

Abstract
Background:
Prostate cancer (PrCa) is one of the most common diseases to affect men worldwide and among the leading causes of cancer-related death. The purpose of this study was to use second-generation sequencing technology to assess the frequency of deleterious mutations in 22 tumour suppressor genes in familial PrCa and estimate the relative risk of PrCa if these genes are mutated.
Methods:
Germline DNA samples from 191 men with 3 or more cases of PrCa in their family were sequenced for 22 tumour suppressor genes using Agilent target enrichment and Illumina technology. Analysis for genetic variation was carried out by using a pipeline consisting of BWA, Genome Analysis Toolkit (GATK) and ANNOVAR. Clinical features were correlated with mutation status using standard statistical tests. Modified segregation analysis was used to determine the relative risk of PrCa conferred by the putative loss-of-function (LoF) mutations identified.
Results:
We discovered 14 putative LoF mutations in 191 samples (7.3%) and these mutations were more frequently associated with nodal involvement, metastasis or T4 tumour stage (P=0.00164). Segregation analysis of probands with European ancestry estimated that LoF mutations in any of the studied genes confer a relative risk of PrCa of 1.94 (95% CI: 1.56–2.42).
Conclusions:
These findings show that LoF mutations in DNA repair pathway genes predispose to familial PrCa and advanced disease and therefore warrants further investigation. The clinical utility of these findings will become increasingly important as targeted screening and therapies become more widespread.
Keywords: familial prostate cancer, DNA repair gene mutations, next-generation sequencing, relative risk
Prostate cancer (PrCa) is one of the most commonly diagnosed cancers in men worldwide (Jemal et al, 2011), representing the second most common cause of male cancer-related deaths in the United States, the third in the European Union and the sixth worldwide with over 250 000 deaths per year (Ferlay et al, 2013). Established risk factors for PrCa include age, family history of PrCa and ethnicity, although many common genetic variants that individually contribute to moderate increased risk have also been identified (Goh et al, 2012). Prostate cancer has also been observed to aggregate with other types of familial cancer, in particular with breast and ovarian cancers (Hemminki and Chen, 2005). Several genes that were initially implicated as high risk for breast or ovarian cancer predisposition, for example, BRCA1, BRCA2, CHEK2 and BRIP1, have subsequently been shown to increase the risk of PrCa as well (Dong et al, 2003; Cybulski et al, 2004; Agalliu et al, 2007; Kote-Jarai et al, 2009). This suggests that shared genetic and/or environmental factors may be causal for multiple cancer types. Further evidence for pleiotropy has come from the recent Collaborative Oncological Gene-environment Study (COGS), a multicancer mega-consortium and previous genome-wide association studies (GWAS), which report sharing of common loci between cancers and especially hormonal-related malignancies (Bojesen et al, 2013; Eeles et al, 2013; Garcia-Closas et al, 2013; Kote-Jarai et al, 2013; Michailidou et al, 2013). Genomic instability is the hallmark of most cancers and therefore the investigation of DNA repair pathways in hereditary cancer risk is widely established, for example, mutations in the DNA mismatch repair (MMR) pathway cause hereditary non-polyposis colon cancer (HNPCC/Lynch syndrome) and mutations in Fanconi anaemia (FA) genes, which include BRCA2, BRIP1 and PALB2, predispose carriers to multiple cancers including PrCa (Silva et al, 2009; Kottemann and Smogorzewska, 2013).
We propose that additional moderate penetrance genes for PrCa have yet to be discovered and these will be facilitated by the recent development of massively parallel sequencing. This allows the candidate gene study to be easily expanded both in terms of depth and breadth, enabling the targeting of multiple genes across multiple samples in a single experiment. In this study, we investigated whether deleterious mutations in a set of DNA repair genes have a role in familial PrCa predisposition. We selected for this study the BROCA tumour suppressor gene set designed by Walsh et al (2010), which comprises known high- and moderate-risk breast/ovarian cancer genes from the BRCA–Fanconi anaemia complex and also genes involved in rare multiorgan cancer syndromes (Table 1); some of these overlap with previously known genes implicated in PrCa predisposition.
Table 1. BROCA 22 tumour suppressor gene set.
| Gene | RefSeq accession | Involvement in rare multiorgan cancer syndromes |
|---|---|---|
|
ATM |
NM_000051.3 |
|
|
BARD1 |
NM_000465.2 |
|
|
BRCA1 |
NM_007294.3 |
|
|
BRCA2 |
NM_000059.3 |
Fanconi anaemia |
|
BRIP1 |
NM_032043.2 |
Fanconi anaemia |
|
CDH1 |
NM_004360.3 |
Hereditary diffuse gastric cancer |
|
CHEK2 |
NM_007194.3 |
|
|
MLH1 |
NM_000249.3 |
HNPCC/Lynch |
|
MLH3a,b |
NM_001040108.1 |
HNPCC/Lynch |
|
MRE11A |
NM_005591.3 |
|
|
MSH2 |
NM_000251.2 |
HNPCC/Lynch |
|
MSH6 |
NM_000179.2 |
HNPCC/Lynch |
|
MUTYH |
NM_001128425.1 |
Familial adenomatous polyposis-2 |
|
NBN |
NM_002485.4 |
Nijmegen breakage syndrome |
|
PALB2 |
NM_024675.3 |
Fanconi anaemia |
|
PMS1b |
NM_000534.4 |
HNPCC/Lynch |
|
PMS2 |
NM_000535.5 |
HNPCC/Lynch |
|
PTEN |
NM_000314.4 |
Cowden |
|
RAD50 |
NM_005732.3 |
|
|
RAD51C |
NM_058216.1 |
Fanconi anaemia |
|
STK11 |
NM_000455.4 |
Peutz–Jeghers |
| TP53 | NM_000546.5 | Li–Fraumeni |
Abbreviations: HNPCC=hereditary non-polyposis colorectal cancer; RefSeq=NCBI Reference Sequence Database.
Added since the study by Walsh et al (2010).
Disputed as a Lynch syndrome gene.
Materials and methods
Study population
We selected a series of men with PrCa from the UK Genetic Prostate Cancer Study (UKGPCS; UKCRN ID 869) (Eeles et al, 1997), based primarily on their PrCa family history. Subjects were eligible if they had two or more relatives affected by PrCa; a total of 191 men were included in this study. Germline DNA was isolated from peripheral whole blood samples using the Nucleon DNA purification system (GE Healthcare Life Sciences, Pittsburgh, PA, USA) or methods described in Edwards et al (1997). The study was approved by the Royal Marsden NHS Trust, Local Research Ethics Committee.
Target capture enrichment and sequencing
A custom Agilent SureSelect bait library (Agilent Technologies, Santa Clara, CA, USA) was used to target capture 22 genes from germline DNA. Capture regions were designed to cover coding, non-coding and intronic sequences with an additional 10 kilobase (kb) genomic sequence flanking each gene. After repetitive DNA elements were masked, the total DNA targeted was 939 kb (Walsh et al, 2010). Sequencing libraries were prepared in batches of 48 and each sample was ‘barcoded' with a 6 base pair (bp) index to allow multiplexed sequencing. An initial batch of 48 libraries was prepared using standard Agilent protocols, while the remaining 3 batches of 48 libraries used a modified Agilent protocol with prehybridisation pooling to allow 3 libraries to be captured at once with a single bait library (Cummings et al, 2010). All libraries were clustered and sequenced on Illumina cBOT and HiSeq 2000 instruments (Illumina, San Diego, CA, USA), using v.2 flowcells and Truseq reagents to produce 2 × 78 bp ‘paired-end' reads and a 6 bp ‘index' read.
Sequencing data analysis and variant annotation
Raw sequencing data were base-called and demultiplexed using Illumina CASAVA software (v.1.8.1, Illumina) and purity filtered reads were removed to produce paired FASTQ files. Each set of paired FASTQ files was aligned using BWA 0.5.8 to ‘The 1000 Genomes Project' Phase 1 reference, human_g1k_v37.fasta (Li and Durbin, 2009). The ‘Best Practice Variant Detection with the Genome Analysis Toolkit (GATK) v.3 for targeted resequencing was implemented with Picard (v.1.52) and the GATK (all tools v.1.0.5216M except Unified genotyper v.1.6-9-g47df7bb) (DePristo et al, 2011) when realigning, recalibrating and genotyping. Variants were annotated using a February 2013 build of ANNOVAR using the summarize_annovar.pl script (Wang et al, 2010). This maps variants to RefSeq genes, known variation from dbSNP137 and annotates the predicted functional consequence of missense variants using six in silico tools (SIFT, PolyPhen-2, LRT, MutationTaster, phyloP and GERP++) from the dbNSFP v.1.3 (Liu et al, 2011). Additional clinical variant annotation was obtained from NCBI ClinVar (last accessed July 2013; http://www.ncbi.nlm.nih.gov/clinvar/).
LoF mutation definition and validation
Putative loss-of-function (LoF) mutations were defined as variants that are protein truncating or result in significant alteration of the protein sequence. This encompasses stop codon gain/loss, insertion/deletion frameshifts or splice site loss variants.
