

Abstract
Bicarbonate (HCO3−) is an abundant anion that regulates extracellular and intracellular pH. Here, we use patch-clamp techniques to assess regulation of hippocampal CA3 pyramidal cell excitability by HCO3− in acute brain slices from C57BL/6 mice. We found that increasing HCO3− levels enhances action potential (AP) generation in both the soma and axon initial segment (AIS) by reducing Kv7/KCNQ channel activity, independent of pH (i.e., at a constant pH of 7.3). Conversely, decreasing intracellular HCO3− leads to attenuation of AP firing. We show that HCO3− interferes with Kv7/KCNQ channel activation by phosphatidylinositol-4,5-biphosphate. Consequently, we propose that, even in the presence of a local depolarizing Cl− gradient, HCO3− efflux through GABAA receptors may ensure the inhibitory effect of axoaxonic cells at the AIS due to activation of Kv7/KCNQ channels.
Introduction
Bicarbonate (HCO3−) is a key product of cellular respiration, resulting from the hydration of CO2 and the subsequent deprotonation of carbonic acid. Thus, HCO3− is one of the most abundant anions in cerebrospinal and intracellular fluids with a typical concentration of ∼26 mm at physiological pH (7.3–7.4) and pCO2 (5%; Jungas, 2006; Casey et al., 2010). Bicarbonate acts as the major pH buffer in most biological systems (under physiological conditions for CO2/HCO3−, pKa is ∼6.1; Jungas, 2006; Casey et al., 2010). Hence, the steady-state intracellular and extracellular HCO3− concentrations ([HCO3−]i and [HCO3−]o, respectively) and fluctuations thereof depend on local proton concentration (i.e., pH). For example, in both vertebrate and crayfish neurons HCO3− flux through GABAA receptor channels controls neuronal excitability by changing local pH (Chesler and Kaila, 1992; Jungas, 2006; Casey et al., 2010). Moreover, disturbance in this signaling mechanism might play an important role in some forms of epilepsy, and manipulation of brain pH may be a promising approach to therapeutic intervention (Pavlov et al., 2013). Apart from its well defined role as a pH buffer, HCO3− also is a weak Ca2+ buffer, with a Kd ∼96 mm (Greenwald, 1941; Nakayama, 1971; Jacobson and Langmuir, 1974; Reardon and Langmuir, 1974; Schaer, 1974; Fry and Poolewilson, 1981; Hablitz and Heinemann, 1987). Despite a relatively low affinity for free Ca2+, we initially hypothesized that the relatively high [HCO3−]i may provide physiologically relevant Ca2+ buffering, resulting in the modulation of neuronal excitability. To test this hypothesis, we examined the effects of changing [HCO3−]i, at a set pH of 7.3, on the firing properties of hippocampal CA3 pyramidal cells (PCs), a cell type that lacks major Ca2+ buffering proteins such as parvalbumin, calbindin, and calretinin (Schwaller, 2010). We found that HCO3− has a significant effect on neuronal excitability, independent of pH. However, contrary to our initial hypothesis, the effects of HCO3− are not due to a direct modulation of Ca2+ signaling, but rather to an inhibition of Kv7/KCNQ channel activity. Moreover, fluctuations in [HCO3−]i may control Kv7/KCNQ channel activation through a phosphatidylinositol-4,5-biphosphate (PIP2)-dependent mechanism.
Materials and Methods
Electrophysiology.
Horizontal hippocampal brain slices (350 μm) were prepared from male C57BL/6J (4–6 weeks) mice according to protocols approved by the UCLA Chancellor's Animal Research Committee. Mice were deeply anesthetized with isofluorane, avoiding all handling and stress to the animals, and were decapitated using a guillotine. Their brains were quickly removed and cooled to 4°C in a sodium-free cutting solution containing the following (in mm): 135 N-methyl-d-glucamine, 26 HEPES, 10 d-glucose, 4 MgCl2, 1.2 KH2PO4, 1 KCl, and 0.5 CaCl2 (bubbled with 100% O2, pH 7.4, 290–300 mOsm). Slices were cut in the same sodium-free solution using a Leica V1200S vibratome and subsequently transferred to an interface holding chamber containing a reduced sodium artificial CSF (aCSF) at 32°C containing the following (in mm): 85 NaCl, 55 sucrose, 25 d-glucose, 26 HEPES, 4 MgCl2, 2.5 KCl, 1.25 NaH2PO4, 0.5 CaCl2 (bubbled with 100% O2, pH 7.4, 290–300 mOsm). After 30 min, the slices were allowed to cool to room temperature (22–23°C) and kept in this solution until used for recordings. Brain slices were transferred to a submerged recording chamber at 34°C and perfused at 5 ml/min with aCSF containing the following (in mm): 126 NaCl, 26 HEPES, 10 d-glucose, 2.5 KCl, 2 MgCl2, 2 CaCl2, 1.25 NaH2PO4, 1.5 Na-pyruvate, and 1 l-glutamine (100% O2, pH 7.4, 290–300 mOsm). All salts were purchased from Sigma-Aldrich. DNQX (25 μm), d-AP5 (50 μm) and gabazine or picrotoxin (20 or 100 μm, respectively) were added to block ionotropic glutamatergic and GABAergic transmission (Tocris Biosciences). Slices were visualized under IR-DIC upright microscope (Olympus BX-51WI, 20× XLUMPlan FL N objective) and whole-cell recordings were obtained from CA3 pyramidal neurons with borosilicate patch pipettes (4–6 MΩ, King Precision Glass) containing a mixture (according to the desired [HCO3−]p, see below) of two internal solutions: (1) 0 mm HCO3− solution (in mm): 135 K-methanesulphonate, 5 KCl, 10 HEPES, 2 MgCl2, 3 NaCl, 0.2 EGTA, 2 Na2ATP, 0.2 NaGTP, pH 7.3–7.35 with KOH; or (2) 26 mm HCO3− solution (in mm): 120 K-methanesulphonate, 15 KHCO3, 5 KCl, 10 HEPES, 2 MgCl2, 3 NaCl, 0.2 EGTA, 2 Na2ATP, 0.2 Na2GTP. The pH was set to 7.3–7.35 while bubbling with 5% CO2/95% O2. Internal solutions were stored at −80°C in one ml aliquots. Before each experiment, intracellular solution aliquots were thawed to room temperature and the 26 mm HCO3− containing intracellular solution (solution 2) was bubbled with 5% CO2/95% O2 for 15–20 min. The pH of both solutions was confirmed to be between 7.3 and 7.35 before each day's experiments. The liquid junction potential (LJP) was +5 mV, but reported Vm values have not been adjusted to account for the LJP. The pH drift of the HCO3− containing intracellular solution was confirmed to be <0.1 pH/h when sealed in an airtight tube. If the HCO3− intracellular solution was exposed to the air, the pH gradually became more alkaline, drifting to pH ∼7.6 as CO2 left solution, but had a very slow time constant of ∼4.5 h. The 26 mm HCO3− intracellular solution was diluted 1:2 or 1:4 with the 0 mm HCO3− solution to produce 13 or 6.5 mm containing pipette solutions, respectively. In a subset of recordings, AlexaFluor 488 dextran (50 μm, Tocris Bioscience) was included in the pipette solution to verify the neuronal morphology of CA3 neurons (Fig. 1A). For PIP2 experiments, PIP2 (Echelon Biosciences) was dissolved in deionized water at a stock solution concentration of 1 μg/μl and stored at −20°C. Before each experiment, one aliquot of PIP2 was thawed and diluted into 0 mm [HCO3−] K-Met pipet solution to a final concentration of 10 or 30 μm.
