

Abstract
Kv2.1 is a major delayed rectifying K+ channel normally localized to highly phosphorylated somatodendritic clusters in neurons. Excitatory stimuli induce calcineurin-dependent dephosphorylation and dispersal of Kv2.1 clusters, with a concomitant hyperpolarizing shift in the channel's activation kinetics. We showed previously that sublethal ischemia, which renders neurons transiently resistant to excitotoxic cell death, can also induce Zn2+-dependent changes in Kv2.1 localization and activation kinetics, suggesting that activity-dependent modifications of Kv2.1 may contribute to cellular adaptive responses to injury. Recently, cyclin-dependent kinase 5 (Cdk5) was shown to phosphorylate Kv2.1, with pharmacological Cdk5 inhibition being sufficient to decluster channels. In another study, cyclin E1 was found to restrict neuronal Cdk5 kinase activity. We show here that cyclin E1 regulates Kv2.1 cellular localization via inhibition of Cdk5 activity. Expression of cyclin E1 in human embryonic kidney cells prevents Cdk5-mediated phosphorylation of Kv2.1, and cyclin E1 overexpression in rat cortical neurons triggers dispersal of Kv2.1 channel clusters. Sublethal ischemia in neurons induces calcineurin-dependent upregulation of cyclin E1 protein expression and cyclin E1-dependent Kv2.1 channel declustering. Importantly, overexpression of cyclin E1 in neurons is sufficient to reduce excitotoxic cell death. These results support a novel role for neuronal cyclin E1 in regulating the phosphorylation status and localization of Kv2.1 channels, a likely component of signaling cascades leading to ischemic preconditioning.
Keywords: Cdk5, cyclin E1, excitotoxicity, ischemia, Kv2.1, preconditioning
Introduction
Voltage-gated Kv2.1 channels mediate a significant component of delayed rectifying K+ currents in neurons. As such, they critically regulate neuronal excitability, particularly during periods of high-frequency synaptic transmission (Murakoshi and Trimmer, 1999; Du et al., 2000; Malin and Nerbonne, 2002; Guan et al., 2013). Approximately half of the Kv2.1 channels present on cortical and hippocampal neuronal cell membranes are maintained in highly phosphorylated, somatodendritic clusters (Fox et al., 2013). Excitatory or injurious stimuli trigger calcineurin-dependent channel dephosphorylation at multiple C-terminal serine residues, which is accompanied by cluster dispersal and a hyperpolarizing shift in channel voltage-gated activation (Misonou et al., 2004; Mulholland et al., 2008; Aras et al., 2009a; Baver and O'Connell, 2012; Shepherd et al., 2012; Shah and Aizenman, 2014). Calcineurin-mediated dephosphorylation at several C-terminal residues is thought to mediate the hyperpolarizing activation shift (Park et al., 2006), which may mitigate neuronal damage and cell death within the context of excitotoxic injury by reducing neuronal excitability (Aras et al., 2009a,b; Mohapatra et al., 2009; Shepherd et al., 2013).
The phosphorylation status of one Kv2.1 C-terminal serine residue in particular, Ser603, is highly sensitive to changes in neuronal activity. Glutamate stimulation triggers a precipitous decrease in Ser603 phosphorylation, whereas acute activity blockade results in Ser603 hyperphosphorylation (Misonou et al., 2006). Ser603 is phosphorylated by cyclin dependent kinase 5 (Cdk5), a kinase highly expressed in postmitotic neurons that regulates many critical physiological functions (Cheung et al., 2006; Cerda and Trimmer, 2011). Pharmacologic inhibition of Cdk5 kinase activity causes Ser603 dephosphorylation and channel declustering, and prevents recovery of Ser603 phosphorylation and channel reclustering following washout of glutamate (Cerda and Trimmer, 2011).
Here, we identify cyclin E1 as a novel regulator of Kv2.1 localization via inhibition of Cdk5 kinase-mediated phosphorylation of the channel. Our studies suggest that cyclin E1 facilitates Kv2.1 channel dephosphorylation and declustering in neurons subjected to sublethal ischemia, and may be critical for cell survival mechanisms following excitotoxic injury.
Materials and Methods
Cell culture, transfection, and ischemic preconditioning.
Cortical neuronal cultures were prepared from embryonic day 16–17 rat embryos of either sex (Aras et al., 2009a) and transfected at 21–25 DIV using Lipofectamine 2000 (Invitrogen; Aras et al., 2009b). Cultures were preconditioned with 3 mm potassium cyanide (KCN) in a glucose-free balanced salt solution [containing (in mm) 150 NaCl, 2.8 KCl, 1 CaCl2, and 10 HEPES, pH 7.2] for 90 min at 37°C (McLaughlin et al., 2003). For viability assays in transfected neurons, 24 h after transfection or preconditioning, neurons were treated with 10 μm glycine (vehicle) ± 75 μm NMDA for 30 min and assayed for viability after 24 h as reported previously (Aras et al., 2009b).
Confocal imaging.
