

Abstract
The Drosophila neuromuscular junction (NMJ) is capable of rapidly budding new presynaptic varicosities over the course of minutes in response to elevated neuronal activity. Using live imaging of synaptic growth, we characterized this dynamic process and demonstrated that rapid bouton budding requires retrograde bone morphogenic protein (BMP) signaling and local alteration in the presynaptic actin cytoskeleton. BMP acts during development to provide competence for rapid synaptic growth by regulating the levels of the Rho-type guanine nucleotide exchange factor Trio, a transcriptional output of BMP–Smad signaling. In a parallel pathway, we find that the BMP type II receptor Wit signals through the effector protein LIM domain kinase 1 (Limk) to regulate bouton budding. Limk interfaces with structural plasticity by controlling the activity of the actin depolymerizing protein Cofilin. Expression of constitutively active or inactive Cofilin in motor neurons demonstrates that increased Cofilin activity promotes rapid bouton formation in response to elevated synaptic activity. Correspondingly, the overexpression of Limk, which inhibits Cofilin, inhibits bouton budding. Live imaging of the presynaptic F-actin cytoskeleton reveals that activity-dependent bouton addition is accompanied by the formation of new F-actin puncta at sites of synaptic growth. Pharmacological disruption of actin turnover inhibits bouton budding, indicating that local changes in the actin cytoskeleton at pre-existing boutons precede new budding events. We propose that developmental BMP signaling potentiates NMJs for rapid activity-dependent structural plasticity that is achieved by muscle release of retrograde signals that regulate local presynaptic actin cytoskeletal dynamics.
Keywords: actin, BMP, Drosophila, neuromuscular junction, synapse formation, synaptic plasticity
Introduction
Activity-dependent changes in synaptic structure play an important role in developmental wiring of the nervous system. The Drosophila larval neuromuscular junction (NMJ) has emerged as a model glutamatergic synapse that is well suited to study activity-dependent structural plasticity. The NMJ can be imaged in vivo during developmental periods of rapid synaptic growth when the axonal terminal expands ∼5- to 10-fold in size over 5 d (Zito et al., 1999). Forward genetic screens to identify mutations that alter synaptic growth have revealed essential roles for retrograde bone morphogenic protein (BMP) signaling (Aberle et al., 2002) mediated by the secreted ligand Glass bottom boat (Gbb; Marqués, 2005). Mutations that disrupt BMP signaling lead to synaptic undergrowth and neurotransmitter release defects (Aberle et al., 2002; Marqués et al., 2002; McCabe et al., 2003; Rawson et al., 2003). Multiple pathways downstream of retrograde BMP signaling through the type II receptor wishful thinking (wit) have been linked to synaptic growth, synapse stability, and homeostatic plasticity in Drosophila. BMP signaling via the Smad transcription factor Mothers against Dpp (Mad) regulates the expression of the Rho-type guanine nucleotide exchange factor (GEF) trio to control normal synaptic growth (Ball et al., 2010). Wit also interacts with LIM domain kinase 1 (Limk) to enhance synaptic stabilization in a pathway parallel to canonical Smad-dependent signaling (Eaton and Davis, 2005). BMP signaling through wit also potentiates synapses for homeostatic plasticity in a pathway that is independent of limk and synaptic growth regulation (Goold and Davis, 2007).
In additional to developmental synapse formation during the larval stages, the NMJ displays acute structural plasticity in the form of rapid presynaptic bouton budding in response to elevated levels of neuronal activity (Ataman et al., 2008). These rapidly generated presynaptic varicosities, referred to as ghost boutons, lack presynaptic and postsynaptic transmission machinery when initially formed. The budding of ghost boutons requires retrograde signaling mediated by the postsynaptic Ca2+-sensitive vesicle trafficking regulator synaptotagmin (Syt) 4 (Korkut et al., 2013). Syt4 also participates in developmental synaptic growth and controls retrograde signaling that mediates enhanced spontaneous release at the NMJ (Yoshihara et al., 2005; Barber et al., 2009). Beyond the role of Syt4 in ghost bouton budding, little is known about the signaling pathways that underlie this rapid form of structural synaptic plasticity. In particular, it is unclear whether pathways that regulate synaptic growth over the longer time scales of larval development also trigger acute structural plasticity. To address these issues, we identified synaptic pathways that are required for rapid structural plasticity at Drosophila NMJs. We find that ghost bouton budding can be locally regulated at the synapse level, occurring in axons that have been severed from the neuronal cell body. In addition, activity-induced ghost bouton formation requires Syt1-mediated neurotransmitter release and postsynaptic glutamate receptor function. Like developmental growth, retrograde BMP signaling is required for ghost bouton budding. BMP signaling functions through a permissive role mediated by developmental Smad and Trio signaling, as well as through a local Wit-dependent modulation of Limk and Cofilin (Twinstar) activity that alters presynaptic actin dynamics.
Materials and Methods
Drosophila genetics and molecular biology.
Flies were cultured on standard medium at 25°C. All stocks were obtained from the Bloomington Stock Center unless otherwise specified. Wild-type flies used in the analysis were Canton S. cDNA encoding the endosomal target SNARE (t-SNARE) syntaxin 13 was modified to add a monomeric RFP (mRFP) tag to the N terminus of the encoded protein. The sequence was subcloned into PUAST and transgenic flies were generated. The following RNA hairpin lines from the Harvard TRiP collection were used: UAS-gbbRNAi (HMS01243), UAS-gluRIIARNAi (JF02647), UAS-gluRIIBRNAi (JF03145), and UAS-Syt4RNAi (JF02272). Wishful thinking mutants were analyzed as the heterozygote allelic combination witA12/witB11. TrioS137203 mutants (Ball et al., 2010) were analyzed as homozygotes. LimkP1 (P[EY08757]; Eaton and Davis, 2005) animals were analyzed as male hemizygotes. Homozygous Syt4BA1 (Yoshihara et al., 2005) mutants were used for Syt4-null animals. Syt 1-null mutants were generated by crossing sytN13 and sytAD4 alleles. The following Gal4 lines were used: elav-Gal4 (C155), c164-Gal4 (Torroja et al., 1999), and 24B-Gal4 (Brand and Perrimon, 1993). UAS-gluRIIARNAi, UAS-gluRIIBRNAi, UAS-gbbRNAi, UAS-dad, UAS-trio, UAS-wit, UAS-witdCT, UAS-limk, UAS-tsrS3E, UAS-tsrS3A, and UAS-ssh were analyzed as transheterozygotes with the indicated Gal4 driver. Live imaging was performed using the following lines: (1) elav-Gal4, UAS-mRFP-syx13; (2) c164-Gal4, UAS-CD8-GFP/+; (3) c164-Gal4, UAS-CD8-RFP/+; UAS-GMA/+; (4) UAS-tsrS3E/+, c164-Gal4/+, UAS-GMA/+; and (5) UAS-tsrS3A+, c164-Gal4/+, UAS-GMA/+. Rescue lines consisted of the following genotypes: (1) wit rescue: c164-Gal4/UAS-wit, witA12/witB11; and (2) witdCT rescue: c164-Gal4/UAS-witdCT, witA12/witB11.
High K+ stimulation of larval NMJs.
The activity-dependent ghost bouton growth protocol was adapted from Ataman et al. (2008). Wandering third instar larvae were dissected in HL3 saline solution as follows (in mm): 70 NaCl, 5 KCl, 0.2 CaCl2, 20 MgCl2, 10 NaHCO3, 5 trehalose, 115 sucrose, and 5 HEPES-NaOH, pH 7.2. Larvae were dissected according to a guide with consistent dissecting pin locations varying in size by 5% increments (schematic representation of guide with pins placed at line termini: -|-|-). Dissecting pins were then moved inward to the same guide shape at 60% of the original size for each larva. Relaxed fillets were subjected to three 2 min incubations in the following 90 mm K+ solution (in mm): 40 NaCl, 90 KCl, 1.5 CaCl2, 20 MgCl2, 10 NaHCO3, 5 Trehalose, 5 sucrose, and 5 HEPES-NaOH, pH 7.2, spaced by 10 min in HL3 solution. After the third 90 mm K+ incubation, larvae were returned to HL3 solution for 2 min and then stretched to the original position by moving the dissecting pins outward according to the original guide. Ghost boutons were identified by the appearance of a bouton that was not previously observed in live imaging, or by the presence of a presynaptic bouton (HRP labeled) that lacked Discs large (DLG) staining in fixed preparations. Muscle 6/7 NMJs from abdominal segments 2 through 5 were included in the analysis. Histograms show the mean ± SEM, and the imbedded text indicates number of replicates (n). Statistical significance in two-way comparisons was determined by a Student's t test, while ANOVA analysis was used when comparing more than two datasets. The p values associated with ANOVA tests were obtained from a Tukey's post-test.
