

Abstract
Alterations in the activity of neural circuits are a common consequence of traumatic brain injury (TBI), but the relationship between single-neuron properties and the aggregate network behavior is not well understood. We recently reported that the GluN2B-containing NMDA receptors (NMDARs) are key in mediating mechanical forces during TBI, and that TBI produces a complex change in the functional connectivity of neuronal networks. Here, we evaluated whether cell-to-cell heterogeneity in the connectivity and aggregate contribution of GluN2B receptors to [Ca2+]i before injury influenced the functional rewiring, spontaneous activity, and network plasticity following injury using primary rat cortical dissociated neurons. We found that the functional connectivity of a neuron to its neighbors, combined with the relative influx of calcium through distinct NMDAR subtypes, together contributed to the individual neuronal response to trauma. Specifically, individual neurons whose [Ca2+]i oscillations were largely due to GluN2B NMDAR activation lost many of their functional targets 1 h following injury. In comparison, neurons with large GluN2A contribution or neurons with high functional connectivity both independently protected against injury-induced loss in connectivity. Mechanistically, we found that traumatic injury resulted in increased uncorrelated network activity, an effect linked to reduction of the voltage-sensitive Mg2+ block of GluN2B-containing NMDARs. This uncorrelated activation of GluN2B subtypes after injury significantly limited the potential for network remodeling in response to a plasticity stimulus. Together, our data suggest that two single-cell characteristics, the aggregate contribution of NMDAR subtypes and the number of functional connections, influence network structure following traumatic injury.
Keywords: functional connectivity, GluN2B, mechanical injury, network activity, synchrony
Introduction
Traumatic brain injury (TBI) is a leading cause of death and disability in the US population. Approximately 80% of these injuries are classified as mild TBI (mTBI), and a large percentage (30–80%) of these patients experience long-term neuropsychiatric symptoms (Alexander, 1995; Kushner, 1998; Hall and Chapman, 2005). In addition, repetitive mTBI is correlated with delayed neurodegenerative changes, and a severe decline in cognitive and psychiatric health (Stern et al., 2011). Although advanced radiological and laboratory tests have led to better early diagnosis of mTBI (Ingebrigtsen et al., 2000; De Kruijk et al., 2001), the lack of a clear consensus on the mechanisms responsible for the deficits after mTBI has limited the progress in developing effective treatments.
An emerging concept explaining the persistent cognitive deficits after mTBI is that there is a broad, chronic disruption of networks throughout the brain. Both diffusion tensor imaging and resting-state functional MRI show reduced connectivity between brain regions involved in supporting cognitive function in TBI patients compared with age-matched control subjects (Hulkower et al., 2013; Pandit et al., 2013). Similarly, scalp EEG recordings of injured subjects show decreased interhemispheric coordinated activity relative to control subjects, and the magnitude of the deficits on neuroimaging corresponds to the severity of inefficient cognitive control (Bonnelle et al., 2011, 2012; Sharp et al., 2011). Due to the loss in cortical functional connectivity after mTBI, cortical circuits lose their ability to synchronize activity patterns across regions, severely affecting the ability to relay information throughout the brain (Buzsáki and Draguhn, 2004; Fries, 2005; Womelsdorf et al., 2007).
This past work, however, does not provide a more precise, cellular-scale window into how changes in the functional connections among neurons will give rise to changes in either the synchrony of an existing network or the steps necessary to rebuild the connectivity of this network. To address this gap, we probed the mechanisms of acute network dysfunction after mTBI in vitro by studying the following three network features: the functional connectivity of the network; the integration of each neuron into this overall connectivity map; and the relative balance of GluN2A- and GluN2B-containing NMDA receptor (NMDAR) signaling within each neuron in the network. These collective properties are difficult to assess in vivo even with recent advances in genetically encoded calcium indicators. Moreover, we focused our work on these two NMDAR subtypes because of their key roles in sensing mechanical signals, influencing neuronal fate, and contributing to synaptic plasticity, all of which would play a significant role in shaping network function after TBI (Hardingham and Bading, 2010; Hunt and Castillo, 2012; Sanz-Clemente et al., 2013). We found that GluN2B-containing NMDARs selectively impair network dynamics following injury. Relatedly, we found that highly connected neurons within the network were relatively resistant to mechanical injury and that GluN2A-containing NMDARs were key to network remodeling early after mechanical injury. This work significantly extends our mechanistic understanding of mTBI, demonstrates the heterogeneity of injury at the single-cell scale, and proves that efforts to rebuild synchrony are key to rebuilding network structure after TBI.
Materials and Methods
Cell culture.
Animal procedures were performed in accordance with the Institutional Animal Care and Use Committee at the University of Pennsylvania. Embryos at embryonic day 18 were surgically removed from a timed pregnant Sprague Dawley rat anesthetized with 5% CO2 and killed via cervical dislocation. Neocortical tissue was dissected from the embryos and dissociated for 15 min at 37°C in trypsin (1.4 mg/ml) and DNase (0.6 mg/ml, Roche Applied Science). After trituration and filtration through Nitex mesh (Crosswire Cloth), cells were resuspended in MEM with Earle's salts and GlataMAX supplemented with 0.6% d-glucose (Sigma-Aldrich), 1% Pen-Strep, and 10% Horse Serum, and plated on poly-d-lysine (0.08 mg/ml, Sigma-Aldrich) and laminin-coated (0.001 mg/ml BD Biosciences) deformable membranes (Sylgard 184 and 186, Dow Corning). Cells were plated at a density of 200,000 cells/ml (∼10,000 cells/mm2). After overnight adhesion, media was replaced with Neurobasal media supplemented with B-27 and 0.4 mm GlutaMAX and grown in a humidified 37°C 5% CO2 incubator. Cells were allowed to mature for days in vitro (DIV) 18–21. The length of time for culturing was used to allow for the full expression of glutamate receptors and to reach stable functional connectivity within neuronal networks (Patel et al., 2012). Double-immunostaining with NeuN and anti-VGAT (1 μg, mouse monoclonal vesicular GABA transporter, Synaptic Systems) identified that 22 ± 7% of neurons in culture were inhibitory (N = 4 cultures, four fields of view per culture, n = 1723 total neurons).
Mechanical injury of cortical neurons.
Primary cortical neurons were plated onto an optically transparent, SILASTIC membrane. Growth media was replaced with controlled saline solution (CSS) containing the following (in mm): 120 NaCl, 5.4 KCl, 1 MgCl2, 1.8 CaCl2, 25 HEPES, 15 glucose, pH 7.3–7.4. Injuries and calcium imaging were performed in CSS.
Mechanical injury to the neural network was delivered by stretching the underlying SILASTIC membrane, per previous reports, to mimic the mechanical deformations that occur during mild traumatic brain injury (Lusardi et al., 2004b). Briefly, cell cultures were placed on a stainless steel plate and covered with a top plate to form a sealed chamber. Increasing the chamber pressure to its peak level in 15 ms caused the compliant silicone membrane to stretch, in turn applying a stretch to the cultured neurons. We designed the supporting stainless steel plate to expose only cells in a defined region (3 × 18 mm rectangular region) to a stretch, providing a uniaxial stretch injury. The membrane in the injured region was stretched 35% beyond its initial width before returning to its original dimension. This stretch level does not cause gross morphological changes or delayed cell death compared with unstretched cultures (Lusardi et al., 2004a).
Calcium imaging.
To measure neural circuit dynamics, we used technology developed in past reports showing that the transient elevations in cytosolic calcium within individual neurons correlate with action potential firing in each neuron, therefore providing a method to approximate the coordinated activation of neurons across a neural network (Yuste et al., 1992; Fetcho et al., 1998; Stosiek et al., 2003). Further, the measurement of spontaneous calcium activity in vitro strongly correlates with electrophysiological measurements using multiple electrode arrays (Choo et al., 2013). Cultures at 10 DIV were transduced with the genetically encoded calcium indicator GCaMP5 (AAV2/1-hSynapsin1) and allowed to mature until DIV 18–21 (Tian et al., 2009; Akerboom et al., 2012). Time-lapse calcium imaging of a neuronal population (150–250 neurons in field of view; 20 Hz framing rate) was measured preinjury and postinjury. The relative effects of GluN2A and GluN2B receptors in altering network activity, functional connectivity, and reintegration of neurons into the postinjury network were assessed using subtype-specific antagonists for GluN1/GluN2ANMDARs [NVP-AAM077 (NVP) 50 nm, Novartis] and GluN1/GluN2B NMDARs (Ro 25–6981 2 μm, Sigma-Aldrich). NVP-AAM077 at 50 nm preferentially antagonizes glutamate-evoked currents at the NR1/NR2A NMDA receptor (Auberson et al., 2002; Berberich et al., 2005; Frizelle et al., 2006), while Ro 25–6981 has >5000-fold selectivity for NR1/NR2B over NR1/NRA (IC50, 0.009 and 52 μm, respectively, for NR1/NR2B and NR1/NR2A; Fischer et al., 1997).
Image processing and data analysis for describing network properties was performed with custom MATLAB scripts per our recent report (Patel et al., 2012). From these algorithms, we extracted both single-cell and network properties, as follows: onset of calcium transients for each neuron and their amplitudes; pairwise synchronization of neurons; local and global synchronization indices; functional connectivity; and metrics of network topology such as clustering coefficient, modularity, and path length. MATLAB source code is available upon request.
Single-cell phenotyping of the relative contribution of NMDAR subtypes to synaptically evoked rise in intracellular calcium.
We developed a noninvasive and rapid method to determine the relative fraction of calcium influx through GluN2B- and GluN2A-NMDA receptors of individual neurons within an intact network. We performed calcium imaging of neuronal networks and measured calcium fluorescence at the soma, as described above. When a neuron fires a single action potential or a train of action potentials, the rise in calcium indicator fluorescence is the product of influx through the NMDARs (all subtypes), calcium-permeable AMPA receptors (AMPARs), and the voltage-gated calcium channels that are opened when the neuron is depolarized with AMPAR activation. We stimulated the excitatory network by blocking GABAA receptors with bicuculline (100 μm). This induced rhythmic oscillations with little variability in calcium amplitudes for a given neuron. The fractional drop in calcium indicator fluorescence amplitude with the application of either GluN2A-selective antagonist (NVP-AAM077) or GluN2B-selective antagonist (Ro25–6981) provided an estimate of the relative contribution of NR2B and NR2A subtypes to a synaptically evoked rise in intracellular calcium. Note that bicuculline was applied only to remove inhibitory currents and to measure the fractional contributions of NMDAR subtypes to calcium influx during maximum excitatory transmission; injuries were done in the absence of bicuculline.
