and its Dox-selected ABCB1-overexpressing derivative MCF-7/adr [29]; the human colon carcinoma cell lines S1 and its mitoxantrone-selected derivative ABCG2-overexpressing S1-M1-80 [30]; the human leukemia cell lines HL60 and its Dox-selected derivative ABCC1-overexpressing cell line HL60/ADR [31] and the human embryonic kidney cell line HEK293 stably-transfected with pcDNA3.1 (HEK293/pcDNA3.1) or ABCB1 (HEK293/ABCB1) (cultured in medium with 2 mg/ml G418) [32]. All cells were grown in drug free culture medium for more than 2 weeks before assay.
Cell cytotoxicity test
The MTT assay was performed to assess the sensitivity of cells to anticancer drugs as described previously [33]. Briefly, cells were seeded in 96-well plates and allowed to attach overnight. Then cells were preincubated with or without UMMS-4 (or the control inhibitor for ABCB1 (verapamil) or ABCG2 (FTC), respectively) for 1 h, and then various concentrations of anticancer drugs were added to the wells. After 68 h of incubation, MTT (5 mg/mL, 20 μL/well) was added into the cells for 4 h (37°C). Afterwards, the medium was discarded, and 200 μL of dimethylsulfoxide (DMSO) was added to dissolve the formazan product from the metabolism of MTT. Optical density was measured at 540 nm with background subtraction at 670 nm by use of the Model 550 Microplate Reader (BIO-RAD, Hercules, CA, USA). The concentration required to inhibit cell growth by 50% (IC50) was calculated from survival curves using the Bliss method. The degree of resistance was estimated by dividing the IC50 for the MDR cells by that of the parental sensitive cells. The fold-reversal factor of MDR was calculated by dividing the IC50 of the anticancer drug in the absence of UMMS-4 by that obtained in the presence of UMMS-4.
Rho 123 and Dox accumulation
The effect of UMMS-4 on the cellular accumulations of Rho 123 and Dox was measured by flow cytometry. Briefly, the cells were seeded in 6-well plates and then incubated with UMMS-4 at desired concentration at 37°C for 3 h. Then 10 μmol/L Dox or 5 mmol/L Rho 123 was added and further incubated for another 3 h or 0.5 h, respectively. Following this incubation, the cells were collected and washed twice with ice-cold PBS. Finally, the cells were resuspended in PBS and intracellular fluorescence was measured by flow cytometric analysis. Verapamil was used as a positive control ABCB1 inhibitor [34]. The relative values were identified by dividing the fluorescence intensity of each measurement by that of control cells.
Western blot analysis
Identical amounts of total cell lysates (50 μg of protein), from cells treated with a range of concentration of UMMS-4, were resolved using SDS-PAGE and transferred onto PVDF membranes. After incubation in a blocking solution containing 5% skim milk in TBST buffer (10 mmol/L Tris-HCl [pH 8.0], 150 mmol/L NaCl, and 0.1% Tween 20) for 1 h at room temperature, the membranes were immunoblotted overnight with primary anti-ABCB1 (1:200 dilution) and anti-GAPDH (1:200 dilution) antibodies at 4°C, and were then incubated overnight at 4°C with HRP-conjugated secondary antibody (1:1000 dilution). Protein-antibody complexes were detected using chemiluminescence.
Reverse transcription-PCR analysis of ABCB1 expression
After treating the ABCB1-overexpressing cells (KBv200 or MCF-7/adr) with a range of different concentrations and for different duration of UMMS-4, total cellular RNA was isolated by Trizol Reagent (Molecular Research Center, USA) RNA extraction kit following manufacturer’s instruction. The first strand cDNA was synthesized by Oligo dT primers. Specific PCR primers used were 5’-GTGGG-GCAAGTCAGTTCATT-3’ (forward) and 5’-TCTTCACCTCCAGGCTCAGT-3’ (reverse) for ABCB1 and 5’-GAGTCAACGGATTTGGTCGT-3’ (forward) and 5’-GATCTCGCTCCTGGAAGATG-3’ (reverse) for GAPDH. Using the GeneAmp PCR system 9700 (PE Applied Biosystems, USA), PCR amplification reactions were carried out at 94°C for 2 min for initial denaturation, and then at 94°C for 30 s, 58°C for 30 s, and 72°C for 1 min. After 35 cycles of amplification, an additional extension was done at 72°C for 10 min. PCR products were resolved and examined by 1.5% agarose gel electrophoresis. Expected reverse transcription-PCR (RT-PCR) products were 222 bp for ABCB1 and 224 bp for GAPDH, respectively.
Real-time PCR was performed by the Bio-Rad CFX96TM Real-Time (Applied Biosystems, USA). The geometric mean of the GAPDH was used as an internal control to normalize the variability in expression levels. The primers used for ABCB1 and GAPDH were the same as described above. The PCR reactions were performed at 50°C for 2 min, 95°C for 5 min and 40 cycles at 95°C for 15 s, 60°C for 30 s. Relative quantification of ABCB1 was performed using the 2–ΔΔCt method [35]. To ensure reproducibility of the results, all real time PCR analyses were performed in triplicate and in three independent experiments.
Dox efflux studies
Dox efflux was determined following a modification of methods described earlier [36]. KB and KBv200 cells were incubated with 10 μmol/L Dox for 3 h at 37°C. Each sample was washed twice with ice-cold PBS, and then resuspended in Dox-free buffer with or without 20 μmol/L UMMS-4. Thereafter, at 0, 15, 30, 60 and 120 min, cells were collected and washed again twice with ice-cold PBS, and analyzed with flow cytometry.
ABCB1 ATPase activity assay
A colorimetric ATPase assay was performed as previously described with minor modification [37]. Briefly, crude membranes isolated from High Five insect cells expressing ABCB1 (20 μg protein/sample) were incubated in ATPase assay buffer (50 mmol/L MES [pH 6.8], 50 mmol/L KCl, 5 mmol/L sodium azide, 2 mmol/L EGTA, 2 mol/L DTT, 1 mmol/L ouabain, 10 mmol/L MgCl2] with or without 0.3 mol/L sodium orthovanadate at 37°C for 5 min and then incubated with different concentrations of UMMS-4 at 37°C for 5 min. The ATPase reaction was induced by the addition of 5 mmol/L Mg-ATP and the total volume was 60 L. After incubation at 37°C for 20 min, the reactions were stopped by loading 30 L of 10% SDS solution. Specific UMMS-4-stimulated ABCB1 ATPase activity (i.e. vanadate-sensitive) was determined as the difference between the amounts of inorganic phosphate (Pi) released from ATP in the absence and presence of sodium orthovanadate. The amount of Pi released was estimated by comparing to a standard curve.