Figure 1.

Intracellular HCO3− regulates intrinsic excitability via KCNQ channel activation. A, Whole-cell current-clamp recordings were made from CA3 PCs. A subset of recorded neurons was filled with AlexaFluor 488 (50 μm) to confirm CA3 PC morphology. B, Current steps (300 ms) evoked fewer APs with 0 mm [HCO3−]p, black traces versus 26 mm [HCO3−]p, gray traces (n = 10, p = 0.01, RM two-way ANOVA; asterisks indicate statistically significant post hoc paired tests). C, Spontaneous AP waveforms recorded with 0 or 26 mm [HCO3−]p revealed significantly enhanced mAHP (CI, CIII) in 0 mm [HCO3−]p, whereas AP amplitude, threshold, and half-width were unaltered (CII). D, Current ramp protocols (F, bottom) also revealed reduced AP generation in 0 versus 26 mm [HCO3−]p, indicating reduced excitability. E, The KCNQ channel activator retigabine (20 μm) reduced the number of APs/ramp in 26 mm [HCO3−]p to levels comparable to 0 mm [HCO3−]p. F, In contrast, the KCNQ channel antagonist XE 991 (10 μm) increased the number of APs in both 0 and 26 mm [HCO3−]p and removed the difference between conditions. Data shown as mean ± SEM.
Recording and analysis.
Recordings were obtained using a MultiClamp 700A amplifier (Molecular Devices), low-pass filtered at 5 kHz (Bessel, 8-pole) and digitized at 20 kHz with a National Instruments data acquisition board (NI USB-6221). All recording protocols and analyses were performed using custom procedures written in Igor Pro (WaveMetrics).
Current-clamp protocol.
After achieving stable whole-cell configuration in voltage-clamp, the amplifier was switched to current-clamp mode. Only recordings with series resistances <20 MΩ were used and bridge balance compensation was applied while in current-clamp. After measuring the resting membrane potential, constant current was injected (no more than ±50 pA) into the cell to keep its membrane potential between −60 and −65 mV. Current steps (300 ms) from −100 to +500 pA were injected every 30 s. To induce steady-state action potential (AP) firing, a steady current was injected through the pipette and was gradually increased until spontaneous action potentials were observed (∼−40 to −30 mV). Spontaneous APs were detected and extracted by calculating the second derivative of the membrane voltage deflection and defining AP initiation as the point at which the second derivative deviated significantly from zero. The second derivative was also used to determine AP threshold and was used as the reference point to measure AP amplitude, fast afterhyperpolarization (fAHP; immediately following the AP) amplitude and medium afterhyperpolarization area. The mAHP was defined as the hyperpolarization immediately following the fAHP and lasting ∼100 ms (Storm, 1987b, 1989). Statistical analysis of spontaneous AP threshold, amplitude, half-width, fAHP, and mAHP was performed by analyzing 10 consecutive APs and comparing 0 and 26 mm ([HCO3−]p conditions with a two-way repeated-measures (RM) ANOVA. Ramp protocols consisted of a −100 pA step for 1 s, followed by a current ramp to +400 pA (250 pA/s), and then a plateau stage for an additional 3 s. Ramps were applied every 60 s.
Antidromic action current protocols.
Whole-cell voltage-clamp recordings were obtained with 0 mm HCO3− pipette solution containing AlexaFluor 488 (50 μm) to visualize the axon. A theta glass electrode was positioned near the labeled axon (∼250 μm away from soma) and antidromic action currents were evoked with 50–100 μs stimuli. Stimulus trains were applied (5 stimuli, 50 Hz) and the stimulus intensity was adjusted so that the probability of evoking an antidromic action current was ∼50%.
Statistical analysis.
Statistics were calculated using GraphPad Prism 6 and statistics procedures in R. Data are presented as mean ± SEM. Nonparametric statistical analysis was used for datasets that deviated from normality (as determined using a QQ-plot) and the respective statistical tests are indicated in the text and figure legends. The median value (± SE of median) was reported in some cases, because it is a better representation of central tendency for skewed distributions compared with the mean. SE of medians was calculated using resampling procedures in R. For ANOVAs with significance levels <0.05, post hoc paired tests were performed and corrected for multiple comparisons. Significance level (α) was set to 0.05.
Results
Intracellular bicarbonate modulates CA3 pyramidal cell excitability
To assess the influence of [HCO3−]i on neuronal excitability, AP firing properties were measured in CA3 PCs in acute hippocampal slices in HCO3−-free (0 mm [HCO3−]o/HEPES buffered, pH 7.4) aCSF containing 25 μm DNQX, 50 μm D-APV, and either 20 μm gabazine or 100 μm PTX to block AMPA, NMDA, and GABAA receptors, respectively. Whole-cell recordings were made using pipette solutions containing 0, 6.5, 13, or 26 mm HCO3− ([HCO3−]p) to set [HCO3−]i. Because the conversion of HCO3− to CO2 is catalyzed by multiple carbonic anhydrases and CO2 readily passes through the membrane (Maren, 1967), it is difficult to set [HCO3−]i to an exact value. However, varying [HCO3−]p will result in correspondingly different values of [HCO3−]i. Importantly, to test the effect of [HCO3−]i independent of pH, the pH was carefully set to 7.3 for each pipette solution directly before each experiment. Separate experiments confirmed that pH drift, due to CO2 dissipation out of the pipette solution, was minimal (<0.1 pH unit) for the typical duration of an experiment.
To measure AP firing, square pulse current steps (300 ms, −100 to +300 pA) were injected into PCs recorded with either 0 or 26 mm [HCO3−]p. PCs patched with 26 mm [HCO3−]p exhibited greater intrinsic excitability, measured by the number of APs, than cells recorded with 0 mm [HCO3−]p (n = 10 each, p = 0.013, R.M. two-way ANOVA; Fig. 1A,B), whereas resting membrane potentials and input resistances were unaffected (Table 1). Steady firing of spontaneous APs was elicited by gradually depolarizing the cells to −40 to −35 mV with constant current injections. AP properties were assessed at these depolarized potentials. We found that the area of the medium post-spike afterhyperpolarization (mAHP) was significantly larger with 0 mm [HCO3−]p compared with 26 mm [HCO3−]p (1.12 ± 0.1 vs 0.68 ± 0.1 mV*s; n = 11 for both [HCO3−]p, p < 0.0001; Fig. 1CI,CIII). However, AP threshold, amplitude, and half-width, were unaffected (Fig. 1CII; Table 2). Bicarbonate can act as a weak Ca2+ buffer (Greenwald, 1941; Nakayama, 1971; Jacobson and Langmuir, 1974; Reardon and Langmuir, 1974; Schaer, 1974; Fry and Poolewilson, 1981; Hablitz and Heinemann, 1987) and if the effect of HCO3− on the mAHP was Ca2+-dependent, adding a strong Ca2+-buffer or reducing Ca2+ entry should abolish this effect. However, the mAHP persisted in the presence of 5 mm intracellular BAPTA or when 50 μm Cd2+ was added to the aCSF (data not shown), confirming earlier observations (Storm, 1987a), and indicating that the reduction of mAHP by HCO3− was not due to fast modulation of Ca2+ signaling. In summary, these data indicate that intracellular HCO3− regulates intrinsic CA3 PC excitability by modulating postspike mAHP, independent of pH and Ca2+.
Table 1.