Loss of function variants were validated by Sanger sequencing in the probands and in other family members if available. Polymerase chain reaction amplicons were designed in Primer-Z (Tsai et al, 2007), except for the PMS2 variant, where published primers were used from De Vos et al (2004) because of the pseudogene PMS2CL (primer sequences available in Supplementary Table S1), sequenced on a ABI3730 Genetic Analyzer using a 1/16th BigDye v.3 protocol (Applied Biosystems, Foster City, CA, USA) and analysed using Mutation surveyor 3.97 (Softgenetics, State College, PA, USA), against the appropriate RefSeq accession sequence (Table 1).
Statistical and segregation analysis
We investigated the correlations between LoF mutation status and clinical features using Fisher's exact test, Mann–Whitney U-test or the Mantel–Haenszel test for linear trend; patients with missing data for a particular clinical feature were excluded from that analysis (Table 3). All statistical analyses were performed using R 2.15.1; ‘stats' and ‘vcdExtra' 0.5-7 packages (R Core Team, 2012; Friendly, 2013).
Table 3. Clinical characteristics of LoF mutation carriers.
| |
LoF (n=14) |
Non-carriers (n=140) |
|
||
|---|---|---|---|---|---|
| Clinical characteristic | No. | % | No. | % | LoF vs non-carriers (P-value) |
|
Age (years) | |||||
| Median | 58.5 | 59 | 0.334 | ||
| Range |
41–71 |
|
47–82 |
|
|
|
PSA at diagnosis (ng ml−1) | |||||
| Median | 11.1 | 8.25 | 0.156 | ||
| Range |
3.09–91.12 |
|
0.04–259 |
|
|
|
Gleason score | |||||
| Gleason ⩽6 | 5 | 35.71 | 62 | 44.29 | 0.312 |
| Gleason 7 | 2 | 14.29 | 27 | 19.29 | |
| Gleason ⩾8 | 3 | 21.43 | 15 | 10.71 | |
| Unknown |
4 |
28.57 |
36 |
25.71 |
|
|
Tumour stage | |||||
| T1 | 3 | 21.43 | 38 | 27.14 | 0.476 |
| T2 | 4 | 28.57 | 45 | 32.14 | |
| T3 | 2 | 14.29 | 24 | 17.14 | |
| T4 | 1 | 7.14 | 2 | 1.43 | |
| TX |
4 |
28.57 |
31 |
22.14 |
|
|
Nodal stage | |||||
| N0 | 4 | 28.57 | 76 | 54.29 | 0.00141 |
| N1 | 3 | 21.43 | 1 | 0.71 | |
| NX |
7 |
50.00 |
63 |
45.00 |
|
|
Metastasis | |||||
| M0 | 7 | 50.00 | 74 | 52.86 | 0.0431 |
| M1 | 3 | 21.43 | 5 | 3.57 | |
| MX |
4 |
28.57 |
61 |
43.57 |
|
|
AJCC prognostic groups | |||||
| I–III | 3 | 21.43 | 68 | 48.57 | 0.00121 |
| IV | 5 | 35.71 | 7 | 5.00 | |
| Unknown | 6 | 42.86 | 75 | 46.43 | |
Abbreviations: AJCC=American Joint Committee on Cancer; LoF=loss-of-function mutation; PSA=prostate-specific antigen.
To obtain an estimate of the average PrCa risk conferred by the LoF mutations identified, we carried out a modified segregation analysis using information on 186 probands of European ancestry. This was implemented in the pedigree analysis software MENDEL (Lange et al, 2013). The analysis was based on PrCa occurrence in male family members. Unaffected male subjects were censored at the age of 85 years, the age at death or last observation, whichever occurred first. As no proband was found to carry LoF mutations in more than one gene, we assumed a genetic model where all identified LoF mutations across all genes represent the alleles of a single genetic locus and assumed that all alleles conferred the same relative risk of PrCa. We assumed that the PrCa incidence depends on the underlying genotype through a model of the form: 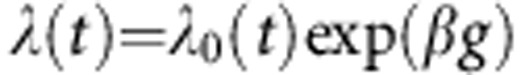 , where
, where 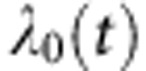 is the baseline incidence at age t in non-mutation carriers, β is the log risk ratio associated with the LoF mutation and g takes value 0 for non-mutation carriers and 1 for LoF mutation carriers. The overall PrCa incidence and overall possible genotypes in the model were constrained to agree with the population incidences for England and Wales in the period 1993–1997 (Parkin et al, 2002). We assumed that the total mutation carrier frequency in the model was equal to the sum of mutation carrier frequencies in the genes, as estimated in previous studies (total mutation frequency=1.4% frequencies obtained from UK studies (European ancestry) where available or NHLBI GO Exome Sequencing Project (ESP); last accessed November 2013; http://evs.gs.washington.edu/EVS/) (Antoniou et al, 2002; The CHEK2 Breast Cancer Case–Control Consortium, 2004; Thompson et al, 2005; Seal et al, 2006; Rahman et al, 2007). The models were parameterised in terms of the log-relative risk ratios for PrCa. Parameters were estimated using maximum-likelihood estimation. To adjust for ascertainment, we modelled the conditional likelihood of all family phenotypes and mutation status of all tested family members (including the index/proband), given the disease phenotypes of all family members.
is the baseline incidence at age t in non-mutation carriers, β is the log risk ratio associated with the LoF mutation and g takes value 0 for non-mutation carriers and 1 for LoF mutation carriers. The overall PrCa incidence and overall possible genotypes in the model were constrained to agree with the population incidences for England and Wales in the period 1993–1997 (Parkin et al, 2002). We assumed that the total mutation carrier frequency in the model was equal to the sum of mutation carrier frequencies in the genes, as estimated in previous studies (total mutation frequency=1.4% frequencies obtained from UK studies (European ancestry) where available or NHLBI GO Exome Sequencing Project (ESP); last accessed November 2013; http://evs.gs.washington.edu/EVS/) (Antoniou et al, 2002; The CHEK2 Breast Cancer Case–Control Consortium, 2004; Thompson et al, 2005; Seal et al, 2006; Rahman et al, 2007). The models were parameterised in terms of the log-relative risk ratios for PrCa. Parameters were estimated using maximum-likelihood estimation. To adjust for ascertainment, we modelled the conditional likelihood of all family phenotypes and mutation status of all tested family members (including the index/proband), given the disease phenotypes of all family members.
Results
Patient characteristics
Of the 191 men with ⩾3 PrCa cases in their family, 128 men also had at least 1 relative affected by breast, ovarian or colon cancer. Ethnicity was known for 72% (137) of our series, with 96% (131) of white European descent, and the remainder of our set also included two men of black African descent, three men of black Caribbean descent and one Ashkenazi Jewish man. The method of diagnosis was available for 69% of patients, with an even split between clinically detected and PSA screened patients (64 and 68 patients, respectively).
Sequencing and variant quality control
All samples reached the required coverage threshold of 20 × read depth across 80% of the target regions. The median value of average target region read depth was 135.85, and STK11 had about half as much median read coverage compared with the rest of the target genes (76.8; Figure 1).
Figure 1.

Average read coverage per gene. The 22 genes are shown along the x axis and the average sequencing coverage is shown along the y axis. All genes except STK11 had over 150x median coverage, and even STK11 coverage was above the required 20x.
After QC filtering, 7138 exonic and splice site variants were genotyped, corresponding to 300 unique variants. Of the initial 14 unique putative LoF mutations identified, a BRCA2 stop-gain K3326X (rs11571833) was classed as ‘non-pathogenic' in ClinVar (Wu et al, 2005) and consequently removed from further analysis. Non-carriers were defined as patients who did not carry an LoF mutation, a predicted deleterious SNV by all six in silico tools in dbNSFP v.1.3 or an SNV classed as ‘pathogenic' in the NCBI ClinVar database for a disease other than PrCa (Table 3 and Supplementary Tables S2 and S3). Therefore 14 LoF mutation carriers and 140 non-carriers were selected for further analysis.
Frequency and type of LoF mutations
Thirteen LoF mutations in eight genes were identified in 14 familial PrCa cases (Figure 2 and Table 2). We found three frameshift mutations and one stop-gain mutation in BRCA2; two stop-gain mutations in ATM; a recurring stop-gain mutation in BRIP1 affecting two families and two frameshift mutations in CHEK2. One mutation was found in each of BRCA1, MUTYH, PALB2 and PMS2. Five of the 13 unique LoF variants found were not listed in dbSNP137, the 1000 Genomes Project (April 2012 data release) and the NHLBI GO ESP (ES6500SI data release). No subject carried more than one LoF mutation; therefore, 7.3% (14 of 191 men) of these familial cases were carriers of a deleterious mutation in 1 of the 22 tumour suppressor genes investigated here.
Figure 2.

Proportion of LoF mutations by gene. Proportion of the genes contributing to the 14 LoF identified in this study.