Statistical analysis
Results are shown as means±SD, unless otherwise indicated. All experiments were repeated at least three times and the differences were determined by using the Student’s t-test. The statistical significance was determined to be P<0.05.
Results
UMMS-4 potentiated the sensitivity of anticancer agents in ABCB1-overexpressing cells but not in ABCG2- or ABCC1-overexpreesing cells
The overexpression of the three major MDR transporters was first verified in the resistant cell lines used in the study by Western blot analysis. ABCB1 was overexpressed in KBv200, MCF-7/adr and HEK293/ABCB1 cells, whereas ABCG2 and ABCC1 were overexpressed in S1-M1-80 and HL60/ADR cells, respectively (Figure 1A). The basal expression of ABCB1, ABCG2 and ABCC1 was undetectable in their respective parental cell lines. MTT assay was then performed to examine the cytotoxic effect of UMMS-4 alone on these cell lines. The results showed that UMMS-4, at up to 20 μmol/L, had no appreciable cytotoxic effect to all cell lines used in this study, and more 90% cells were survived (Figure 1C-G). Therefore, 20 μmol/L was used as the highest MDR reversal concentration of UMMS-4 in all subsequent analyses.
To investigate the possible reversal effect of UMMS-4 on ABCB1-, ABCG2- and ABCC1-mediated MDR, the cytotoxic effect of several chemotherapeutic agents in various pairs of sensitive and resistant cells was tested with or without the addition of a range of different concentration of UMMS-4 (5, 10, or 20 μmol/L). A control ABCB1 inhibitor (verapamil, 10 μmol/L) was used for comparison. As expected, the five MDR cell lines are highly resistant to to the various chemotherapeutic agents than their parental cell lines. With the addition of UMMS-4, only ABCB1-mediated resistant cells were found to be sensitized. The mean IC50 values of chemotherapeutic agents in different cell lines in the absence or presence of different concentration of UMMS-4 are summarized in Tables 1 and 2. Interestingly, only in ABCB1-overexpressing cells, UMMS-4 was found to enhance the cytotoxicity of Dox (1.98, 7.88, and 3.50-fold), VCR (2.27, 6.76, and 17.53-fold) and paclitaxel (1.58, 3.40, and 8.93-fold) in a concentration-dependent manner in KBv200 cells or Dox (1.33, 2.44, and 7.28-fold) in MCF-7/adr cells (Table 1). Moreover, UMMS-4 was also found to remarkably decrease ABCB1-mediated resistance to Dox (1.68, 4.31, and 9.13-fold) in the ABCB1-transfected HEK293/ABCB1 cell line in a concentration-dependent manner (Table 2). On the other hand, UMMS-4 did not alter the cytotoxicity of non-ABCB1 substrates (cisplatin) in either MDR cells or their parental sensitive cells (Tables 1 and 2). In addition, UMMS-4 did not affect the sensitivity of the drug-sensitive parental cells to any antineoplastic drugs used in our study (Tables 1 and 2). Furthermore, as illustrated in Table 1, UMMS-4 also did not affect the anticancer drug activity in ABCG2- (S1-M1-80) or ABCC1-overexpressing (HL60/ADR) cells, respectively. Taken together, our results suggest that UMMS-4 selectively sensitized ABCB1-overexpressing cells to antineoplastic drugs that are ABCB1 substrates.
Table 1.
Effect of UMMS-4 on reversing ABCB1-, ABCC1- and ABCG2-mediated MDR in pairs of sensitive and drug-resistant cell lines
| Compounds | IC50±SD (μM) (fold-reversal) | |||
|
| ||||
| KB | KBv200 (ABCB1-overexpressing) | |||
|
| ||||
| Doxorubicin | 0.05349±0.0085 | (1.00) | 3.6186±0.0196 | (1.00) |
| +5 μM UMMS-4 | 0.0467±0.0077 | (1.14) | 1.8249±0.0331** | (1.98) |
| +10 μM UMMS-4 | 0.0358±0.0058 | (1.49) | 1.0338±0.0261** | (3.50) |
| +20 μM UMMS-4 | 0.0332±0.0065 | (1.61) | 0.4589±0.0198** | (7.88) |
| +10 μM Verapamil | 0.0360±0.0085 | (1.48) | 0.2324±0.0162** | (15.57) |
| Vincristine | 0.0026±0.0002 | (1.00) | 0.2227±0.0073 | (1.00) |
| +5 μM UMMS-4 | 0.0019±0.0006 | (1.36) | 0.0979±0.0093** | (2.27) |
| +10 μM UMMS-4 | 0.0020±0.0005 | (1.30) | 0.0329±0.0035** | (6.76) |
| +20 μM UMMS-4 | 0.0013±0.0001 | (2.00)* | 0.0127±0.0008** | (17.53) |
| +10 μM Verapamil | 0.0012±0.0004 | (2.16)* | 0.0070±0.0010 | (31.81) |
| Paclitaxel | 0.0018±0.0001 | (1.00) | 0.1277±0.01166 | (1.00) |
| +5 μM UMMS-4 | 0.0017±0.0002 | (1.05) | 0.0806±0.0033* | (1.58) |
| +10 μM UMMS-4 | 0.0017±0.0001 | (1.05) | 0.0376±0.0039** | (3.40) |
| +20 μM UMMS-4 | 0.0017±0.0001 | (1.05) | 0.0143±0.0025** | (8.93) |
| +10 μM Verapamil | 0.0012±0.0001 | (1.50)* | 0.0100±0.0012** | (12.77) |