NMDA agonist stimulation.
We measured the change in [Ca2+]I in response to NMDA agonist stimulus using the fluorescent Ca2+ indicator fura-2 (5 μm; Invitrogen). Control neurons and stretch-injured neurons (1 h poststretch) were loaded with 5 μm fura-2 for 45 min at 37°C in buffered saline solution. Immediately before imaging, neuronal cultures were rinsed in buffered saline solution (containing 2 mm Mg2+) and placed on a Nikon TE300 microscope (Optical Apparatus) for calcium fluorescence imaging. To measure only the contribution of NMDA receptors to elevation in [Ca2+]i, other sources of calcium influx were pharmacologically blocked with tetrodotoxin (TTX; 1 μm), bicuculline (100 μm), and nimodipine (10 μm). Cells were alternately excited at 340 and 380 nm using an excitation shutter filter wheel (Sutter Instruments), and the corresponding emission images (510 nm) were collected using a 14-bit Hamamatsu camera (Optical Apparatus) at a rate of approximately every 3 s. The fluorescence ratio from excitation at the two wavelengths (F340, F380) was used to calculate the fura-2 ratio (r = F340/F380). In a subset of cultures, we blocked either the NR2B-containing NMDARs (Ro25–6981; 2 μm) or the NR2A-containing NMDARs (NVP-AAM077; 50 nm). To determine the response of neurons to NMDA stimulation, a baseline set of images was acquired for 30 s before the addition of 200 μm NMDA. Immediately following NMDA treatment, the response was captured for 2.5 min in the presence of NMDA. In each cell, we normalized the response to NMDA stimulation to its baseline fluorescence ratio. Cells with abnormally high baseline ratios, defined as a fluorescence ratio >1 or <0.3, were excluded from analysis. The excluded cells were typically <5% of the observed cells. All cells in the field of view, averaging ∼150 cells per well, were imaged across three to four wells for each experimental condition studied. The fluorescence ratio was converted to [Ca2+]i using the following equation: [Ca2+] = Kd × (R − Rmin)/(Rmax − R), where Kd for fura-2 = 145 nm, Rmax was the measured fluorescence ratio of cells bathed in 5 μm ionomycin, and Rmin was the measured fura-2 fluorescence ratio of neurons bathed in Ca2+-free saline solution in TTX (1 μm).
A nested ANOVA, with the culture well for each cell as the nesting variable, was used to detect significant differences across experimental conditions. Tukey's post hoc procedures were used for planned multiple comparisons following the ANOVA. For all statistical conditions, p values of <0.05 were considered significant.
Electrophysiology.
Whole-cell patch-clamp recordings were performed on 10 neurons each from the uninjured and injured groups. Neurons selected had a pyramid-like morphology, and recordings were performed using an Axoclamp-2B amplified (Molecular Devices) at room temperature. None of the wells used for electrophysiology were used either for immunocytochemistry or calcium imaging measurements. Patch pipettes were fabricated from borosilicate glass capillaries (1.5 mm OD; World Precision Instruments), and were drawn to a resistance between 4 and 6 MΩ using a pipette puller (Sutter Instrument) and then filled with a solution containing the following (in mm): 135 CsAsp, 4 KCl, 2 NaCl, 10 EGTA, 0.2 CaCl2, 2 MgATP, 0.6 Na2GTP, and 10 HEPES, pH 7.2 (290–295 mOsm).
Cells were bathed in external recording solution containing the following (in mm): 130 NaCl, 4 KCl, 3 CaCl2, 2 MgCl2, 10 HEPES, 11 glucose, 0.01 glycine, 0.001 TTX, and 100 μm bicuculline methobromide, pH 7.3 (290–310 mOsm). The current–voltage relationship (I–V) was determined by applying a voltage ramp (−100 to 40 mV, 16.7 mV/s) in the presence and absence of NMDA (100 μm), and with or without Ro 25–6981 (2 μm). NMDA-elicited currents and the contribution of GluN2B-containing NMDA receptors were measured in the same cell. Cells with leak currents less than −100 pA were not used for analysis. NMDA-elicited currents were completed blocked by coadministration of the nonselective NMDA receptor antagonist APV (25 μm), and repeated application of NMDA did not result in significant current rundown (data not shown).
Immunocytochemistry.
Three hours following washout of 15 min bicuculline stimulus, neurons were fixed with 4% paraformaldehyde and probed for nuclear c-fos and NeuN using rabbit polyclonal c-fos antibody and mouse monoclonal NeuN antibody, respectively, per standard protocol (Abcam). Briefly, cells were permeabilized with 0.2% Triton X for 10 min, blocked in 5% normal goat serum (NGS) for 30 min, incubated with c-fos and NeuN primary antibodies in NGS overnight and stained with Alexa Fluor goat anti-rabbit 594 (c-fos) and Alexa Fluor goat anti-mouse 488 (NeuN) secondary antibodies. Nuclei were labeled with Hoechst 33342 stain (Invitrogen) 15 min before imaging. Images were acquired on an upright Leica SP5 confocal microscope with a 25× objective (numerical aperture 0.95). NeuN allowed us to distinguish neurons from glia. Of the labeled neurons, the Hoechst stain image was binarized to create a mask of neuronal nuclei, and the mean c-fos intensity within the mask was computed.
Statistical analysis.
Data were analyzed using Student's t test or ANOVA with Tukey's post hoc test using the statistical software JMP (SAS Institute). At least 10 cultures from three different isolations were tested for each experimental condition. For calcium imaging experiments, each field of view consisted of N = 100–150 neurons. All values are reported as the mean ± SEM.
Results
Our approach was based on using an established in vitro model of traumatic mechanical injury (Lusardi et al., 2004b), newly developed methods for estimating functional connections among neurons within an in vitro network (Patel et al., 2012), and pharmacological manipulations to identify the relative balance of GluN2A- and GluN2B-containing NMDAR signaling within each neuron in a cortical microcircuit in vitro.
Traumatic mechanical injury in vitro leads to a loss in functional connectivity mediated by NMDAR activation
Using newly developed methods to estimate the functional connectivity of neurons within a dissociated microcircuit (Fig. 1A; Patel et al., 2012), we first measured the initial reduction and early recovery in the circuit connectivity after stretch injury. Cortical neurons were cultured on a SILASTIC membrane, and a small region of the membrane was stretched uniaxially to 35 ± 5% peak stretch within 15 ms before returning to its original dimensions (Fig. 1B). At this level of stretch, we observed no significant neuronal death 1 d following injury, consistent with a relative lack of neuronal death observed in models of acceleration-induced mTBI (Meaney et al., 1995) and percussion-induced mTBI (Dixon et al., 1987). Mechanically injured cortical neuron networks showed an immediate but transient increase in cytosolic calcium that returned to prestretch baseline levels with 10 min of injury (Fig. 1B). Consistent with previous reports detailing the mechanosensitivity of the NMDA receptor (Paoletti and Ascher, 1994; Kloda et al., 2007; Singh et al., 2012), this calcium transient was eliminated completely by inhibiting the NMDA receptor population (25 μm APV) before stretch insult. At this injury level, we saw both an immediate (10 min) and more persisting impairment (1 h) in functional connectivity of the microcircuit after injury (Fig. 1C). To quantify the change in network connectivity, we define functional connectivity index (CI) as the average number of functional connections (edges) per neuron (node), normalized by the number of neurons in the network. Hence, functional connectivity index ranges from 0 to 1, where 1 signifies that every node is connected to every other node in the network and 0 signifies that all nodes are disconnected from all other nodes. The overall functional connectivity index and spontaneous activity of the circuits were significantly reduced 10 min and 1 h following injury (Fig. 1D,E). Once again, stretch-induced reduction of connectivity and activity was blocked by pretreating cultures with APV (Fig. 1D,E).
Figure 1.

Traumatic mechanical injury in vitro leads to a loss in functional connectivity mediated by NMDAR activation. A, The spontaneous activity of a population of neurons in vitro was recorded using a virally transduced fluorescent calcium indicator (GCaMP5). Middle, Fluorescence time series of three example neurons is depicted; the dot denotes the onset of calcium transient. Phase synchronization index of each pairwise neuron reflects the degree of temporal similarity in spontaneous activity. Statistically significant synchronized interactions are defined as functional connections and yield a functional connectivity map for the network (right). B, For stretch injury, neurons were plated and cultured on a SILASTIC membrane, and a small area of the membrane was dynamically stretched to peak at 35% strain within 15 ms. This level of stretch injury resulted in the immediate influx of Ca2+and could be prevented by pretreating neurons with the NMDA receptor antagonist APV. The solid black line is the average somatic calcium fluorescence trace of 122 neurons, with the 95% confidence interval shown in gray. C, Stretch injury causes an immediate drop in functional connectivity by 10 min that slowly recovers by 1 h. D, Functional connectivity of the network, defined as the average number of functional connections per neuron normalized by the number of neurons, was significantly reduced with injury by 10 min and 1 h. The effect of injury on network functional connectivity was dependent on NMDAR activation since APV pretreatment abolished the injury response. E, The frequency of calcium activity was also significantly reduced by 10 min and 1 h poststretch, and was dependent on NMDAR activation. Scale bar, 50 μm.
Single-neuron NMDAR-mediated calcium signaling shows a broad distribution within neurons from the same functionally integrated network
Our methodology also provides the ability to measure the relative contribution of different calcium influx pathways within individual neurons during action potential bursts, potentially linking these single-neuron measures to the resulting changes in functional connectivity after injury. We first blocked inhibitory GABAA receptors with bicuculline, removing the contribution of fast inhibitory synaptic transmission from network activity and creating a repeating pattern of calcium transients, previously shown to correlate with a regular pattern of action potential bursts (Chen et al., 1996). The transient increases in intracellular calcium during either the spontaneous activity phase or synchronized activity phase were due primarily to influx through the NMDA receptor (65 ± 7.3% of total), calcium-permeable AMPA receptors (4.2 ± 2.3% of total), and l-type voltage-gated calcium channels (32 ± 10.1% of total; Fig. 2A–C). The order of antagonist application did not change our estimate of receptor contribution, and these results were reproducible on the same set of neurons, suggesting that this brief pharmacological manipulation does not alter the source of calcium signaling.
Figure 2.