Table 2. List of loss-of-function mutations and characteristics of proband and family with the mutations.
| ID | Gene | HGVS nomenclature | Mutation outcome | Age of onset (years) | Tumour stagea | Nodal stagea | Meta-stasesa | Gleason score | PSA (ng mL−1) | Years to death | Family history of PrCab | Mutation segregationb,c | Family history of other cancerb |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Prs1 |
ATM |
NM_000051.3: c.7327C>T: p.(Arg2443a) |
Stop-gain |
59 |
TX |
NX |
MX |
NA |
NA |
Alive (19) |
Brother (61), father (80), grandfather (m) |
Brother +(61) |
Sister CoCa, uncle (p) CoCa |
| Prs2 |
ATM |
NM_000051.3: c.7777C>T: p.(Gln2593a) |
Stop-gain |
65 |
T1b |
NX |
M0 |
3+3 |
NA |
Alive (20) |
3 × brothers (68, 69, NA) |
Brother +(68) |
Father CoCa |
| Prs3 |
BRCA1 |
NM_007294.3: c.4065_4068del: p.(Asn1355Lysfsa10) |
Frameshift |
58 |
T2a |
NX |
MX |
3+4 |
6.34 |
Alive (6) |
Father and 3 uncles (p) |
NA |
4 × Cousins (p) BrCa |
| Prs4 |
BRCA2 |
NM_000059.3: c.4876_4877del: p.(Asn1626Serfsa12) |
Frameshift |
54 |
T3b |
N1 |
M1 |
4+4 |
70.1 |
Alive (11) |
Father (67), uncle (m;63), 4 × cousins (m; 51, 55, 56, 57) |
Cousin −(55) |
Cousin (m) BrCa |
| Prs5 |
BRCA2 |
NM_000059.3: c.4981de l: p.(Tyr1661Ilefsa9) |
Frameshift |
62 |
TX |
NX |
M0 |
3+3 |
13.3 |
Alive (17) |
2 × brothers (67, 69) |
Brother +(62), brother −(67) |
Sister, aunt (m), grandmother (m) BrCa; aunt (m) CoCa |
| Prs6 |
BRCA2 |
NM_000059.3: c.5909C>A: p.(Ser1970a) |
Stop-gain |
71 |
T2b |
NX |
MX |
NA |
11.1 |
12 |
Father, 2 × brothers |
NA |
Sister (30) and aunt (p) BrCa |
| Prs7 |
BRCA2 |
NM_000059.3: c.9382C>T: p.(Arg3128a) |
Stop-gain |
41 |
T2a |
N0 |
M0 |
3+4 |
3.09 |
Alive (6) |
Uncle (p), uncle (m) |
NA |
Mother (41), aunt (m; 48), aunt (p) BrCa; aunt (m) OvCa (51) |
| Prs8 |
BRIP1 |
NM_032043.2: c.2392C>T: p.(Arg798a) |
Stop-gain |
59 |
T1c |
N0 |
M0 |
3+3 |
10.5 |
Alive (10) |
Father, grandfather (p) |
NA |
NA |
| Prs9 |
BRIP1 |
NM_032043.2: c.2392C>T: p.(Arg798a) |
Stop-gain |
56 |
T1 |
N0 |
M1 |
NA |
NA |
6 |
3 × half-brothers (m) |
Unaffected brother + 2 × half-brothers +(m; 59, NA) |
Half-brother (m) StCa; uncle (p) BoCa |
| Prs10 |
CHEK2 |
NM_007194.3: c.1263de l: p.(Ser422Valfsa15) |
Frameshift |
65 |
T2 |
N0 |
M0 |
3+3 |
11 |
Alive (16) |
3 × brothers (67, 72, 74), 2 × cousins (p; 67, 68) |
Brother +(72), brother −(74), unaffected brother + and unaffected brother − |
Father melanoma; sister BrCa |
| Prs11 |
CHEK2 |
NM_007194.3: c.869del: p.(Asn290Thrfsa14) |
Frameshift |
53 |
TX |
N1 |
M0 |
4+5 |
14.2 |
Alive (8) |
Father (68), grandfather (p) |
Father − (68) |
Mother, grandfather (m), grandmother (p) CoCa; aunt (p) BrCa |
| Prs12 |
MUTYH |
NM_001128425.1: c.940C>T: p.(Gln314a) |
Stop-gain |
55 |
TX |
NX |
MX |
3+3 |
7 |
Alive (6) |
2 × brothers (69, NA), uncle |
Brother +(69) |
Aunt (m) BrCa |
| Prs13 |
PALB2 |
NM_024675.3: c.3507_3508del: p.(His1170Phefsa19) |
Frameshift |
58 |
T3 |
NX |
M1 |
NA |
91.1 |
2 |
2 × brothers (63, NA), uncle (m) |
Brother +(63) |
Mother BoCa; sister, grandmother (m) BrCa |
| Prs14 | PMS2 | NM_000535.5: c.2186_2187del: p.(Leu729Glnfsa6) | Frameshift | 51 | T4 | N1 | M0 | 4+5 | 20.9 | 13 | Father, 2 × half uncles (p), grandfather (p) | NA | Aunt (m) BrCa |
Abbreviations: BoCa=bone cancer; BrCa=breast cancer; CoCa=colon cancer; HGVS=Human Genome Variation Society; m=maternal; NA=not available; OvCa=ovarian cancer; p=paternal; PrCa=prostate cancer; PSA=prostate-specific antigen; StCa=stomach cancer.
TX, NX or MX is used if tumour, nodal or metastases status is unknown.
Age of onset is marked within parentheses where available.
Positive and negative mutation status marked by + or − respectively.
LoF mutations and clinical characteristics
Table 3 shows clinical characteristics of LoF carriers vs non-carriers. Median age at diagnosis in LoF carriers was very similar to non-carriers: 58.5 and 59.0 years, respectively (P=0.334), and median presenting PSA was higher in carriers compared with non-carriers but the difference was not significant (11.10 vs 8.25 ng ml−1, P=0.156). Gleason scores were categorised into three groups: ⩽6, 7 and ⩾8. There was no significant association between LoF carrier status and the Gleason grade groups (P=0.312), or when analysed against high grades only (Gleason ⩾8; P=0.193). There was also no significant association between LoF carrier status and tumour stage trend (P=0.476), or when analysed against high tumour stage (T3–T4; P=0.704). However, there was a significant association between LoF carrier status and the presence of nodal involvement (42.9% vs 1.3% P=0.0014) and metastasis (30.0% vs 6.3% P=0.043).
LoF mutations and advanced PrCa
To further investigate the associations seen with nodal involvement and metastasis, we applied the AJCC Stage IV prognostic grouping for advanced disease, as defined either by nodal involvement, metastasis or primary tumour grade of T4 (Edge et al, 2010). We performed a logistic regression on LoF carrier status vs Stage IV status, controlling for the effect of age at PrCa diagnosis. This showed that LoF mutation carriers have significantly higher odds of having advanced disease (OR 15.09, 95% CI: 2.95–95.81, P=0.00164) (Table 3). Even after excluding the BRCA2 mutations (as these have been shown to be associated with poorer prognosis; Castro et al, 2013), the association remained significant (P=0.00285), indicating that this correlation is independent of BRCA2 LoF status.
LoF mutations and family history of cancer
We also investigated the role of the proband's family history on the odds of having an LoF mutation; however, no association was found with the total number of PrCa cases within a family (P=0.808). We then used a modified risk prediction algorithm developed by Macinnis et al (2011) to assess the association between LoF mutations and family history of PrCa with and without 25 common risk SNPs, but no significant association was found with either (P=0.456 and P=0.856, respectively). However, this is not unexpected as all study cases were selected for having multiple relatives diagnosed with PrCa and only a subset of DNA repair genes were tested. On the other hand, LoF carriers were more likely to have a family history of breast cancer than non-carriers (OR 3.94, 95% CI: 1.07–18.10, P=0.023) and there was also a significant trend with the increase in the total number of breast cancer cases within a family (P=0.0035). Table 4 shows the 191 families grouped by family history of cancers other than PrCa and the percentage of LoF mutations found in each group.
Table 4. LoF mutations by family history of PrCa and other cancers.
| |
Family history of other cancersa |
|
|||||
|---|---|---|---|---|---|---|---|
| No. of PrCa in family | 1 × BrCa/OvCa | 2 × BrCa/OvCa | 3+ BrCa/OvCa | 1+ CoCa | BrCa/OvCa and CoCa | PrCa only | Total |
|
3+ | |||||||
| No. of families |
24 |
11 |
15 |
47 |
31 |
63 |
191 |
| LoF genes | CHEK2, BRCA2, PMS23 (12.5%) | BRCA21 (9.1%) | BRCA1, BRCA2, PALB23 (20%) | 2 × ATM2 (4.2%) | CHEK2, BRCA2, MUTYH2 (6.5%) | BRIP12 (3.2%) | 14 (7.3%) |
Abbreviations: BrCa=breast cancer; CoCa=colon cancer; LoF=loss of function; OvCa=ovarian cancer; PrCa=prostate cancer.
Percentage of LoF mutation in the given family history group is shown within parentheses.
LoF mutations and familial segregation
Of the 13 unique LoF mutations, 8 were frameshift and 5 were stop-gain. The eight affected genes represent four DNA damage repair or response pathways; homologous recombination (HR) and FA, ataxia–telangiectasia-mutated signalling (ATM), MMR and base excision repair (BER). As might be expected, mutations in the ATM and MMR pathways were observed in families where PrCa coaggregated with colon cancer cases (three of the four families), whereas mutations affecting the HR/FA pathway were found mainly in families with breast cancer, and to a lesser extent in families with ovarian cancer in addition to PrCa (six of the families) (Table 2). The only mutation unique to families with only PrCa reported was a previously known (rs137852986) stop-gain mutation c.2392C>T (p.(Arg798*)) in the BRIP1 gene that was present in two families.