Basic membrane properties of CA3 pyramidal neurons
| [HCO3−]o (mm) | 0 | 0 | 0 | 0 | ||
| [HCO3−]p (mm) | 0 | 6.5 | 13 | 26 | 0 | p value |
| Resting membrane potential (mV) | −63 ± 1 | −57 ± 2 | −60 ± 1 | −63 ± 2 | −66 ± 1 | 0.208 |
| Input resistance (MΩ) | 181 ± 12 | 185 ± 11 | 222 ± 30 | 199 ± 19 | 202 ± 26 | 0.638 |
| n | 23 | 5 | 8 | 14 | 5 |
Resting membrane potentials and input resistances were not different across all HCO3− conditions. Values are mean ± SEM. Statistical comparisons were calculated using one-way ANOVA.
Table 2.
Spontaneous AP properties
| [HCO3−]o (mm) | 0 | 0 | |
| [HCO3−]p (mm) | 0 | 26 | p value |
| Threshold (mV)a | −29 ± 0.2 | −31 ± 0.1 | 0.40 |
| AP amplitude (mV)a | 49 ± 0.6 | 53 ± 0.4 | 0.12 |
| Half-width (μs) | 784 ± 55 | 768 ± 39 | 0.65 |
| fAHP amplitude (mV)a | 8.8 ± 0.1 | 10.5 ± 0.1 | 0.02* |
| mAHP area (mV · s)a | −1.12 ± 0.01 | −0.68 ± 0.01 | <0.0001* |
| n | 11 | 11 |
Fast AP properties were not significantly affected by [HCO3−]p. However, the area of the mAHP was significantly enhanced in 0 mM [HCO3−]p. Values are mean ± SEM.
aMeasured relative to AP threshold.
*Indicates statistical significance, two-way RM ANOVA.
Intracellular bicarbonate regulates action potential generation by modulating KCNQ channel activation
Previous reports suggest that a major component of the mAHP in CA1 PCs is mediated by Kv7/KCNQ K+ channels, regulating intrinsic neuronal excitability (Gu et al., 2008; Tzingounis and Nicoll, 2008; Cooper, 2011; Klinger et al., 2011). Because the major effect of lowering [HCO3−]i (0 mm [HCO3−]p) was a reduction in intrinsic excitability and an enhancement of the mAHP, we hypothesized that [HCO3−]i may regulate KCNQ channel activation. Similar to previous reports in CA1 PCs (Gu et al., 2005), bath-application of the KCNQ selective antagonist XE 991 (10 μm) abolished the mAHP in both 0 and 26 mm [HCO3−]p (data not shown), confirming that KCNQ currents constitute a major component of the mAHP in CA3 PCs.
To establish whether [HCO3−]i can regulate PC excitability by modulating KCNQ channels, we used a current ramp protocol to elicit APs that are sensitive to KCNQ channel activation (Hu et al., 2007). When these ramp protocols were performed in 0 mm [HCO3−]o with either 0 or 26 mm [HCO3−]p, we found that PCs recorded with 0 mm [HCO3−]p generated significantly fewer APs/ramp (median = 12 ± 4 APs/ramp, n = 23) compared with PCs recorded with 26 mm [HCO3−]p (median = 36 ± 4 APs/ramp, n = 14, p = 0.001, Mann–Whitney; Fig. 1D). Bath application of the KCNQ channel activator retigabine (20 μm) reduced the number of elicited APs/ramp with 26 mm [HCO3−]p (median = 4 ± 3 APs/ramp, n = 4) to levels comparable to 0 mm [HCO3−]p plus retigabine (median = 1 ± 1 AP/ramp, n = 4, p = 0.36, Mann–Whitney; Fig. 1E). In contrast, XE 991 (10 μm) dramatically enhanced excitability in both 0 and 26 mm [HCO3−]p, resulting in depolarization block, and rendered AP firing comparable between conditions (0 mm: 19 ± 4 APs/ramp, 26 mm: 22 ± 11 APs/ramp, median ± SEM, n = 4 each, p = 0.65, Mann–Whitney; Fig. 1F). Given these data, it is possible that [HCO3−]i regulates PC excitability by modulating KCNQ channel activation such that a decrease in [HCO3−]i increases KCNQ channel activity.
Kv7/KCNQ channels are enriched at the axon initial segment (AIS) of pyramidal cells, where they associate with NaV channels via the scaffolding protein ankyrin-G and regulate AP initiation (Gu et al., 2005, 2008; Hu et al., 2007; Cooper, 2011). Because the AIS is the predominant site of AP generation in cortical PCs and has lower AP threshold than the soma (Meeks et al., 2005; Meeks and Mennerick, 2007; Kole and Stuart, 2012), we investigated whether AP generation is particularly sensitive to [HCO3−]i regulation via KCNQ channels at the AIS. Previous studies have demonstrated APs generated in the soma or AIS exhibit subtle but measurable differences in AP waveforms (Meeks and Mennerick, 2007). To determine the origin of APs generated during current ramp protocols, the second derivative was calculated for each AP and was categorized as being either somatic or AIS in origin based on its distinct waveform (Fig. 2A). In our experiments, somatically generated APs preceded AIS generated APs during the ramp protocol and the number of APs/ramp remained stable during successive ramps (Fig. 2B,C). When current ramps were performed with 26, 13, 6.5, and 0 mm [HCO3−]p, we found that somatic and AIS AP generation was reduced in a dose-dependent manner in (p < 0.001, two-way ANOVA; Fig. 3A–C, Fig. 4). Interestingly, decreasing the [HCO3−]p by half (26 to 13 mm) dramatically reduced the median number of AIS APs per ramp (26 mm [HCO3−]p: 13 ± 1 AIS APs/ramp vs 13 mm [HCO3−]p: 4 ± 4 AIS APs/ramp, median, p = 0.036, Mann–Whitney; Fig. 4), whereas somatically generated APs were unaffected (26 mm [HCO3−]p: 22 ± 4 somatic APs/ramp vs 13 mm [HCO3−]p: 19 ± 5 somatic APs/ramp, median, p = 0.788, Mann–Whitney; Fig. 4). These data provide evidence that [HCO3−]i modulates AP generation in both cellular compartments and that AIS-generated APs are particularly sensitive to variations in [HCO3−]i.
Figure 2.
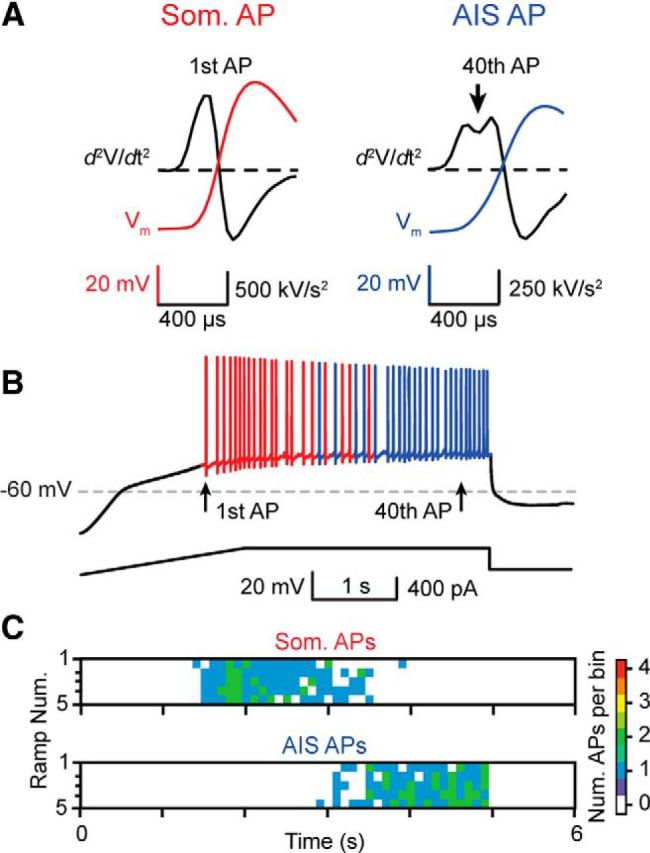
Detection and classification of somatic or axon initial segment generated APs. A, Somatic (Som.; red) or AIS (blue) generated APs were determined by calculating the second derivative of each AP. As shown by previous studies (Meeks and Mennerick, 2007), the second derivative of the voltage traces of somatically generated APs exhibit a biphasic waveform during the rising phase, whereas AIS-generated APs exhibit a pronounced biphasic dip (arrow). B, The site of AP generation transitioned from the soma to AIS during current ramps, most likely due to the large somatic voltage change caused by the current ramp injection protocol, and was consistent and reproducible over multiple ramps (C). The color scale indicates the number of APs in 100 ms bins.