| Cisplatin | 1.9952±0.0588 | (1.00) | 2.1979±0.2002 | (1.00) |
| +20 μM UMMS-4 | 2.0639±0.2491 | (0.97) | 2.0432±0.3123 | (1.07) |
| +10 μM Verapamil | 2.1256±0.1013 | (0.94) | 1.9593±0.2829 | (1.12) |
|
| ||||
| MCF-7 | MCF-7/adr (ABCB1-overexpressing) | |||
|
| ||||
| Doxorubicin | 0.1895±0.0043 | (1.00) | 9.5213± 0.7970 | (1.00) |
| +5 μM UMMS-4 | 0.2037±0.0072 | (0.93) | 7.1759±0.1970* | (1.33) |
| +10 μM UMMS-4 | 0.1730±0.0065 | (1.09) | 3.9058±0.0422** | (2.44) |
| +20 μM UMMS-4 | 0.1413±0.0228 | (1.34) | 1.3508±0.032** | (7.28) |
| +10 μM Verapamil | 0.1812±0.0129 | (1.05) | 0.8076±0.0196** | (11.80) |
| Cisplatin | 15.3195±0.0652 | (1.00) | 15.3670±0.0542 | (1.00) |
| +20 μM UMMS-4 | 16.2365±0.0433 | (0.94) | 15.9191±0.0493 | (0.97) |
| +10 μM Verapamil | 14.7376±0.1182 | (1.03) | 17.3571±0.0615 | (0.89) |
|
| ||||
| HL60 | HL60/ADR (ABCC1-overexpressing) | |||
|
| ||||
| Doxorubicin | 0.0338±0.0004 | (1.00) | 2.4398±0.3303 | (1.00) |
| +5 μM UMMS-4 | 0.0319±0.0021 | (1.06) | 2.4095±0.2344 | (1.01) |
| +10 μM UMMS-4 | 0.0387±0.0017 | (0.87) | 2.5466±0.5358 | (0.96) |
| +20 μM UMMS-4 | 0.0334±0.0044 | (1.01) | 2.2553±0.4540 | (1.08) |
| +40 μM MK571 | 0.0248±0.0021 | (1.36) | 0.2590±0.0498** | (9.42) |
|
| ||||
| S1 | S1-M1-80 (ABCG2-overexpressing) | |||
|
| ||||
| Topotecan | 0.2112±0.0281 | (1.00) | 11.127±0.1310 | (1.00) |
| +5 μM UMMS-4 | 0.1772±0.0323 | (1.19) | 11.068±0.2126 | (1.05) |
| +10 μM UMMS-4 | 0.2199±0.0261 | (0.96) | 9.534±0.3519 | (1.16) |
| +20 μM UMMS-4 | 0.1849±0.0372 | (1.40) | 9.382±0.3394 | (1.18) |
| +2.5 μM fumitremorgin C | 0.1817±0.09 | (1.16) | 0.608±0.0843** | (18.30) |
Drug cytotoxicity was evaluated by MTT assay as described in Materials and Methods. Data are shown as the mean±standard deviation (SD) of at least three independent experiments performed in triplicate. The fold-reversal of MDR (values given in parentheses) was calculated by dividing the IC50 for cells with the chemotherapeutic drugs in the absence of inhibitor by that obtained in the presence of inhibitor.
p<0.05;
p<0.01, for values versus that obtained in the absence of UMMS-4, verapamil,MK571 or FTC.
Table 2.
Effect of UMMS-4 on reversing ABCB1- mediated MDR in transfected cell lines
| Compounds | IC50±SD (μM) (fold-reversal) | |||
|---|---|---|---|---|
|
| ||||
| HEK293/pcDNA3.1 | HEK293/ABCB1 | |||
| Doxorubicin | 0.05916±0.0084 | (1.00) | 2.2052±0.2501 | (1.00) |
| +5 μM UMMS-4 | 0.0490±0.0069 | (1.20) | 1.3077±0.0492** | (1.68) |
| +10 μM UMMS-4 | 0.0542±0.0053 | (1.09) | 0.5115±0.0306** | (4.31) |
| +20 μM UMMS-4 | 0.0486±0.0031 | (1.21) | 0.2415±0.0087** | (9.13) |
| +10 μM Verapamil | 0.0545±0.008 | (1.08) | 0.1242±0.0249** | (17.69) |
| Cisplatin | 4.3149±0.1850 | (1.00) | 6.4096±0.2586 | (1.00) |
| +20 μM UMMS-4 | 6.1039±0.3488 | (0.71) | 6.3202±0.3918 | (1.01) |
| +10 μM Verapamil | 2.9149±0.1246 | (1.47) | 6.0898±0.2288 | (1.05) |
Drug cytotoxicity was evaluated by MTT assay as described in Materials and Methods. Data are presented as means±SD (μM) from at least three independent experiments performed in triplicate. The fold reversal of MDR (values given in parentheses) was calculated by dividing the IC50 for cells with the anticancer drugs in the absence of inhibitor by that obtained in the presence of inhibitor.
P<0.01, significantly different from those obtained in the absence of inhibitor.
UMMS-4 increased the accumulation of Rho 123 and Dox in ABCB1-overexpressing cells
The results above indicated that UMMS-4 could enhance the sensitivity of ABCB1-overexpressing cells to ABCB1 substrate chemotherapeutic agents. To understand the underlying mechanism, we measured the effect of UMMS-4 on the accumulation of a ABCB1 substrate Dox in ABCB1-overexpressing cells and their parental cells in the absence or presence of UMMS-4. UMMS-4, at 5, 10, and 20 μmol/L, concentration-dependently increased the intracellular accumulation of Dox in all ABCB1-overexpressing cells (Figure 2). It is noted that the increase in Dox accumulation by UMMS-4 at 20 μmol/L was similar to that achieved by verapamil (a known ABCB1 inhibitor) at 10 μmol/L. The fluorescence index of Dox in the presence of UMMS-4 was increased by 1.35-, 1.87-, and 2.35-fold in KBv200 cells; 1.09-, 1.66-, and 2.07-fold in MCF-7/adr cells, respectively (Figure 2B, 2D). However, UMMS-4 did not significantly alter the intracellular accumulation of Dox in parental cells.
Figure 2.