The most frequently mutated gene in this study was BRCA2, with four protein truncating mutations. Three of these were in exon 11 (two frameshifts and one stop-gain), and a stop-gain in exon 25. Of the four men affected, we had additional DNA available from other family members in the two men with exon 11 frameshifts. We found partial segregation of the c.4981del (p.(Tyr1661Ilefs*9); Supplementary Figure S5) mutation, where the proband had two brothers with PrCa: one diagnosed (Dx) at 67 years who did not carry the mutation, whereas the other did and died of PrCa 4 years after diagnosis (69 years) at 73 years. The second exon 11 frameshift c.4876_4877del (p.(Asn1626Serfs*12); Supplementary Figure S6) was not present in the proband's brother with PrCa.
Two mutations were found in the ATM gene. An exon 50 stop-gain c.7327C>T (p.(Arg2443*); Supplementary Figure S1) was found in a young proband (Dx 59 years) and his affected brother also carried the same mutation (Dx 61 years); furthermore, the proband had an additional MSH2 (NM_000251.2) c.1275A>G substitution at the −2 position in the 3′ end of exon 7, which has been characterised as causing partial exon skipping at the RNA level (Pagenstecher et al, 2006). The second ATM mutation, a stop-gain c.7777C>T (p.(Gln2593*)) in exon 52 was present in a family (Supplementary Figure S2) where the father had colon cancer; four out of eight brothers had PrCa and a sister was reported to have had an unspecified leukaemia. We confirmed the mutation in the proband (Dx 65 years) and in a brother (Dx 68 years, the only available sample). It is also worth noting that both brothers also had a secondary cancer diagnosis of the colon.
We identified two mutations in the CHEK2 gene. An exon 12 frameshift c.1263del (p.(Ser422Valfs*15)) was found in a family with multiple PrCa cases. DNA samples were available for five brothers, two of the three PrCa cases carried the mutation (Dx 65 years and 72 years and non-carrier Dx 74 years). Of the two unaffected brothers, one was also a carrier of this mutation (Supplementary Figure S10). The second CHEK2 mutation was an exon 8 frameshift c.869del (p.(Asn290Thrfs*14)), which was present in a patient diagnosed with PrCa at the age of 53 years, but did not segregate with his father, the only available sample (Dx 68 years); therefore, it is likely that this mutation was inherited from the maternal line, which contains two colon cancers (in the proband's mother and grandfather; Supplementary Figure S11).
Only one variant was observed in more than one family: the BRIP1 stop-gain mutation c.2392C>T (p.(Arg798*)), first discovered in FA and later described in breast cancer and PrCa families (Levran et al, 2005; Seal et al, 2006; Kote-Jarai et al, 2009). This mutation resides in exon 17 and has an MAF of 0.02% in 4300 European Americans from the ESP variant server. In the first family this variant was present in two brothers, both diagnosed young at 59 and 56 years (Supplementary Figure S9). In the second family there were three PrCa cases, but the only DNA available was from the proband that was diagnosed at 59 years (Supplementary Figure S8).
In four other genes, a single mutation was found: the BRCA1 mutation c.4065_4068del (p.(Asn1355Lysfs*10), Dx 58 years) was identified in a family with multiple (4) breast cancers and also had four PrCas (Supplementary Figure S3). The PALB2 frameshift mutation c.3507_3508del (p.(His1170Phefs*19), Dx 58 years) carrier had an affected brother (Dx 63 years) who also carried the mutation (Supplementary Figure S13) and similarly the MUTYH stop-gain mutation c.940C>T (p.(Gln314*), Dx 55 years) carrier had an affected brother (Dx 69 years) with the same mutation (Supplementary Figure S12). The only homozygous LoF mutation found was a PMS2 frameshift mutation c.2186_2187del (p.(Leu729Glnfs*6), Dx 51 years), this has a ∼2% MAF in African-American data from ESP but occurs as a homozygote ∼0.14% (3 of 2121). The carrier of this homozygous mutation in our study was a man of black African ancestry with a family history of four additional PrCa cases; however, we had no other DNA available from his family to test for segregation (Supplementary Figure S14).
LoF mutations and risk of PrCa
We carried out a modified segregation analysis of the seven genes found to contain putative LoF mutations in probands of European ancestry using estimated mutation frequencies from previous UK studies, except for MUTYH where UK data was unavailable, and therefore we used the European ancestry data from the NHLBI GO ESP NHLBI GO Exome Sequencing Project (last accessed November 2013; http://evs.gs.washington.edu/EVS/) (Antoniou et al, 2002; The CHEK2 Breast Cancer Case–Control Consortium, 2004; Thompson et al, 2005; Seal et al, 2006; Rahman et al, 2007).
Loss-of-function mutations in the DNA repair genes investigated in the present study were estimated to confer a relative risk of PrCa of 1.94 (95% CI 1.56–2.42). After excluding the families found to carry BRCA1 and BRCA2 mutations, the relative risk was estimated to be slightly lower but still elevated at 1.80 (95% CI: 1.38–2.35).
As the mutation frequencies of these LoF mutations were based on sparse published data, we repeated the analysis by assuming that the mutation frequencies were 50% greater or smaller than in the studies listed above. This resulted in small changes to the estimated relative risks for all LoF mutations ranging from 1.67 (95% CI: 1.34–2.09) to 2.42 (95% CI: 1.97–2.99). Much larger series of screened families would be required to obtain cancer risk estimates associated with LoF mutations in specific genes.
Discussion
We have analysed the coding sequences of 22 tumour suppressor genes in 191 familial PrCa cases in the United Kingdom and found that 7.3% (14 of 191) of these cases were carriers of a putative LoF mutation. These mutations showed partial segregation with PrCa within the families, which is consistent with previous observations of moderately penetrant genes mutated within families. The eight affected genes represent four DNA damage repair or response pathways: HR and FA, ATM, MMR and BER. The most frequently mutated gene was BRCA2, which is in concordance with previous studies showing BRCA2 as the most strongly associated PrCa predisposition gene identified to date (Kote-Jarai et al, 2011). There is currently an international targeted PrCa screening study in men with germline mutations in BRCA1 and BRCA2 (IMPACT; Mitra et al, 2011). Our study provides further evidence that ATM, CHEK2, BRCA1 and BRIP1 are also involved in familial PrCa predisposition (Dong et al, 2003; Angèle et al, 2004; Kote-Jarai et al, 2009; Leongamornlert et al, 2012).
Most importantly, we have shown that LoF mutation carriers were more likely to have advanced disease, defined by nodal involvement, metastasis or primary tumour grade of T4 (OR 15.09, 95% CI: 2.95–95.81, P=0.00164). We have previously shown that BRCA2 LoF mutation carriers have significantly reduced survival and present with a more aggressive disease (Castro et al, 2013); however, even when excluding the BRCA2 LoF mutations, this significant association was preserved. This finding could have important clinical implications as men with deleterious germline mutations in these genes should be considered for more intensive screening and treatment. Furthermore, some of the genes studied here are in the HR repair pathway, in which targeted agents such as poly (ADP-ribose) polymerase inhibitors (PARPi) can be considered.
In addition to the clearly deleterious mutations, we identified several missense variants; 13 of these are predicted deleterious by a consensus of six in silico tools (Supplementary Table S2). Therefore, some of these may be classified in the future as deleterious, and accordingly, our estimated frequency of deleterious mutation presented here is likely to be an underestimate. Previous studies have shown that common SNPs identified by GWAS can be used to stratify cumulative risk of PrCa. We were able to calculate a 25 SNP risk score using the algorithm developed by Macinnis et al (2011); the distribution of the median scores between of LoF carriers vs non-carriers were 0.223 vs 0.194, respectively, but this was not statistically significant (P=0.456). This would seem to suggest that, in our sample set, there is no correlation between putative LoF carrier status and PrCa risk score using the common risk SNPs and family history data. This perhaps is not surprising as all our subjects had strong family history and the non-carriers may have additional rare LoF mutations in genes not tested here.
To further investigate the association of LoF carrier status with additional breast cancer family history, we applied the BOADICEA model, commonly used in clinical practice to predict the probability that a proband carries a BRCA1 or BRCA2 mutation. BOADICEA models the genetic susceptibility to PrCa only in terms of the effects of BRCA1 and BRCA2. This model predicted that 8.6% of our probands (15 of the 175 cases that were possible to be scored) had a combined BRCA1 and BRCA2 mutation carrier probability ⩾10% and therefore would have been recommended to have genetic testing in the United Kingdom under the upcoming (Q3 2013) National Institute for Health and Care Excellence (NICE) guidelines for familial breast cancer. Of the 14 men with an LoF mutation, only three (PRS4, 6 and 7; Table 2) of the five BRCA1/2 mutation carriers would have been eligible for genetic testing in the United Kingdom based on these new guidelines (Supplementary Figures S1–14). On the basis of our results, we would therefore recommend that in the future a panel of DNA repair genes should be tested in PrCa families with 3 or more PrCa cases.