Figure 3.

Intracellular HCO3− modulates somatic and axon initial segment action potential generation. A, CA3 PCs exhibit a [HCO3−]p dose-dependent reduction in the number of somatic (som, red) and AIS (blue) APs/ramp. B, Median spike time histograms (100 ms/bin) for all cells indicated in C illustrate the evolution of som to AIS generated APs during the ramp. AI–AIV, Decreasing [HCO3−]p dramatically reduced the median number of som and AIS APs/bin. C, Evolution of AP firing during the ramps is displayed as heat maps in each recorded neuron for different [HCO3−]p. Arrowheads indicate the representative neurons shown in A.
Figure 4.

Intracellular HCO3− regulates somatic and AIS AP generation. A, Two-way ANOVA shows a significant effect of [HCO3−]p on both somatic and AIS AP generation in 0 mm [HCO3−]o (F(3,92) = 8.40, p < 0.0001), and an interaction between som. and AIS generated APs (F(3,92) = 2.76, p = 0.046). AIS generated APs were particularly sensitive to a reduction of [HCO3−]p from 26 to 13 mm. Values given as median ± SE of median.
Extracellular bicarbonate regulates action potential generation by modulating KCNQ channel activation
CO2 diffuses freely through the lipid bilayer according to the CO2/HCO3− concentration gradient (Maren, 1967). Consequently, it should be possible to (locally) achieve an [HCO3−]i > 0 mm even with 0 mm [HCO3−]p by changing [HCO3−]o. This gives us the ability to modulate [HCO3−]i during an experiment simply by changing [HCO3−]o. To address this, whole-cell recordings were made from CA3 PCs with 26 mm [HCO3−]o, and 0 mm [HCO3−]p and were held for ∼10 min before beginning each experiment to dialyze each cell with the pipette solution. Under these conditions, current ramp protocols generated robust APs (median = 60 ± 8 APs/ramp; Fig. 5A), with most APs generated at the AIS (blue colored APs). Subsequent [HCO3−]o wash out significantly reduced the number of AIS APs/ramp (26 mm [HCO3−]o: 48 ± 15 AIS APs/ramp; 0 mm [HCO3−]o: 19 ± 7 AIS APs/ramp, mean ± SEM, p < 0.001, R.M. two-way ANOVA; Fig. 5B,C) while the number of somatically generated APs was unaffected (26 mm [HCO3−]o: 16 ± 7 somatic APs/ramp; 0 mm [HCO3−]o: 12 ± 7 somatic APs/ramp, mean ± SEM, p > 0.999, RM two-way ANOVA; Fig. 5B,C). No effect of [HCO3−]o was observed when the experiments were repeated in the presence of XE 991 (data not shown).
Figure 5.

Wash out of extracellular HCO3− regulate somatic and AIS AP generation. AI, A sample CA3 PCs exhibited robust AP generation under 0 mm [HCO3−]p and 26 mm [HCO3−]o conditions, likely due to diffusion of extracellular CO2 through the membrane. AII, Washing out [HCO3−]o with HEPES-buffered aCSF significantly reduced the number of APs generated in response to the same current ramp protocol. B, PCs exhibit robust som and AIS AP generation in 0 mm [HCO3−]p and 26 mm [HCO3−]o, (time = 0 min). B, C, AIS APs were significantly reduced after 10 min. of [HCO3−]o wash out (minimum not reached after 10 min), whereas som AP generation remained unchanged (B, AIS: p = 0.02, som: p = 0.25, n = 5, two-way ANOVA; C, AIS: p = 0.52, som: p = 0.01, n = 5, paired t test).
To further evaluate the effect of reduced [HCO3−]i on AP generation at the AIS, we performed voltage-clamp recordings on CA3 PCs with 0 mm [HCO3−]p and 26 mm [HCO3−]o. Recorded neurons were visualized by filling with AlexaFluor 488, and antidromic action currents were generated by local axon stimulation (∼250 μm away from the soma; Fig. 6A). Stimulation intensity was adjusted to produce ∼0.5 probability of antidromic action currents (pAC) in response to stimulus trains (5 stimuli, 50 Hz; Fig. 6C). When [HCO3−]o was washed out with HEPES-buffered aCSF, pH 7.4, the pAC was dramatically reduced from 0.56 ± 0.10 in 26 mm [HCO3−]o to 0.16 ± 0.05 in 0 mm [HCO3−]o (mean ± SEM, p = 0.009, n = 5, one-way ANOVA; Fig. 6B,C,E,F,H). The pAC recovered to 0.77 ± 0.16 upon subsequent XE 991 (10 μm) application after [HCO3−]o washout (n = 3, p = 0.142, one-way ANOVA, Fig. 6D,G,H). These data support the hypothesis that reductions in [HCO3−]i can reduce the probability of AP generation at the AIS though a KCNQ channel-dependent mechanism.
Figure 6.

Wash out of extracellular HCO3− reduces back-propagation of antidromic APs. A, Antidromic ACs were elicited by local axon stimulation (80 μs pulse width, ∼250 μm away from the soma) of visually identified CA3 PCs. B, E, Trains of stimuli (5 stimuli, 50 Hz, asterisks) evoked antidromic ACs recorded with 0 mm [HCO3−]p and 26 mm [HCO3−]o and stimulus intensities were adjusted so that the pAC ≈ 0.5 over 10 successive stimulus trains (raster plot, E). C, F, Wash out of [HCO3−]o significantly reduced the pAC, illustrated with raster plots of ACs during 10 successive stimulus trains after 10–12 min (n = 5, p = 0.009, one-way ANOVA, Dunnet's test). D, G, The pAC recovered upon subsequent XE 991 wash in (n = 3, p = 0.143, one-way ANOVA, Dunnet's test). H, Individual recordings indicated with gray lines, p = 0.001, one-way ANOVA. Black lines represent mean pAC ± SEM.
Previous studies have demonstrated that changes in [HCO3−]o can alter intracellular pH and alter neuronal excitability (Schwiening and Boron, 1994; Bonnet et al., 1998). In our experiments, the intracellular solution used in our recordings was buffered to pH 7.3–7.35 with 10 mm HEPES. Although 10 mm HEPES is typically used to set the pH of pipette solutions during whole-cell recordings, previous reports have demonstrated that 10 mm HEPES may not provide sufficient buffering capacity during periods of high neuronal activity or in the absence of HCO3−-dependent intraneuronal buffering (Schwiening and Boron, 1994; Trapp et al., 1996). Therefore, we sought to determine to what extent fluctuations in intraneuronal pH may contribute to the excitability of PCs in response to our current ramp protocol. We recorded from CA3 PCs in 0 mm [HCO3−]o using intracellular solutions buffered with 50 mm HEPES and the pH set to either 7.0 or 7.6. Current ramp protocols performed at these halved or doubled [H+] relative to our controls (pH = 7.3) revealed no significant effect of intraneuronal pH on the number of APs generated per ramp (pH 7.0: 14 ± 3 vs pH 7.6: 14 ± 4 APs/ramp, median, n = 6 and 5, respectively, p > 0.99, Mann–Whitney). These data demonstrate that pH fluctuations between 7.0 and 7.6 do not significantly affect CA3 PC excitability in our experimental conditions, and therefore the observed changes in excitability are likely to be directly caused by changes in HCO3−.