24 h following transfection with GFP-tagged Kv2.1, cyclin E1, Cdk5-DN, or corresponding vector, live imaging of transfected neurons was performed on a Nikon A1+ confocal microscope at 60× magnification. Five to 10 optical sections (0.5 μm) were acquired to generate a maximum intensity projection image that was analyzed using NIH image processing software (ImageJ). Following background subtraction, a plot displaying a three-dimensional graph of pixel intensity over the neuronal soma was used to show Kv2.1 localization (Fig. 1B). Channel clusters appearing as orange-red peaks in pixel intensity were counted. In several control cells, there were large-density peaks that likely represented multiple channel clusters; these peaks were counted as one cluster to preclude any possibility of overcounting for these cells. Therefore, we may have underestimated the cluster number under control conditions. Nikon Instruments Software (NIS)-Elements Advanced Research was used to measure cluster surface area and to confirm channel cluster counts.
Figure 1.
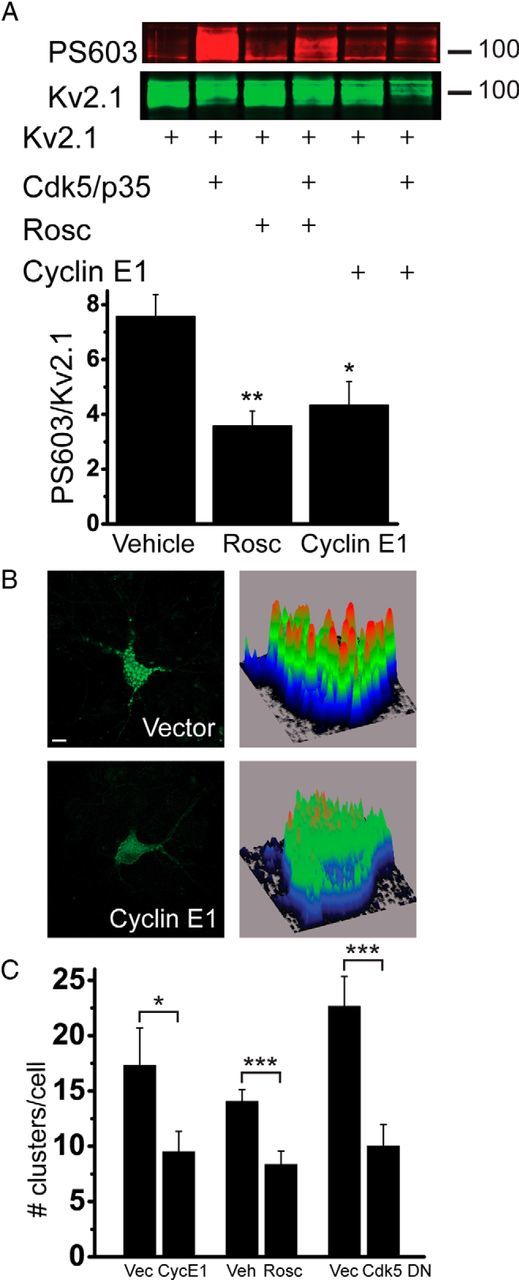
Cyclin E1 overexpression blocks Cdk5/p35-mediated Kv2.1 phosphorylation and induces channel declustering. A, HEK293T cells were cotransfected with Kv2.1, Cdk5/p35 or vector, and cyclin E1 or vector. Twenty-one hours after transfection, cells were treated with vehicle (DMSO) or roscovitine (rosc), and proteins were harvested immediately after exposure. Top, Representative immunoblot. Duplicate membranes were probed with phosphorylated Ser603 Kv2.1 antibody (PS603) or Kv2.1 antibody. Numbers to the right indicate the mobility of molecular mass standard (in kilodaltons). Bottom, Summary of six independent experiments. Values represent Cdk5/p35-mediated increases in PS603 normalized to total Kv2.1, expressed as the ratio of PS603/total Kv2.1 (+Cdk5/p35) to corresponding PS603/total Kv2.1 (no Cdk5/p35), e.g., the first column in the bar graph is the ratio of PS603/total Kv2.1 (lane 2) to PS603/total Kv2.1 (lane 1; mean ± SEM). *p < 0.05, **p < 0.01 (ANOVA/Dunnett's test vs vehicle). B, Neuronal cultures were transfected with GFP-Kv2.1 and cyclin E1, dominant-negative Cdk5 (Cdk5-DN), or vector, or transfected with GFP-Kv2.1 and treated with DMSO or roscovitine 23 h after transfection. Neurons were imaged 24 h after transfection. Shown are representative neurons and their associated background-subtracted surface maps, which show relative Kv2.1 staining intensity values plotted along the cell body area. Scale bar, 10 μm. C, Clusters that appeared as orange-red peaks in pixel intensity were counted for 24–42 cells per group from four to seven independent experiments. Data points represent the average number of clusters per cell (mean ± SEM). *p < 0.05; ***p < 0.001 (two-tailed unpaired t test vs vector or vehicle). Vec, Vector; CycE1, cyclin E1; Veh, vehicle; Rosc, roscovitine.
Immunoblotting.