Profiling the contributions of NMDA receptor subtypes to intracellular calcium oscillations. We extended the calcium imaging methodology to profile the source of calcium for each neuron in the network. Inhibitory currents were blocked with bicuculline. The contribution of synaptically activated NMDA receptors to [Ca2+]I oscillations was assessed by comparing calcium oscillation amplitude in bicuculline to that in bicuculline plus APV. Similarly, the sensitivity to naspm and nimodipine was used to assess the contributions of calcium-permeable AMPA receptors and l-type voltage-gated channels to [Ca2+]i oscillations. An example of calcium receptor profiling of two neurons from the same field of view is illustrated in A and B. C, In general, the contribution of NMDARs and voltage-gated Ca2+ channels (VGCCs) to [Ca2+]I varied with developmental maturation of neurons. The NMDA receptor component of [Ca2+]i oscillations was further subdivided in fractional contribution from GluN2A and GluN2B subtypes with Ro25–6981 and NVP-AAM077, respectively (D). E, The cumulative distribution of NMDA receptor subtype-specific source of calcium influx varied across neurons from the same network but did not differ among networks from the same DIV. Immature neurons at DIV 13 had significantly different relative contributions of GluN2A and GluN2BN subtypes to [Ca2+]i oscillations (average cumulative distribution at DIV 13 and DIV 20, n = 10 wells each; N = 843 neurons at DIV 13 and N = 964 neurons at DIV 20; Kolmogorov–Smirnov test, p < 0.001). Scale bar, 50 μm. CP-AMPAR, Calcium-permeable AMPA receptors.
We focused our analysis by measuring the relative contribution of GluN2A-containing and GluN2B-containing NMDARs to the overall total NMDAR calcium influx. We measured the percentage reduction in peak amplitude with a GluN2A NMDAR antagonist (NVP-AAM077; 50 nm) and a GluN2B NMDAR antagonist (Ro25–6981; 2 μm; Fig. 2D) applied sequentially to a culture, and saw that the GluN2A subtypes contributed 64 ± 7.3% to the overall NMDAR-mediated rise in [Ca2+]i during the oscillations, while GluN2B NMDARs contributed 28 ± 8.4%. Within the same network, the fractional calcium influx from both NMDAR subtypes varied broadly. Moreover, this broad distribution of NMDAR calcium influx appeared consistently in cultures maintained for the same number of days in vitro, and the relative fraction of GluN2A NMDAR calcium influx increased in networks cultured for longer periods of time (21 DIV; Fig. 2E). These aggregate developmental changes in NMDA receptor-mediated calcium influx correlates with electrophysiological and Western blot-based estimates of subunit composition in cultured neurons at this developmental stage (Stocca and Vicini, 1998; Tovar and Westbrook, 1999; Liu et al., 2004).
NMDAR subtype composition and connectivity influence functional rewiring of individual neurons following injury
We next tested directly whether these two single-cell-based features—the functional connectivity of a neuron to its neighbors and the relative influx of calcium through distinct NMDAR subtypes (estimated as the ratio of GluN2B/GluN2A calcium peak) for each neuron—contributed to the individual neuronal response to trauma.
We first computed a CI for each neuron, which was defined as the ratio of total functional targets to the total number of neurons. CI ranges from 0 (completely deintegrated neuron) to 1 (neuron is functionally connected to all other neurons in the network). We classified neurons in our cortical cultures before stretch as highly integrated (CI, >0.7), moderately connected (CI, ≥0.3 and ≤0.7), or sparsely connected (CI, <0.3). Neurons that remained in the network after stretch injury (CI, >0.05) showed a significant shift in their functional connectivity across these broad connectivity groupings (Fig. 3A). By comparing the functional connectivity before and following injury, we found that sparsely connected neurons showed a significant increase in functional connectivity (CI, <0.3; ratio = 2.64 ± 0.28) compared with neurons in either the moderate (CI, >0.3 and <0.7; ratio = 0.82 ± 0.03) or highly connected group (CI, >0.7; ratio = 0.71 ± 0.03; Fig. 3A,B; n = 221 neurons, N = 5 experiments; ANOVA, p < 0.01). As expected, blocking the activation of NMDARs before injury eliminated the loss in functional connections caused by stretch injury regardless of how well a neuron was connected in the network (data not shown), again demonstrating the role that NMDAR mechanosensitivity plays in altering network structure.
Figure 3.

NMDAR subtype composition and connectivity influence functional rewiring of individual neurons following injury. A, The normalized functional connectivity of a neuron (i.e., its level of integration in the network) before stretch influenced post-stretch connectivity. B, Sparsely connected neurons (normalized CI, <0.3) showed a significant increase in functional connectivity following stretch injury compared with moderately connected neurons (CI, >0.3 and <0.7) or highly connected neurons (CI, >0.7). C, D, As a result of injury, a neuron could undergo one of three changes in its functional connectivity. Of the set of all functional targets, a fraction of connections may be lost as a result of injury, a fraction of connections may remain stable following injury, or there may be some newly formed functional connections that were not present in the uninjured network. The “lost,” “stable,” and “new” fractions for a given neuron compactly describe its extent of functional rewiring in the injured network. E, For each neuron, we plotted the fraction of total connections that were lost, remained stable, or appeared new as a result of injury on three independent axes. The contributions of GluN2A- and GluN2B-containing NMDARs to somatic calcium oscillations for each neuron before injury were projected on the 3D plot of its functional rewiring. F, We found a significant positive correlation between prestretch GluN2B contribution and deintegration from the network following injury. Additionally, highly connected neurons retained many of their functional targets following stretch injury, whereas sparsely connected neurons participated in network remodeling by forming new functional targets (F).
We next segregated the functional connections for each neuron after injury as a lost fraction of the initial prestretch connections, a stable fraction of connections maintained after injury, and a new fraction of functional connections that extended to new neuron targets (Fig. 3C,D). In sham cultures, the lost and new fraction for nearly all neurons was <5% over 1 h, indicating very little change in the functional connectivity in uninjured cultures over our monitoring period. In contrast, we found a large number of functional connections were lost and a smaller number of newly formed functional connections appeared 1 h following stretch injury (Fig. 3E).
By overlaying the prestretch GluN2B and GluN2A calcium influx measures of each neuron with its functional connectivity following injury, we found a significant positive correlation between prestretch GluN2B contribution and deintegration of the neuron from the network after injury (Fig. 3E,F). These data showed that individual neurons whose [Ca2+]i oscillations were most sensitive to Ro25–6981 before stretch lost many of their functional targets at 1 h following stretch, consistent with our earlier work showing that the sensitivity of the NMDA receptor to mechanical stretch injury is linked primarily to the GluN2B subunit (Singh et al., 2012). There was no correlation between the connectivity index of a given neuron and GluN2A or GluN2B contribution before stretch, indicating that NMDAR subtypes do not influence functional connectivity in an uninjured network.
Manipulating GluN2B NMDAR signaling and NMDAR mechanosensitivity influence the network structure after injury
Since the presence of mechanosensitive GluN1/GluN2B NMDARs increases the likelihood for a neuron to deintegrate from the network after stretch injury, we expected that experimental manipulations both to reduce the relative GluN2B:GluN2A receptor signaling before stretch or to attenuate the mechanosensitivity of the NMDAR would protect against neuronal deintegration after injury. We tested this hypothesis in three experimental conditions. During development, GluN2B-containing receptors are expressed in immature neurons, whereas GluN2A-containing receptors become more prominent at later stages of development (Carmignoto and Vicini, 1992; Hestrin, 1992; Monyer et al., 1994; Kew et al., 1998; Roberts and Ramoa, 1999). Although there was very little spontaneous activity in cultures younger than DIV 8, the frequency of spontaneous calcium activity reached a level to accurately infer functional connectivity by DIV 13 (Patel et al., 2012). We confirmed that the GluN2B contribution to [Ca2+]i oscillations was greater in DIV 13 neurons compared with more mature neurons at DIV 20 (DIV 13 GluN2B contribution, 38 ± 3.9%; DIV 20 GluN2B contribution, 19 ± 3.8%; p < 0.05; Fig. 4A). Stretch injury to DIV 13 cultures caused a significant reduction in functional connectivity relative to injury to DIV 20 cultures (Fig. 4B; DIV 13, 72 ± 5% reduction in total number of functional connections; DIV 20, 51 ± 6% reduction in total number of functional connections; p < 0.01).
Figure 4.
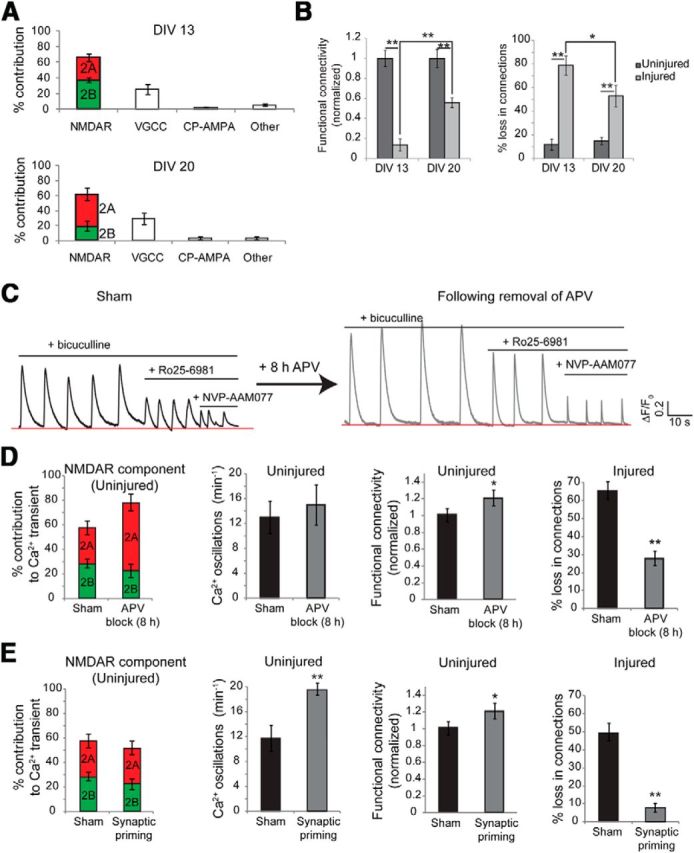
Manipulating GluN2B NMDAR signaling and NMDAR mechanosensitivity influences the network structure after injury. A, The contribution of NMDAR subtypes to somatic calcium oscillations shifted from GluN2B dominant at DIV 13 to GluN2A dominant at DIV 21. B, The same level of stretch injury yielded a greater deficit in functional connectivity in GluN2B-dominant young (DIV 13) neurons relative to the GluN2A-dominant mature neurons (DIV 20). C, Chronic inhibition of NMDA receptors (25 μm APV for 8 h) led to homeostatic enhancement of the GluN2A contribution to calcium oscillations in the young DIV 13 network upon release from inhibition. The frequency of spontaneous calcium activity and the functional connectivity of the network did not change as a result of APV treatment. D, However, stretch injury attenuated the loss in functional connectivity in DIV 13 neurons treated with APV relative to age-matched untreated cultures, suggesting that single-cell prestretch GluN2B contribution influences deintegration from the network. E, The mechanosensitivity of GluN2B receptors was reduced by inducing a brief period of heightened synaptic activity and acute synaptic priming. The relative contributions of GluN2A and GluN2B subtypes did not change as a result of synaptic priming. Compared with age-matched untreated cultures, cultures treated with bicuculline were protected against stretch-induced loss in network functional connectivity.