The limitations of this study include the lack of events to allow overall survival analysis and the lack of functional evidence to enable the characterisation of the potentially pathogenic missense mutations. Nonetheless, our results suggest that mutations in a wider range of DNA repair genes, other than BRCA2, predispose to PrCa and that such cases are more likely to have advanced disease. Therefore, this warrants further investigation of an expanded set of genes within these pathways in a larger sample set. Also, we highlight that current genetic testing criteria would only have identified 3 of the 14 LoF carriers with breast cancer family history, and therefore current PrCa testing guidelines are likely to be inadequate in the era of personalised genomic medicine.
To our knowledge, this is the first study to apply second-generation sequencing to screen for germline mutations in multiple DNA repair genes in a familial PrCa cohort. We identified frequent deleterious mutations in these genes and the mutation carriers were more likely to present with advanced disease. These findings present strong evidence that genes in DNA repair pathways are good candidates for PrCa predisposition. The clinical utility of these and future findings within these pathways should become increasingly important as targeted screening (such as is undertaken in IMPACT; Mitra et al, 2011) and targeted therapies such as PARPi (Sandhu et al, 2013) become more widespread. If we can more effectively screen these men, clinicians can potentially offer more tailored screening, staging and treatment pathways.
Acknowledgments
We would like to acknowledge the NCRN nurses and Consultants for their work in the UKGPCS study. We thank all the patients who took part in this study. We would also like to thank the following for funding support: The Prostate Cancer Research Foundation (now Prostate Cancer UK), Prostate Research Campaign UK (now Prostate Cancer UK), The Institute of Cancer Research and The Everyman Campaign, The National Cancer Research Network UK, The National Cancer Research Institute (NCRI) UK. We are grateful for support of NIHR funding to the NIHR Biomedical Research Centre at The Institute of Cancer Research and The Royal Marsden NHS Foundation Trust. This work was also supported by Cancer Research UK Grants C5047/A15007 C12292/A11174. ACA is a Cancer Research UK Senior Cancer Research Fellow.
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies this paper on British Journal of Cancer website (http://www.nature.com/bjc)
Supplementary Material
References
- Agalliu I, Karlins E, Kwon EM, Iwasaki LM, Diamond A, Ostrander EA, Stanford JL. Rare germline mutations in the BRCA2 gene are associated with early-onset prostate cancer. BrJ Cancer. 2007;97:826–831. doi: 10.1038/sj.bjc.6603929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angèle S, Falconer A, Edwards SM, Dörk T, Bremer M, Moullan N, Chapot B, Muir K, Houlston R, Norman AR, Bullock S, Hope Q, Meitz J, Dearnaley D, Dowe A, Southgate C, Ardern-Jones A, Easton DF, Eeles RA, Hall J. ATM polymorphisms as risk factors for prostate cancer development. Br J Cancer. 2004;91:783–787. doi: 10.1038/sj.bjc.6602007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoniou AC, Pharoah PDP, McMullan G, Day NE, Stratton MR, Peto J, Ponder BJ, Easton DF. A comprehensive model for familial breast cancer incorporating BRCA1, BRCA2 and other genes. Br J Cancer. 2002;86:76–83. doi: 10.1038/sj.bjc.6600008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bojesen SE, Pooley KA, Johnatty SE, Beesley J, Michailidou K, Tyrer JP, Edwards SL, Pickett HA, Shen HC, Smart CE, Hillman KM, Mai PL, Lawrenson K, Stutz MD, Lu Y, Karevan R, Woods N, Johnston RL, French JD, Chen X, Weischer M, Nielsen SF, Maranian MJ, Ghoussaini M, Ahmed S, Baynes C, Bolla MK, Wang Q, Dennis J, McGuffog L, Barrowdale D, Lee A, Healey S, Lush M, Tessier DC, Vincent D, Bacot F, Vergote I, Lambrechts S, Despierre E, Risch HA, González-Neira A, Rossing MA, Pita G, Doherty Ja, Alvarez N, Larson MC, Fridley BL, Schoof N, Chang-Claude J, Cicek MS, Peto J, Kalli KR, Broeks A, Armasu SM, Schmidt MK, Braaf LM, Winterhoff B, Nevanlinna H, Konecny GE, Lambrechts D, Rogmann L, Guénel P, Teoman A, Milne RL, Garcia JJ, Cox A, Shridhar V, Burwinkel B, Marme F, Hein R, Sawyer EJ, Haiman CA, Wang-Gohrke S, Andrulis IL, Moysich KB, Hopper JL, Odunsi K, Lindblom A, Giles GG, Brenner H, Simard J, Lurie G, Fasching PA, Carney ME, Radice P, Wilkens LR, Swerdlow A, Goodman MT, Brauch H, Garcia-Closas M, Hillemanns P, Winqvist R, Dürst M, Devilee P, Runnebaum I, Jakubowska A, Lubinski J, Mannermaa A, Butzow R, Bogdanova NV, Dörk T, Pelttari LM, Zheng W, Leminen A, Anton-Culver H, Bunker CH, Kristensen V, Ness RB, Muir K, Edwards R, Meindl A, Heitz F, Matsuo K, Bois A, du, Wu AH, Harter P, Teo S-H, Schwaab I, Shu X-O, Blot W, Hosono S, Kang D, Nakanishi T, Hartman M, Yatabe Y, Hamann U, Karlan BY, Sangrajrang S, Kjaer SK, Gaborieau V, Jensen A, Eccles D, Høgdall E, Shen C-Y, Brown J, Woo YL, Shah M, Azmi MAN, Luben R, Omar SZ, Czene K, Vierkant RA, Nordestgaard BG, Flyger H, Vachon C, Olson JE, Wang X, Levine DA, Rudolph A, Weber RP, Flesch-Janys D, Iversen E, Nickels S, Schildkraut JM, Silva IDS, Cramer DW, Gibson L, Terry KL, Fletcher O, Vitonis AF, van der Schoot CE, Poole EM, Hogervorst FBL, Tworoger SS, Liu J, Bandera EV, Li J, Olson SH, Humphreys K, Orlow I, Blomqvist C, Rodriguez-Rodriguez L, Aittomäki K, Salvesen HB, Muranen TA, Wik E, Brouwers B, Krakstad C, Wauters E, Halle MK, Wildiers H, Kiemeney LA, Mulot C, Aben KK, Laurent-Puig P, Altena AM, Truong T, Massuger LF, Benitez J, Pejovic T, Perez JIA, Hoatlin M, Zamora MP, Cook LS, Balasubramanian SP, Kelemen LE, Schneeweiss A, Le ND, Sohn C, Brooks-Wilson A, Tomlinson I, Kerin MJ, Miller N, Cybulski C, Henderson BE, Menkiszak J, Schumacher F, Wentzensen N, Le Marchand L, Yang HP, Mulligan AM, Glendon G, Engelholm SA, Knight JA, Høgdall CK, Apicella C, Gore M, Tsimiklis H, Song H, Southey MC, Jager A, den Ouweland AMW, Brown R, Martens JWM, Flanagan JM, Kriege M, Paul J, Margolin S, Siddiqui N, Severi G, Whittemore AS, Baglietto L, McGuire V, Stegmaier C, Sieh W, Müller H, Arndt V, Labrèche F, Gao Y-T, Goldberg MS, Yang G, Dumont M, McLaughlin JR, Hartmann A, Ekici AB, Beckmann MW, Phelan CM, Lux MP, Permuth-Wey J, Peissel B, Sellers TA, Ficarazzi F, Barile M, Ziogas A, Ashworth A, Gentry-Maharaj A, Jones M, Ramus SJ, Orr N, Menon U, Pearce CL, Brüning T, Pike MC, Ko Y-D, Lissowska J, Figueroa J, Kupryjanczyk J, Chanock SJ, Dansonka-Mieszkowska A, Jukkola-Vuorinen A, Rzepecka IK, Pylkäs K, Bidzinski M, Kauppila S, Hollestelle A, Seynaeve CM, Tollenaar RA, Durda K, Jaworska K, Hartikainen JM, Kosma V-M, Kataja V, Antonenkova NN, Long J, Shrubsole M, Deming-Halverson S, Lophatananon A, Siriwanarangsan P, Stewart-Brown S, Ditsch N, Lichtner P, Schmutzler RK, Ito H, Iwata H, Tajima K, Tseng C-C, Stram DO, van den Berg D, Yip CH, Ikram MK, Teh Y-C, Cai H, Lu W, Signorello