PIP2 prevents HCO3−-mediated increase in PC excitability
KCNQ channels are subject to a number of regulatory pathways and can be controlled via Ca2+, diacylgycerol, and Src tyrosine kinase, though primary control runs through the phosphoinositide-phospholipase C cycle via PIP2 (Delmas and Brown, 2005; Suh and Hille, 2008; Andrade et al., 2012; Telezhkin et al., 2012). Intracellular PIP2 facilitates KCNQ channel opening in a dose-dependent manner and recent findings demonstrated that PIP2 is required to couple the voltage sensing domain to pore opening (Zhang et al., 2003; Telezhkin et al., 2012; Zaydman et al., 2013). Consequently, we tested the hypothesis that HCO3− regulates KCNQ channel activation through intracellular PIP2.
We first tested to see whether CA3 PCs would exhibit an increase in excitability when switching from 0 to 26 mm [HCO3−]o, analogous to the previous [HCO3−]o wash out experiments. Whole-cell current-clamp recordings from CA3 PCs were performed in 0 mm [HCO3−]o and 0 mm [HCO3−]p and each recording was held for ∼10 min before beginning the experiment to allow adequate dialysis of the PC with the pH buffered (7.3) internal solution. Wash in of 26 mm [HCO3−]o aCSF elicited a robust increase in PC excitability from 17 ± 3 APs/ramp to 53 ± 6 APs/ramp after 10 min of wash in (mean ± SEM, n = 6, p < 0.01, two-way RM ANOVA; Fig. 7A, red trace). When [HCO3−]o wash in experiments were repeated in the presence of 10 μm XE 991, PCs exhibited strong AP generation in 0 mm [HCO3−]o aCSF, with an average of 49 ± 7 APs/ramp (Fig. 7A, gray trace). Wash in of 26 mm [HCO3−]o aCSF, did not significantly increase CA3 PC excitability (0 mm [HCO3−]o: 49 ± 7 APs/ramp to 26 mm [HCO3−]o: 40 ± 3 APs/ramp, n = 5, p = 0.943, two-way RM ANOVA, Fig. 7A, gray trace).
Figure 7.

[HCO3−]o wash in increases PC excitability and does not affect the fast components of APs. A, The wash in of 26 mm [HCO3−]o under conditions of 0 mm [HCO3−]p increases AP firing (red, No Drug; p < 0.001, RM two-way ANOVA), but no such effect is seen when the KCNQ selective antagonist XE 991 is present (gray, +10 μm XE 991; p = 0.943, RM two-way ANOVA). B, Wash in of 26 mm [HCO3−]o aCSF does not alter the fast AP components (AP amplitude, half-width, and fAHP amplitude) both in the absence (No Drug) and in the presence of the XE 991 (+10 μm XE 991, p > 0.05, RM two-way ANOVA).
To confirm that [HCO3−]o does not affect other conductances underlying AP generation, fast AP waveform properties (i.e., amplitude, half-width, and fAHP) were assessed before and after 26 mm [HCO3−]o wash in and in the presence of XE 991 (Fig. 7B; Table 3). No significant effects were observed on AP amplitude, half-width, or fHAP before or after 26 mm [HCO3−]o wash in with or without XE 991, suggesting that no other major conductances underlying AP generation are affected by HCO3− other than KCNQ channels (p > 0.05 for [HCO3−]o effect, XE 991 effect, and interaction, RM two-way ANOVA; Fig. 7B; Table 3). Interestingly, AP threshold was ∼2 mV higher in 26 mm [HCO3−]o whether or not XE 991 was present, but the biological significance of this finding is unclear at this time (p values: [HCO3−]o effect = 0.04, XE 991 effect = 0.11, interaction = 0.85, RM two-way ANOVA; Fig. 7B; Table 3).
Table 3.
Fast AP properties
| [HCO3−]o (mm) | 0 | 26 | 0 | 26 |
| XE 991 (10 μm) | − | − | + | + |
| Threshold (mV) | −40 ± 2 | −38 ± 2* | −43 ± 1 | −41 ± 1* |
| AP amplitude (mV) | 75 ± 3 | 71 ± 3 | 70 ± 2 | 67 ± 3 |
| Half-width (μs) | 800 ± 33 | 858 ± 47 | 866 ± 55 | 878 ± 68 |
| fAHP amplitude (mV)a | 7.8 ± 0.5 | 8.4 ± 0.5 | 6.3 ± 1.4 | 6.1 ± 1.5 |
| n | 6 | 5 | ||
AP amplitude, half-width, and fAHP properties were unaffected by wash-in of [HCO3−]o or KCNQ channel block by XE 991 (p > 0.05 for HCO3− effect, XE 991 effect and interaction, two-way RM ANOVA). AP thresholds were ∼2 mV higher in 26 mM [HCO3−]o both with and without XE 991 (p = 0.04 for HCO3−; effect and p > 0.05 for XE 991 effect and interaction, two-way RM ANOVA). Values are mean ± SEM.
afAHP amplitude was measured relative to AP threshold.
*Indicates statistical significance, two-way RM ANOVA.
Having established that 26 mm [HCO3−]o wash in could dramatically increase the excitability of CA3 PCs and that this effect was dependent on KCNQ channels, we sought to determine whether the HCO3− dependent increase in neuronal excitability could be due to modulation of intracellular PIP2. Whole-cell current-clamp recordings from CA3 PCs were performed in 0 mm [HCO3−]o and 0 mm [HCO3−]p containing 0, 10 or 30 μm PIP2 ([PIP2]p) and current ramps were used to assess PC excitability in 0 mm [HCO3−]o and in 26 mm [HCO3−]o. Wash in of 26 mm [HCO3−]o with 0 μm [PIP2]p significantly increased the number of APs/ramp (+40 ± 4 APs/ramp, n = 6; p < 0.001, two-way ANOVA; Fig. 8A,D). In contrast, including 10 or 30 μm [PIP2] in the pipet solution dampened or completely occluded the increase in PC excitability during 26 mm [HCO3−]o wash in, respectively (10 μm [PIP2]p: +19 ± 6 APs/ramp, n = 6, 30 μm [PIP2]p: −2 ± 5 APs/ramp, n = 4; p < 0.001, two-way ANOVA; Fig. 8B–D). Therefore, [HCO3−] appears to inhibit KCNQ channel activation by interfering with the actions of PIP2 on the channel, resulting in increased neuronal excitability.
Figure 8.