In our second manipulation, we used a chronic blockade of NMDA receptors (8 h APV treatment) to stimulate an increase in synaptic surface expression of GluN2A-containing NMDARs, but not GluN2B-containing NMDARs (von Engelhardt et al., 2009). We confirmed that GluN2A contribution to [Ca2+]i oscillations increased significantly following an 8 h APV block in DIV 13 cultures relative to no treatment (DIV 13 GluN2A contribution, 53 ± 4.3%; post-chronic APV block GluN2A contribution, 72 ± 7%; p < 0.01; Fig. 4C,D). The decrease in functional connectivity following injury was significantly attenuated in age-matched cultures preconditioned with chronic APV block (loss in functional connections in DIV 13 cultures treated with 8 h APV, 28 ± 4%; loss in functional connections in untreated DIV 13 cultures, 67 ± 5%; p < 0.01; Fig. 4D). Since injury to young neurons following washout from chronic APV block significantly prevented changes in network connectivity compared with no drug treatment, the more severe loss of functional connectivity in young cultures versus mature cultures is not likely due to other age-related factors.
For our third manipulation, we decreased the mechanosensitivity of the NMDAR subtypes by inducing [Ca2+]i oscillations with bicuculline, a treatment that results in PKC-mediated uncoupling of NMDARs from spectrin, rendering neurons insensitive to mechanically induced [Ca2+]i transients (Geddes-Klein et al., 2006). Neurons at DIV 20 were exposed to bicuculline for 15 min (synaptic priming) and immediately stretch injured, as before. This brief stimulation with bicuculline did not acutely alter the sensitivity to Ro25–65981 or NVP-AAM077, hence the relative contribution of GluN2A and GluN2B subtypes remained unchanged. Network disinhibition with bicuculline increased the frequency of synchronized calcium oscillations, increasing the apparent functional connectivity of the network (Fig. 4E). However, the bicuculline treatment led to a significant protection from injury-induced functional rewiring (acute synaptic priming 8 ± 3% loss in functional connections vs 49 ± 6% in untreated, injured cultures; p < 0.01).
GluN2B-containing NMDA receptors undergo stretch-induced reduction in Mg2+ block, which results in a selective enhancement of GluN2B NMDAR calcium influx
Traumatic injury both in vitro and in vivo causes a reduction in the voltage-dependent Mg2+ block of the NMDARs (Zhang et al., 1996; Furukawa et al., 2000). NMDA-activated currents recorded from uninjured control neurons under voltage-clamp in the presence of 2 mm extracellular Mg2+ showed a typical “J” shape when plotted against membrane command potentials (Fig. 5A). Consistent with previous reports (Zhang et al., 1996), the I–V relationship was more linear in stretch-injured neurons 1 h postinjury (Fig. 5A, inset, raw trace of representative single injured and uninjured neuron), showing significant differences from uninjured neurons at normal resting membrane potential (−62 mV; normalized current in stretched neurons, −1.12 ± 0.16; normalized current in uninjured neurons, −0.13 ± 0.17; n = 16; p < 0.001). These changes in the NMDA I–V relationship occurred with no difference in either the input resistance (uninjured neuron, 321.2 ± 12.2 MΩ; injured neuron, 305.7 ± 16.3 MΩ; n = 16) or the resting membrane potential (uninjured neuron, −64.6 ± 3.4 mV; injured neuron, −61.6 ± 4.1 mV). In the presence of the GluN2B-selective antagonist Ro25–6981, injured neurons no longer displayed a shift in their I–V relationship relative to uninjured neurons (control; normalized current in stretched neurons, −1.12 ± 0.16; stretched neurons plus Ro25–6981, −0.42 ± 0.21; p < 0.01), suggesting that the GluN1/GluN2B NMDA receptors undergo a reduction in Mg2+ block following traumatic injury.
Figure 5.
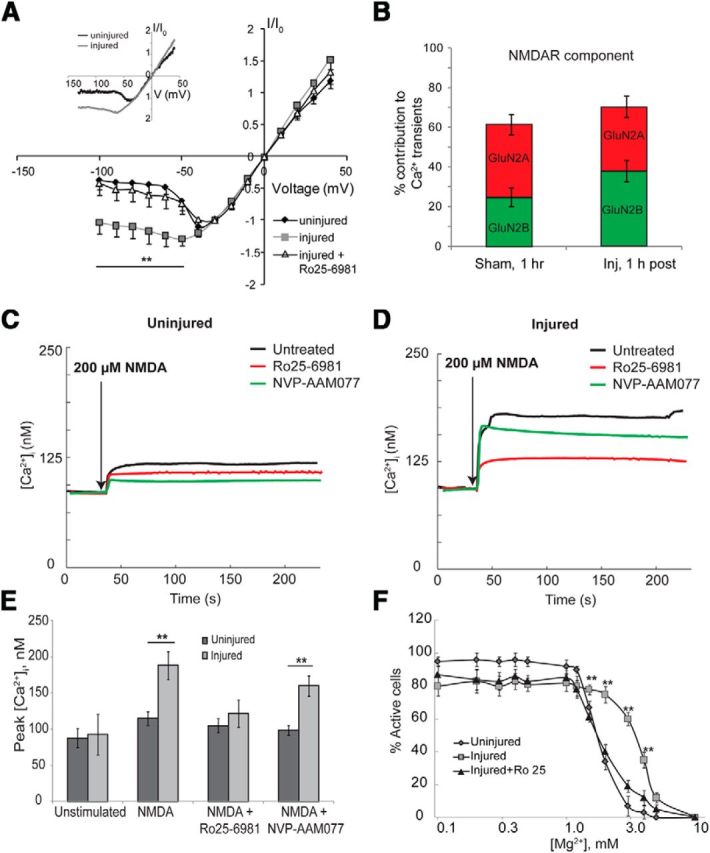
GluN2B-containing NMDA receptors undergo stretch induced reduction in Mg2+ block and results in a selective enhancement of GluN2B NMDAR calcium influx. NMDA-activated I–V relationship of stretch-injured neurons was more linear compared with the typical J shape of uninjured neurons. A, Blocking the GluN2B subtypes (Ro25–6981) restored the J curve, suggesting that injury specifically reduces the voltage-dependent Mg2+ block of GluN2B-containing NMDA receptors (average response of n = 10 neurons; inset, raw traces of representative single uninjured and injured neurons). B, Consistent with partial relief of GluN2B Mg2+ block, stretch-injured neurons also had a significantly greater calcium influx from GluN2B receptors during spontaneous network activity. C, D, To further quantify this change, all sources of calcium except the NMDAR were blocked, and calcium entry in response to NMDA (200 μm) was measured with the fluorescent ratiometric calcium indicator fura-2. NMDA stimulation yielded a significantly greater peak [Ca2+]i in injured neurons compared with age-matched uninjured samples (102% rise over baseline injured vs 36% uninjured). E, Blocking the GluN2B subtypes in injured neurons significantly attenuated the NMDA-induced increase in [Ca2+]i to uninjured levels. F, In a separate set of experiments, the spontaneous activity of cultures was recorded in increasing [Mg2+]e ranging from 0 to 10 mm and the percentage of active neurons at each [Mg2+]e value computed. In uninjured cultures, raising [Mg2+]e from physiologic 0.8 to 3.0 mm almost completely abolished spontaneous activity, whereas the effect of [Mg2+]e block was significantly reduced in injured cultures. Sensitivity to [Mg2+]e was restored in injured cultures by blocking GluN2B subtypes. **p < 0.01.
A selective reduction of voltage-dependent Mg2+ block of GluN2B-containing NMDARs suggests that increased Ca2+ influx would occur selectively through this subpopulation of NMDARs in mechanically injured neurons. In cultures showing spontaneous activity 1 h following injury, we observed a significant relative increase in GluN2B-mediated NMDAR calcium influx relative to sham controls (Fig. 5B). We further measured the change in [Ca2+]i in uninjured and stretch-injured neurons in response to direct NMDA treatment (200 μm) using the fluorescent ratiometric Ca2+ indicator fura-2 AM (Fig. 5C,D). To eliminate other sources of calcium influx, spontaneous activity and synaptic transmission were reduced with TTX (1 μm), bicuculline (100 μm), and nimodipine (10 μm). We observed that NMDA treatment (200 μm) increased [Ca2+]i by 36% over basal levels in uninjured neurons (Fig. 5C,E; basal [Ca2+]I, 88 ± 13 nm; NMDA-induced [Ca2+]I, 115 ± 9 nm), whereas [Ca2+]I increased by 102% in injured neurons (Fig. 5D,E; basal [Ca2+]I, 93 ± 28 nm; NMDA-induced [Ca2+]I, 188 ± 19 nm). Moreover, pretreating injured neurons with Ro25–6981, but not NVP-AAM077, significantly attenuated NMDA-induced increase in [Ca2+]i relative to no treatment (injured plus Ro25–6981 peak [Ca2+]I, 122 ± 19 nm; injured plus NVP-AAM077 peak [Ca2+]I, 160 ± 14 nm; p < 0.01; Fig. 5E).