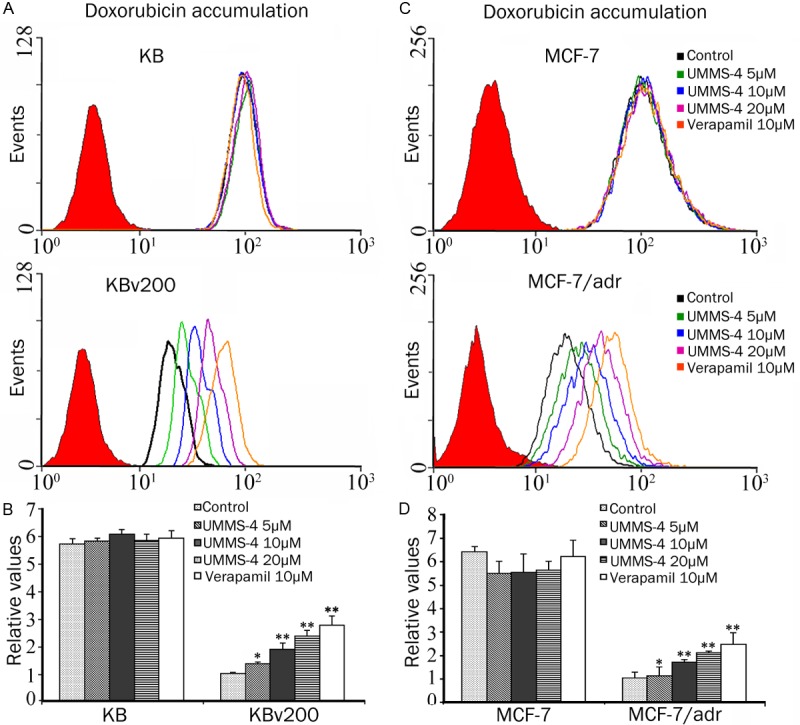
Effect of UMMS-4 on the cellular accumulation of Dox. The accumulation of Dox in KB, KBv200 (A) and in MCF-7, MCF-7/adr (C) was measured by flow cytometric analysis as described in materials and methods. The results are summarized by plotting in histograms the fold change in fluorescence intensity relative to that in untreated (control) MDR cells (B and D). Columns, means of triplicate determinations; bars, SD. *, P<0.05; **, P<0.01, versus the MDR control group, respectively.
Subsequently, we examined the intracellular accumulation of Rho 123 (a fluorescent probe ABCB1 substrate) in the same set of ABCB1-overexpressing cell lines. As depicted in Figure 3, after incubating with 5, 10, or 20 μmol/L of UMMS-4, Rho 123 accumulation was significantly enhanced by 2.00-, 3.30-, and 8.31-fold in KBv200 cells; and 1.87-, 5.59-, and 9.03-fold in MCF-7/adr cells, respectively. However, in the drug sensitive cells, UMMS-4 did not appreciably alter the intracellular accumulation of Rho 123 (Figure 3). The increase in Rho 123 accumulation by the reference ABCB1 inhibitor (verapamil, 10 μmol/L) was found to be 17.81 and 17.90-fold, respectively, in KBv200 and MCF-7/adr cells. Taken together, these data suggested that UMMS-4 was able to directly inhibit the drug efflux function of ABCB1, resulting in the increase of the intracellular accumulation of Rho 123 and Dox.
Figure 3.
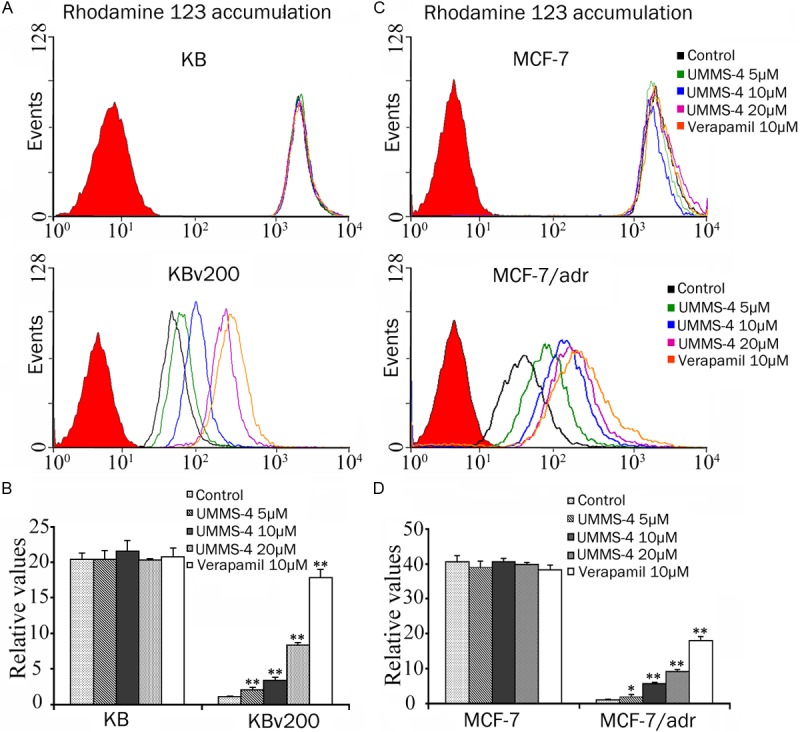
Effect of UMMS-4 on the cellular accumulation of a fluorescent ABCB1 substrate probe compound (Rho 123). The accumulation of Rho 123 was measured by flow cytometric analysis in KB, KBv200 (A) and in MCF-7, MCF-7/adr (C). The results are summarized as histograms in (B) and (D), respectively. Columns, means of triplicate determinations; bars, SD. *, P<0.05; **, P<0.01, versus the control group. Experiments were performed at least three times independently. Flow cytometric histograms from a representative experiment are shown in (A) and (C).
UMMS-4 inhibited the efflux of Dox in MDR cells overexpressing ABCB1
To further investigate whether UMMS-4 inhibited the transport function of ABCB1, the efflux of Dox was examined by flow cytometry in ABCB1-overexpressing MDR cells and their parental cells. The efflux of Dox after an initial 3 h drug loading was traced over 2 h and the result was shown in Figure 4A. As expected, the extent of Dox efflux from the ABCB1-overexpressing KBv200 cells was remarkably greater than that in the parental KB cells (at 60 min, 63±4.8% efflux in KBv200 versus only 15±4.1% efflux in KB cells). Importantly, UMMS-4 was found to significantly inhibit Dox efflux from KBv200 cells but it had no effect on the parental KB cells. At 120 min, 47±4.6% of accumulated Dox was pumped out of KBv200 cells in the presence of 20 μmol/L UMMS-4, whereas 69±3.2% of accumulated Dox was lost from KBv200 cells in the absence of UMMS-4. On the other hand, approximately the same extent of Dox efflux was observed in KB cells in the absence (23±6.6%) and in the presence (18±4.8%) of 20 μmol/L UMMS-4. These results indicated that UMMS-4 specifically inhibited the efflux function of ABCB1.