Live imaging of NMJ growth.
Wandering third instar larvae expressing UAS-mRFP-syx13, UAS-CD8-GFP, or UAS-GMA were dissected in HL3 saline solution. For experiments involving high K+ stimulation, after initial imaging, larvae were subjected to the high K+ protocol as described above and imaged again after one, two, or three 2 min 90 mm K+ solution incubations. Latrunculin A (Sigma) and jasplakinolide (Invitrogen) were prepared as 1 mm stocks in DMSO, and diluted in HL3 and 90 mm K+ solutions. Drug treatments were performed by pretreating dissected larvae in HL3 solution containing 10 μm latrunculin A or 10 μm jasplakinolide for 15 min. Stimulation using HL3 and 90 mm K+ solutions containing either 10 μm latrunculin A or 10 μm jasplakinolide was then used as described above. Images were acquired with a PerkinElmer Ultraview Vox spinning disc confocal microscope equipped with a Hamamatsu C9100–13 ImagEM EM CCD at 8–35 Hz with a 40× 0.8 numerical aperture (NA) water-immersion objective (Carl Zeiss).
Immunostaining.
Larvae were fixed for 40 min in HL3.1 solution containing 4% formaldehyde. Following washes in PBS and PBS containing 1% Triton X-100, larvae were incubated overnight with primary antibody at 4°C, incubated with secondary antibodies for 4 h at room temperature the following day, and mounted in 70% glycerol in PBS for imaging. Antibodies were diluted as follows: mouse anti-DLG (1:500; Developmental Studies Hybridoma Bank at the University of Iowa), mouse anti-Trio (1:250; Developmental Studies Hybridoma Bank at the University of Iowa), TRITC-conjugated anti-HRP (1:500; Jackson ImmunoResearch), and Alexa Fluor 488 goat anti-mouse (1:1000) (Invitrogen). Images were acquired with a 40× 1.3 NA oil-immersion objective (Carl Zeiss) and analyzed with Velocity Software.
Results
Rapid synaptic growth at Drosophila NMJs requires local activity-dependent signaling
Prior studies at the Drosophila NMJ identified patterns of developmental synaptic growth by imaging through the cuticle of intact larvae genetically expressing fluorescent synaptic markers at multiple time points separated by several days (Zito et al., 1999). To more acutely analyze patterns of synaptic growth over a shorter time interval, we performed live imaging of NMJs of dissected larvae genetically expressing fluorescently tagged synaptic proteins. For our initial analysis, transgenic animals were generated that expressed UAS-mRFP-Syntaxin 13. Syntaxin 13 is a t-SNARE protein that decorates endosomal compartments and the presynaptic membrane when expressed in motor neurons with elav-GAL4 (Fig. 1A), and proved to be an effective marker for visualizing synaptic growth dynamics. mRFP-syntaxin 13 expressing third instar larvae were dissected, and selected NMJs were imaged at 1 Hz over 30 min in live preparations with the brain intact. Using this approach, rapid presynaptic budding events were observed that triggered formation of a new bouton in <1 min during normal central pattern generated muscle contraction (Fig. 1A, Movie 1). These budding events occurred very rarely and generated extremely round boutons with thin axonal connections to the parent bouton. The newly formed presynaptic varicosities morphologically resembled ghost boutons that have been previously described (Ataman et al., 2006). Ghost boutons lack postsynaptic specializations, including the PSD-95 homolog DLG and glutamate receptor clusters (Fig. 1B). Undifferentiated boutons have been previously observed developmentally in the absence of external stimulation (Ataman et al., 2006; Fuentes-Medel et al., 2009; Korkut et al., 2009; Mosca and Schwarz, 2010), indicating that they may contribute to normal synaptic development. Muscle and surrounding glia have also been shown to engulf a subset of these immature boutons (Fuentes-Medel et al., 2009), indicating that some ghost boutons are normally eliminated during development. In unstimulated preparations, we observed that ghost boutons represented ∼1% of all synaptic boutons at muscle 6/7 NMJs, with their frequency positively correlated with overall NMJ size (n = 68, r2 = 0.3002, p < 0.0001; Fig. 1C). In several cases, we observed ghost bouton-like presynaptic structures that were surrounded by trace amounts of DLG (Fig. 1D), suggesting a subset of these varicosities may be maturing into functional connections.
Figure 1.

Rapid synaptic growth occurs spontaneously and in response to elevated activity. A, Live confocal imaging of a dissected larval NMJ presynaptically expressing mRFP-Syx13. The arrow indicates the site of a new bouton spontaneously budding and stabilizing (arrow) over the course of a minute. A second bouton can also be seen to emerge from the same budding site, but later collapses. Scale bar, 5 μm. B, Ghost boutons can be detected in fixed tissue by staining for the presynaptic neuronal membrane (anti-HRP, red) and the postsynaptic scaffold protein DLG (green), appearing as presynaptic varicosities that lack DLG staining. Scale bar, 11 μm. Arrowheads indicate ghost boutons. C, Histogram of ghost bouton frequency observed at unstimulated NMJs. N = 68 NMJs, 12 animals. D, Putative ghost boutons identified by morphology in a fixed preparation can display faint DLG accumulation (arrowheads), suggesting some newly formed varicosities are likely to be undergoing synaptic maturation. Scale bar, 5 μm.
Rapid presynaptic bouton budding imaged at muscle 12/13. Visualization of a presynaptic arbor expressing mRFP-syntaxin 13 at muscle 12/13 in a dissected third instar larva with the nervous system intact. The parent bouton swells at a restricted site (∼2 μm across), producing two discernable boutons within 20 s. The new bouton (labeled A) then separates from the parent bouton at the budding site, but remains connected by a thin axon. A second, smaller bouton (labeled B) buds from the same initial site, but collapses rapidly and is not maintained. The first newly budded bouton is stabilized within 1 min at ∼4 μm from the parent bouton and has a strikingly round morphology. The parent bouton appears incrementally smaller after budding. The movie was acquired at a rate of 1 Hz, with video speed set at 10 frames/s, 10× real time.
During live imaging of normal larval synaptic dynamics, we observed two general patterns by which new synaptic boutons appeared. In the first, an existing presynaptic bouton would bud off membrane in a relatively rapid fashion over 10–30 s, appearing to split following the emergence of the new bouton (Movie 1, Fig. 1A). These events were often associated with muscle contraction. Given that the presynaptic arbor extends into the muscle at the Drosophila NMJ, we hypothesize that adhesive interactions between the muscle and the presynaptic bouton at particular attachment points may participate in “pulling” out a new bouton from an existing varicosity, with force for the process generated during muscle contraction. In a second pathway, often observed in preparations with less muscle contraction, presynaptic membrane would slowly flow from an existing bouton into a small bud over several minutes to form a bigger structure that would take on the shape of a new bouton and eventually separate from the main arbor. This budding mechanism appeared to rely more on presynaptic force generation than the postsynaptic “pulling” events shown in Movie 1. Syntaxin 13-positive compartments were often observed near these budding sites, suggesting that local endosomal dynamics may contribute to membrane addition or to the recycling of cell adhesion proteins at budding sites. In both cases, newly formed varicosities were highly dynamic, often moving tens of micrometers within the muscle while pulling on small axons that connected the bouton to the main arbor. Since we were only able to image over 30 min in dissected preparations before signs of tissue damage, it is unclear how many newly formed boutons mature into functional connections or are disassembled by neighboring glia or muscle.
Due to the rarity in capturing synaptic budding events with live imaging in unstimulated larvae, we proceeded to examine the molecular mechanisms that underlie rapid synaptic growth using a modified high K+ stimulation protocol in dissected larvae that has been previously shown to rapidly induce presynaptic bouton budding (Ataman et al., 2008). We dissected third instar wandering larvae such that the resulting fillets were relaxed enough to allow for muscle contraction. Dissected preps were stimulated by washing in high K+ solution three times for 2 min, with each spaced by 10 min in HL3 dissecting solution. After the third incubation with high K+ solution, larvae were allowed to rest in HL3 solution for 2 min before being stretched and fixed. Ghost boutons were identified either by live imaging of NMJs in animals presynaptically expressing UAS-CD8-GFP, or by staining with DLG and HRP antibodies to identify ghost boutons that were not surrounded by a postsynaptic specialization in fixed animals. This short protocol improved upon prior efforts to promote synaptic growth and robustly induced budding of new presynaptic varicosities in live animals at ∼12% of existing boutons within 30 min, providing an easily quantifiable assay for rapid activity-induced synaptic growth.