We also demonstrate that stretch injury reduces GluN2B Mg2+ sensitivity by estimating the [Mg2+]e required to block spontaneous activity. In uninjured cultures, spontaneous calcium activity could be almost completely blocked by increasing [Mg2+]e from physiologic 0.8 to 3 mm (92 ± 4% spontaneously active neurons at [Mg2+]e = 0.8 mm; 7 ± 6% active neurons at [Mg2+]e = 3 mm). In contrast, nearly 60 ± 4% of injured neurons were active at [Mg2+]e = 3 mm and required [Mg2+]e = 5 mm to quench spontaneous activity. Pretreating injured neurons with the GluN2B antagonist Ro25–6981 rescued [Mg2+] sensitivity (Fig. 5F). Together with electrophysiological recordings, these data strongly support a GluN2B-selective reduction in voltage-dependent Mg2+ block following mechanical stretch injury.
Stretch-induced reduction in GluN2B NMDAR Mg2+ block causes asynchronous circuit activity
Coincidence detection is a fundamental feature of neurons that allows temporal control over postsynaptic action potential initiation following an appropriate pattern of afferent inputs (Regehr and Tank, 1990; Gasparini et al., 2004; Losonczy and Magee, 2006). Given that injury selectively reduces GluN2B NMDAR Mg2+ block, little evidence exists to predict how this selective change to an NMDAR subpopulation would influence the temporal patterns of network activity. Blocking either the GluN2A NMDAR or the GluN2B NMDAR subtypes did not alter the pattern of synchronous calcium oscillations in uninjured neurons (Fig. 6A). In contrast, the activity of a mechanically injured network was significantly desynchronized (synchronization index normalized to uninjured mean value, 1.0 ± 0.13; synchronization index normalized to injured mean value, 0.38 ± 0.09; paired t test, p < 0.01). Although the global synchronization of the network was reduced after injury, several clusters of modestly synchronized oscillations within smaller neuronal ensembles emerged with the reduction of overall synchronization. Antagonizing GluN1/GluN2A NMDARs with NVP-AAM077 caused a further reduction in the synchrony and frequency of calcium oscillations 1 h after injury, but these changes were not significantly different from untreated, injured networks (Fig. 6B,C; mean frequency of calcium oscillations normalized to uninjured, untreated networks, 1.0 ± 0.047; mean frequency of calcium oscillations normalized to injured, untreated networks, 0.58 ± 0.11; p < 0.05; injured plus NVP, 0.37 ± 0.14; p > 0.05 relative to injured, untreated networks). However, selective blockade of GluN2B-containing subtypes significantly rescued synchronization of injured neuronal networks (normalized Synchronization Index injured networks, 0.38 ± 0.09; injured plus Ro25–6981 networks, 0.81 ± 0.14; p < 0.05). Together, these data suggest that the underlying synchronous oscillations in an injured network are masked by the activity of GluN2B NMDARs, and this synchronization can be recovered by selectively antagonizing GluN1/GluN2B-containing NMDARs.
Figure 6.

Stretch-induced reduction in GluN2B NMDAR Mg2+ block causes asynchronous circuit activity. A–C, The pattern of spontaneous calcium activity in mature cultures was characterized by rhythmic, synchronized oscillations and was unaffected by antagonism of either GluN2A or GluN2B subtypes. In representative raster plots, unique temporal patterns of calcium activity are color coded for visualization; a pairwise synchronization matrix depicts the level of temporal correlation between the calcium activities of neuronal pairs across the network. Blocking either GluN2A or GluN2B subtypes separately did not change either the frequency or the synchronization of calcium activity in uninjured cultures. In contrast, 1 h following stretch injury, both the frequency of calcium activity of individual neurons and the temporal pattern of activity across the network were significantly reduced. B, C, Inhibiting GluN2B subtypes in an injured network further reduced the amount of calcium activity (B); however, the remaining spontaneous activity was more synchronized (C), suggesting that the activation of GluN2B subtypes underlies asynchronous firing. Antagonism of GluN2A subtypes reduced both the frequency and the synchronization; however, they were not significantly different from untreated injured cultures (p > 0.05).
Activation of GluN2B NMDARs in injured neurons reduces network augmentation induced with synchronization
Past work shows that pharmacological blockade of inhibitory neurotransmission induces a period of induced synchronization that can, over a period of hours, change the activity pattern of in vitro networks and increase global functional connectivity (Arnold et al., 2005). Although this model of “recurrent network excitability” is not a classic model of long-term potentiation (LTP), this adaptation response of the microcircuit shares many mechanisms with different models of LTP, as follows: reshaping the microcircuit depends on protein translation, NMDA receptor activation, and the early transcriptional changes occurring during the induced synchronization (Arnold et al., 2005). Therefore, we selected this as a method to explore how the function of injured microcircuits can be recovered after injury in vitro.
We explored the plasticity of an injured circuit by measuring changes in global functional connectivity, single-cell somatic Ca2+ amplitudes, and nuclear c-fos localization 2 h following an induced synchronization period (Fig. 7). To maintain consistency with prior experiments, we introduced this synchronization period 1 h after injury and evaluated the network 3 h after injury. In uninjured cultures, 15 min of bicuculline treatment led to persistent, synchronous calcium oscillations and significantly increased global functional connectivity 2 h later (Fig. 7A–C; increase in functional connectivity following bicuculline stimulus of culture, 89 ± 9.2%; functional connectivity in untreated culture, −12 ± 5.4%; t test, p < 0.001, df = 4). In addition, the peak increase in somatic cytosolic calcium following bicuculline treatment—an index of excitability (Frick et al., 2004)—was significantly higher than before treatment (Fig. 7E; unstimulated change in amplitude, 8.5 ± 2.2%; change in amplitude following bicuculline stimulus, 58.4 ± 9.1%; p < 0.001). This change in functional connectivity and calcium signaling was dependent on GluN2A-containing, but not GluN2B-containing, receptor activation during bicuculline treatment (Fig. 7C). Although we could induce synchronous oscillations in an injured network with bicuculline treatment, we did not see either an increase in functional connectivity or a potentiation of individual somatic calcium transients following bicuculline washout 2 h later (Fig. 7D; change in functional connectivity following injury, −62 ± 7.3%; change in functional connectivity following bicuculline washout, −44 ± 8.5%; p > 0.05). Blocking GluN1/GluN2B NMDARs during the period of induced synchronization successfully recovered most of the increases in global connectivity and somatic calcium increases that would occur in uninjured cultures (Fig. 7D,E; 37 ± 4.1% change in functional connectivity with bicuculline plus Ro25 in injured cultures and 37.7 ± 7.7% rise in somatic calcium amplitude). In comparison, blocking GluN1/GluN2A NMDARs led to a further reduction in global connectivity following the induced synchronization (Fig. 7E). Finally, since the long-term changes in excitability following bicuculline stimulus are dependent on transcription (Arnold et al., 2005), we measured the nuclear localization of the activity-dependent transcription factor c-fos in uninjured and stretch-injured neurons (3 h postinjury and 2 h post-bicuculline stimulus). Uninjured neurons showed significantly elevated nuclear c-fos levels following bicuculline stimulus compared with untreated cultures, whereas this response was greatly reduced in injured neurons (Fig. 7F; normalized nuclear c-fos intensity in injured neurons, 0.77 ± 0.04; normalized nuclear c-fos intensity in uninjured neurons, 1.0 ± 0.03; p < 0.01). Similar to the changes in functional connectivity, targeted blockade of GluN2B-containing NMDARs led to a significant recovery of c-fos nuclear localization (0.91 ± 0.05). Together, these data support an impairment in network plasticity following stretch injury, consistent with previous reports of altered excitability and impaired expression of LTP following TBI (Reeves et al., 1995; Sick et al., 1998; Schwarzbach et al., 2006), and highlight the role that NMDAR subtypes play in recovering microcircuit function after traumatic injury in vitro.
Figure 7.
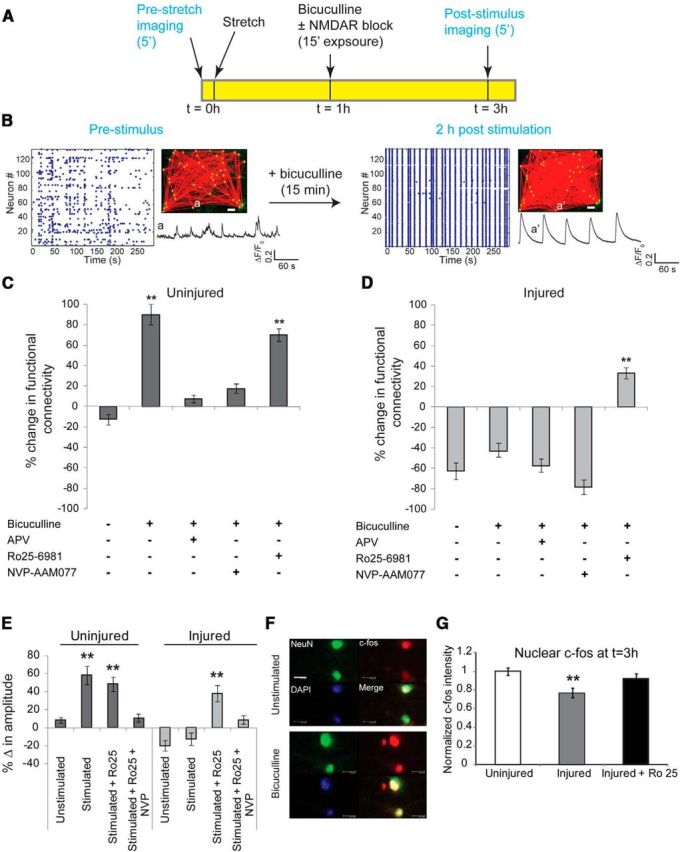
Activation of GluN2B NMDARs in injured neurons reduces network augmentation induced with synchronization. A, Plasticity through recurrent excitation was induced by blocking inhibitory currents with bicuculline for 15 min. Calcium activity of the network was recorded before injury, the network was subjected to stretch injury and stimulated with bicuculline, and the calcium activity of the same field of view was recorded again 2 h after bicuculline washout. B, In uninjured cultures, a brief period of pharmacological blockade of inhibitory neurons resulted in a persistent increase in synchronized activity and functional connectivity, and an augmentation in single-cell somatic calcium amplitude. Inset, GCaMP5 trace of the same neuron before (a) and 2 h after bicuculline stimulus (a′), illustrating the emergence of rhythmic, large-amplitude calcium oscillations in a′ compared with a. Scale bar, 50 μm. C, We used the percentage change in functional connectivity as one of three metrics to explore the plasticity of the circuit [change in somatic calcium amplitude (E) and nuclear localization of c-fos (G) are the other two metrics]. In uninjured cultures, 15 min of bicuculline treatment led to a significant increase in functional connectivity relative to no stimulus (change in functional connectivity following bicuculline stimulus, 89 ± 9.2%; change in functional connectivity untreated, −12 ± 5.4%; t test, p < 0.001, df = 4). Enhancement in connectivity was abolished by blocking either the activation of NMDARs or specifically targeting GluN2A-containing NMDARs (bicuculline plus APV, 8.2 ± 3.1%; bicuculline plus NVP-AAM077, 17.8 ± 4.7%). D, Injury resulted in a significant decrease in network connectivity (injured, −62 ± 7.3%; uninjured, −12 ± 5.4%; p < 0.001), which could not be restored with bicuculline stimulus (−44 ± 8.5%). However, blocking GluN2B NMDARs during the period induced synchronization led to a significant enhancement in global connectivity (treatment with bicuculline plus Ro25–6981, 37 ± 4.1%; treatment with bicuculline, −44 ± 8.5%; p < 0.001). E, Amplitude of somatic calcium transients increased significantly following washout of bicuculline stimulus in uninjured cultures, which is indicative of enhanced excitability (unstimulated change in amplitude, 8.5 ± 2.2%; change in amplitude following bicuculline stimulus, 58.4 ± 9.1%; p < 0.001). This was dependent on activation of GluN2A-containing NMDARs since exposure to NVP-AAM077 during the period of bicuculline stimulus abolished the change in somatic calcium amplitude (10.2 ± 4.7%). In comparison, the amplitude of somatic calcium transients was significantly lower following injury (−20.2 ± 5.5%) and did not recover following bicuculline treatment (−13.8 ± 6.2%). However, antagonism of GluN2B-containing NMDARs during bicuculline stimulus recovered somatic amplitude to near the levels observed in uninjured cultures (change after bicuculline plus Ro25 stimulus, 37.7 ± 7.7%; change after bicuculline stimulus, −13.8 ± 6.2%; p < 0.001). F, G, We probed the localization of the immediate early transcription factor c-fos and found increased nuclear localization within 2 h of bicuculline treatment in uninjured cultures. Nuclear localization of c-fos was significantly reduced in injured cultures (normalized injured c-fos intensity, 0.77 ± 0.04; normalized uninjured c-fos intensity, 1.0 ± 0.03; p < 0.01) but could be restored by blocking GluN2B-containing NMDARs during the period of bicuculline stimulation (0.91 ± 0.05). Scale bar, 100 μm.