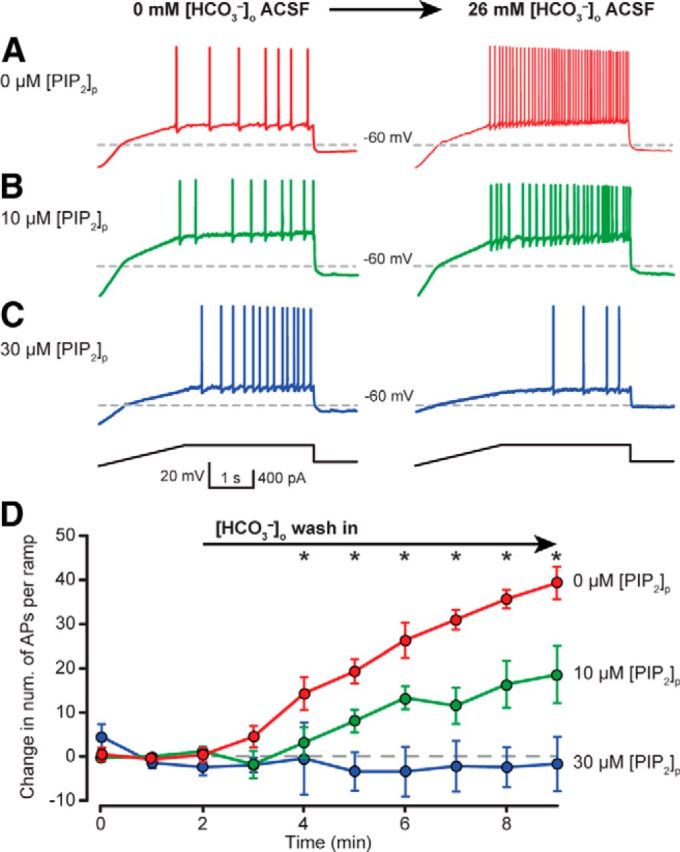
[HCO3−] regulates neuronal excitability by modulating intracellular PIP2 concentration. PCs exhibited dampened excitability in HEPES-buffered (0 mm [HCO3−]o) aCSF, and wash in of 26 mm [HCO3−]o significantly increased the number of APs/ramp (A, D, red). Increase in PC excitability during 26 mm [HCO3−]o wash in was dampened or absent when 10 μm (B, D, green) or 30 μm PIP2 (C, D, blue) was included in the patch solution, respectively. D, Current ramp protocol assessed relative neuronal excitability during transition from 0 to 26 mm [HCO3−]o. Recordings made with 0 mm [HCO3−]p pipette solution and 0 μm PIP2 (red) showed a robust increase in the number of APs/ramp during 26 mm [HCO3−]o wash in. Including 10 (green) or 30 (blue) μm PIP2 to the patch solution dampened or blocked the increase in neuronal excitability, respectively, upon 26 mm [HCO3−]o wash in (mean ± SEM). *Indicates statistically significant post hoc paired tests with correction for multiple comparisons, RM two-way ANOVA.
Discussion
Here we describe a novel mechanism whereby [HCO3−]i profoundly modulates the excitability of hippocampal CA3 pyramidal neurons through KCNQ channel regulation. Moreover, HCO3− may regulate KCNQ channels by preventing PIP2 from activating the channels, a fundamental mechanism previously shown to exert a strong control over KCNQ channel opening. Our findings are the first demonstration of a direct regulation of neuronal excitability by [HCO3−]i, independent of pH.
Discovering the molecular identity of the HCO3− sensor and of the precise pathway leading to interference with the PIP2-dependent KCNQ channel modulation will require further extensive studies. To date, only two signaling molecules are known to be directly activated by HCO3− independently of pH: guanylyl cyclase-D (GC-D; Guo et al., 2009; Luo et al., 2009) and a soluble form of adenylyl cyclase (sAC; Chen et al., 2000; Tresguerres et al., 2010). GC-D has been described to play a role in the olfactory detection of CO2 in some species and has only been found in olfactory bulb neurons (Fülle et al., 1995; Scott, 2011). Therefore, GC-D is unlikely to play a role in regulating KCNQ channels in our results. Soluble adenylyl cyclase has been shown to directly sense HCO3− in a pH-independent manner, resulting in an increase in local cAMP levels and protein kinase A (PKA) activation (Chen et al., 2000). Recent studies have demonstrated strong expression of sAC in astrocytes, where it modulates astrocyte-neuron metabolic coupling, and immunoelectron microscopy has also revealed the presence of sAC in neurons (Choi et al., 2012; Chen et al., 2013). Importantly, the rate-limiting enzyme for PIP2 generation, PIP5 kinase, is strongly inhibited by phosphorylated PKA (Park et al., 2001; Delmas and Brown, 2005). Thus, sAC may be an attractive candidate as the molecular detector that couples HCO3− to KCNQ channel modulation in CA3 PCs. However, activation of PKA has also been shown to have a direct long-lasting potentiating effect on Kv7/KCNQ channels (Wu et al., 2008). Considering these dual and potentially opposing effects of PKA activation on Kv7/KCNQ channels, the identification of PKA as the downstream effector of HCO3− will require extensive future studies.
The reduction in KCNQ channel activity by HCO3− could be significant in the context of GABAergic innervation of the AIS by chandelier or axoaxonic cells. The excitatory or inhibitory nature of this innervation is highly controversial. Anatomical studies have demonstrated the absence of the Cl− extruding transporter KCC2 and the presence of the Cl− importing transporter NKCC1 at the AIS, leading to a depolarizing EGABA (by ∼20 mV) relative to somatic and dendritic compartments (Khirug et al., 2008; Woodruff et al., 2009; Báldi et al., 2010) and possibly resulting GABAergic excitation (Fig. 9; Szabadics et al., 2006). Other studies, however, have provided evidence for GABAergic inhibition by AIS-innervating interneurons (Glickfeld et al., 2009). The inhibitory nature of the GABAergic input onto the AIS, regardless of the relationship between EGABA and the membrane potential, may be further supported by our present findings. Synaptic GABAA receptors are permeable to HCO3−, with a permeability ratio of PHCO3−/PCl− = 0.2 (Kaila, 1994). At the synapse, EHCO3 ≈ −10 mV due to active intracellular HCO3− regulation (Kaila, 1994). Consequently, GABAA receptor activation at normal resting membrane potential (∼−70 mV) results in HCO3− efflux through the channel and local decreases in [HCO3−]i. However, during heavy synaptic transmission, the outward driving force for HCO3− may be even larger than ∼60 mV due to acidification of the synaptic cleft. The luminal pH of synaptic vesicles is ∼5.5 (Miesenböck et al., 1998), and even single-vesicle release (e.g., miniature IPSCs) has been shown to acidify the synaptic cleft at GABAergic synapses (Dietrich and Morad, 2010). Therefore, periods of heavy GABAergic activity (Dugladze et al., 2012) may cause considerable synaptic cleft acidification at axoaxonic synapses, further decreasing the local [HCO3−]o in the cleft and increasing the HCO3− driving force (ΔEHCO3 ∼60 mV/pH unit). Given the large surface-to-volume ratio of the AIS and its low intracellular volume, the rapid outflow of HCO3− could conceivably cause a sharp drop in local [HCO3−]i.
Figure 9.

Proposed function of HCO3−-dependent regulation of KCNQ channels at the AIS. A, Schematic representation of CA3 PC axon initial segment and axo-axonic GABAergic synapse. B, With a depolarized EGABA, activation of synaptic GABAA receptors (GABAARs) during synaptic GABA release would result in membrane depolarization leading to an increased AP firing probability at the AIS (gray trace). However, local depletion of [HCO3−]i through GABAARs facilitates PIP2-KCNQ channel interactions and consequently KCNQ channel activity (green arrow). Enhanced KCNQ channel activation dampens excitability at the AIS and reduces AP probability, thus ensuring an inhibitory effect of axoaxonic cell activation despite a depolarized EGABA (red trace).