Using live imaging before and after stimulation, we observed that new ghost boutons can be generated within a single 2 min incubation with 90 mm K+ solution (Fig. 2A, 3′). NMJs continue to bud new boutons throughout the synaptic terminal with repeated exposures to high K+ solution (Fig. 2A, 15′). We did not observe budding by live imaging in mock-treated animals that were incubated with HL3 solution in place of high K+ solution (Fig. 2D; mock treated, n = 26). In all instances in which synapses were continually monitored throughout the duration of the K+ stimulation protocol, ghost boutons emerged during periods of high K+ incubation. Following three rounds of K+ stimulation, the number of ghost boutons at an NMJ was no longer correlated with the baseline bouton number, indicating that this form of rapid activity-dependent growth is not dependent on the prior size of the synaptic field (Fig. 2B; n = 123, r2 = 0.0031, p = 0.54). Ghost bouton budding was observed from both type 1b and 1s boutons. However, boutons were less likely to bud from terminal boutons compared with all other boutons (p = 1.17e-7, binomial test), suggesting that the terminal bouton is not a favored site for a new bouton addition. In addition, we never observed budding of new varicosities from an axon segment that lacked a pre-existing bouton, suggesting that budding events are always initiated from previously formed synaptic varicosities. A similar conclusion was made based on prior in vivo imaging of developmental synaptic growth (Zito et al., 1999), suggesting that pre-existing boutons likely contain important molecular components for bouton addition that are not concentrated in nonsynaptic regions of the axon.
Figure 2.

Ghost bouton budding induced by high K+ stimulation is a rapid local signaling event. A, Live imaging of bouton budding (arrowheads) in response to 2 min incubations in high K+ spaced 10 min apart. New varicosity formation was visualized by presynaptic expression of membrane-tethered CD8-GFP. Scale bar, 7 μm. B, Quantification of ghost boutons in relation to existing bouton number at the NMJ following high K+ stimulation. N = 123 NMJs, 18 animals. C, New bouton budding (arrowheads) in response to high K+ stimulation is strongly dependent upon external Ca2+ but is not changed when axons are severed from motor neuron cell bodies. Scale bar, 14 μm. D, Quantification of ghost bouton budding detected by live imaging of animals presynaptically expressing membrane-tethered CD8-GFP at the indicated conditions. N (NMJs, animals): control = 38, 7; mock treated = 26, 4; 0 mm Ca2+ = 25, 4; 0.5 mm EGTA = 13, 4; axon cut = 21, 4. Error bars indicate SEM.
Further analysis of the conditions permitting ghost bouton budding in response to elevated activity revealed that budding is a local signaling event that requires Ca2+. When Ca2+ was removed from the HL3 and 90 mm K+ solution, the number of ghost bouton budding events fell significantly (Fig. 2C,D; mean ± SD: control = 6.605 ± 5.998, n = 38; 0 mm Ca2+ = 0.2 ± 0.5, n = 25, p < 0.0001, ANOVA), but were not completely eliminated. The addition of 0.5 mm EGTA to the 0.0 mm Ca2+ solution completely eliminated ghost bouton budding events following stimulation (n = 13). The formation of ghost boutons also did not require an axonal connection to the cell body (Fig. 2C,D; axon cut = 6.667 ± 5.073, n = 21), indicating that the signaling events that initiate bouton budding, as well as the machinery that physically drives new bouton addition, are unlikely to acutely require transcription or translation. Additionally, some boutons were observed to bud within seconds of exposure to high K+, indicating that a subset of synaptic terminal sites are likely to be prepotentiated for budding in response to elevated neuronal activity. We conclude that ghost bouton budding in response to elevated activity is a local signaling event that does not acutely require function of the neuronal cell body.
Ghost bouton budding requires synaptic transmission and retrograde BMP signaling
It has been demonstrated previously that ghost bouton budding in response to K+ stimulation requires muscle depolarization (Korkut et al., 2013). To further characterize the requirements for synaptic transmission in ghost bouton budding, we examined mutants in the presynaptic Ca2+ sensor syt1. Mutations in syt1 decrease neurotransmitter release at Drosophila NMJs by specifically disrupting the synchronous component of evoked fusion (Yoshihara and Littleton, 2002; Jorquera et al., 2012). We found that ghost bouton budding was substantially reduced in syt1AD4/N13 mutants (Fig. 3A,B; wild-type = 8.361 ± 7.403, n = 57; syt1AD4/N13 = 0.9778 ± 1.5, n = 45; p < 0.0001, ANOVA). We also tested the postsynaptic requirement for glutamate receptor subunits in ghost bouton budding. The expression of RNAi directed against the glutamate receptor subunits DGluRIIA or DGluRIIB in muscle using the 24B-Gal4 driver caused a reduction in ghost bouton budding frequency (Fig. 3A,B; 24B, gluRIIARNAi = 0.8065 ± 1.276, n = 31, p < 0.0001, ANOVA; 24B, gluRIIBRNAi = 2.586 ± 3.978, n = 29, p < 0.0001, ANOVA). Knockdown of DGluRIIA reduced budding frequency significantly more than knockdown of DGluRIIB (p = 0.0214, Student's t test). Given the prominent role of DGluRIIA-containing glutamate receptor complexes in mediating postsynaptic Ca2+ entry, these findings suggest that a potential Ca2+-dependent postsynaptic process may initiate rapid structural plasticity. A prime candidate for such a role would be the release of growth-promoting factors following activity-triggered fusion of postsynaptic vesicles containing the Ca2+ sensor Syt4 (Yoshihara et al., 2005; Barber et al., 2009). Indeed, the mammalian Syt4 homolog has been shown to regulate BDNF release (Dean et al., 2012), a key modulator of structural plasticity at mammalian synapses. We therefore tested the role of Syt4 in regulating rapid activity-dependent structural plasticity at Drosophila NMJs using null mutations in the locus that we previously generated. The loss of Syt4 substantially reduced ghost bouton budding in response to K+ stimulation (Fig. 3A,B; syt4BA1 = 4.585 ± 5.08; n = 65; p = 0.0037, ANOVA), similar to observations made by Korkut et al. (2013). Postsynaptic knockdown of Syt4 using a UAS-Syt4 RNAi driven by 24B-GAL phenocopied syt4BA1 mutants, indicating a postsynaptic source of Syt4 contributes to this effect (Fig. 3A,B; 24B, syt4RNAi = 3.548 ± 2.694; n = 31; p = 0.0016, ANOVA). These data indicate that ghost bouton budding is sensitive to the levels of both presynaptic neurotransmitter release and postsynaptic glutamate receptor and Syt4 function, rather than being triggered only by changes in presynaptic membrane depolarization induced by high K+.
Figure 3.
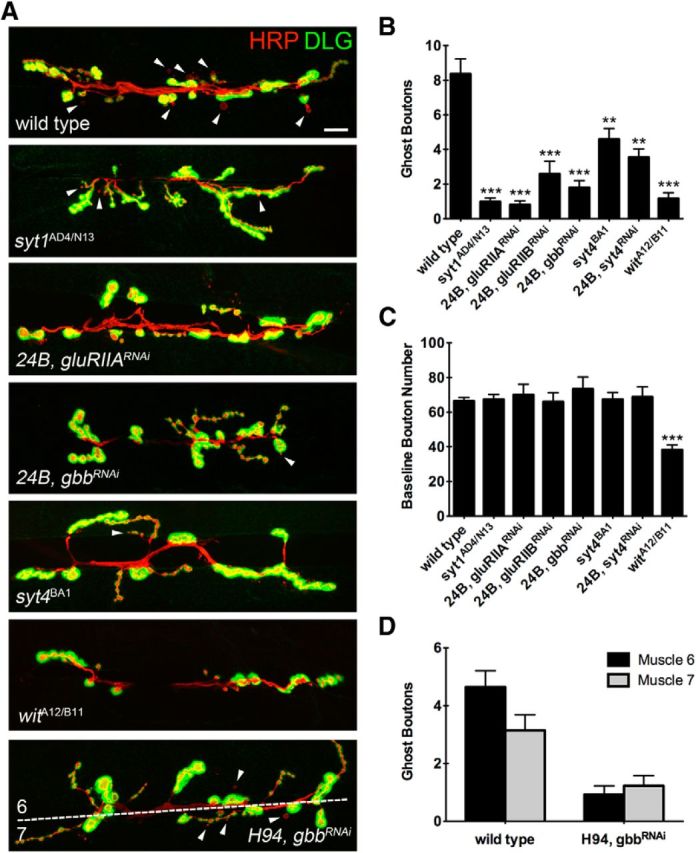
Ghost bouton budding requires normal synaptic transmission and local retrograde BMP signaling. A, Wandering third instar animals were fixed in formaldehyde after high K+ stimulation and were stained with anti-HRP and anti-DLG to identify ghost boutons. Loss of Syt1 and postsynaptic knockdown of GluRIIA and GluRIIB reduce activity-dependent budding. Likewise, loss of the postsynaptic Ca2+ sensor Syt4, or postsynaptic knockdown of Gbb with the muscle driver 24B-Gal4, reduces ghost bouton budding. Knockdown of Gbb with the muscle 6-specific H94-Gal4 preferentially reduces budding at muscle 6. Scale bar, 12 μm. Arrowheads indicate ghost boutons. B, Quantification of ghost boutons per NMJ in the indicated genetic backgrounds. N (NMJs, animals): wild-type = 57, 11; syt1AD4/N13 = 45, 6; 24B, gluRIIARNAi = 31, 4; 24B, gluRIIBRNAi = 29, 4; 24B, gbbRNAi = 20, 3; syt4BA1 = 65, 11; 24B, syt4RNAi = 3.548 ± 2.694, n = 31, 3; witA12/B11 = 37, 6. C, Quantification of baseline bouton number in the indicated genetic backgrounds. N, same as in B. D, The average number of ghost boutons that bud onto muscle 6 or muscle 7 is quantified for Gbb knockdown by the muscle 6-specific driver H94-Gal4. N (NMJs, animals): wild-type = 52, 7; H94, gbbRNAi = 40, 6. **p < 0.01; ***p < 0.001; ANOVA. Error bars indicate SEM.