Discussion
In this study, we explored how properties of connectivity and calcium signaling at the single-neuron scale relate to the disruption in network structure and function that occurs following mechanical injury. Our main finding was that individual neurons undergo complex functional rewiring following injury; this rewiring response is influenced strongly by the initial neuronal connectivity within the microcircuit, the relative balance of GluN2B and GluN2A NMDARs in each neuron, and the subsequent synchronization of the circuit after injury. Together, these data point to dual roles for the GluN2B subunit in influencing outcome after injury. First, by acting as primary mechanosensors, the GluN2B NMDARs confer varying degrees of individual neuronal susceptibility to mechanical injury. Second, by contributing to uncorrelated activation after injury, the activation of GluN1/GluN2B NMDARs further impairs network activity and reduces network plasticity.
Although many reports highlight the detrimental effects of excessive Ca2+ influx through the NMDAR acutely following traumatic brain injuries, relatively little is known about how the NMDAR may shape the network structure after injury. Our data show that stretch activation of the NMDA receptor is critical in disrupting the wiring of circuitry following mechanical trauma, as inhibition completely blocks any stretch-induced changes in functional connectivity of the microcircuit. Our findings move one step further, showing that traumatically induced changes in network activity that we and others have reported (Cullen and LaPlaca, 2006; Goforth et al., 2011; Ferrario et al., 2013) are now predictable from features of individual neurons within a microcircuit. Perhaps most intriguing, our data show that neurons with relatively large calcium influx through GluN2B-containing NMDARs during spontaneous activity have the most dramatic changes in their integration within the microcircuit following injury. We are careful not to presume that a larger proportion of calcium influx through either GluN2A- or GluN2B-containing NMDARs is a direct measure of the distribution of synaptic NMDARs of either type, as the relative activation of these receptors depends on the incoming stimulus frequency, the number of glutamate vesicles released, and the spatial location of these receptors on the postsynaptic surface (Santucci and Raghavachari, 2008; Singh et al., 2011). However, it is known that the activation of postsynaptic GluN2A NMDARs is more likely than the activation of GluN2B NMDARs (Franks et al., 2002; Singh et al., 2011), and we would suggest that neurons showing a significant calcium influx through GluN2B NMDARs are likely to contain an even higher actual GluN1/GluN2B NMDAR ratio. Our data showing that individual neurons with relatively prominent GluN2B NMDAR calcium signaling are most likely to change their integration within the circuit is consistent with past work demonstrating the selective mechanosensitivity conferred by the GluN2B subunit (Singh et al., 2012; Ferrario et al., 2013) and highlights how mechanical injury may lead to a heterogeneous response for individual neurons in a given circuit.
These connectivity-dependent changes in the network wiring—with some neurons losing relatively few connections, and other neurons both losing existing and gaining new connections—suggests that the network may undergo a distinct shift in the fundamental properties. From a network controllability standpoint, nodes with a high degree of connectivity, or hub nodes, are essential for maintaining the rhythm or a particular state of the network, whereas nodes with a low degree of connectivity are required to change the network state (Liu et al., 2011). Our results suggest that the modest loss of connections among the hub neurons will help retain some rhythm to the network, consistent with our data showing that some synchronization in the network is retained after mechanical injury. However, as neurons with a low degree of connectivity are removed from the network following injury, the intrinsic ability to shape the overall network state is compromised (Liu et al., 2011) and is one contributing reason to the emergence of smaller, uncoordinated modules of activity within the broader circuit after injury. These network changes are reminiscent of recent work showing the degeneration of synchronized network behavior in the chronically epileptic dentate gyrus (Feldt Muldoon et al., 2013), but extend these by developing predictive measures for determining neuronal integration into the circuitry after mechanical injury. With our current analysis, we cannot distinguish the number of autaptic connections relative to the number of connections with neighboring neurons. However, if neurons enriched in autapses appeared in large number, this may introduce a group of neurons with low connectivity that lose most of their connections after stretch injury but still retain some integration and activity within the network. In future work, studying the effects of injury in culture conditions that favor autaptic connections may reveal deeper interpretation of the presented data.
Perhaps the most significant impact of these microcircuit changes is on the synchronization that can be achieved by the network. Rhythmic, synchronized activity is a natural property of neuronal networks and is conserved across species, from Caenorhabditis elegans to humans. Mounting evidence suggests that transient synchronization of neuronal discharges dynamically binds widely distributed sets of neurons into functionally coherent ensembles, allowing these networks to communicate effectively (Singer, 1993; Womelsdorf et al., 2007). For example, synchronization in the β-band and γ-band is involved in a variety cognitive functions, such as perceptual grouping, attention-dependent stimulus selection, routing of signals across distributed cortical networks, working memory, and perceptual awareness in mice and humans (Varela et al., 2001; Fries, 2009). Factors that directly affect circuit synchronization in pathological brain states include axonal demyelination, traumatic axonal injury, or early signs of axonal protein aggregates in α-synucleinopathies and tauopathies (Babiloni et al., 2004; Arrondo et al., 2009; Marquez de la Plata et al., 2011; Volpicelli-Daley et al., 2011; Patel et al., 2012). Here, we report an entirely new mechanism for network desynchronization: the reduction in voltage-dependent blockade of GluN2B-containing NMDARs following mechanical injury, leading to increased, uncorrelated network activity. We propose that presynaptic GluN2B NMDARs likely play an important role in this uncorrelated activity, as their role in facilitating presynaptic glutamate release is well known (Pinheiro and Mulle, 2008). However, pharmacologically isolating this receptor population is difficult. Future work will attempt to identify this potential source of modulation for circuit desynchronization, as controlling the level of synchrony is a key parameter to consider when rebuilding injured networks.
One consequence of loss in synchronization is a corresponding impairment in the injured network to change or augment its network structure following injury. In general, the NMDAR Mg+2 block is important for the induction of plasticity (Miyashita et al., 2012). With a selective reduction in the voltage-sensitive Mg+2 block of GluN2B-containingNMDARs, the resulting calcium influx during normal network-bursting events significantly shifts further toward GluN2B-mediated signaling. Influx through GluN1/GluN2B NMDARs is frequently associated with neuronal degeneration pathways, and additional evidence suggests that it may be a key mechanism of long-term synaptic depression (Massey et al., 2004). Furthermore, GluN2B NMDAR activation will limit nuclear CREB signaling, a feature of transcriptionally dependent LTP (Hardingham et al., 2002). The restoration of network augmentation by selective inhibition of the GluN2B NMDARs indicates that this NMDA receptor subpopulation not only is important for the initial disruption of network structure, but is also a key mediator in the rebuilding of the circuitry within the acute period following injury. Future work can identify the specific mechanisms responsible for this GluN2B-mediated improvement in network function, and could point toward new therapeutic manipulation points beyond the receptor that could facilitate the rebuilding of networks after mechanical injury.
In closing, our work identifies new features embedded within neural circuitry that dictate whether individual neurons will deintegrate from a microcircuit following injury, how the structure of the network is reshaped after injury, and how selective changes to the properties of individual NMDAR subtypes mediate the impairment of plasticity in networks after injury. Although the exact mechanisms to reintegrate neurons into the remaining microcircuit after injury are the source of future study, our current work identifies important properties of individual neurons that lead to the complex rewiring of circuitry that occurs following traumatic injury, and may provide insights into how the circuitry within different brain regions can change and recover following traumatic brain injury.
Footnotes
This work was funded by National Institutes of Health Grants RO1 NS35712, HD41699, and PO1 NS056202; and Department of Defense Grant W911NF-10-1-0526.
The authors declare no competing financial interests.