Protein samples harvested from transfected human embryonic kidney (HEK) 293T cells or neuronal cultures were incubated with mouse monoclonal anti-Kv2.1 antibody (1:1000; NeuroMab), rabbit polyclonal anti-phosphorylated Ser603 Kv2.1 antibody (1:500; gift from Dr. James Trimmer, University of California Davis, Davis, CA), rabbit polyclonal anti-cyclin E1 antibody (1:500; Santa Cruz Biotechnology), or mouse monoclonal anti-GAPDH antibody (1:1000; Novus Biologicals) as a loading control. Blots were quantified by infrared fluorimetry (Li-Cor).
Electrophysiology.
Whole-cell recordings from cortical neurons were obtained with 2–3 MΩ electrodes (Aras et al., 2009a). The extracellular solution contained the following (in mm): 115 NaCl, 2.5 KCl, 2.0 MgCl2, 1.0 CaCl2, 10 HEPES, 10 d-glucose, and 0.25 μm tetrodotoxin, pH 7.2. The electrode contained the following (in mm): 100 K-gluconate, 10 KCl, 1 MgCl2, 1 CaCl2, 2.2 Mg2 · ATP, 0.33 GTP, 11 EGTA, and 10 HEPES, pH 7.2. Measurements were obtained under voltage clamp with an Axopatch 200B amplifier and pClamp software (Molecular Devices). Eighty percent compensation for series resistance was provided. Currents were filtered at 2 kHz and digitized at 10 kHz. K+ currents were evoked with a series of 200 ms voltage steps from a holding potential of −80 to +80 mV in 10 mV increments. Before depolarization, a single 30 ms prepulse to +10 mV was used to inactivate A-type K+ currents. Peak conductance (G) was calculated from peak steady-state current amplitudes (I) using the equation G = I/(V − EK) (EK = Nerst K+ equilibrium potential) and plotted against the potential (V) and fitted to a single Boltzmann function, G = Gmax/(1 + exp[−(V − V1/2)/k]), where Gmax is the maximum conductance, V1/2 is the potential at half-maximal conductance, and k is the slope of activation curve.
Results
Cyclin E1 inhibits Cdk5-mediated phosphorylation and clustering of Kv2.1 channels
Cyclin E1 was shown previously to influence dendritic spine density and synaptic function by regulating the kinase activity of Cdk5 (Odajima et al., 2011). We first determined whether cyclin E1 expression could inhibit Cdk5-facilitated Kv2.1 Ser603 phosphorylation. HEK293T cells were transfected with Kv2.1 and Cdk5, together with Cdk5 kinase coactivator p35, which yielded an increase in Ser603 phosphorylation that was significantly reduced by cyclin E1 coexpression (Fig. 1A). Similarly, as shown previously (Cerda and Trimmer, 2011), the Cdk5 inhibitor roscovitine (30 μm, 3 h) blocked Cdk5-mediated Kv2.1 Ser603 phosphorylation.
Previous studies have demonstrated that injury-induced Kv2.1 dephosphorylation is accompanied by dispersal of channel clusters (Misonou et al., 2004, 2006; Mulholland et al., 2008; Aras et al., 2009a; Shepherd et al., 2012, 2013). We hypothesized that cyclin E1 overexpression, by blocking Cdk5-mediated Kv2.1 Ser603 phosphorylation, would be sufficient to induce channel declustering in cortical neurons. We transfected rat cortical neurons with a GFP-tagged Kv2.1 construct, which exhibits somatodendritic clustering similar to endogenous Kv2.1 channels (O'Connell et al., 2006; Fig. 1B), along with cyclin E1 or empty vector. Neurons overexpressing cyclin E1 displayed a significantly reduced number of channel clusters (Fig. 1B,C), which we verified using Nikon image analysis software. We also observed reduced cluster surface area in cyclin E1-expressing cells (vector, 4.0 ± 1.2 μm2, n = 23; cyclin E1, 1.6 ± 0.5 μm2, n = 30; p < 0.05, two-tailed unpaired t test). To confirm that Cdk5 kinase activity inhibition promotes channel declustering, we determined that overexpression of Cdk5-DN, a kinase-inactive, dominant-negative Cdk5 mutant (Nikolic et al., 1996), declustered Kv2.1 channels, and verified that roscovitine treatment triggered dispersal of channel clusters (Fig. 1C; Cerda and Trimmer, 2011).
Stimuli that activate Kv2.1 dephosphorylation and declustering induce a concomitant hyperpolarizing shift in the channel's voltage-gated activation. Cotreatment with calcineurin inhibitors in neurons (Misonou et al., 2004; Mohapatra and Trimmer, 2006; Aras et al., 2009a; Mohapatra et al., 2009; Shepherd et al., 2013) and mutational analysis of calcineurin dephosphorylation-dependent residues in HEK cells (Park et al., 2006) strongly suggest that channel dephosphorylation is closely associated with the activation shift. Therefore, we explored whether cyclin E1 overexpression or roscovitine exposure would promote a hyperpolarizing channel activation shift. We measured whole-cell K+ currents in neurons overexpressing cyclin E1 or exposed to roscovitine (30 μm, 1 h, followed by removal from roscovitine-containing media, as it has been shown to directly block Kv2.1 channels; Buraei et al., 2007). We found, however, that neither condition shifted Kv2.1 channel voltage-gated activation (V1/2, vector, 13.7 ± 1.1 mV; cyclin E1, 10.5 ± 2.3 mV; vehicle, 14.2 ± 1.5 mV; roscovitine, 18.5 ± 1.5 mV; n = 9, 10, 5, and 7 cells, respectively). We also noted no changes in current density (current densities at +10 mV in pA/pF, vector, 284.5 ± 22.9; cyclin E1, 228.4 ± 37.0; vehicle, 181.3 ± 19.0; roscovitine, 145.2 ± 27.3; n = 6, 7, 3, and 6 cells, respectively).