Retrograde BMP signaling from muscle to motor neuron has been well characterized for its role in normal developmental synaptic growth. To determine whether BMP signaling is also required for ghost bouton budding at larval NMJs, we manipulated components of the BMP signaling pathway and assayed their potential for rapid activity-induced presynaptic growth. Postsynaptic reduction of the BMP ligand Gbb in muscle by RNAi significantly reduced ghost bouton budding (Fig. 3A,B; 24B, gbbRNAi = 1.8 ± 1.795; n = 20; p < 0.0001, ANOVA), although baseline bouton number remained unchanged (in contrast to gbb mutants; Fig. 3C). These results suggest that partial knockdown of Gbb is sufficient to disrupt activity-induced ghost bouton budding, but does not alter normal developmental synaptic growth. This observation indicates that developmental versus acutely triggered synaptic growth is likely to have different sensitivity or distinct molecular components for driving new synapse formation. To examine whether ghost bouton budding is locally regulated at individual synapses by BMPs or is instead controlled mainly by a BMP-dependent developmental transcriptional signal that would affect all synapses of a given motor neuron, we drove RNAi against Gbb using H94-Gal4, which is expressed predominantly in muscle 6, but not in muscle 7 (Davis et al., 1997). Muscles 6 and 7 are innervated by two motor neurons that branch onto both muscle fibers. If BMP signaling solely functioned in a developmental role to allow NMJs to express the potential to undergo structural plasticity, we would expect Gbb expression from either muscle fiber to be sufficient to promote normal ghost bouton budding. However, if BMP signaling plays a more acute instructive role in structural plasticity, we would expect to see preferential defects in new synaptic budding events at muscle 6, which would have reduced local Gbb output due to preferential RNAi expression in this muscle driven by the H94-Gal4 promoter. In wild-type animals, there is a slight bias for ghost bouton budding onto muscle 6, as this is the larger of the two muscles. This bias is eliminated when Gbb levels are reduced in muscle 6 and not in muscle 7 (Fig. 3A,D; p = 0.0058, Fisher's exact test), indicating that bouton budding is likely to require in part a local postsynaptic source of Gbb given the comparatively enhanced ability of muscle 7 to support budding. However, total bouton budding events were also decreased at muscle 7 compared with controls, indicating that a developmental role for Gbb also contributes to this form of structural plasticity.
Wishful Thinking controls ghost bouton budding through multiple signaling pathways
To further analyze the requirement for BMP signaling in rapid structural plasticity, we examined how disruptions of additional components of the signaling pathway would alter developmental versus acute synaptic growth. Null mutants for the BMP type II receptor wit (witA12/B11) displayed a reduction in K+-stimulated bouton budding (Fig. 3A,B; witA12/B11 = 1.162 ± 2.089; n = 37; p < 0.0001, ANOVA). Wit mutant animals also strongly reduced developmental synaptic growth. However, bouton budding as a fraction of baseline bouton number was still significantly reduced (wild-type = 0.1369 ± 0.1327 ghost boutons/baseline boutons, n = 66; witA12/B11 = 0.0353 ± 0.0662, n = 37; p < 0.0001, Student's t test). Given that Wit signals through multiple pathways, we sought to determine how wit contributes to ghost bouton budding. Canonical BMP receptor signaling leads to Smad phosphorylation and, together with cofactors, translocation to the nucleus to act as a transcription factor (Bayat et al., 2011). To assay Smad signaling in ghost bouton formation, we overexpressed the inhibitory Smad, daughters against dpp (dad; Tsuneizumi et al., 1997; Kamiya et al., 2008). Overexpression of UAS-dad in motor neurons has previously been shown to reduce levels of phosphorylated Mad and cause synaptic undergrowth (Eaton and Davis, 2005; Dudu et al., 2006). Overexpression of UAS-dad in motor neurons inhibited ghost bouton formation (Fig. 4A,D; c164, dad = 2.633 ± 3.944; n = 60; p < 0.0001, ANOVA), indicating that Wit signals through Mad to developmentally regulate ghost bouton formation.
Figure 4.

Ghost bouton budding requires Smad signaling and is modulated by Limk activity. A, Wandering third instar larvae were fixed in formaldehyde after high K+ stimulation, and stained with anti-HRP and anti-DLG to identify ghost boutons. The overexpression of the inhibitory Smad dad causes synaptic undergrowth and a reduction in ghost bouton budding frequency. Trio protein levels correlate with ghost bouton budding frequency. Overexpression of full-length UAS-wit causes a reduction in bouton budding, while the overexpression of truncated UAS-witdCT does not. Motor neuron rescue with full-length UAS-wit did not completely rescue ghost bouton budding frequency, while rescue with UAS-witdCT rescued ghost bouton budding to a significantly greater extent. Scale bar, 12 μm. Arrowheads indicate ghost boutons. B, c164, UAS-trio animals were stimulated, fixed, and stained with anti-Trio antibody. Scale bar, 12 μm. Arrowheads indicate ghost boutons identified by morphology. C, Normalized fluorescence intensity of Trio antisera staining within ghost boutons was normalized to average fluorescence intensity of all other normal boutons at the same NMJ. N (ghost boutons, NMJs) = 77, 9. Solid line indicates mean; dashed line indicates the average normal bouton fluorescence intensity. D, Quantification of ghost boutons per NMJ in the indicated genetic background. N (NMJs, animals): wild-type = 57, 11; c164, dad = 70, 8; trioS137203 = 37, 5; c164, trio = 27, 4; witA12/B11 = 37, 6; wit rescue = 45, 7; witdCT rescue = 56, 7; c164, wit = 55, 8; c164 witdCT = 55, 8. E, Quantification of baseline bouton number. N, same as in D. F, Wit and Tsr show genetic interactions for defective ghost bouton budding. Quantification of ghost boutons per NMJ in the indicated genetic background is shown. N (NMJs, animals): wild-type = 57, 11; tsr1/+ = 24, 3; witB11/+ = 45, 6; tsr1/+;witB11/+ = 37, 5. *p < 0.05; **p < 0.01; ***p < 0.001; ANOVA. Error bars indicate SEM.
One well characterized transcriptional target of Mad that regulates synaptic growth in Drosophila motor neurons is the Rho-type GEF trio (Ball et al., 2010). We assayed for a requirement for Trio in rapid activity-dependent growth by performing K+ stimulation in trioS137203 mutant animals. We observed a quantitatively similar reduction in ghost bouton budding in trioS137203 animals compared with wit mutants (Fig. 4A,D; trioS137203 = 1.486 ± 1.995; n = 37; p < 0.0001, ANOVA). Conversely, the overexpression of trio caused an increase in ghost bouton budding well above wild-type levels (Fig. 4A,D; c164, trio = 15.41 ± 11.39; n = 27; p < 0.0001, ANOVA). These data indicate that Trio may play a key role in the execution of activity-induced synaptic growth given this bidirectional modulation. We therefore sought to determine whether Trio protein preferentially targeted to sites of new synaptic growth. Antisera against Trio poorly detected the protein at NMJs in wild-type animals, but robustly detected overexpressed protein. As such, we performed immunostaining for Trio in larvae presynaptically expressing UAS-trio. Strikingly, Trio immunoreactivity was enriched in ghost boutons following high K+ stimulation (Fig. 4B). Average fluorescence intensity detected in ghost boutons was significantly greater than that in normal boutons throughout the terminal (Fig. 4B,C; normalized ghost bouton fluorescence intensity = 1.419 ± 0.363; ghost boutons, n = 77; NMJs, n = 9; p < 0.0001, paired t test). We hypothesize that Trio may be enriched at sites primed for bouton budding, generating a higher concentration of the protein that later becomes trapped in newly formed boutons following K+ stimulation. Together, these data indicate that Wit signaling through a canonical Smad transcriptional pathway is likely to mediate the developmental role for BMP signaling in ghost bouton budding. In terms of Smad-dependent transcriptional targets, rapid structural plasticity at the NMJ is bidirectionally correlated with levels of the Rho-type GEF trio.