References
- Akerboom J, Chen TW, Wardill TJ, Tian L, Marvin JS, Mutlu S, Calderón NC, Esposti F, Borghuis BG, Sun XR, Gordus A, Orger MB, Portugues R, Engert F, Macklin JJ, Filosa A, Aggarwal A, Kerr RA, Takagi R, Kracun S, et al. Optimization of a GCaMP calcium indicator for neural activity imaging. J Neurosci. 2012;32:13819–13840. doi: 10.1523/JNEUROSCI.2601-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander MP. Mild traumatic brain injury: pathophysiology, natural history, and clinical management. Neurology. 1995;45:1253–1260. doi: 10.1212/WNL.45.7.1253. [DOI] [PubMed] [Google Scholar]
- Arnold FJ, Hofmann F, Bengtson CP, Wittmann M, Vanhoutte P, Bading H. Microelectrode array recordings of cultured hippocampal networks reveal a simple model for transcription and protein synthesis-dependent plasticity. J Physiol. 2005;564:3–19. doi: 10.1113/jphysiol.2004.077446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrondo G, Alegre M, Sepulcre J, Iriarte J, Artieda J, Villoslada P. Abnormalities in brain synchronization are correlated with cognitive impairment in multiple sclerosis. Mult Scler. 2009;15:509–516. doi: 10.1177/1352458508101321. [DOI] [PubMed] [Google Scholar]
- Auberson YP, Allgeier H, Bischoff S, Lingenhoehl K, Moretti R, Schmutz M. 5-Phosphonomethylquinoxalinediones as competitive NMDA receptor antagonists with a preference for the human 1A/2A, rather than 1A/2B receptor composition. Bioorg Med Chem Lett. 2002;12:1099–1102. doi: 10.1016/S0960-894X(02)00074-4. [DOI] [PubMed] [Google Scholar]
- Babiloni C, Ferri R, Moretti DV, Strambi A, Binetti G, Dal Forno G, Ferreri F, Lanuzza B, Bonato C, Nobili F, Rodriguez G, Salinari S, Passero S, Rocchi R, Stam CJ, Rossini PM. Abnormal fronto-parietal coupling of brain rhythms in mild Alzheimer's disease: a multicentric EEG study. Eur J Neurosci. 2004;19:2583–2590. doi: 10.1111/j.0953-816X.2004.03333.x. [DOI] [PubMed] [Google Scholar]
- Berberich S, Punnakkal P, Jensen V, Pawlak V, Seeburg PH, Hvalby Ø, Köhr G. Lack of NMDA receptor subtype selectivity for hippocampal long-term potentiation. J Neurosci. 2005;25:6907–6910. doi: 10.1523/JNEUROSCI.1905-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnelle V, Leech R, Kinnunen KM, Ham TE, Beckmann CF, De Boissezon X, Greenwood RJ, Sharp DJ. Default mode network connectivity predicts sustained attention deficits after traumatic brain injury. J Neurosci. 2011;31:13442–13451. doi: 10.1523/JNEUROSCI.1163-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnelle V, Ham TE, Leech R, Kinnunen KM, Mehta MA, Greenwood RJ, Sharp DJ. Salience network integrity predicts default mode network function after traumatic brain injury. Proc Natl Acad Sci U S A. 2012;109:4690–4695. doi: 10.1073/pnas.1113455109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsáki G, Draguhn A. Neuronal oscillations in cortical networks. Science. 2004;304:1926–1929. doi: 10.1126/science.1099745. [DOI] [PubMed] [Google Scholar]
- Carmignoto G, Vicini S. Activity-dependent decrease in NMDA receptor responses during development of the visual cortex. Science. 1992;258:1007–1011. doi: 10.1126/science.1279803. [DOI] [PubMed] [Google Scholar]
- Chen G, Trombley PQ, van den Pol AN. Excitatory actions of GABA in developing rat hypothalamic neurones. J Physiol. 1996;494:451–464. doi: 10.1113/jphysiol.1996.sp021505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choo AM, Miller WJ, Chen YC, Nibley P, Patel TP, Goletiani C, Morrison B, 3rd, Kutzing MK, Firestein BL, Sul JY, Haydon PG, Meaney DF. Antagonism of purinergic signalling improves recovery from traumatic brain injury. Brain. 2013;136:65–80. doi: 10.1093/brain/aws286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen DK, LaPlaca MC. Neuronal response to high rate shear deformation depends on heterogeneity of the local strain field. J Neurotrauma. 2006;23:1304–1319. doi: 10.1089/neu.2006.23.1304. [DOI] [PubMed] [Google Scholar]
- De Kruijk JR, Twijnstra A, Leffers P. Diagnostic criteria and differential diagnosis of mild traumatic brain injury. Brain Inj. 2001;15:99–106. doi: 10.1080/02699050119160. [DOI] [PubMed] [Google Scholar]
- Dixon CE, Lyeth BG, Povlishock JT, Findling RL, Hamm RJ, Marmarou A, Young HF, Hayes RL. A fluid percussion model of experimental brain injury in the rat. J Neurosurg. 1987;67:110–119. doi: 10.3171/jns.1987.67.1.0110. [DOI] [PubMed] [Google Scholar]
- Feldt Muldoon S, Soltesz I, Cossart R. Spatially clustered neuronal assemblies comprise the microstructure of synchrony in chronically epileptic networks. Proc Natl Acad Sci U S A. 2013;110:3567–3572. doi: 10.1073/pnas.1216958110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrario CR, Ndukwe BO, Ren J, Satin LS, Goforth PB. Stretch injury selectively enhances extrasynaptic, GluN2B-containing NMDA receptor function in cortical neurons. J Neurophysiol. 2013;110:131–140. doi: 10.1152/jn.01011.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fetcho JR, Cox KJ, O'Malley DM. Monitoring activity in neuronal populations with single-cell resolution in a behaving vertebrate. Histochem J. 1998;30:153–167. doi: 10.1023/A:1003243302777. [DOI] [PubMed] [Google Scholar]
- Fischer G, Mutel V, Trube G, Malherbe P, Kew JN, Mohacsi E, Heitz MP, Kemp JA. Ro 25–6981, a highly potent and selective blocker of N-methyl-D-aspartate receptors containing the NR2B subunit. Characterization in vitro. J Pharmacol Exp Ther. 1997;283:1285–1292. [PubMed] [Google Scholar]
- Franks KM, Bartol TM, Jr, Sejnowski TJ. A Monte Carlo model reveals independent signaling at central glutamatergic synapses. Biophys J. 2002;83:2333–2348. doi: 10.1016/S0006-3495(02)75248-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frick A, Magee J, Johnston D. LTP is accompanied by an enhanced local excitability of pyramidal neuron dendrites. Nat Neurosci. 2004;7:126–135. doi: 10.1038/nn1178. [DOI] [PubMed] [Google Scholar]
- Fries P. A mechanism for cognitive dynamics: neuronal communication through neuronal coherence. Trends Cogn Sci. 2005;9:474–480. doi: 10.1016/j.tics.2005.08.011. [DOI] [PubMed] [Google Scholar]
- Fries P. Neuronal gamma-band synchronization as a fundamental process in cortical computation. Annu Rev Neurosci. 2009;32:209–224. doi: 10.1146/annurev.neuro.051508.135603. [DOI] [PubMed] [Google Scholar]
- Frizelle PA, Chen PE, Wyllie DJ. Equilibrium constants for (R)-[(S)-1-(4-bromo-phenyl)-ethylamino]-(2,3-dioxo-1,2,3,4-tetrahydroquinoxalin-5-yl)-methyl]-phosphonic acid (NVP-AAM077) acting at recombinant NR1/NR2A and NR1/NR2B N-methyl-D-aspartate receptors: implications for studies of synaptic transmission. Mol Pharmacol. 2006;70:1022–1032. doi: 10.1124/mol.106.024042. [DOI] [PubMed] [Google Scholar]
- Furukawa Y, Okada M, Akaike N, Hayashi T, Nabekura J. Reduction of voltage-dependent magnesium block of N-methyl-D-aspartate receptor-mediated current by in vivo axonal injury. Neuroscience. 2000;96:385–392. doi: 10.1016/S0306-4522(99)00553-9. [DOI] [PubMed] [Google Scholar]
- Gasparini S, Migliore M, Magee JC. On the initiation and propagation of dendritic spikes in CA1 pyramidal neurons. J Neurosci. 2004;24:11046–11056. doi: 10.1523/JNEUROSCI.2520-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geddes-Klein DM, Serbest G, Mesfin MN, Cohen AS, Meaney DF. Pharmacologically induced calcium oscillations protect neurons from increases in cytosolic calcium after trauma. J Neurochem. 2006;97:462–474. doi: 10.1111/j.1471-4159.2006.03761.x. [DOI] [PubMed] [Google Scholar]
- Goforth PB, Ren J, Schwartz BS, Satin LS. Excitatory synaptic transmission and network activity are depressed following mechanical injury in cortical neurons. J Neurophysiol. 2011;105:2350–2363. doi: 10.1152/jn.00467.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall RC, Chapman MJ. Definition, diagnosis, and forensic implications of postconcussional syndrome. Psychosomatics. 2005;46:195–202. doi: 10.1176/appi.psy.46.3.195. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Bading H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci. 2010;11:682–696. doi: 10.1038/nrn2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci. 2002;5:405–414. doi: 10.1038/nn835. [DOI] [PubMed] [Google Scholar]
- Hestrin S. Developmental regulation of NMDA receptor-mediated synaptic currents at a central synapse. Nature. 1992;357:686–689. doi: 10.1038/357686a0. [DOI] [PubMed] [Google Scholar]
- Hulkower MB, Poliak DB, Rosenbaum SB, Zimmerman ME, Lipton ML. A decade of DTI in traumatic brain injury: 10 years and 100 articles later. AJNR Am J Neuroradiol. 2013;34:2064–2074. doi: 10.3174/ajnr.A3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt DL, Castillo PE. Synaptic plasticity of NMDA receptors: mechanisms and functional implications. Curr Opin Neurobiol. 2012;22:496–508. doi: 10.1016/j.conb.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingebrigtsen T, Romner B, Marup-Jensen S, Dons M, Lundqvist C, Bellner J, Alling C, Børgesen SE. The clinical value of serum S-100 protein measurements in minor head injury: a Scandinavian multicentre study. Brain Inj. 2000;14:1047–1055. doi: 10.1080/02699050050203540. [DOI] [PubMed] [Google Scholar]