Figure 4.
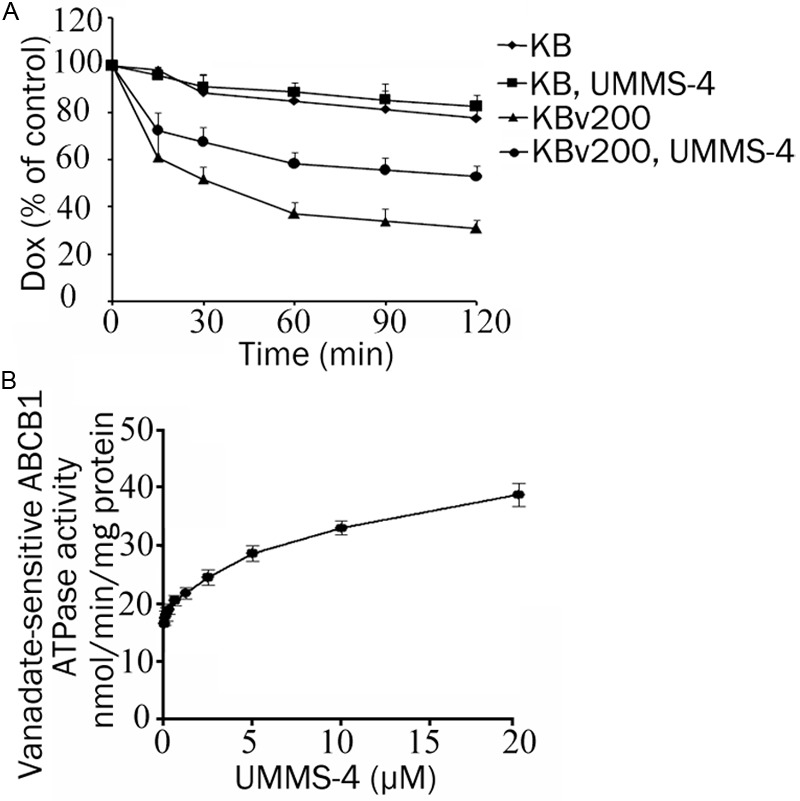
Effect of UMMS-4 on the efflux of Dox and on ABCB1 ATPase activity. Time course of DOX efflux was measured in KB and KBv200 cells, with or without 20 μM UMMS-4 (A). Vanadate-sensitive ATPase activity of ABCB1 (B). The ATP hydrolysis in membrane vesicles was determined with different concentrations of UMMS-4, as described in “Materials and Methods”. All experiments were repeated at least three times. Data shown are means±SD for independent determinations in triplicate.
UMMS-4 activated the ATPase activity of ABCB1
When ABC transporters interact with their modulators, their ATPases are known to be stimulated in order to generate energy from ATP hydrolysis to mediate drug efflux. To provide evidence as to whether UMMS-4 interacts with ABCB1, we measured ABCB1 ATPase activity after incubating crude membrane from ABCB1-overexpressing sf6 cells with a range of concentrations of UMMS-4. Sodium orthovanadate was added in our assay to suppress other major membrane ATPases, therefore our measurement should be specific for ABCB1. As shown in Figure 4B, UMMS-4 stimulated ABCB1 ATPase activity in a concentration-dependent manner. The data suggests that UMMS-4 interacts with ABCB1.
UMMS-4 did not affect the expression of ABCB1 at mRNA or protein level
The reversal of ABCB1-mediated MDR can be achieved either by inhibiting the transport function or by lowering the expression of the transporter. Therefore, we determined the effect of UMMS-4 on the protein and mRNA expression of ABCB1 by Western blot and RT-PCR analyses, respectively. The results showed that UMMS-4, at 5, 10, 20, and 40 μmol/L, did not appreciably alter the protein expression of ABCB1 in KBv200 and MCF-7/adr cells (Figure 5A, 5B). Moreover, quantitative real-time PCR results indicated that there was no significant difference in the expression of ABCB1 mRNA in both KBv200 and MCF-7/adr cells after treatment with UMMS-4 at up to 40 μmol/L (Figure 5C, 5D). These data suggested that the reversal of MDR is most likely obtained by inhibition of the efflux function of ABCB1 as opposed to the downregulation of ABCB1 mRNA or protein expression.
Figure 5.
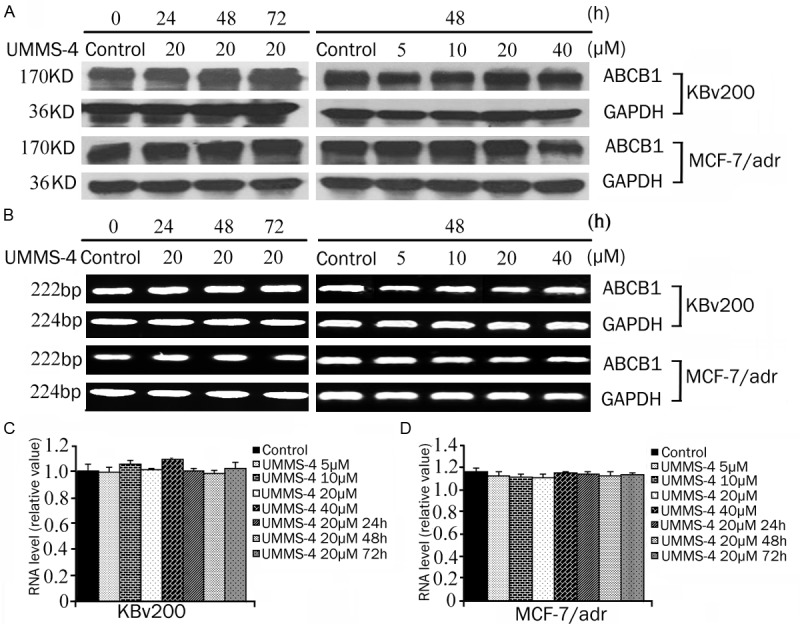
Effect of UMMS-4 on mRNA and protein expression of ABCB1 in MDR cells. KBv200 (A and B) and MCF-7/adr (C and D) were treated with UMMS-4 at a range of concentrations for 48 h or for various time up to 72 h at 20 μmol/L. Equal amounts of total cell lysates were loaded and detected by Western blot analysis. A representative set of images from at least three independent experiments is shown. The mRNA level of ABCB1 was determined by PCR and RT-PCR as described in Materials and Methods.