These results suggest that, as noted previously (Aras et al., 2009a; Baver and O'Connell, 2012), Kv2.1 dephosphorylation and declustering may not be unequivocally linked to changes in channel activation kinetics in all cases.
Neuronal ischemic preconditioning in vitro induces calcineurin-dependent upregulation of cyclin E1 expression
We have shown previously that chemical ischemic preconditioning, which mitigates subsequent excitotoxic neuronal injury, induces Kv2.1 channel dephosphorylation and declustering in cortical neurons (Aras et al., 2009a,b). Rapid Ser603 dephosphorylation is observed following ischemic or epileptic injury in vivo, and after glutamate treatment in cultured hippocampal neurons (Misonou et al., 2006; Cerda and Trimmer, 2011). We found that ischemic preconditioning in cortical neurons also reduced Ser603 phosphorylation in native Kv2.1 channels by 82% (Fig. 2A). We thus hypothesized that preconditioning-stimulated signaling pathways may lead to upregulation of cyclin E1 protein expression, which would contribute to Ser603 dephosphorylation and channel declustering by reducing Cdk5 kinase activity. Therefore, we measured cyclin E1 protein expression immediately following and 24 h after preconditioning, at which point Kv2.1 channels revert to the phosphorylated and clustered state (Aras et al., 2009a). Ischemic preconditioning triggered an increase in cyclin E1 protein expression immediately following treatment (Fig. 2B). Importantly, we also observed a return to control cyclin E1 levels 24 h after preconditioning. Thus, ischemia-induced changes in cyclin E1 coincide temporally with the modifications in phosphorylation and localization of Kv2.1 channels in neurons.
Figure 2.

Chemical ischemia induces calcineurin activity-dependent transient increase in neuronal cyclin E1 expression. Neurons were exposed to 3 mm KCN with vehicle (Veh) or FK520 (FK; 5 μm) for 90 min. Cell lysates were harvested immediately (0′) or 24 h following exposure. A, Membranes were probed with anti-PS603 Kv2.1 or anti-Kv2.1 antibody. A representative blot from one of three independent experiments is shown; phosphorylated Ser603 Kv2.1 normalized to total Kv2.1 expression is 0.18 ± 0.03 relative units (r.u.; vehicle), compared to 0.03 ± 0.02 r.u. (KCN; n = 3; p < 0.05, two-tailed paired t test). B, Membranes were probed with anti-cyclin E1 or anti-GAPDH antibody. Top, Representative immunoblots are shown. Bottom, Summaries of four (left) and five (right) independent experiments. Cyclin E1 values are normalized to loading control GAPDH (mean ± SEM). *p < 0.05 versus KCN; **p < 0.01 versus vehicle at 0′ after treatment; ***p < 0.001 versus KCN at 0′ after treatment (ANOVA/Bonferroni's test).
Kv2.1 channel dephosphorylation and declustering following ischemic or epileptic stimuli are dependent on the calcium-activated phosphatase calcineurin, which has been suggested to directly dephosphorylate Kv2.1 channels (Misonou et al., 2004; Park et al., 2006; but see Mulholland et al., 2008; Aras et al., 2009a; Shepherd et al., 2012). We explored the possibility that calcineurin may also be required for the preconditioning-mediated rise in cyclin E1 expression, as restricting calcineurin activity blocks growth factor-stimulated upregulation of cyclin E in fibroblasts (Tomono et al., 1998). Accordingly, we found that cotreatment with the calcineurin inhibitor FK520 (C43H69NO12; 5 μm) blocked the increase in cyclin E1 protein expression in preconditioned neurons (Fig. 2B). We confirmed that calcineurin activation occurs upstream of cyclin E1 upregulation by measuring cyclin E1 expression in neurons exposed to the calcium ionophore A23187 (1 μm, 10 min) with or without FK520: cyclin E1 expression normalized to loading control increases 1.5 ± 0.18-fold with A23187 treatment, compared with 0.78 ± 0.15-fold in A23187-exposed cells cotreated with FK520 (n = 3, p < 0.01, two-tailed paired t test).
p35 overexpression blocks ischemic preconditioning-mediated Kv2.1 channel declustering
Cyclin E1 restricts Cdk5 kinase activity by outcompeting binding of Cdk5 to p35 and forming a catalytically inactive complex with Cdk5 (Odajima et al., 2011). If ischemic preconditioning-triggered Kv2.1 declustering is dependent on cyclin E1-mediated inhibition of Cdk5 kinase activity, then overexpressing p35 should restore channel clustering in preconditioned neurons. Indeed, we found that p35-overexpressing preconditioned neurons retain Kv2.1 channel clusters (Fig. 3), strongly suggesting that increased cyclin E1 expression and the consequent inhibition of Cdk5 kinase activity contribute significantly to preconditioning-induced cluster dispersal.