Previous reports have demonstrated that HCO3− efflux through the GABA receptor promotes the accumulation of intracellular Cl−, resulting in decreased GABAergic efficacy during periods of high GABAergic activation (Staley et al., 1995; Kaila et al., 1997). Additionally, the activity of neuronal carbonic anhydrases (II and VII) was recently shown to be required for this HCO3−-dependent intracellular Cl− accumulation (Ruusuvuori et al., 2013), providing more evidence for a strong interplay between HCO3− and GABA receptor efficacy. Given the data presented in this study, it is possible that a large drop in local [HCO3−]i at the AIS during heavy GABAergic activity could facilitate local KCNQ channel activity and greatly reduce AP generation despite the accumulation of intracellular Cl− and the reduction of GABAergic efficacy (Fig. 9). Although this hypothesis will require further investigation, such a mechanism would ensure that the GABAergic input to the AIS is predominantly inhibitory regardless of EGABA.
Footnotes
This work was supported by the US National Institutes of Health Grant 5T32NS058280 to R.T.J., R21AG037151 to G.C.F., and R01NS075429 and the Coelho Endowment to I.M.
The authors declare no competing financial interests.
References
- Andrade R, Foehring RC, Tzingounis AV. The calcium-activated slow AHP: cutting through the Gordian knot. Front Cell Neurosci. 2012;6:47. doi: 10.3389/fncel.2012.00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Báldi R, Varga C, Tamás G. Differential distribution of KCC2 along the axo-somato-dendritic axis of hippocampal principal cells. Eur J Neurosci. 2010;32:1319–1325. doi: 10.1111/j.1460-9568.2010.07361.x. [DOI] [PubMed] [Google Scholar]
- Bonnet U, Wiemann M, Bingmann D. CO2/HCO3−-withdrawal from the bath medium of hippocampal slices: biphasic effect on intracellular pH and bioelectric activity of CA3-neurons. Brain Res. 1998;796:161–170. doi: 10.1016/S0006-8993(98)00341-2. [DOI] [PubMed] [Google Scholar]
- Casey JR, Grinstein S, Orlowski J. Sensors and regulators of intracellular pH. Nat Rev Mol Cell Biol. 2010;11:50–61. doi: 10.1038/nrm2820. [DOI] [PubMed] [Google Scholar]
- Chen J, Martinez J, Milner TA, Buck J, Levin LR. Neuronal expression of soluble adenylyl cyclase in the mammalian brain. Brain Res. 2013;1518:1–8. doi: 10.1016/j.brainres.2013.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Cann MJ, Litvin TN, Iourgenko V, Sinclair ML, Levin LR, Buck J. Soluble adenylyl cyclase as an evolutionarily conserved bicarbonate sensor. Science. 2000;289:625–628. doi: 10.1126/science.289.5479.625. [DOI] [PubMed] [Google Scholar]
- Chesler M, Kaila K. Modulation of pH by neuronal activity. Trends Neurosci. 1992;15:396–402. doi: 10.1016/0166-2236(92)90191-A. [DOI] [PubMed] [Google Scholar]
- Choi HB, Gordon GR, Zhou N, Tai C, Rungta RL, Martinez J, Milner TA, Ryu JK, McLarnon JG, Tresguerres M, Levin LR, Buck J, MacVicar BA. Metabolic communication between astrocytes and neurons via bicarbonate-responsive soluble adenylyl cyclase. Neuron. 2012;75:1094–1104. doi: 10.1016/j.neuron.2012.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper EC. Made for “anchorin”: Kv7.2/7.3 (KCNQ2/KCNQ3) channels and the modulation of neuronal excitability in vertebrate axons. Semin Cell Dev Biol. 2011;22:185–192. doi: 10.1016/j.semcdb.2010.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmas P, Brown DA. Pathways modulating neural KCNQ/M (Kv7) potassium channels. Nat Rev Neurosci. 2005;6:850–862. doi: 10.1038/nrn1785. [DOI] [PubMed] [Google Scholar]
- Dietrich CJ, Morad M. Synaptic acidification enhances GABAA signaling. J Neurosci. 2010;30:16044–16052. doi: 10.1523/JNEUROSCI.6364-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugladze T, Schmitz D, Whittington MA, Vida I, Gloveli T. Segregation of axonal and somatic activity during fast network oscillations. Science. 2012;336:1458–1461. doi: 10.1126/science.1222017. [DOI] [PubMed] [Google Scholar]
- Fry CH, Poole-Wilson PA. Effects of acid-base changes on excitation-contraction coupling in guinea-pig and rabbit cardiac ventricular muscle. J Physiol-London. 1981;313:141–160. doi: 10.1113/jphysiol.1981.sp013655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fülle HJ, Vassar R, Foster DC, Yang RB, Axel R, Garbers DL. A receptor guanylyl cyclase expressed specifically in olfactory sensory neurons. Proc Natl Acad Sci U S A. 1995;92:3571–3575. doi: 10.1073/pnas.92.8.3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glickfeld LL, Roberts JD, Somogyi P, Scanziani M. Interneurons hyperpolarize pyramidal cells along their entire somatodendritic axis. Nat Neurosci. 2009;12:21–23. doi: 10.1038/nn.2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwald I. The dissociation of calcium and magnesium carbonates and bicarbonates. J Biol Chem. 1941;141:789–796. [Google Scholar]
- Gu N, Vervaeke K, Hu H, Storm JF. Kv7/KCNQ/M and HCN/h, but not KCa2/SK channels, contribute to the somatic medium after-hyperpolarization and excitability control in CA1 hippocampal pyramidal cells. J Physiol. 2005;566:689–715. doi: 10.1113/jphysiol.2005.086835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu N, Hu H, Vervaeke K, Storm JF. SK (KCa2) channels do not control somatic excitability in CA1 pyramidal neurons but can be activated by dendritic excitatory synapses and regulate their impact. J Neurophysiol. 2008;100:2589–2604. doi: 10.1152/jn.90433.2008. [DOI] [PubMed] [Google Scholar]
- Guo D, Zhang JJ, Huang XY. Stimulation of guanylyl cyclase-D by bicarbonate. Biochemistry. 2009;48:4417–4422. doi: 10.1021/bi900441v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hablitz JJ, Heinemann U. Extracellular K+ and Ca2+ changes during epileptiform discharges in the immature rat neocortex. Brain Res. 1987;433:299–303. doi: 10.1016/0165-3806(87)90036-8. [DOI] [PubMed] [Google Scholar]
- Hu H, Vervaeke K, Storm JF. M-channels (Kv7/KCNQ channels) that regulate synaptic integration, excitability, and spike pattern of CA1 pyramidal cells are located in the perisomatic region. J Neurosci. 2007;27:1853–1867. doi: 10.1523/JNEUROSCI.4463-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson RL, Langmuir D. Dissociation-constants of calcite and Cahco3+ from 0 to 50°C. Geochim Cosmochim Acta. 1974;38:301–318. doi: 10.1016/0016-7037(74)90112-4. [DOI] [Google Scholar]
- Jungas RL. Best literature values for the pK of carbonic and phosphoric acid under physiological conditions. Anal Biochem. 2006;349:1–15. doi: 10.1016/j.ab.2005.08.020. [DOI] [PubMed] [Google Scholar]
- Kaila K. Ionic basis of GABAA receptor channel function in the nervous system. Prog Neurobiol. 1994;42:489–537. doi: 10.1016/0301-0082(94)90049-3. [DOI] [PubMed] [Google Scholar]