- Kew JN, Richards JG, Mutel V, Kemp JA. Developmental changes in NMDA receptor glycine affinity and ifenprodil sensitivity reveal three distinct populations of NMDA receptors in individual rat cortical neurons. J Neurosci. 1998;18:1935–1943. doi: 10.1523/JNEUROSCI.18-06-01935.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloda A, Lua L, Hall R, Adams DJ, Martinac B. Liposome reconstitution and modulation of recombinant N-methyl-D-aspartate receptor channels by membrane stretch. Proc Natl Acad Sci U S A. 2007;104:1540–1545. doi: 10.1073/pnas.0609649104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushner D. Mild traumatic brain injury: toward understanding manifestations and treatment. Arch Intern Med. 1998;158:1617–1624. doi: 10.1001/archinte.158.15.1617. [DOI] [PubMed] [Google Scholar]
- Liu L, Wong TP, Pozza MF, Lingenhoehl K, Wang Y, Sheng M, Auberson YP, Wang YT. Role of NMDA receptor subtypes in governing the direction of hippocampal synaptic plasticity. Science. 2004;304:1021–1024. doi: 10.1126/science.1096615. [DOI] [PubMed] [Google Scholar]
- Liu YY, Slotine JJ, Barabási AL. Controllability of complex networks. Nature. 2011;473:167–173. doi: 10.1038/nature10011. [DOI] [PubMed] [Google Scholar]
- Losonczy A, Magee JC. Integrative properties of radial oblique dendrites in hippocampal CA1 pyramidal neurons. Neuron. 2006;50:291–307. doi: 10.1016/j.neuron.2006.03.016. [DOI] [PubMed] [Google Scholar]
- Lusardi TA, Wolf JA, Putt ME, Smith DH, Meaney DF. Effect of acute calcium influx after mechanical stretch injury in vitro on the viability of hippocampal neurons. J Neurotrauma. 2004a;21:61–72. doi: 10.1089/089771504772695959. [DOI] [PubMed] [Google Scholar]
- Lusardi TA, Rangan J, Sun D, Smith DH, Meaney DF. A device to study the initiation and propagation of calcium transients in cultured neurons after mechanical stretch. Ann Biomed Eng. 2004b;32:1546–1558. doi: 10.1114/B:ABME.0000049038.75368.75. [DOI] [PubMed] [Google Scholar]
- Marquez de la Plata CD, Garces J, Shokri Kojori E, Grinnan J, Krishnan K, Pidikiti R, Spence J, Devous MD, Sr, Moore C, McColl R, Madden C, Diaz-Arrastia R. Deficits in functional connectivity of hippocampal and frontal lobe circuits after traumatic axonal injury. Arch Neurol. 2011;68:74–84. doi: 10.1001/archneurol.2010.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massey PV, Johnson BE, Moult PR, Auberson YP, Brown MW, Molnar E, Collingridge GL, Bashir ZI. Differential roles of NR2A and NR2B-containing NMDA receptors in cortical long-term potentiation and long-term depression. J Neurosci. 2004;24:7821–7828. doi: 10.1523/JNEUROSCI.1697-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meaney DF, Smith DH, Shreiber DI, Bain AC, Miller RT, Ross DT, Gennarelli TA. Biomechanical analysis of experimental diffuse axonal injury. J Neurotrauma. 1995;12:689–694. doi: 10.1089/neu.1995.12.689. [DOI] [PubMed] [Google Scholar]
- Miyashita T, Oda Y, Horiuchi J, Yin JC, Morimoto T, Saitoe M. Mg(2+) block of Drosophila NMDA receptors is required for long-term memory formation and CREB-dependent gene expression. Neuron. 2012;74:887–898. doi: 10.1016/j.neuron.2012.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron. 1994;12:529–540. doi: 10.1016/0896-6273(94)90210-0. [DOI] [PubMed] [Google Scholar]
- Pandit AS, Expert P, Lambiotte R, Bonnelle V, Leech R, Turkheimer FE, Sharp DJ. Traumatic brain injury impairs small-world topology. Neurology. 2013;80:1826–1833. doi: 10.1212/WNL.0b013e3182929f38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoletti P, Ascher P. Mechanosensitivity of NMDA receptors in cultured mouse central neurons. Neuron. 1994;13:645–655. doi: 10.1016/0896-6273(94)90032-9. [DOI] [PubMed] [Google Scholar]
- Patel TP, Ventre SC, Meaney DF. Dynamic changes in neural circuit topology following mild mechanical injury in vitro. Ann Biomed Eng. 2012;40:23–36. doi: 10.1007/s10439-011-0390-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinheiro PS, Mulle C. Presynaptic glutamate receptors: physiological functions and mechanisms of action. Nat Rev Neurosci. 2008;9:423–436. doi: 10.1038/nrn2379. [DOI] [PubMed] [Google Scholar]
- Reeves TM, Lyeth BG, Povlishock JT. Long-term potentiation deficits and excitability changes following traumatic brain injury. Exp Brain Res. 1995;106:248–256. doi: 10.1007/BF00241120. [DOI] [PubMed] [Google Scholar]
- Regehr WG, Tank DW. Postsynaptic NMDA receptor-mediated calcium accumulation in hippocampal CA1 pyramidal cell dendrites. Nature. 1990;345:807–810. doi: 10.1038/345807a0. [DOI] [PubMed] [Google Scholar]
- Roberts EB, Ramoa AS. Enhanced NR2A subunit expression and decreased NMDA receptor decay time at the onset of ocular dominance plasticity in the ferret. J Neurophysiol. 1999;81:2587–2591. doi: 10.1152/jn.1999.81.5.2587. [DOI] [PubMed] [Google Scholar]
- Santucci DM, Raghavachari S. The effects of NR2 subunit-dependent NMDA receptor kinetics on synaptic transmission and CaMKII activation. PLoS Comput Biol. 2008;4:e1000208. doi: 10.1371/journal.pcbi.1000208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz-Clemente A, Nicoll RA, Roche KW. Diversity in NMDA receptor composition: many regulators, many consequences. Neuroscientist. 2013;19:62–75. doi: 10.1177/1073858411435129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzbach E, Bonislawski DP, Xiong G, Cohen AS. Mechanisms underlying the inability to induce area CA1 LTP in the mouse after traumatic brain injury. Hippocampus. 2006;16:541–550. doi: 10.1002/hipo.20183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp DJ, Beckmann CF, Greenwood R, Kinnunen KM, Bonnelle V, De Boissezon X, Powell JH, Counsell SJ, Patel MC, Leech R. Default mode network functional and structural connectivity after traumatic brain injury. Brain. 2011;134:2233–2247. doi: 10.1093/brain/awr175. [DOI] [PubMed] [Google Scholar]
- Sick TJ, Pérez-Pinzón MA, Feng ZZ. Impaired expression of long-term potentiation in hippocampal slices 4 and 48 h following mild fluid-percussion brain injury in vivo. Brain Res. 1998;785:287–292. doi: 10.1016/S0006-8993(97)01418-2. [DOI] [PubMed] [Google Scholar]
- Singer W. Synchronization of cortical activity and its putative role in information processing and learning. Annu Rev Physiol. 1993;55:349–374. doi: 10.1146/annurev.ph.55.030193.002025. [DOI] [PubMed] [Google Scholar]
- Singh P, Hockenberry AJ, Tiruvadi VR, Meaney DF. Computational investigation of the changing patterns of subtype specific NMDA receptor activation during physiological glutamatergic neurotransmission. PLoS Comput Biol. 2011;7:e1002106. doi: 10.1371/journal.pcbi.1002106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh P, Doshi S, Spaethling JM, Hockenberry AJ, Patel TP, Geddes-Klein DM, Lynch DR, Meaney DF. N-methyl-D-aspartate receptor mechanosensitivity is governed by C terminus of NR2B subunit. J Biol Chem. 2012;287:4348–4359. doi: 10.1074/jbc.M111.253740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern RA, Riley DO, Daneshvar DH, Nowinski CJ, Cantu RC, McKee AC. Long-term consequences of repetitive brain trauma: chronic traumatic encephalopathy. PM R. 2011;3(10 Suppl 2):S460–S467. doi: 10.1016/j.pmrj.2011.08.008. [DOI] [PubMed] [Google Scholar]
- Stocca G, Vicini S. Increased contribution of NR2A subunit to synaptic NMDA receptors in developing rat cortical neurons. J Physiol. 1998;507:13–24. doi: 10.1111/j.1469-7793.1998.013bu.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stosiek C, Garaschuk O, Holthoff K, Konnerth A. In vivo two-photon calcium imaging of neuronal networks. Proc Natl Acad Sci U S A. 2003;100:7319–7324. doi: 10.1073/pnas.1232232100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian L, Hires SA, Mao T, Huber D, Chiappe ME, Chalasani SH, Petreanu L, Akerboom J, McKinney SA, Schreiter ER, Bargmann CI, Jayaraman V, Svoboda K, Looger LL. Imaging neural activity in worms, flies and mice with improved GCaMP calcium indicators. Nat Methods. 2009;6:875–881. doi: 10.1038/nmeth.1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovar KR, Westbrook GL. The incorporation of NMDA receptors with a distinct subunit composition at nascent hippocampal synapses in vitro. J Neurosci. 1999;19:4180–4188. doi: 10.1523/JNEUROSCI.19-10-04180.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varela F, Lachaux JP, Rodriguez E, Martinerie J. The brainweb: phase synchronization and large-scale integration. Nat Rev Neurosci. 2001;2:229–239. doi: 10.1038/35067550. [DOI] [PubMed] [Google Scholar]
- Volpicelli-Daley LA, Luk KC, Patel TP, Tanik SA, Riddle DM, Stieber A, Meaney DF, Trojanowski JQ, Lee VM. Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron. 2011;72:57–71. doi: 10.1016/j.neuron.2011.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Engelhardt J, Doganci B, Seeburg PH, Monyer H. Synaptic NR2A- but not NR2B-containing NMDA receptors increase with blockade of ionotropic glutamate receptors. Front Mol Neurosci. 2009;2:19. doi: 10.3389/neuro.02.019.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Womelsdorf T, Schoffelen JM, Oostenveld R, Singer W, Desimone R, Engel AK, Fries P. Modulation of neuronal interactions through neuronal synchronization. Science. 2007;316:1609–1612. doi: 10.1126/science.1139597. [DOI] [PubMed] [Google Scholar]
- Yuste R, Peinado A, Katz LC. Neuronal domains in developing neocortex. Science. 1992;257:665–669. doi: 10.1126/science.1496379. [DOI] [PubMed] [Google Scholar]
- Zhang L, Rzigalinski BA, Ellis EF, Satin LS. Reduction of voltage-dependent Mg2+ blockade of NMDA current in mechanically injured neurons. Science. 1996;274:1921–1923. doi: 10.1126/science.274.5294.1921. [DOI] [PubMed] [Google Scholar]