LB, Cai Q, Noh D-Y, Yoo K-Y, Miao H, PT-C Iau, Teo YY, McKay J, Shapiro C, Ademuyiwa F, Fountzilas G, Hsiung C-N, Yu J-C, Hou M-F, Healey CS, Luccarini C, Peock S, Stoppa-Lyonnet D, Peterlongo P, Rebbeck TR, Piedmonte M, Singer CF, Friedman E, Thomassen M, Offit K, Hansen TVO, Neuhausen SL, Szabo CI, Blanco I, Garber J, Narod Sa, Weitzel JN, Montagna M, Olah E, Godwin AK, Yannoukakos D, Goldgar DE, Caldes T, Imyanitov EN, Tihomirova L, Arun BK, Campbell I, Mensenkamp AR, van Asperen CJ, van Roozendaal KEP, Meijers-Heijboer H, Collée JM, Oosterwijk JC, Hooning MJ, Rookus MA, van der Luijt RB, Os TA, Evans DG, Frost D, Fineberg E, Barwell J, Walker L, Kennedy MJ, Platte R, Davidson R, Ellis SD, Cole T, Bressac-de Paillerets B, Buecher B, Damiola F, Faivre L, Frenay M, Sinilnikova OM, Caron O, Giraud S, Mazoyer S, Bonadona V, Caux-Moncoutier V, Toloczko-Grabarek A, Gronwald J, Byrski T, Spurdle AB, Bonanni B, Zaffaroni D, Giannini G, Bernard L, Dolcetti R, Manoukian S, Arnold N, Engel C, Deissler H, Rhiem K, Niederacher D, Plendl H, Sutter C, Wappenschmidt B, Borg A, Melin B, Rantala J, Soller M, Nathanson KL, Domchek SM, Rodriguez GC, Salani R, Kaulich DG, Tea M-K, Paluch SS, Laitman Y, Skytte A-B, Kruse Ta, Jensen UB, Robson M, Gerdes A-M, Ejlertsen B, Foretova L, Savage Sa, Lester J, Soucy P, Kuchenbaecker KB, Olswold C, Cunningham JM, Slager S, Pankratz VS, Dicks E, Lakhani SR, Couch FJ, Hall P, Monteiro AN, Gayther SA, Pharoah PDP, Reddel RR, Goode EL, Greene MH, Easton DF, Berchuck A, Antoniou AC, Chenevix-Trench G, Dunning AM. Multiple independent variants at the TERT locus are associated with telomere length and risks of breast and ovarian cancer. Nat Genet. 2013;45 (371–84:384e1–384e2. doi: 10.1038/ng.2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro E, Goh C, Olmos D, Saunders E, Leongamornlert D, Tymrakiewicz M, Mahmud N, Dadaev T, Govindasami K, Guy M, Sawyer E, Wilkinson R, Ardern-Jones A, Ellis S, Frost D, Peock S, Evans DG, Tischkowitz M, Cole T, Davidson R, Eccles D, Brewer C, Douglas F, Porteous ME, Donaldson A, Dorkins H, Izatt L, Cook J, Hodgson S, Kennedy MJ, Side LE, Eason J, Murray A, Antoniou AC, Easton DF, Kote-Jarai Z, Eeles R. Germline BRCA mutations are associated with higher risk of nodal involvement, distant metastasis, and poor survival outcomes in prostate cancer. J Clin Oncol. 2013;31:1748–1757. doi: 10.1200/JCO.2012.43.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings N, King R, Rickers A, Kaspi A, Lunke S, Haviv I, Jowett JBM. Combining target enrichment with barcode multiplexing for high throughput SNP discovery. BMC Genom. 2010;11:641. doi: 10.1186/1471-2164-11-641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cybulski C, Górski B, Debniak T, Gliniewicz B, Mierzejewski M, Masojć B, Jakubowska A, Matyjasik J, Złowocka E, Sikorski A, Narod SA, Lubiński J. NBS1 is a prostate cancer susceptibility gene. Cancer Res. 2004;64:1215–1219. doi: 10.1158/0008-5472.can-03-2502. [DOI] [PubMed] [Google Scholar]
- da Silva FCC, Valentin MD, De Oliveira Ferreira O, Carraro DM, Rossi BM. Mismatch repair genes in Lynch syndrome: a review. Sao Paulo Med J. 2009;127:46–51. doi: 10.1590/S1516-31802009000100010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vos M, Hayward BE, Picton S, Sheridan E, Bonthron DT. Novel PMS2 pseudogenes can conceal recessive mutations causing a distinctive childhood cancer syndrome. Am J Hum Genet. 2004;74:954–964. doi: 10.1086/420796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MAa, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X, Wang L, Taniguchi K, Wang X, Cunningham JM, McDonnell SK, Qian C, Marks AF, Slager SL, Peterson BJ, Smith DI, Cheville JC, Blute ML, Jacobsen SJ, Schaid DJ, Tindall DJ, Thibodeau SN, Liu W. Mutations in CHEK2 associated with prostate cancer risk. Am J Hum Genet. 2003;72:270–280. doi: 10.1086/346094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edge S, Bryd D, Compton C, Fritz A, Greene F, Trotti A.(eds) (2010Prostate in AJCC Cancer Staging Manual Springer: New York, NY, USA; 457–468. [Google Scholar]
- Edwards SM, Dearnaley DP, Ardern-Jones AT, Hamoudi RA, Easton DF, Ford D, Shearer R, Dowe A, Eeles RA, The CRC/BPG UK Familial Prostate Cancer Study Collaborators No germline mutations in the dimerization domain of MXI1 in prostate cancer clusters. Br J Cancer. 1997;76:992–1000. doi: 10.1038/bjc.1997.498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eeles RA, Dearnaley DP, Ardern-Jones AT, Shearer RJ, Easton DF, Ford D, Edwards SM, Dowe A. Familial prostate cancer: the evidence and the Cancer Research Campaign/British Prostate Group (CRC/BPG) UK Familial Prostate Cancer Study. Br J Urol. 1997;79 (Suppl 1:8–14. doi: 10.1111/j.1464-410x.1997.tb00795.x. [DOI] [PubMed] [Google Scholar]
- Eeles RA, Al Olama AA, Benlloch S, Saunders EJ, Leongamornlert DA, Tymrakiewicz M, Ghoussaini M, Luccarini C, Dennis J, Jugurnauth-Little S, Dadaev T, Neal DE, Hamdy FC, Donovan JL, Muir K, Giles GG, Severi G, Wiklund F, Gronberg H, Haiman CA, Schumacher F, Henderson BE, Le Marchand L, Lindstrom S, Kraft P, Hunter DJ, Gapstur S, Chanock SJ, Berndt SI, Albanes D, Andriole G, Schleutker J, Weischer M, Canzian F, Riboli E, Key TJ, Travis RC, Campa D, Ingles SA, John EM, Hayes RB, Pharoah PDP, Pashayan N, Khaw K-T, Stanford JL, Ostrander EA, Signorello LB, Thibodeau SN, Schaid D, Maier C, Vogel W, Kibel AS, Cybulski C, Lubinski J, Cannon-Albright L, Brenner H, Park JY, Kaneva R, Batra J, Spurdle AB, Clements JA, Teixeira MR, Dicks E, Lee A, Dunning AM, Baynes C, Conroy D, Maranian MJ, Ahmed S, Govindasami K, Guy M, Wilkinson RA, Sawyer EJ, Morgan A, Dearnaley DP, Horwich A, Huddart RA, Khoo VS, Parker CC, Van As NJ, Woodhouse CJ, Thompson A, Dudderidge T, Ogden C, Cooper CS, Lophatananon A, Cox A, Southey MC, Hopper JL, English DR, Aly M, Adolfsson J, Xu J, Zheng SL, Yeager M, Kaaks R, Diver WR, Gaudet MM, Stern MC, Corral R, Joshi AD, Shahabi A, Wahlfors T, Tammela TLJ, Auvinen A, Virtamo J, Klarskov P, Nordestgaard BG, Røder MA, Nielsen SF, Bojesen SE, Siddiq A, Fitzgerald LM, Kolb S, Kwon EM, Karyadi DM, Blot WJ, Zheng W, Cai Q, McDonnell SK, Rinckleb AE, Drake B, Colditz G, Wokolorczyk D, Stephenson RA, Teerlink C, Muller H, Rothenbacher D, Sellers TA, Lin H-Y, Slavov C, Mitev V, Lose F, Srinivasan S, Maia S, Paulo P, Lange E, Cooney KA, Antoniou AC, Vincent D, Bacot F, Tessier DC, Kote-Jarai Z, Easton DF.2013Identification of 23 new prostate cancer susceptibility loci using the iCOGS custom genotyping array Nat Genet 45385–391.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferlay J, Steliarova-Foucher E, Lortet-Tieulent J, Rosso S, Coebergh JWW, Comber H, Forman D, Bray F. Cancer incidence and mortality patterns in Europe: estimates for 40 countries in 2012. Eur J Cancer. 2013;49:1374–1403. doi: 10.1016/j.ejca.2012.12.027. [DOI] [PubMed] [Google Scholar]
- Friendly M.2013. vcdExtra: vcd extensions and additions. R package version 0.5-10. Available at http://CRAN.R-project.org/package=vcdExtra .