Figure 3.
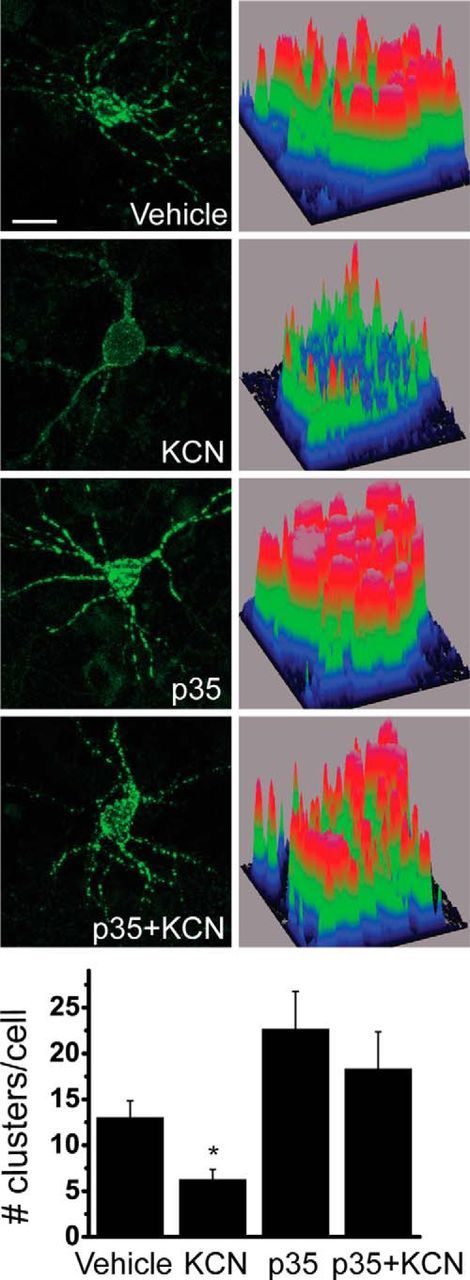
Overexpression of Cdk5 coactivator p35 blocks KCN-induced Kv2.1 channel declustering in cortical neurons. Top, Neurons were transfected with GFP-Kv2.1 and p35 or vector, preconditioned 24 h after transfection, and imaged immediately following preconditioning. Scale bar, 10 μm. Bottom, Number of clusters per cell was counted and averaged for 15–39 cells per group from six independent experiments (mean ± SEM). *p < 0.05 (ANOVA/Bonferroni's test vs vehicle).
Cyclin E1 overexpression reduces excitotoxic cell death
Finally, we investigated whether cyclin E1 overexpression, which produces similar changes in Kv2.1 channel phosphorylation status and clustering as ischemic preconditioning (Figs. 1, 2; Aras et al., 2009a), could alone promote neuronal tolerance to excitotoxic cell death. As shown in Figure 4, overexpressing cyclin E1 in cortical neurons mitigates excitotoxicity in NMDA-treated neurons at levels highly comparable to the neuronal tolerance elicited by ischemic preconditioning. Moreover, we confirmed that limiting Cdk5 kinase activity with the Cdk5-DN construct similarly reduces excitotoxic injury.
Figure 4.

Cyclin E1 overexpression decreases NMDA receptor-mediated excitotoxicity. Neurons were transfected with cyclin E1 or vector (first two columns) or Cdk5-DN or vector (last two columns) and a luciferase reporter gene, or transfected with vector and luciferase reporter gene and treated with KCN 24 h after transfection (second two columns). Twenty-four hours after transfection or preconditioning treatment, neurons were exposed to 10 μm glycine ± 75 μm NMDA (30 min), 24 h before viability assay. Mean (±SEM) values expressed as the percentage of toxicity of NMDA-treated relative to vehicle-treated neurons is shown. *p < 0.05; **p = 0.01 (vs corresponding vector or vehicle, two-tailed paired t test); n = 5, 6, and 4 for vector/cycE1, vehicle/preconditioning, and vector/Cdk5-DN, respectively. Note that slight differences in NMDA toxicity between vectors for cyclin E1 and for Cdk5-DN are due to differences in the inherent toxicity of transfected vectors. Precond, Preconditioning.
Discussion
Modulation of Kv2.1 channel phosphorylation, localization, and function, elicited by a range of injurious stimuli, may be a critical component of endogenous neuroprotective signaling pathways that reduce neuronal damage and cell death (Aras et al., 2009a; Mohapatra et al., 2009; Shepherd et al., 2013). We have identified cyclin E1 as a key regulator of Kv2.1 channel phosphorylation and localization in neuronal ischemia. Cyclin E1 blocks Cdk5-mediated Kv2.1 Ser603 phosphorylation and promotes the dispersal of Kv2.1 channel clusters in cortical neurons.