In addition to Smad-dependent transcriptional changes, Wit can signal locally through Limk to promote synapse stabilization (Eaton and Davis, 2005). Limk phosphorylates and inactivates Cofilin [Drosophila Twinstar (Tsr)] at serine 3 and can induce changes in actin cytoskeleton structure to restrain normal developmental synaptic growth (Ohashi et al., 2000; Ang et al., 2006). Limk has been shown to interact directly with BMP type II receptors, which increases its kinase activity (Foletta et al., 2003; Lee-Hoeflich et al., 2004). To test whether the Wit–Limk interaction also regulates ghost bouton budding, we rescued witA12/B11 animals with motor neuron expression of full-length UAS-wit or a truncated copy of the gene UAS-witdCT, which lacks the Limk binding domain (Eaton and Davis, 2005). Expression of either full-length or truncated wit rescued developmental synaptic growth (Fig. 4D), indicating that the Limk-binding domain of Wit is not likely to significantly contribute to normal developmental synaptic growth. In contrast, we observed only a partial rescue of ghost bouton budding frequency in full-length wit rescue animals (Fig. 4A,C; wit rescue = 3.733 ± 3.875; n = 45; p = 0.0004, ANOVA), although budding frequency was significantly higher than in witA12/B11 mutant animals (p = 0.0414, ANOVA). WitdCT rescued animals showed significantly greater bouton budding frequencies than full-length wit rescue animals (Fig. 4A,C; witdCT rescue = 7.821 ± 5.769; p = 0.0054, ANOVA). To further examine the Wit–Limk link, we overexpressed full-length wit or truncated witdCT in wild-type motor neurons. The overexpression of full-length UAS-wit reduced ghost bouton budding (Fig. 4C; c164, wit = 3.891 ± 3.629; n = 55; p = 0.0003, ANOVA), while overexpression of UAS-witdCT did not alter budding (c164, witdCT = 6.364 ± 4.507; n = 55; p > 0.999, ANOVA). These observations suggest that the Limk binding domain of Wit normally functions to inhibit activity-dependent ghost bouton budding events.
Given the link between Wit signaling and Limk, we sought to determine whether Cofilin might function in the same pathway for ghost bouton budding by assaying whether wit and tsr showed genetic interactions. Heterozygote witB11/+ and tsr1/+ animals both show similar deficits in ghost bouton budding (Fig. 4E; tsr1/+ = 4.29 ± 3.95, n = 24; witB11/+ = 3.82 ± 3.30, n = 45; p > 0.999, ANOVA), suggesting that this form of rapid structural plasticity is highly sensitive to incremental disruptions in BMP signaling and partial loss of Tsr function. Analysis of double-heterozygous tsr1/+; witB11/+ animals revealed a significantly greater reduction in activity-dependent plasticity than either single heterozygote (Fig. 4E; tsr1/+; witB11/+ = 1.25 ± 1.65 (n = 37); tsr1/+ vs tsr1/+; witB11/+, p = 0.0242; witB11/+ vs tsr1/+; witB11/+, p = 0.0206, ANOVA). This dosage-dependent interaction is consistent with a model in which Wit and Tsr participate in a similar pathway to regulate ghost bouton budding.
Limk regulation of presynaptic Cofilin activity controls rapid activity-dependent synaptic growth
To determine how Limk activity modulated Cofilin and ghost bouton budding, we assayed acute synaptic growth in strains with altered Limk function or disrupted Cofilin regulation by Limk. Motor neuron overexpression of UAS-limk strongly inhibited activity-dependent bouton budding (Fig. 5A,B; c164, limk = 1.675 ± 2.702; n = 40; p < 0.0001, ANOVA), indicating that Limk suppresses ghost bouton formation, potentially through phosphorylation-mediated inhibition of Cofilin (Tsr). To examine whether Cofilin activity regulates ghost bouton budding, we overexpressed either a constitutively inactive (UAS-tsrS3E) or a constitutively active (UAS-tsrS3A) twinstar transgene bearing a phosphomimetic or phosphoincompetent Limk phosphorylation site, respectively. A decrease in bouton budding frequency was observed when the inactive transgene was overexpressed (Fig. 5A,B; c164, tsrS3E = 3.107 ± 3.059; n = 28; p = 0.0006, ANOVA), while a strong increase in bouton budding frequency was found upon overexpression of the active transgene (Fig. 5A,B; c164, tsrS3A = 13.39 ± 7.958; n = 36; p = 0.0003, ANOVA). As predicted based on the ability of Cofilin to disassemble actin filaments, the expression of either transgene altered the presynaptic actin cytoskeleton when visualized by the expression of the F-actin marker GMA. GMA contains the actin-binding domain of Moesin fused to GFP (Dutta et al., 2002). In addition to labeling dynamic actin puncta within synaptic boutons, GMA normally labeled more stable actin structures in axons and interbouton regions (Fig. 5D). In contrast to controls, GMA formed puncta in axons and extended interbouton regions of animals expressing UAS-tsrS3E or UAS-tsrS3A (Fig. 5D). We occasionally observed boutons that lacked discernable F-actin puncta in wild-type animals, whereas boutons lacking F-actin puncta occurred more frequently in tsrS3E-expressing animals (Fig. 5D). We also observed large and bright GMA-positive clusters in tsrS3E and tsrS3A animals that were not observed at wild-type NMJs (Fig. 5D). These findings suggest that BMP signaling through Limk is likely to alter Cofilin activity, with subsequent effects on the presynaptic actin cytoskeleton and its ability to support activity-induced bouton formation.
Figure 5.
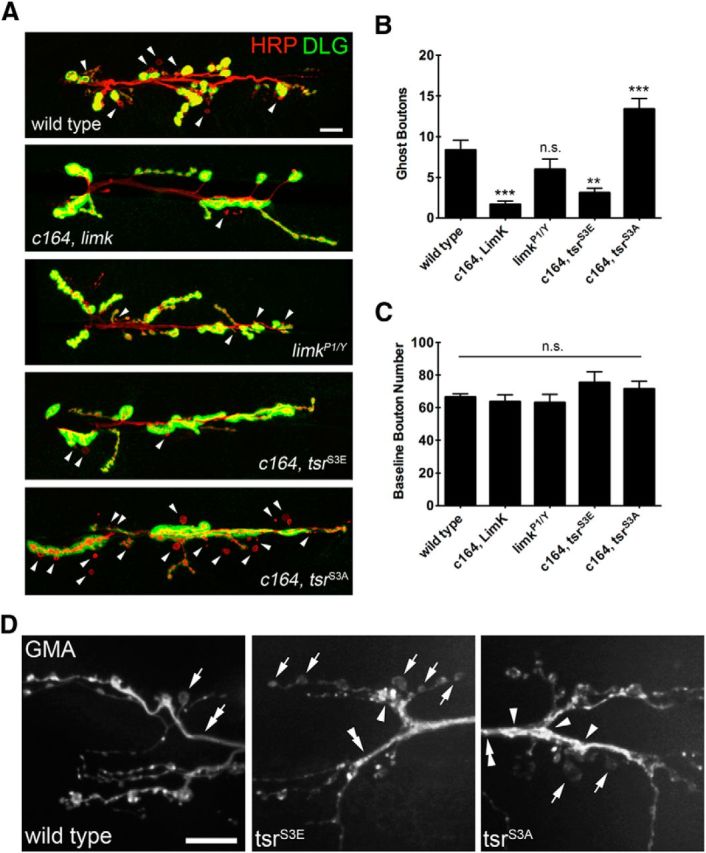
Ghost bouton budding is regulated by Limk and Cofilin activity. A, Presynaptic overexpression of limk strongly reduces activity-dependent bouton budding. Presynaptic overexpression of constitutively inactive tsrS3E reduces ghost bouton budding, while presynaptic overexpression of constitutively active tsrS3A increases ghost bouton budding above wild-type levels. Scale bar, 12 μm. Arrowheads indicate ghost boutons. B, Quantification of ghost bouton budding frequency in the indicated genetic background. N (NMJs, animals): wild-type = 57, 11; c164, limk = 40, 5; limkP1/Y = 36, 5; c164, tsrS3E = 28, 4; c164, tsrS3A = 36, 6. C, Quantification of baseline bouton number in the indicated genetic background. D, Live confocal imaging of GMA-GFP at NMJs driven by c164-Gal4. F-actin labeled by GMA appears as dynamic puncta with relatively even size and spacing in wild-type animals. GMA labeling in axons and extended interbouton regions is stable and uniform at wild-type NMJs (double arrow), but is interrupted by puncta and appears less uniform in tsrS3E and tsrS3E NMJs (double arrowheads). Boutons lacking discernable F-actin puncta occurred rarely at wild-type NMJs and more frequently at tsrS3E NMJs (arrows). Large and bright GMA labeling was observed in some boutons in tsrS3E and tsrS3A NMJs that was not observed in wild-type (arrowheads). Scale bar, 12 μm. *p < 0.05; **p < 0.01; ***p < 0.001; ANOVA. Error bars indicate SEM.