- Garcia-Closas M, Couch FJ, Lindstrom S, Michailidou K, Schmidt MK, Brook MN, Orr N, Rhie SK, Riboli E, Feigelson HS, Le Marchand L, Buring JE, Eccles D, Miron P, Fasching PA, Brauch H, Chang-Claude J, Carpenter J, Godwin AK, Nevanlinna H, Giles GG, Cox A, Hopper JL, Bolla MK, Wang Q, Dennis J, Dicks E, Howat WJ, Schoof N, Bojesen SE, Lambrechts D, Broeks A, Andrulis IL, Guénel P, Burwinkel B, Sawyer EJ, Hollestelle A, Fletcher O, Winqvist R, Brenner H, Mannermaa A, Hamann U, Meindl A, Lindblom A, Zheng W, Devillee P, Goldberg MS, Lubinski J, Kristensen V, Swerdlow A, Anton-Culver H, Dörk T, Muir K, Matsuo K, Wu AH, Radice P, Teo SH, Shu X-O, Blot W, Kang D, Hartman M, Sangrajrang S, Shen C-Y, Southey MC, Park DJ, Hammet F, Stone J, Veer LJV, Rutgers EJ, Lophatananon A, Stewart-Brown S, Siriwanarangsan P, Peto J, Schrauder MG, Ekici AB, Beckmann MW, Dos Santos Silva I, Johnson N, Warren H, Tomlinson I, Kerin MJ, Miller N, Marme F, Schneeweiss A, Sohn C, Truong T, Laurent-Puig P, Kerbrat P, Nordestgaard BG, Nielsen SF, Flyger H, Milne RL, Perez JIA, Menéndez P, Müller H, Arndt V, Stegmaier C, Lichtner P, Lochmann M, Justenhoven C, Ko Y-D, Muranen TA, Aittomäki K, Blomqvist C, Greco D, Heikkinen T, Ito H, Iwata H, Yatabe Y, Antonenkova NN, Margolin S, Kataja V, Kosma V-M, Hartikainen JM, Balleine R, Tseng C-C, Van Den Berg D, Stram DO, Neven P, Dieudonné A-S, Leunen K, Rudolph A, Nickels S, Flesch-Janys D, Peterlongo P, Peissel B, Bernard L, Olson JE, Wang X, Stevens K, Severi G, Baglietto L, McLean C, Coetzee GA, Feng Y, Henderson BE, Schumacher F, Bogdanova NV, Labrèche F, Dumont M, Yip CH, Taib NAM, Cheng C-Y, Shrubsole M, Long J, Pylkäs K, Jukkola-Vuorinen A, Kauppila S, Knight JA, Glendon G, Mulligan AM, Tollenaar RAEM, Seynaeve CM, Kriege M, Hooning MJ, van den Ouweland AMW, van Deurzen CHM, Lu W, Gao Y-T, Cai H, Balasubramanian SP, Cross SS, Reed MWR, Signorello L, Cai Q, Shah M, Miao H, Chan CW, Chia KS, Jakubowska A, Jaworska K, Durda K, Hsiung C-N, Wu P-E, Yu J-C, Ashworth A, Jones M, Tessier DC, González-Neira A, Pita G, Alonso MR, Vincent D, Bacot F, Ambrosone CB, Bandera EV, John EM, Chen GK, Hu JJ, Rodriguez-Gil JL, Bernstein L, Press MF, Ziegler RG, Millikan RM, Deming-Halverson SL, Nyante S, Ingles SA, Waisfisz Q, Tsimiklis H, Makalic E, Schmidt D, Bui M, Gibson L, Müller-Myhsok B, Schmutzler RK, Hein R, Dahmen N, Beckmann L, Aaltonen K, Czene K, Irwanto A, Liu J, Turnbull C, Rahman N, Meijers-Heijboer H, Uitterlinden AG, Rivadeneira F, Olswold C, Slager S, Pilarski R, Ademuyiwa F, Konstantopoulou I, Martin NG, Montgomery GW, Slamon DJ, Rauh C, Lux MP, Jud SM, Bruning T, Weaver J, Sharma P, Pathak H, Tapper W, Gerty S, Durcan L, Trichopoulos D, Tumino R, Peeters PH, Kaaks R, Campa D, Canzian F, Weiderpass E, Johansson M, Khaw K-T, Travis R, Clavel-Chapelon F, Kolonel LN, Chen C, Beck A, Hankinson SE, Berg CD, Hoover RN, Lissowska J, Figueroa JD, Chasman DI, Gaudet MM, Diver WR, Willett WC, Hunter DJ, Simard J, Benitez J, Dunning AM, Sherman ME, Chenevix-Trench G, Chanock SJ, Hall P, Pharoah PDP, Vachon C, Easton DF, Haiman CA, Kraft P. Genome-wide association studies identify four ER negative-specific breast cancer risk loci. Nat Genet. 2013;45:392–398. doi: 10.1038/ng.2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh CL, Schumacher FR, Easton DF, Muir K, Henderson BE, Kote-Jarai Z, Eeles RA. Genetic variants associated with predisposition to prostate cancer and potential clinical implications. J Intern Med. 2012;271 (4:353–365. doi: 10.1111/j.1365-2796.2012.02511.x. [DOI] [PubMed] [Google Scholar]
- Hemminki K, Chen B. Familial association of prostate cancer with other cancers in the Swedish Family-Cancer Database. Prostate. 2005;65:188–194. doi: 10.1002/pros.20284. [DOI] [PubMed] [Google Scholar]
- Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- Kote-Jarai Z, Jugurnauth SK, Mulholland S, Leongamornlert DA, Guy M, Edwards SM, Tymrakiewicz M, O'Brien L, Hall AL, Wilkinson R, Al Olama AA, Morrison J, Muir K, Neal D, Donovan JL, Hamdy F, Easton DF, Eeles RA, Tymrakiewitcz M. A recurrent truncating germline mutation in the BRIP1/FANCJ gene and susceptibility to prostate cancer. Br J Cancer. 2009;100:426–430. doi: 10.1038/sj.bjc.6604847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kote-Jarai Z, Leongamornlert DA, Saunders E, Tymrakiewicz M, Castro E, Mahmud N, Guy M, Edwards S, O'Brien L, Sawyer E, Hall A, Wilkinson R, Dadaev T, Goh C, Easton D, Goldgar D, Eeles R. BRCA2 is a moderate penetrance gene contributing to young-onset prostate cancer: implications for genetic testing in prostate cancer patients. Br J Cancer. 2011;105:1230–1234. doi: 10.1038/bjc.2011.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kote-Jarai Z, Saunders EJ, Leongamornlert DA, Tymrakiewicz M, Dadaev T, Jugurnauth-Little S, Ross-Adams H, AA AlOlama, Benlloch S, Halim S, Russel R, Dunning AM, Luccarini C, Dennis J, Neal DE, Hamdy FC, Donovan JL, Muir K, Giles GG, Severi G, Wiklund F, Gronberg H, Haiman Ca, Schumacher F, Henderson BE, Le Marchand L, Lindstrom S, Kraft P, Hunter DJ, Gapstur S, Chanock S, Berndt SI, Albanes D, Andriole G, Schleutker J, Weischer M, Canzian F, Riboli E, Key TJ, Travis RC, Campa D, Ingles SA, John EM, Hayes RB, Pharoah P, Khaw K-T, Stanford JL, Ostrander EA, Signorello LB, Thibodeau SN, Schaid D, Maier C, Vogel W, Kibel AS, Cybulski C, Lubinski J, Cannon-Albright L, Brenner H, Park JY, Kaneva R, Batra J, Spurdle A, Clements JA, Teixeira MR, Govindasami K, Guy M, Wilkinson RA, Sawyer EJ, Morgan A, Dicks E, Baynes C, Conroy D, Bojesen SE, Kaaks R, Vincent D, Bacot F, Tessier DC, Easton DF, Eeles RA. Fine-mapping identifies multiple prostate cancer risk loci at 5p15, one of which associates with TERT expression. Hum Mol Genet. 2013;22:2520–2528. doi: 10.1093/hmg/ddt086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kottemann MC, Smogorzewska A. Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature. 2013;493:356–363. doi: 10.1038/nature11863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange K, Papp JC, Sinsheimer JS, Sripracha R, Zhou H, Sobel EM. Mendel: the Swiss army knife of genetic analysis programs. Bioinformatics. 2013;29 (12:1568–1570. doi: 10.1093/bioinformatics/btt187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leongamornlert DA, Mahmud N, Tymrakiewicz M, Saunders E, Dadaev T, Castro E, Goh C, Govindasami K, Guy M, O'Brien L, Sawyer E, Hall A, Wilkinson R, Easton D, Goldgar D, Eeles R, Kote-Jarai Z. Germline BRCA1 mutations increase prostate cancer risk. Br J Cancer. 2012;106:1697–1701. doi: 10.1038/bjc.2012.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levran O, Attwooll C, Henry RT, Milton KL, Neveling K, Rio P, Batish SD, Kalb R, Velleuer E, Barral S, Ott J, Petrini J, Schindler D, Hanenberg H, Auerbach AD. The BRCA1-interacting helicase BRIP1 is deficient in Fanconi anemia. Nat Genet. 2005;37:931–933. doi: 10.1038/ng1624. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Jian X, Boerwinkle E. dbNSFP: a lightweight database of human nonsynonymous SNPs and their functional predictions. Hum Mutat. 2011;32:894–899. doi: 10.1002/humu.21517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macinnis RJ, Antoniou AC, Eeles RA, Severi G, Al Olama AA, McGuffog L, Kote-Jarai Z, Guy M, O'Brien LT, Hall AL, Wilkinson RA, Sawyer E, Ardern-Jones AT, Dearnaley DP, Horwich A, Khoo VS, Parker CC, Huddart RA, Van As N, McCredie MR, English DR, Giles GG, Hopper JL, Easton DF. A risk prediction algorithm based on family history and common genetic variants: application to prostate cancer with potential clinical impact. Genet Epidemiol. 