Interestingly, neither cyclin E1 overexpression nor exposure to roscovitine produced hyperpolarizing changes in Kv2.1 channel voltage-gated activation. In contrast, a previous study showed that Kv2.1 voltage-gated activation was closely linked to the phosphorylation status of Ser603 (Park et al., 2006). However, Park et al. (2006) used recombinant expression of a single population of channel mutants, which may not be completely representative of native Kv2.1 channels with varying degrees of phosphorylation in each of the four subunits that assemble to form functional ion channels in neurons. In fact, in neurons, injury-induced Kv2.1 channel activation shifts span a wide range of half-maximal activation voltages, from near 9 to 30 mV, depending on the stimulus and neuronal cell type under study (Misonou et al., 2004, 2008; Mulholland et al., 2008; Aras et al., 2009a; Mohapatra et al., 2009; Shepherd et al., 2013). Additionally, neuronal signaling pathways that are activated by excitatory or ischemic stimuli, but not associated with reducing Cdk5 kinase activity alone, may be required to alter the channel's function, including zinc-activated processes (Aras et al., 2009a), calcineurin activity-independent channel dephosphorylation (Mulholland et al., 2008), or posttranslational modifications at channel regions other than the C terminus (Baver and O'Connell, 2012).
We demonstrate here that ischemic preconditioning induces calcineurin activity-dependent upregulation of cyclin E1 protein expression in cortical neurons. The mechanism of calcineurin-driven elevated cyclin E1 levels is unlikely to involve NFAT (nuclear factor of activated T-cells)-mediated transcriptional upregulation, as the cyclin E1 gene does not contain NFAT consensus binding sites in its promoter region (UCSC Genome Browser). Calcineurin may dephosphorylate cyclin E1 at ubiquitin ligase-binding sites, preventing its degradation (Hwang and Clurman, 2005), although this potential mechanism remains to be fully characterized.
Our results point to a calcineurin activity-driven mechanism that ensures Kv2.1 dephosphorylation in preconditioned neurons, both by dephosphorylating the channel (Misonou et al., 2004; Park et al., 2006; Shepherd et al., 2012, 2013) and by promoting cyclin E1-mediated inhibition of Cdk5 kinase activity, as we have demonstrated in this study. We postulate that neuroprotection through increased cyclin E1 levels, as reported here, occurs at least in part through Kv2.1 dephosphorylation and declustering in preconditioned neurons, phenomena that have been closely tied to cell survival in a wide range of injury models (Aras et al., 2009a,b; Mohapatra et al., 2009; Shepherd et al., 2012, 2013). Naturally, other mechanisms in addition to Kv2.1 channel modifications may aid in this process, such as modulation of NMDA receptors by Cdk5 (Wang et al., 2003; Rashidian et al., 2009).
The specific contribution of Kv2.1 declustering to limiting neuronal hyperactivity in ischemia is unknown. Channel cluster localization adjacent to astrocytic processes (Du et al., 1998) may enable them to sense ischemic glial dysfunction due to compromised glutamatergic uptake, leading to excessive neuronal glutamate signaling and calcium influx, which would facilitate calcineurin-dependent dephosphorylation and the hyperpolarizing shift in channel activation (Misonou et al., 2008; Mulholland et al., 2008; Mohapatra et al., 2009). Subsequent channel declustering would remove the channels from the site of calcium release, initiating recovery. Alternatively, it was reported previously that the majority of clustered Kv2.1 channels are nonconducting (O'Connell et al., 2010; Fox et al., 2013), and may play roles in depolarization-driven vesicle trafficking (Feinshreiber et al., 2009). In fact, Kv2.1 channel clusters may serve as insertion platforms for targeting of new channels to the cell surface (Deutsch et al., 2012). In cortical neurons, an oxidative injury-triggered K+ current surge, mediated by newly inserted Kv2.1 channels at the plasma membrane, leads to apoptotic cell death (Pal et al., 2003, 2006; McCord and Aizenman, 2013). This cell death mechanism may also be critical in promoting NMDA receptor-mediated excitotoxicity (Yao et al., 2009). Cyclin E1-mediated dispersal of channel clusters may thus prevent proapoptotic insertion of new Kv2.1 channels at the cell surface, reducing neuronal damage and cell death in excitotoxic injury.
Footnotes
This work was supported by National Institutes of Health Grant NS043277 (E.A.) and American Heart Association Predoctoral Fellowship 12PRE11070001 (N.H.S.). We thank Karen Hartnett for performing the preconditioning assays and Junko Odajima for valuable advice.
The authors declare no competing financial interests.