Discussion
Cancer has become an emerging public health problem for its high morbidity and mortality [38,39]. Overexpression of the efflux transporter ABCB1 in cancer cells is well known as a major obstacle to successful chemotherapy because of its contribution to multidrug resistance during anticancer treatment [40,41]. The basal expression of ABCB1 in some solid tumors is known to be relatively high, which cause intrinsic resistance to anticancer drugs. Moreover, it has also been reported that numerous anticancer drugs can induce the mdr1 gene, subsequently leading to acquired resistance. As a molecular prognostic marker, ABCB1 is also implicated in treatment failure, poor outcome, short survival time and low CR rate. Research efforts to overcome multidrug resistance have primarily focused on the inhibition of the ABCB1 transporter function in resistant cancer cells. Therefore, the combination of anticancer drugs with a non-toxic and potent ABCB1 inhibitor may be a promising approach to solve the MDR problem. Since the discovery of verapamil and cyclosporin A as ABCB1 inhibitors and MDR reversing agents 30 years ago, significant efforts have been made to search and design specific ABCB1 inhibitors for MDR reversal. However, to date, none of the reported ABCB1 inhibitors have been approved for clinical use because of intolerable toxicity or unpredictable pharmacokinetic drug-drug interactions. Therefore, the development of more efficacious, non-toxic and less expensive ABCB1 inhibitors is still warranted to help reverse MDR in cancer cells [42].
UMMS-4, which shows low cytotoxic activity in our study, is a hydrophobic compound with multiple amine groups, which fits well in our structure activity relationship characteristic for ABCB1 inhibitors. Therefore, in present study, we investigated the MDR reversing effect of UMMS-4 on ABCB1-, ABCG2- and ABCC1-overexpressing cells. Our result showed that UMMS-4 significantly and specifically sensitized ABCB1-overexpressing cells to doxorubicin and paclitaxel. At concentration up to 20 μmol/L, UMMS-4 did not appreciably affect the cytotoxicity of anticancer drugs in the parental sensitive KB, MCF-7, S1 or HEK293/pcDNA3.1 cells used in this study. Moreover, UMMS-4 also did not affect the cytotoxicity of cisplatin, which is not a substrate of ABCB1, in both sensitive and resistant cells. In addition, UMMS-4 had no significant effect on sensitivity of ABCG2 and ABCC1-overexpressing cells to their respective substrate anticancer drugs. These data indicated that the reversing ability of UMMS-4 was specific to ABCB1. The drug accumulation and efflux assay using the ABCB1 substrates (Rho 123 and Dox) also demonstrated that UMMS-4 is specifically inhibiting the efflux activity of ABCB1. Since ABCB1 mRNA and protein expressions were not affected by UMMS-4 treatment (up to 72 h), the MDR reversing effect of UMMS-4 is not related to the regulation of the transporter expression. ABCB1 is known to utilize energy from ATP hydrolysis to mediate its drug efflux activity. Therefore, ABCB1 ATPase activity, measured by the rate of ATP hydrolysis, is directly proportional to its transport activity [9]. Our result revealed that UMMS-4 stimulated ABCB1 ATPase activity in a concentration-dependent manner, suggesting an interaction between UMMS-4 and ABCB1. UMMS-4 may potentially be a substrate of ABCB1 (Figure 6).
Figure 6.
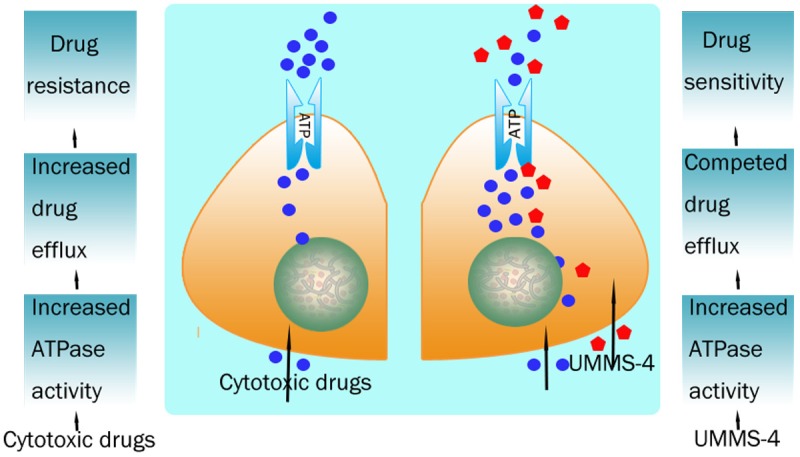
A schematic overview of the effect of UMMS-4 on inhibiting function of ABCB1 transporter.
In summary, UMMS-4 was found to inhibit the drug efflux activity of ABCB1 at non-toxic concentrations. The combination of UMMS-4 with other anticancer drugs that are substrates of ABCB1 may be useful in surmounting clinical drug resistance in cancer chemotherapy.
Acknowledgements
We thank S Bates (National Cancer Institute, NIH) for the ABCG2-overexpressing cell lines; This work was supported by grants from the National High Technology Research and Development Program of China (863 Program) (NO. 2012AA02A303 for L.W. Fu); the Major State Basic Research Development Program of China (973 Program) (NO. 2012CB967000 for L.W. Fu, NO. 2010CB833603 for J.Y. Cai) and Scientific and technological foundation of Guangzhou (NO. 12C32061587 for L.W. Fu).
Disclosure of conflict of interest
The authors have no conflict of interests for this manuscript.