2011;35:549–556. doi: 10.1002/gepi.20605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michailidou K, Hall P, Gonzalez-Neira A, Ghoussaini M, Dennis J, Milne RL, Schmidt MK, Chang-Claude J, Bojesen SE, Bolla MK, Wang Q, Dicks E, Lee A, Turnbull C, Rahman N, Fletcher O, Peto J, Gibson L, Dos Santos Silva I, Nevanlinna H, Muranen TA, Aittomäki K, Blomqvist C, Czene K, Irwanto A, Liu J, Waisfisz Q, Meijers-Heijboer H, Adank M, van der Luijt RB, Hein R, Dahmen N, Beckman L, Meindl A, Schmutzler RK, Müller-Myhsok B, Lichtner P, Hopper JL, Southey MC, Makalic E, Schmidt DF, Uitterlinden AG, Hofman A, Hunter DJ, Chanock SJ, Vincent D, Bacot F, Tessier DC, Canisius S, Wessels LFA, Haiman CA, Shah M, Luben R, Brown J, Luccarini C, Schoof N, Humphreys K, Li J, Nordestgaard BG, Nielsen SF, Flyger H, Couch FJ, Wang X, Vachon C, Stevens KN, Lambrechts D, Moisse M, Paridaens R, Christiaens M-R, Rudolph A, Nickels S, Flesch-Janys D, Johnson N, Aitken Z, Aaltonen K, Heikkinen T, Broeks A, Veer LJV, van der Schoot CE, Guénel P, Truong T, Laurent-Puig P, Menegaux F, Marme F, Schneeweiss A, Sohn C, Burwinkel B, Zamora MP, Perez JIA, Pita G, Alonso MR, Cox A, Brock IW, Cross SS, Reed MWR, Sawyer EJ, Tomlinson I, Kerin MJ, Miller N, Henderson BE, Schumacher F, Le Marchand L, Andrulis IL, Knight JA, Glendon G, Mulligan AM, Lindblom A, Margolin S, Hooning MJ, Hollestelle A, van den Ouweland AMW, Jager A, Bui QM, Stone J, Dite GS, Apicella C, Tsimiklis H, Giles GG, Severi G, Baglietto L, Fasching PA, Haeberle L, Ekici AB, Beckmann MW, Brenner H, Müller H, Arndt V, Stegmaier C, Swerdlow A, Ashworth A, Orr N, Jones M, Figueroa J, Lissowska J, Brinton L, Goldberg MS, Labrèche F, Dumont M, Winqvist R, Pylkäs K, Jukkola-Vuorinen A, Grip M, Brauch H, Hamann U, Brüning T, Radice P, Peterlongo P, Manoukian S, Bonanni B, Devilee P, Tollenaar RAEM, Seynaeve C, van Asperen CJ, Jakubowska A, Lubinski J, Jaworska K, Durda K, Mannermaa A, Kataja V, Kosma V-M, Hartikainen JM, Bogdanova NV, Antonenkova NN, Dörk T, Kristensen VN, Anton-Culver H, Slager S, Toland AE, Edge S, Fostira F, Kang D, Yoo K-Y, Noh D-Y, Matsuo K, Ito H, Iwata H, Sueta A, Wu AH, Tseng C-C, Van Den Berg D, Stram DO, Shu X-O, Lu W, Gao Y-T, Cai H, Teo SH, Yip CH, Phuah SY, Cornes BK, Hartman M, Miao H, Lim WY, Sng J-H, Muir K, Lophatananon A, Stewart-Brown S, Siriwanarangsan P, Shen C-Y, Hsiung C-N, Wu P-E, Ding S-L, Sangrajrang S, Gaborieau V, Brennan P, McKay J, Blot WJ, Signorello LB, Cai Q, Zheng W, Deming-Halverson S, Shrubsole M, Long J, Simard J, Garcia-Closas M, Pharoah PDP, Chenevix-Trench G, Dunning AM, Benitez J, Easton DF. Large-scale genotyping identifies 41 new loci associated with breast cancer risk. NatGenet. 2013;45 (353–361:361–362. doi: 10.1038/ng.2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra AV, Bancroft EK, Barbachano Y, Page EC, Foster CS, Jameson C, Mitchell G, Lindeman GJ, Stapleton A, Suthers G, Evans DG, Cruger D, Blanco I, Mercer C, Kirk J, Maehle L, Hodgson S, Walker L, Izatt L, Douglas F, Tucker K, Dorkins H, Clowes V, Male A, Donaldson A, Brewer C, Doherty R, Bulman B, Osther PJ, Salinas M, Eccles D, Axcrona K, Jobson I, Newcombe B, Cybulski C, Rubinstein WS, Buys S, Townshend S, Friedman E, Domchek S, Ramon Y, Cajal T, Spigelman A, Teo SH, Nicolai N, Aaronson N, Ardern-Jones AT, Bangma C, Dearnaley DP, Eyfjord J, Falconer A, Grönberg H, Hamdy FC, Johannsson O, Khoo VS, Kote-Jarai Z, Lilja H, Lubiński J, Melia J, Moynihan C, Peock S, Rennert G, Schröder F, Sibley P, Suri M, Wilson P, Bignon YJ, Strom S, Tischkowitz M, Liljegren A, Ilencikova D, Abele A, Kyriacou K, van Asperen C, Kiemeney L, Easton DF, Eeles RA. Targeted prostate cancer screening in men with mutations in BRCA1 and BRCA2 detects aggressive prostate cancer: preliminary analysis of the results of the IMPACT study. BJU Int. 2011;107:28–39. doi: 10.1111/j.1464-410X.2010.09648.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagenstecher C, Wehner M, Friedl W, Rahner N, Aretz S, Friedrichs N, Sengteller M, Henn W, Buettner R, Propping P, Mangold E. Aberrant splicing in MLH1 and MSH2 due to exonic and intronic variants. Hum Genet. 2006;119:9–22. doi: 10.1007/s00439-005-0107-8. [DOI] [PubMed] [Google Scholar]
- Parkin DM, Whelan SL, Ferlay J, Teppo L, Thomas DB.eds (2002Cancer Incidence in Five Continents Vol. VIII.International Agency for Research on Cancer: Lyon, France [Google Scholar]
- R Core Team . R Foundation for Statistical Computing: Vienna, Austria; 2012. R: A Language and Environment for Statistical Computing. [Google Scholar]
- Rahman N, Seal S, Thompson D, Kelly P, Renwick A, Elliott A, Reid S, Spanova K, Barfoot R, Chagtai T, Jayatilake H, McGuffog L, Hanks S, Evans DG, Eccles D, Easton DF, Stratton MR. PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nat Genet. 2007;39:165–167. doi: 10.1038/ng1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandhu SK, Omlin A, Hylands L, Miranda S, Barber LJ, Riisnaes R, Reid AH, Attard G, Chen L, Kozarewa I, Gevensleben H, Campbell J, Fenwick K, Assiotis I, Olmos D, Yap TA, Fong P, Tunariu N, Koh D, Molife LR, Kaye S, Lord CJ, Ashworth A, Bono Jde. Poly (ADP-ribose) polymerase (PARP) inhibitors for the treatment of advanced germline BRCA2 mutant prostate cancer. Ann Oncol. 2013;24:1416–1418. doi: 10.1093/annonc/mdt074. [DOI] [PubMed] [Google Scholar]
- Seal S, Thompson D, Renwick A, Elliott A, Kelly P, Barfoot R, Chagtai T, Jayatilake H, Ahmed M, Spanova K, North B, McGuffog L, Evans DG, Eccles D, Easton DF, Stratton MR, Rahman N. Truncating mutations in the Fanconi anemia J gene BRIP1 are low-penetrance breast cancer susceptibility alleles. Nat Genet. 2006;38:1239–1241. doi: 10.1038/ng1902. [DOI] [PubMed] [Google Scholar]
- The CHEK2 Breast Cancer Case-Control Consortium CHEK2*1100delC and susceptibility to breast cancer: a collaborative analysis involving 10 860 breast cancer cases and 9065 controls from 10 studies. Am J Hum Genet. 2004;74:1175–1182. doi: 10.1086/421251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson D, Duedal S, Kirner J, McGuffog L, Last J, Reiman A, Byrd P, Taylor M, Easton DF. Cancer risks and mortality in heterozygous ATM mutation carriers. J Natl Cancer Inst. 2005;97:813–822. doi: 10.1093/jnci/dji141. [DOI] [PubMed] [Google Scholar]
- Tsai M-F, Lin Y-J, Cheng Y-C, Lee K-H, Huang C-C, Chen Y-T, Yao A. PrimerZ: streamlined primer design for promoters, exons and human SNPs. Nucleic Acids Res. 2007;35:W63–W65. doi: 10.1093/nar/gkm383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh T, Lee MK, Casadei S, Thornton AM, Stray SM, Pennil C, Nord AS, Mandell JB, Swisher EM, King M-C. Detection of inherited mutations for breast and ovarian cancer using genomic capture and massively parallel sequencing. Proc Natl Acad Sci USA. 2010;107:12629–12633. doi: 10.1073/pnas.1007983107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu K, Hinson SR, Ohashi A, Farrugia D, Wendt P, Tavtigian SV, Deffenbaugh A, Goldgar D, Couch FJ. Functional evaluation and cancer risk assessment of BRCA2 unclassified variants unclassified variants. Cancer Res. 2005;65:417–426. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.