References
- Aras MA, Saadi RA, Aizenman E. Zn2+ regulates Kv2.1 voltage-dependent gating and localization following ischemia. Eur J Neurosci. 2009a;30:2250–2257. doi: 10.1111/j.1460-9568.2009.07026.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aras MA, Hara H, Hartnett KA, Kandler K, Aizenman E. Protein kinase C regulation of neuronal zinc signaling mediates survival during preconditioning. J Neurochem. 2009b;110:106–117. doi: 10.1111/j.1471-4159.2009.06106.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baver SB, O'Connell KM. The C-terminus of neuronal Kv2.1 channels is required for channel localization and targeting but not for NMDA-receptor-mediated regulation of channel function. Neuroscience. 2012;217:56–66. doi: 10.1016/j.neuroscience.2012.04.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buraei Z, Schofield G, Elmslie KS. Roscovitine differentially affects CaV2 and Kv channels by binding to the open state. Neuropharmacology. 2007;52:883–894. doi: 10.1016/j.neuropharm.2006.10.006. [DOI] [PubMed] [Google Scholar]
- Cerda O, Trimmer JS. Activity-dependent phosphorylation of neuronal Kv2. 1 potassium channels by CDK5. J Biol Chem. 2011;286:28738–28748. doi: 10.1074/jbc.M111.251942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung ZH, Fu AK, Ip NY. Synaptic roles of Cdk5: implications in higher cognitive functions and neurodegenerative diseases. Neuron. 2006;50:13–18. doi: 10.1016/j.neuron.2006.02.024. [DOI] [PubMed] [Google Scholar]
- Deutsch E, Weigel AV, Akin EJ, Fox P, Hansen G, Haberkorn CJ, Loftus R, Krapf D, Tamkun MM. Kv2.1 cell surface clusters are insertion platforms for ion channel delivery to the plasma membrane. Moll Biol Cell. 2012;23:2917–2929. doi: 10.1091/mbc.E12-01-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Tao-Cheng JH, Zerfas P, McBain CJ. The K+ channel, Kv2.1, is apposed to astrocytic processes and is associated with inhibitory postsynaptic membranes in hippocampal and cortical principal neurons and inhibitory interneurons. Neuroscience. 1998;84:37–48. doi: 10.1016/S0306-4522(97)00519-8. [DOI] [PubMed] [Google Scholar]
- Du J, Haak LL, Phillips-Tansey E, Russell JT, McBain CJ. Frequency-dependent regulation of rat hippocampal somato-dendritic excitability by the K+ channel subunit Kv2.1. J Physiol. 2000;522:19–31. doi: 10.1111/j.1469-7793.2000.t01-2-00019.xm. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinshreiber L, Singer-Lahat D, Ashery U, Lotan I. Voltage-gated potassium channel as a facilitator of exocytosis. Ann N Y Acad Sci. 2009;1152:87–92. doi: 10.1111/j.1749-6632.2008.03997.x. [DOI] [PubMed] [Google Scholar]
- Fox PD, Loftus RJ, Tamkun MM. Regulation of Kv2.1 K+ conductance by cell surface channel density. J Neurosci. 2013;33:1259–1270. doi: 10.1523/JNEUROSCI.3008-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan D, Armstrong WE, Foehring RC. Kv2 channels regulate firing rate in pyramidal neurons from rat sensorimotor cortex. J Physiol. 2013;591:4807–4825. doi: 10.1113/jphysiol.2013.257253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang HC, Clurman BE. Cyclin E in normal and neoplastic cell cycles. Oncogene. 2005;24:2776–2786. doi: 10.1038/sj.onc.1208613. [DOI] [PubMed] [Google Scholar]
- Malin SA, Nerbonne JM. Delayed rectifier K+ currents, IK, are encoded by Kv2 α-subunits and regulate tonic firing in mammalian sympathetic neurons. J Neurosci. 2002;22:10094–10105. doi: 10.1523/JNEUROSCI.22-23-10094.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCord MC, Aizenman E. Convergent Ca2+ and Zn2+ signaling regulates apoptotic Kv2. 1 K+ currents. Proc Natl Acad Sci U S A. 2013;110:13988–13993. doi: 10.1073/pnas.1306238110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin B, Hartnett KA, Erhardt JA, Legos JJ, White RF, Barone FC, Aizenman E. Caspase 3 activation is essential for neuroprotection in preconditioning. Proc Natl Acad Sci U S A. 2003;100:715–720. doi: 10.1073/pnas.0232966100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misonou H, Mohapatra DP, Park EW, Leung V, Zhen D, Misonou K, Anderson AE, Trimmer JS. Regulation of ion channel localization and phosphorylation by neuronal activity. Nat Neurosci. 2004;7:711–718. doi: 10.1038/nn1260. [DOI] [PubMed] [Google Scholar]