References
- 1.Perez-Tomas R. Multidrug resistance: retrospect and prospects in anti-cancer drug treatment. Curr Med Chem. 2006;13:1859–1876. doi: 10.2174/092986706777585077. [DOI] [PubMed] [Google Scholar]
- 2.Donnenberg VS, Donnenberg AD. Multiple drug resistance in cancer revisited: the cancer stem cell hypothesis. J Clin Pharmacol. 2005;45:872–877. doi: 10.1177/0091270005276905. [DOI] [PubMed] [Google Scholar]
- 3.Sparreboom A, Danesi R, Ando Y, Chan J, Figg WD. Pharmacogenomics of ABC transporters and its role in cancer chemotherapy. Drug Resist Updat. 2003;6:71–84. doi: 10.1016/s1368-7646(03)00005-0. [DOI] [PubMed] [Google Scholar]
- 4.Vasiliou V, Vasiliou K, Nebert DW. Human ATP-binding cassette (ABC) transporter family. Hum Genomics. 2009;3:281–290. doi: 10.1186/1479-7364-3-3-281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Choi CH. ABC transporters as multidrug resistance mechanisms and the development of chemosensitizers for their reversal. Cancer Cell Int. 2005;5:30. doi: 10.1186/1475-2867-5-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tiwari AK, Sodani K, Dai CL, Ashby CR Jr, Chen ZS. Revisiting the ABCs of multidrug resistance in cancer chemotherapy. Curr Pharm Biotechnol. 2011;12:570–594. doi: 10.2174/138920111795164048. [DOI] [PubMed] [Google Scholar]
- 7.Walker JE, Saraste M, Runswick MJ, Gay NJ. Distantly related sequences in the alpha and beta-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J. 1982;1:945–951. doi: 10.1002/j.1460-2075.1982.tb01276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Germann UA. P-glycoprotein--a mediator of multidrug resistance in tumour cells. Eur J Cancer. 1996;32A:927–944. doi: 10.1016/0959-8049(96)00057-3. [DOI] [PubMed] [Google Scholar]
- 9.Ambudkar SV, Dey S, Hrycyna CA, Ramachandra M, Pastan I, Gottesman MM. Biochemical cellular, and pharmacological aspects of the multidrug transporter. Annu Rev Pharmacol Toxicol. 1999;39:361–398. doi: 10.1146/annurev.pharmtox.39.1.361. [DOI] [PubMed] [Google Scholar]
- 10.Malayeri R, Filipits M, Suchomel RW, Zochbauer S, Lechner K, Pirker R. Multidrug resistance in leukemias and its reversal. Leuk Lymphoma. 1996;23:451–458. doi: 10.3109/10428199609054853. [DOI] [PubMed] [Google Scholar]
- 11.Krishna R, Mayer LD. Multidrug resistance (MDR) in cancer. Mechanisms, reversal using modulators of MDR and the role of MDR modulators in influencing the pharmacokinetics of anticancer drugs. Eur J Pharm Sci. 2000;11:265–283. doi: 10.1016/s0928-0987(00)00114-7. [DOI] [PubMed] [Google Scholar]
- 12.Sikic BI. Pharmacologic approaches to reversing multidrug resistance. Semin Hematol. 1997;34:40–47. [PubMed] [Google Scholar]
- 13.Fojo AT, Ueda K, Slamon DJ, Poplack DG, Gottesman MM, Pastan I. Expression of a multidrug- resistance gene in human tumors and tissues. Proc Natl Acad Sci U S A. 1987;84:265–269. doi: 10.1073/pnas.84.1.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Teodori E, Dei S, Martelli C, Scapecchi S, Gualtieri F. The functions and structure of ABC transporters: implications for the design of new inhibitors of Pgp and MRP1 to control multidrug resistance (MDR) Curr Drug Targets. 2006;7:893–909. doi: 10.2174/138945006777709520. [DOI] [PubMed] [Google Scholar]
- 15.Leonard GD, Fojo T, Bates SE. The role of ABC transporters in clinical practice. Oncologist. 2003;8:411–424. doi: 10.1634/theoncologist.8-5-411. [DOI] [PubMed] [Google Scholar]
- 16.Krishna R, Mayer LD. Modulation of P-glycoprotein (PGP) mediated multidrug resistance (MDR) using chemosensitizers: recent advances in the design of selective MDR modulators. Curr Med Chem Anticancer Agents. 2001;1:163–174. doi: 10.2174/1568011013354705. [DOI] [PubMed] [Google Scholar]
- 17.Coley HM. Overcoming multidrug resistance in cancer: clinical studies of p-glycoprotein inhibitors. Methods Mol Biol. 2010;596:341–358. doi: 10.1007/978-1-60761-416-6_15. [DOI] [PubMed] [Google Scholar]
- 18.Darby RA, Callaghan R, McMahon RM. P-glycoprotein inhibition: the past, the present and the future. Curr Drug Metab. 2011;12:722–731. doi: 10.2174/138920011798357006. [DOI] [PubMed] [Google Scholar]
- 19.Freshney RI. ZD6474 reverses multidrug resistance by directly inhibiting the function of P-glycoprotein. Br J Cancer. 2007;97:1714. doi: 10.1038/sj.bjc.6604077. author reply 1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tan B, Piwnica-Worms D, Ratner L. Multidrug resistance transporters and modulation. Curr Opin Oncol. 2000;12:450–458. doi: 10.1097/00001622-200009000-00011. [DOI] [PubMed] [Google Scholar]
- 21.Roe M, Folkes A, Ashworth P, Brumwell J, Chima L, Hunjan S, Pretswell I, Dangerfield W, Ryder H, Charlton P. Reversal of P-glycoprotein mediated multidrug resistance by novel anthranilamide derivatives. Bioorg Med Chem Lett. 1999;9:595–600. doi: 10.1016/s0960-894x(99)00030-x. [DOI] [PubMed] [Google Scholar]
- 22.Dantzig AH, Shepard RL, Cao J, Law KL, Ehlhardt WJ, Baughman TM, Bumol TF, Starling JJ. Reversal of P-glycoprotein-mediated multidrug resistance by a potent cyclopropyldibenzosuberane modulator, LY335979. Cancer Res. 1996;56:4171–4179. [PubMed] [Google Scholar]