Local actin turnover is required for bouton budding
Manipulations to the actin cytoskeleton regulators limk and tsr resulted in changes to ghost bouton budding frequency and in the case of tsr observable changes to the presynaptic actin cytoskeleton. To directly examine the role of presynaptic actin in bouton budding, we performed live imaging in animals expressing GMA and the membrane marker CD8-RFP expressed in motor neurons. Third instar larvae were dissected and imaged, and then subjected to a single 2 min incubation in high K+ and imaged again. We then identified sites of ghost bouton budding to observe how K+ stimulation affected the local actin cytoskeleton at sites of new bouton formation by comparing before and after images. We consistently observed the emergence of new F-actin puncta localized at the sites of ghost bouton budding from previously existing boutons (Fig. 6A), suggesting that local actin rearrangements occur at regions where new boutons form. To test whether local actin rearrangements are required for ghost bouton budding, we directly interfered with actin turnover through bath application of the F-actin-depolymerizing drug latrunculin A or of the F-actin-stabilizing drug jasplakinolide (Spector et al., 1999). The application of 10 μm latrunculin A caused dispersal of F-actin puncta within minutes as well as a reduction in the number of puncta (Fig. 6B). The application of 10 μm jasplakinolide stabilized existing F-actin puncta and caused the formation of new puncta within minutes (Fig. 6B). The effects of the drugs were still observed 15 min after washout. To examine how these manipulations regulate bouton budding, we incubated dissected preps in HL3 solution containing 10 μm drug for 15 min and then proceeded with the K+ stimulation protocol with solutions containing 10 μm drug. Disruption of normal actin turnover with either drug resulted in a decrease in bouton budding compared with controls (Fig. 6C: no drug = 8.767 ± 7.035, n = 30; latrunculin A = 4.829 ± 6.046, n = 35, p = 0.0155, ANOVA; Jasplakinolide = 1.933 ± 2.586, n = 30, p < 0.0001, ANOVA). Some F-actin puncta were still observed in animals treated with 10 μm latrunculin A, indicating that this treatment may allow the formation of a limited number of new puncta. In contrast, the actin cytoskeleton appeared highly stable in jasplakinolide-treated animals, and caused a much greater reduction in ghost bouton budding. These data indicate that local actin rearrangement occurs during ghost bouton budding, and that actin turnover contributes to the rapid formation of new synaptic varicosities in an activity-dependent manner.
Figure 6.

Ghost bouton budding is accompanied by local rearrangements of the presynaptic actin cytoskeleton. A, Animals presynaptically expressing the membrane marker CD8-RFP and the F-actin marker GMA-GFP were imaged before and after high K+ stimulation. New F-actin puncta (arrowheads) are observed at sites of budding (arrows) where newly formed ghost boutons (double arrowheads) attach to the main axonal arbor. Scale bar, 6 μm. B, Application of 10 μm latrunculin A to the bath solution rapidly disperses F-actin puncta, while application of 10 μm jasplakinolide causes formation and stabilization of F-actin puncta. Scale bar, 6 μm. C, Wild-type animals pretreated with latrunculin A or jasplakinolide for 15 min before high K+ stimulation display a reduction in ghost bouton budding frequency. N (NMJs, animals): no drug = 30, 4; latrunculin A = 35, 5; jasplakinolide = 30, 4. *p < 0.05; ***p < 0.001; ANOVA. Error bars indicate SEM.
Discussion
Our experimental analysis of ghost bouton budding at the Drosophila NMJ indicates that rapid activity-dependent synaptic growth requires retrograde BMP signaling at this synapse. The current data support a model in which BMP signaling through the type II receptor Wit is required developmentally to potentiate synapses for budding in response to elevated synaptic activity. This pathway requires Smad-dependent expression of the Rho-type GEF trio (Fig. 7), and parallels a requirement for BMP signaling and Trio in developmental synaptic growth that occurs during the larval stages. In a parallel pathway, Wit interaction with Limk inhibits bouton budding through regulation of Cofilin activity. Both pathways regulate the synaptic actin cytoskeleton and may converge on similar actin regulatory molecules such as Limk and Cofilin via Rac1 or RhoA. Manipulating Cofilin activity levels by the overexpression of Limk or the expression of constitutively active/inactive Cofilin demonstrates that high Cofilin activity favors bouton budding, while low Cofilin activity inhibits budding. Local changes in the actin cytoskeleton that accompany activity-dependent bouton budding were also observed at sites of new synaptic growth. In addition, pharmacological disruption of normal actin turnover inhibits budding, suggesting that increased actin turnover mediated by Cofilin potentiates rapid activity-dependent synaptic plasticity.
Figure 7.

Model for ghost bouton formation through parallel signaling pathways involving Gbb and Wit. BMP signaling through Wit is predicted to both promote and inhibit changes to the actin cytoskeleton that regulate ghost bouton formation. Gbb signaling developmentally through the phosphorylation and nuclear translocation of Mad potentiates synaptic terminals for activity-induced bouton budding by promoting transcription of the Rho GEF trio. Trio activity may also be regulated locally and acutely by synaptic activity. Wit also signals locally through Limk to inhibit Cofilin (Tsr), thereby suppressing ghost bouton formation.
BMP signaling in ghost bouton budding
Multiple genetic perturbations of BMP signaling were identified that altered the frequency of activity-dependent bouton budding at the NMJ. Although several of these mechanisms are shared with those previously characterized to control BMP-mediated developmental synaptic growth, several manipulations separated rapid activity-dependent BMP-mediated bouton budding from the slower forms of developmental growth. In the case of wit mutants or motor neuron overexpression of dad, a reduction in baseline bouton number was observed that showed varying degrees of severity. Wit mutants displayed strongly undergrown synapses, while dad overexpression animals had only modest synaptic undergrowth. In contrast, both these manipulations strongly suppressed ghost bouton budding. Additionally, synaptic undergrowth with partial knockdown of Gbb using postsynaptic RNAi was not observed, while this manipulation caused a strong reduction in ghost bouton budding. These observations indicate that rapid ghost bouton budding is more sensitive to modest perturbations in BMP signaling compared with developmental synaptic growth. One explanation for this differential sensitivity is that BMP signaling potentiates NMJs for activity-dependent bouton budding via transcriptional regulation of molecular components that are not required for normal synaptic growth. Alternatively, similar molecular pathways are required, but at different levels of output. In particular, trio mutants display a less severe synaptic undergrowth phenotype than wit mutants, but show similarly severe defects in ghost bouton budding. Because trio expression is strongly dependent on BMP signaling (Ball et al., 2010), a modest reduction in BMP output could reduce Trio levels such that ghost bouton budding is significantly reduced, while normal synaptic growth is less affected. It will be interesting to determine in future studies whether the developmental role for BMP signaling for acute structural plasticity shares a critical period as has recently been found for BMP function during developmental synaptic growth (Berke et al., 2013).
Given the requirement of the postsynaptic Ca2+ sensor Syt4 for normal levels of ghost bouton budding, an attractive model is that BMP is released acutely in response to elevated activity through the fusion of Syt4-positive postsynaptic vesicles. However, our analysis indicates that retrograde BMP signaling through trio transcriptional upregulation is unlikely to be an instructive cue for bouton budding, as the severing of axons and the inhibition of retrograde trafficking of P-Mad before stimulation does not reduce budding in response to elevated activity. It is possible that synaptic P-Mad may play an instructional role in ghost bouton budding, as a local decrease in budding frequency was observed when Gbb expression was specifically reduced in muscle 6. Neuronal overexpression of dad also reduced synaptic P-Mad, as has been previously observed (data not shown; Dudu et al., 2006). Therefore, dad overexpression could inhibit ghost bouton budding by decreasing synaptic P-Mad signaling, in addition to decreasing nuclear Smad signaling. However, we did not observe dosage-dependent genetic interactions between syt4 and wit (data not shown), suggesting that Syt4 may participate in a separate pathway to regulate ghost bouton budding. Activity-dependent fusion of Syt4 postsynaptic vesicles (Yoshihara et al., 2005) could release a separate unidentified retrograde signal that provides an instructive cue for budding that would function in parallel to a developmental requirement for retrograde BMP signaling.