- Misonou H, Menegola M, Mohapatra DP, Guy LK, Park KS, Trimmer JS. Bidirectional activity-dependent regulation of neuronal ion channel phosphorylation. J Neurosci. 2006;26:13505–13514. doi: 10.1523/JNEUROSCI.3970-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misonou H, Thompson SM, Cai X. Dynamic regulation of the Kv2.1 voltage-gated potassium channel during brain ischemia through neuroglial interaction. J Neurosci. 2008;28:8529–8538. doi: 10.1523/JNEUROSCI.1417-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohapatra DP, Trimmer JS. The Kv2.1 C terminus can autonomously transfer Kv2.1-like phosphorylation-dependent localization, voltage-dependent gating, and muscarinic modulation to diverse Kv channels. J Neurosci. 2006;26:685–695. doi: 10.1523/JNEUROSCI.4620-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohapatra DP, Misonou H, Pan SJ, Held JE, Surmeier DJ, Trimmer JS. Regulation of intrinsic excitability in hippocampal neurons by activity-dependent modulation of the KV2.1 potassium channel. Channels. 2009;3:46–56. doi: 10.4161/chan.3.1.7655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulholland PJ, Carpenter-Hyland EP, Hearing MC, Becker HC, Woodward JJ, Chandler LJ. Glutamate transporters regulate extrasynaptic NMDA receptor modulation of Kv2.1 potassium channels. J Neurosci. 2008;28:8801–8809. doi: 10.1523/JNEUROSCI.2405-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakoshi H, Trimmer JS. Identification of the Kv2.1 K+ channel as a major component of the delayed rectifier K+ current in rat hippocampal neurons. J Neurosci. 1999;19:1728–1735. doi: 10.1523/JNEUROSCI.19-05-01728.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolic M, Dudek H, Kwon YT, Ramos YF, Tsai LH. The cdk5/p35 kinase is essential for neurite outgrowth during neuronal differentiation. Genes Dev. 1996;10:816–825. doi: 10.1101/gad.10.7.816. [DOI] [PubMed] [Google Scholar]
- O'Connell KM, Rolig AS, Whitesell JD, Tamkun MM. Kv2.1 potassium channels are retained within dynamic cell surface microdomains that are defined by a perimeter fence. J Neurosci. 2006;26:9609–9618. doi: 10.1523/JNEUROSCI.1825-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell KM, Loftus R, Tamkun MM. Localization-dependent activity of the Kv2.1 delayed-rectifier K+ channel. Proc Natl Acad Sci U S A. 2010;107:12351–12356. doi: 10.1073/pnas.1003028107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odajima J, Wills ZP, Ndassa YM, Terunuma M, Kretschmannova K, Deeb TZ, Geng Y, Gawrzak S, Quadros IM, Newman J, Das M, Jecrois ME, Yu Q, Li N, Bienvenu F, Moss SJ, Greenberg ME, Marto JA, Sicinski P. Cyclin E constrains Cdk5 activity to regulate synaptic plasticity and memory formation. Dev Cell. 2011;21:655–668. doi: 10.1016/j.devcel.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal S, Hartnett KA, Nerbonne JM, Levitan ES, Aizenman E. Mediation of neuronal apoptosis by Kv2.1-encoded potassium channels. J Neurosci. 2003;23:4798–4802. doi: 10.1523/JNEUROSCI.23-12-04798.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal SK, Takimoto K, Aizenman E, Levitan ES. Apoptotic surface delivery of K+ channels. Cell Death Differ. 2006;13:661–667. doi: 10.1038/sj.cdd.4401792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park KS, Mohapatra DP, Misonou H, Trimmer JS. Graded regulation of the Kv2.1 potassium channel by variable phosphorylation. Science. 2006;313:976–979. doi: 10.1126/science.1124254. [DOI] [PubMed] [Google Scholar]
- Rashidian J, Rousseaux MW, Venderova K, Qu D, Callaghan SM, Phillips M, Bland RJ, During MJ, Mao Z, Slack RS, Park DS. Essential role of cytoplasmic cdk5 and Prx2 in multiple ischemic injury models, in vivo. J Neurosci. 2009;29:12497–12505. doi: 10.1523/JNEUROSCI.3892-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah NH, Aizenman E. Voltage-gated potassium channels at the crossroads of neuronal function, ischemic tolerance, and neurodegeneration. Trans Stroke Res. 2014;5:38–58. doi: 10.1007/s12975-013-0297-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd AJ, Loo L, Gupte RP, Mickle AD, Mohapatra DP. Distinct modifications in Kv2.1 channel via chemokine receptor CXCR4 regulate neuronal survival-death dynamics. J Neurosci. 2012;32:17725–17739. doi: 10.1523/JNEUROSCI.3029-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd AJ, Loo L, Mohapatra DP. Chemokine co-receptor CCR5/CXCR4-dependent modulation of Kv2.1 channel confers acute neuroprotection to HIV-1 glycoprotein gp120 exposure. PLoS ONE. 2013;8:e76698. doi: 10.1371/journal.pone.0076698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomono M, Toyoshima K, Ito M, Amano H, Kiss Z. Inhibitors of calcineurin block expression of cyclins A and E induced by fibroblast growth factor in Swiss 3T3 fibroblasts. Arch Biochem Biophys. 1998;353:374–378. doi: 10.1006/abbi.1998.0667. [DOI] [PubMed] [Google Scholar]
- Wang J, Liu S, Fu Y, Wang JH, Lu Y. Cdk5 activation induces hippocampal CA1 cell death by directly phosphorylating NMDA receptors. Nat Neurosci. 2003;6:1039–1047. doi: 10.1038/nn1119. [DOI] [PubMed] [Google Scholar]
- Yao H, Zhou K, Yan D, Li M, Wang Y. The Kv2.1 channels mediate neuronal apoptosis induced by excitotoxicity. J Neurochem. 2009;108:909–919. doi: 10.1111/j.1471-4159.2008.05834.x. [DOI] [PubMed] [Google Scholar]