- 23.van Zuylen L, Sparreboom A, van der Gaast A, van der Burg ME, van Beurden V, Bol CJ, Woestenborghs R, Palmer PA, Verweij J. The orally administered P-glycoprotein inhibitor R101933 does not alter the plasma pharmacokinetics of docetaxel. Clin Cancer Res. 2000;6:1365–1371. [PubMed] [Google Scholar]
- 24.Newman MJ, Rodarte JC, Benbatoul KD, Romano SJ, Zhang C, Krane S, Moran EJ, Uyeda RT, Dixon R, Guns ES, Mayer LD. Discovery and characterization of OC144-093, a novel inhibitor of P-glycoprotein-mediated multidrug resistance. Cancer Res. 2000;60:2964–2972. [PubMed] [Google Scholar]
- 25.Hyafil F, Vergely C, Du Vignaud P, Grand-Perret T. In vitro and in vivo reversal of multidrug resistance by GF120918, an acridonecarboxamide derivative. Cancer Res. 1993;53:4595–4602. [PubMed] [Google Scholar]
- 26.Germann UA, Shlyakhter D, Mason VS, Zelle RE, Duffy JP, Galullo V, Armistead DM, Saunders JO, Boger J, Harding MW. Cellular and biochemical characterization of VX-710 as a chemosensitizer: reversal of P-glycoprotein-mediated multidrug resistance in vitro. Anticancer Drugs. 1997;8:125–140. doi: 10.1097/00001813-199702000-00004. [DOI] [PubMed] [Google Scholar]
- 27.Aslam SAM, Athar MM, Ashfaq UA, Gardiner JM, Montero C, Detorio M, Parvez M, Schinazi RF. Synthesis, molecular docking and antiviral screening of novel N’1(1):-substitutedbenzylidene-2-(4-methyl-5,5-dioxido-3-phenylbenzo[e] pyrazolo[4,3-c] [1,2] thiazin-1(4H)-yl)acetohydrazides. Medicinal Chemistry Research. 2013 DOI 10.1007/s00044-013-0879-7. [Google Scholar]
- 28.Zhang JY, Wu HY, Xia XK, Liang YJ, Yan YY, She ZG, Lin YC, Fu LW. Anthracenedione derivative 1403P-3 induces apoptosis in KB and KBv200 cells via reactive oxygen species-independent mitochondrial pathway and death receptor pathway. Cancer Biol Ther. 2007;6:1413–1421. doi: 10.4161/cbt.6.9.4543. [DOI] [PubMed] [Google Scholar]
- 29.Fu L, Liang Y, Deng L, Ding Y, Chen L, Ye Y, Yang X, Pan Q. Characterization of tetrandrine, a potent inhibitor of P-glycoprotein-mediated multidrug resistance. Cancer Chemother Pharmacol. 2004;53:349–356. doi: 10.1007/s00280-003-0742-5. [DOI] [PubMed] [Google Scholar]
- 30.Litman T, Brangi M, Hudson E, Fetsch P, Abati A, Ross DD, Miyake K, Resau JH, Bates SE. The multidrug-resistant phenotype associated with overexpression of the new ABC half-transporter, MXR (ABCG2) J Cell Sci. 2000;113:2011–2021. doi: 10.1242/jcs.113.11.2011. [DOI] [PubMed] [Google Scholar]
- 31.Tang R, Faussat AM, Majdak P, Perrot JY, Chaoui D, Legrand O, Marie JP. Valproic acid inhibits proliferation and induces apoptosis in acute myeloid leukemia cells expressing P-gp and MRP1. Leukemia. 2004;18:1246–1251. doi: 10.1038/sj.leu.2403390. [DOI] [PubMed] [Google Scholar]
- 32.Robey RW, Shukla S, Finley EM, Oldham RK, Barnett D, Ambudkar SV, Fojo T, Bates SE. Inhibition of P-glycoprotein (ABCB1)- and multidrug resistance-associated protein 1 (ABCC1)-mediated transport by the orally administered inhibitor, CBT-1((R)) Biochem Pharmacol. 2008;75:1302–1312. doi: 10.1016/j.bcp.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yan YY, Zheng LS, Zhang X, Chen LK, Singh S, Wang F, Zhang JY, Liang YJ, Dai CL, Gu LQ, Zeng MS, Talele TT, Chen ZS, Fu LW. Blockade of Her2/neu binding to Hsp90 by emodin azide methyl anthraquinone derivative induces proteasomal degradation of Her2/neu. Mol Pharm. 2011;8:1687–1697. doi: 10.1021/mp2000499. [DOI] [PubMed] [Google Scholar]
- 34.Ford JM, Hait WN. Pharmacology of drugs that alter multidrug resistance in cancer. Pharmacol Rev. 1990;42:155–199. [PubMed] [Google Scholar]
- 35.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 36.Dai CL, Tiwari AK, Wu CP, Su XD, Wang SR, Liu DG, Ashby CR Jr, Huang Y, Robey RW, Liang YJ, Chen LM, Shi CJ, Ambudkar SV, Chen ZS, Fu LW. Lapatinib (Tykerb, GW572016) reverses multidrug resistance in cancer cells by inhibiting the activity of ATP-binding cassette subfamily B member 1 and G member 2. Cancer Res. 2008;68:7905–7914. doi: 10.1158/0008-5472.CAN-08-0499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ambudkar SV. Drug-stimulatable ATPase activity in crude membranes of human MDR1-transfected mammalian cells. Methods Enzymol. 1998;292:504–514. doi: 10.1016/s0076-6879(98)92039-0. [DOI] [PubMed] [Google Scholar]
- 38.Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer. 2002;2:48–58. doi: 10.1038/nrc706. [DOI] [PubMed] [Google Scholar]
- 39.Shi Z, Tiwari AK, Patel AS, Fu LW, Chen ZS. Roles of sildenafil in enhancing drug sensitivity in cancer. Cancer Res. 2011;71:3735–3738. doi: 10.1158/0008-5472.CAN-11-0375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Giacomini KM, Huang SM, Tweedie DJ, Benet LZ, Brouwer KL, Chu X, Dahlin A, Evers R, Fischer V, Hillgren KM, Hoffmaster KA, Ishikawa T, Keppler D, Kim RB, Lee CA, Niemi M, Polli JW, Sugiyama Y, Swaan PW, Ware JA, Wright SH, Yee SW, Zamek-Gliszczynski MJ, Zhang L. Membrane transporters in drug development. Nat Rev Drug Discov. 2010;9:215–236. doi: 10.1038/nrd3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hever-Szabo A, Pirity M, Szathmari M, Venetianer A. P-glycoprotein is overexpressed and functional in severely heat-shocked hepatoma cells. Anticancer Res. 1998;18:3045–3048. [PubMed] [Google Scholar]
- 42.Szakacs G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nat Rev Drug Discov. 2006;5:219–234. doi: 10.1038/nrd1984. [DOI] [PubMed] [Google Scholar]