Synaptic actin cytoskeleton regulation via Trio
In addition to instructive cues from the postsynaptic compartment that trigger ghost bouton budding, the presynaptic nerve terminal must have molecular machines in place to read out these signals and execute the budding event. The regulation of Rho GTPases via Rho GEFs and GAPs downstream of extracellular cues is an attractive mechanism, as these proteins play critical roles in the regulation of neuronal morphology and axonal guidance (Luo, 2000; Dickson, 2001). Several studies have shown that retrograde synaptic signaling regulates Rho GTPase activity to alter synaptic function and growth in Drosophila (Tolias et al., 2011). Ghost bouton budding mediated by developmental BMP signaling also shares some similarities with mechanisms underlying homeostatic plasticity at Drosophila NMJs. The Eph receptor is required for synaptic homeostasis at the NMJ, and it interfaces with developmental BMP signaling via Wit (Goold and Davis, 2007; Frank et al., 2009). While Eph receptor-mediated homeostatic plasticity predominantly requires the downstream RhoA-type GEF Ephexin, the Eph receptor may also signal through Rac1 (Frank et al., 2009). Drosophila VAP-33A may also act as a ligand for synaptic Eph receptors, as it has been shown to regulate NMJ morphology and growth, while preferentially localizing to sites of bouton budding (Pennetta et al., 2002; Tsuda et al., 2008). Our analysis indicates that the levels of Trio, which functions as a Rho-type GEF, are bidirectionally correlated with ghost bouton budding activity and that overexpressed Trio is localized to ghost boutons after budding. As such, acute Trio regulation represents another attractive pathway for rapidly modifying bouton budding activity.
Limk regulation of Cofilin controls ghost bouton budding
Rho GTPase signaling can produce distinct effects in differing systems and cell types depending on the presence or absence of downstream effectors, although most of these pathways ultimately impinge on regulation of the actin cytoskeletal (Luo, 2000). Indeed, we have found a key role for Limk regulation of Cofilin activity in the control of ghost bouton budding. The current findings indicate that Limk activity normally functions to inhibit the formation of ghost boutons, as neuronal overexpression of Limk strongly suppressed activity-dependent bouton budding. Consistent with an inhibitory role for Limk, Cofilin activity promotes budding, while the overexpression of an inactive Cofilin inhibited budding. The expression of mutant Cofilin transgenes resulted in visible changes to the presynaptic actin cytoskeleton at NMJs, indicating that these manipulations likely alter rapid budding events by changing local actin dynamics at sites of potential growth. Using live imaging of F-actin dynamics before and after bouton budding, the formation of new F-actin puncta was observed at sites of bouton budding. Elevated Cofilin activity is sufficient to increase ghost bouton budding frequency, and is predicted to increase actin turnover and the formation of F-actin structures (Michelot and Drubin, 2011). Pharmacological disruption of actin polymerization dynamics also disrupts rapid bouton addition in response to elevated activity.
These findings support a model whereby Wit has opposing signaling roles with respect to bouton budding (Fig. 7). Providing a permissive role via Smad signaling and an inhibitory role via Limk activation may provide for a system in which increased potential for rapid synaptic expansion is directly coupled to enhanced synaptic stability. This coupling could set a threshold for ghost bouton budding downstream of synaptic activity. In the background of moderate or low synaptic activity, Limk prevents ghost bouton budding. When synaptic activity is elevated, additional signaling events promote new synaptic growth by either reducing or outcompeting Limk activity, with a concurrent activation of Cofilin. Decreased Limk activity downstream of extracellular cues has been shown to regulate cell morphology in other systems as well (Sparrow et al., 2012), providing an attractive mechanism for rapid activity-dependent regulation of synaptic structure at Drosophila NMJs.
Notes
Supplemental material for this article is available at web.mit.edu/flybrain/littletonlab. Supplemental movie: multiple budding events and subsequent bouton relocalization at a Drosophila muscle 4 neuromuscular junction. This material has not been peer reviewed.
Footnotes
This work was supported by National Institutes of Health Grant NS40296 to J.T.L. We thank Graeme Davis for the UAS-witdCT fly stock, and Brian McCabe for UAS-dad and UAS-wit fly stocks.
The authors declare no competing financial interests.
References
- Aberle H, Haghighi AP, Fetter RD, McCabe BD, Magalhães TR, Goodman CS. wishful thinking encodes a BMP type II receptor that regulates synaptic growth in Drosophila. Neuron. 2002;33:545–558. doi: 10.1016/S0896-6273(02)00589-5. [DOI] [PubMed] [Google Scholar]
- Ang LH, Chen W, Yao Y, Ozawa R, Tao E, Yonekura J, Uemura T, Keshishian H, Hing H. Lim kinase regulates the development of olfactory and neuromuscular synapses. Dev Biol. 2006;293:178–190. doi: 10.1016/j.ydbio.2006.01.030. [DOI] [PubMed] [Google Scholar]
- Ataman B, Ashley J, Gorczyca D, Gorczyca M, Mathew D, Wichmann C, Sigrist SJ, Budnik V. Nuclear trafficking of Drosophila Frizzled-2 during synapse development requires the PDZ protein dGRIP. Proc Natl Acad Sci U S A. 2006;103:7841–7846. doi: 10.1073/pnas.0600387103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ataman B, Ashley J, Gorczyca M, Ramachandran P, Fouquet W, Sigrist SJ, Budnik V. Rapid activity-dependent modifications in synaptic structure and function require bidirectional Wnt signaling. Neuron. 2008;57:705–718. doi: 10.1016/j.neuron.2008.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball RW, Warren-Paquin M, Tsurudome K, Liao EH, Elazzouzi F, Cavanagh C, An BS, Wang TT, White JH, Haghighi AP. Retrograde BMP signaling controls synaptic growth at the NMJ by regulating trio expression in motor neurons. Neuron. 2010;66:536–549. doi: 10.1016/j.neuron.2010.04.011. [DOI] [PubMed] [Google Scholar]
- Barber CF, Jorquera RA, Melom JE, Littleton JT. Postsynaptic regulation of synaptic plasticity by synaptotagmin 4 requires both C2 domains. J Cell Biol. 2009;187:295–310. doi: 10.1083/jcb.200903098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayat V, Jaiswal M, Bellen HJ. The BMP signaling pathway at the Drosophila neuromuscular junction and its links to neurodegenerative diseases. Curr Opin Neurobiol. 2011;21:182–188. doi: 10.1016/j.conb.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berke B, Wittnam J, McNeill E, Van Vactor DL, Keshishian H. Retrograde BMP signaling at the synapse: a permissive signal for synapse maturation and activity-dependent plasticity. J Neurosci. 2013;33:17937–17950. doi: 10.1523/JNEUROSCI.6075-11.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Davis GW, Schuster CM, Goodman CS. Genetic analysis of the mechanisms controlling target selection: target-derived Fasciclin II regulates the pattern of synapse formation. Neuron. 1997;19:561–573. doi: 10.1016/S0896-6273(00)80372-4. [DOI] [PubMed] [Google Scholar]
- Dean C, Liu H, Staudt T, Stahlberg MA, Vingill S, Bückers J, Kamin D, Engelhardt J, Jackson MB, Hell SW, Chapman ER. Distinct subsets of Syt-IV/BDNF vesicles are sorted to axons versus dendrites and recruited to synapses by activity. J Neurosci. 2012;32:5398–5413. doi: 10.1523/JNEUROSCI.4515-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson BJ. Rho GTPases in growth cone guidance. Curr Opin Neurobiol. 2001;11:103–110. doi: 10.1016/S0959-4388(00)00180-X. [DOI] [PubMed] [Google Scholar]
- Dudu V, Bittig T, Entchev E, Kicheva A, Jülicher F, González-Gaitán M. Postsynaptic mad signaling at the Drosophila neuromuscular junction. Curr Biol. 2006;16:625–635. doi: 10.1016/j.cub.2006.02.061. [DOI] [PubMed] [Google Scholar]