- Kaila K, Lamsa K, Smirnov S, Taira T, Voipio J. Long-lasting GABA-mediated depolarization evoked by high-frequency stimulation in pyramidal neurons of rat hippocampal slice is attributable to a network-driven, bicarbonate-dependent K+ transient. J Neurosci. 1997;17:7662–7672. doi: 10.1523/JNEUROSCI.17-20-07662.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khirug S, Yamada J, Afzalov R, Voipio J, Khiroug L, Kaila K. GABAergic depolarization of the axon initial segment in cortical principal neurons is caused by the Na-K-2Cl cotransporter NKCC1. J Neurosci. 2008;28:4635–4639. doi: 10.1523/JNEUROSCI.0908-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinger F, Gould G, Boehm S, Shapiro MS. Distribution of M-channel subunits KCNQ2 and KCNQ3 in rat hippocampus. Neuroimage. 2011;58:761–769. doi: 10.1016/j.neuroimage.2011.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kole MH, Stuart GJ. Signal processing in the axon initial segment. Neuron. 2012;73:235–247. doi: 10.1016/j.neuron.2012.01.007. [DOI] [PubMed] [Google Scholar]
- Luo M, Sun L, Hu J. Neural detection of gases-carbon dioxide, oxygen-in vertebrates and invertebrates. Curr Opin Neurobiol. 2009;19:354–361. doi: 10.1016/j.conb.2009.06.010. [DOI] [PubMed] [Google Scholar]
- Maren TH. Carbonic anhydrase: chemistry, physiology, and inhibition. Physiol Rev. 1967;47:595–781. doi: 10.1152/physrev.1967.47.4.595. [DOI] [PubMed] [Google Scholar]
- Meeks JP, Mennerick S. Action potential initiation and propagation in CA3 pyramidal axons. J Neurophysiol. 2007;97:3460–3472. doi: 10.1152/jn.01288.2006. [DOI] [PubMed] [Google Scholar]
- Meeks JP, Jiang X, Mennerick S. Action potential fidelity during normal and epileptiform activity in paired soma-axon recordings from rat hippocampus. J Physiol. 2005;566:425–441. doi: 10.1113/jphysiol.2005.089086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miesenböck G, De Angelis DA, Rothman JE. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature. 1998;394:192–195. doi: 10.1038/28190. [DOI] [PubMed] [Google Scholar]
- Nakayama FS. Magnesium complex and ion-pair in Mgco3-Co2 solution system. J Chem Eng Data. 1971;16:178–181. doi: 10.1021/je60049a023. [DOI] [Google Scholar]
- Park SJ, Itoh T, Takenawa T. Phosphatidylinositol 4-phosphate 5-kinase type I is regulated through phosphorylation response by extracellular stimuli. J Biol Chem. 2001;276:4781–4787. doi: 10.1074/jbc.M010177200. [DOI] [PubMed] [Google Scholar]
- Pavlov I, Kaila K, Kullmann DM, Miles R. Cortical inhibition, pH and cell excitability in epilepsy: what are optimal targets for antiepileptic interventions? J Physiol. 2013;591:765–774. doi: 10.1113/jphysiol.2012.237958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reardon EJ, Langmuir D. Thermodynamic properties of ion-pairs Mgco30 and Caco30 from 10 to 50°C. Am J Sci. 1974;274:599–612. doi: 10.2475/ajs.274.6.599. [DOI] [Google Scholar]
- Ruusuvuori E, Huebner AK, Kirilkin I, Yukin AY, Blaesse P, Helmy M, Kang HJ, El Muayed M, Hennings JC, Voipio J, ŠSestan N, Hubner CA, Kaila K. Neuronal carbonic anhydrase VII provides GABAergic excitatory drive to exacerbate febrile seizures. EMBO J. 2013;32:2275–2286. doi: 10.1038/emboj.2013.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaer H. Decrease in ionized calcium by bicarbonate in physiological solutions. Pflugers Arch. 1974;347:249–254. doi: 10.1007/BF00592601. [DOI] [PubMed] [Google Scholar]
- Schwaller B. Cytosolic Ca2+ buffers. Cold Spring Harb Perspect Biol. 2010;2:a004051. doi: 10.1101/cshperspect.a004051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwiening CJ, Boron WF. Regulation of intracellular pH in pyramidal neurones from the rat hippocampus by Na(+)-dependent Cl−–HCO3− exchange. J Physiol. 1994;475:59–67. doi: 10.1113/jphysiol.1994.sp020049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott K. Out of thin air: sensory detection of oxygen and carbon dioxide. Neuron. 2011;69:194–202. doi: 10.1016/j.neuron.2010.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staley KJ, Soldo BL, Proctor WR. Ionic mechanisms of neuronal excitation by inhibitory GABAA receptors. Science. 1995;269:977–981. doi: 10.1126/science.7638623. [DOI] [PubMed] [Google Scholar]
- Storm JF. Intracellular injection of a Ca-2+ chelator inhibits spike repolarization in hippocampal-neurons. Brain Res. 1987a;435:387–392. doi: 10.1016/0006-8993(87)91631-3. [DOI] [PubMed] [Google Scholar]
- Storm JF. Action-potential repolarization and a fast after-hyperpolarization in rat hippocampal pyramidal cells. J Physiol. 1987b;385:733–759. doi: 10.1113/jphysiol.1987.sp016517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storm JF. An after-hyperpolarization of medium duration in rat hippocampal pyramidal cells. J Physiol. 1989;409:171–190. doi: 10.1113/jphysiol.1989.sp017491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh BC, Hille B. PIP2 is a necessary cofactor for ion channel function: how and why? Annu Rev Biophys. 2008;37:175–195. doi: 10.1146/annurev.biophys.37.032807.125859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabadics J, Varga C, Molnár G, Oláh S, Barzó P, Tamás G. Excitatory effect of GABAergic axo-axonic cells in cortical microcircuits. Science. 2006;311:233–235. doi: 10.1126/science.1121325. [DOI] [PubMed] [Google Scholar]
- Telezhkin V, Brown DA, Gibb AJ. Distinct subunit contributions to the activation of M-type potassium channels by PI(4,5)P2. J Gen Physiol. 2012;140:41–53. doi: 10.1085/jgp.201210796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp S, Lückermann M, Kaila K, Ballanyi K. Acidosis of hippocampal neurones mediated by a plasmalemmal Ca2+/H+ pump. Neuroreport. 1996;7:2000–2004. doi: 10.1097/00001756-199608120-00029. [DOI] [PubMed] [Google Scholar]
- Tresguerres M, Buck J, Levin LR. Physiological carbon dioxide, bicarbonate, and pH sensing. Pflugers Arch. 2010;460:953–964. doi: 10.1007/s00424-010-0865-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzingounis AV, Nicoll RA. Contribution of KCNQ2 and KCNQ3 to the medium and slow afterhyperpolarization currents. Proc Natl Acad Sci U S A. 2008;105:19974–19979. doi: 10.1073/pnas.0810535105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff A, Xu Q, Anderson SA, Yuste R. Depolarizing effect of neocortical chandelier neurons. Front Neural Circuits. 2009;3:15. doi: 10.3389/neuro.04.015.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu WW, Chan CS, Surmeier DJ, Disterhoft JF. Coupling of L-type Ca2+ channels to KV7/KCNQ channels creates a novel, activity-dependent, homeostatic intrinsic plasticity. J Neurophysiol. 2008;100:1897–1908. doi: 10.1152/jn.90346.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaydman MA, Silva JR, Delaloye K, Li Y, Liang H, Larsson HP, Shi J, Cui J. Kv7.1 ion channels require a lipid to couple voltage sensing to pore opening. Proc Natl Acad Sci U S A. 2013;110:13180–13185. doi: 10.1073/pnas.1305167110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Craciun LC, Mirshahi T, Rohács T, Lopes CM, Jin T, Logothetis DE. PIP(2) activates KCNQ channels, and its hydrolysis underlies receptor-mediated inhibition of M currents. Neuron. 2003;37:963–975. doi: 10.1016/S0896-6273(03)00125-9. [DOI] [PubMed] [Google Scholar]