- Dutta D, Bloor JW, Ruiz-Gomez M, VijayRaghavan K, Kiehart DP. Real-time imaging of morphogenetic movements in Drosophila using Gal4-UAS-driven expression of GFP fused to the actin-binding domain of moesin. Genesis. 2002;34:146–151. doi: 10.1002/gene.10113. [DOI] [PubMed] [Google Scholar]
- Eaton BA, Davis GW. LIM Kinase1 controls synaptic stability downstream of the type II BMP receptor. Neuron. 2005;47:695–708. doi: 10.1016/j.neuron.2005.08.010. [DOI] [PubMed] [Google Scholar]
- Foletta VC, Lim MA, Soosairajah J, Kelly AP, Stanley EG, Shannon M, He W, Das S, Massague J, Bernard O, Soosairaiah J. Direct signaling by the BMP type II receptor via the cytoskeletal regulator LIMK1. J Cell Biol. 2003;162:1089–1098. doi: 10.1083/jcb.200212060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank CA, Pielage J, Davis GW. A presynaptic homeostatic signaling system composed of the Eph receptor, ephexin, Cdc42, and CaV2.1 calcium channels. Neuron. 2009;61:556–569. doi: 10.1016/j.neuron.2008.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuentes-Medel Y, Logan MA, Ashley J, Ataman B, Budnik V, Freeman MR. Glia and muscle sculpt neuromuscular arbors by engulfing destabilized synaptic boutons and shed presynaptic debris. PLoS Biol. 2009;7:e1000184. doi: 10.1371/journal.pbio.1000184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goold CP, Davis GW. The BMP ligand Gbb gates the expression of synaptic homeostasis independent of synaptic growth control. Neuron. 2007;56:109–123. doi: 10.1016/j.neuron.2007.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorquera RA, Huntwork-Rodriguez S, Akbergenova Y, Cho RW, Littleton JT. Complexin controls spontaneous and evoked neurotransmitter release by regulating the timing and properties of synaptotagmin activity. J Neurosci. 2012;32:18234–18245. doi: 10.1523/JNEUROSCI.3212-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiya Y, Miyazono K, Miyazawa K. Specificity of the inhibitory effects of Dad on TGF-beta family type I receptors, Thickveins, Saxophone, and Baboon in Drosophila. FEBS Lett. 2008;582:2496–2500. doi: 10.1016/j.febslet.2008.05.052. [DOI] [PubMed] [Google Scholar]
- Korkut C, Ataman B, Ramachandran P, Ashley J, Barria R, Gherbesi N, Budnik V. Trans-synaptic transmission of vesicular Wnt signals through Evi/Wntless. Cell. 2009;139:393–404. doi: 10.1016/j.cell.2009.07.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korkut C, Li Y, Koles K, Brewer C, Ashley J, Yoshihara M, Budnik V. Regulation of postsynaptic retrograde signaling by presynaptic exosome release. Neuron. 2013;77:1039–1046. doi: 10.1016/j.neuron.2013.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee-Hoeflich ST, Causing CG, Podkowa M, Zhao X, Wrana JL, Attisano L. Activation of LIMK1 by binding to the BMP receptor, BMPRII, regulates BMP-dependent dendritogenesis. EMBO J. 2004;23:4792–4801. doi: 10.1038/sj.emboj.7600418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo L. Rho GTPases in neuronal morphogenesis. Nat Rev Neurosci. 2000;1:173–180. doi: 10.1038/35044547. [DOI] [PubMed] [Google Scholar]
- Marqués G. Morphogens and synaptogenesis in Drosophila. J Neurobiol. 2005;64:417–434. doi: 10.1002/neu.20165. [DOI] [PubMed] [Google Scholar]
- Marqués G, Bao H, Haerry TE, Shimell MJ, Duchek P, Zhang B, O'Connor MB. The Drosophila BMP type II receptor Wishful Thinking regulates neuromuscular synapse morphology and function. Neuron. 2002;33:529–543. doi: 10.1016/S0896-6273(02)00595-0. [DOI] [PubMed] [Google Scholar]
- McCabe BD, Marqués G, Haghighi AP, Fetter RD, Crotty ML, Haerry TE, Goodman CS, O'Connor MB. The BMP homolog Gbb provides a retrograde signal that regulates synaptic growth at the Drosophila neuromuscular junction. Neuron. 2003;39:241–254. doi: 10.1016/S0896-6273(03)00426-4. [DOI] [PubMed] [Google Scholar]
- Michelot A, Drubin DG. Building distinct actin filament networks in a common cytoplasm. Curr Biol. 2011;21:R560–R569. doi: 10.1016/j.cub.2011.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosca TJ, Schwarz TL. The nuclear import of Frizzled2-C by Importins-beta11 and alpha2 promotes postsynaptic development. Nat Neurosci. 2010;13:935–943. doi: 10.1038/nn.2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi K, Hosoya T, Takahashi K, Hing H, Mizuno K. A Drosophila homolog of LIM-kinase phosphorylates cofilin and induces actin cytoskeletal reorganization. Biochem Biophys Res Commun. 2000;276:1178–1185. doi: 10.1006/bbrc.2000.3599. [DOI] [PubMed] [Google Scholar]
- Pennetta G, Hiesinger PR, Fabian-Fine R, Meinertzhagen IA, Bellen HJ. Drosophila VAP-33A directs bouton formation at neuromuscular junctions in a dosage-dependent manner. Neuron. 2002;35:291–306. doi: 10.1016/S0896-6273(02)00769-9. [DOI] [PubMed] [Google Scholar]
- Rawson JM, Lee M, Kennedy EL, Selleck SB. Drosophila neuromuscular synapse assembly and function require the TGF-beta type I receptor saxophone and the transcription factor Mad. J Neurobiol. 2003;55:134–150. doi: 10.1002/neu.10189. [DOI] [PubMed] [Google Scholar]
- Sparrow N, Manetti ME, Bott M, Fabianac T, Petrilli A, Bates ML, Bunge MB, Lambert S, Fernandez-Valle C. The actin-severing protein cofilin is downstream of neuregulin signaling and is essential for Schwann cell myelination. J Neurosci. 2012;32:5284–5297. doi: 10.1523/JNEUROSCI.6207-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spector I, Braet F, Shochet NR, Bubb MR. New anti-actin drugs in the study of the organization and function of the actin cytoskeleton. Microsc Res Tech. 1999;47:18–37. doi: 10.1002/(SICI)1097-0029(19991001)47:1<18::AID-JEMT3>3.0.CO%3B2-E. [DOI] [PubMed] [Google Scholar]
- Tolias KF, Duman JG, Um K. Control of synapse development and plasticity by Rho GTPase regulatory proteins. Prog Neurobiol. 2011;94:133–148. doi: 10.1016/j.pneurobio.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torroja L, Packard M, Gorczyca M, White K, Budnik V. The Drosophila β-amyloid precursor protein homolog promotes synapse differentiation at the neuromuscular junction. J Neurosci. 1999;19:7793–7803. doi: 10.1523/JNEUROSCI.19-18-07793.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuda H, Han SM, Yang Y, Tong C, Lin YQ, Mohan K, Haueter C, Zoghbi A, Harati Y, Kwan J, Miller MA, Bellen HJ. The amyotrophic lateral sclerosis 8 protein VAPB is cleaved, secreted, and acts as a ligand for Eph receptors. Cell. 2008;133:963–977. doi: 10.1016/j.cell.2008.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuneizumi K, Nakayama T, Kamoshida Y, Kornberg TB, Christian JL, Tabata T. Daughters against dpp modulates dpp organizing activity in Drosophila wing development. Nature. 1997;389:627–631. doi: 10.1038/39362. [DOI] [PubMed] [Google Scholar]
- Yoshihara M, Littleton JT. Synaptotagmin I functions as a calcium sensor to synchronize neurotransmitter release. Neuron. 2002;36:897–908. doi: 10.1016/S0896-6273(02)01065-6. [DOI] [PubMed] [Google Scholar]
- Yoshihara M, Adolfsen B, Galle KT, Littleton JT. Retrograde signaling by Syt 4 induces presynaptic release and synapse-specific growth. Science. 2005;310:858–863. doi: 10.1126/science.1117541. [DOI] [PubMed] [Google Scholar]
- Zito K, Parnas D, Fetter RD, Isacoff EY, Goodman CS. Watching a synapse grow: noninvasive confocal imaging of synaptic growth in Drosophila. Neuron. 1999;22:719–729. doi: 10.1016/S0896-6273(00)80731-X. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Rapid presynaptic bouton budding imaged at muscle 12/13. Visualization of a presynaptic arbor expressing mRFP-syntaxin 13 at muscle 12/13 in a dissected third instar larva with the nervous system intact. The parent bouton swells at a restricted site (∼2 μm across), producing two discernable boutons within 20 s. The new bouton (labeled A) then separates from the parent bouton at the budding site, but remains connected by a thin axon. A second, smaller bouton (labeled B) buds from the same initial site, but collapses rapidly and is not maintained. The first newly budded bouton is stabilized within 1 min at ∼4 μm from the parent bouton and has a strikingly round morphology. The parent bouton appears incrementally smaller after budding. The movie was acquired at a rate of 1 Hz, with video speed set at 10 frames/s, 10× real time.