

Abstract
BRAFV600E-inhibitors (BRAFi; e.g., vemurafenib) and modern immune-based therapies such as PD-1/PD-L1 and CTLA-4 checkpoints blockade and adoptive cell transfer (ACT) have significantly improved the care of melanoma patients. Having these two effective (BRAFi and immunotherapy) therapies raises the question whether there is a rational biological basis for using them in combination. We developed an in vitro model to determine whether tumor resistance mechanisms to a small molecule inhibitor of a driver oncogene, and to cytotoxic T lymphocyte (CTL)- and natural killer (NK) cell-delivered apoptotic death signals were exclusive or intersecting. We generated melanoma sublines resistant to BRAFi vemurafenib and to CTL recognizing the MART-1 melanoma antigen. Vemurafenib-resistant (VemR) sublines were cross-resistant to MART CTL and NK cells indicating that a common apoptotic pathway governing tumor response to both modalities was disrupted. Pretreatment of VemR melanomas with a histone deacetylase inhibitor (HDACi) restored sensitivity to MART CTL and NK apoptosis by skewing the apoptotic gene programs towards a proapoptotic phenotype. Our in vitro findings suggest that during the course of acquisition of BRAFi resistance, melanomas develop cross-resistance to CTL- and NK-killing. Further, aberrant apoptotic pathways, amenable by an FDA-approved chromatin remodeling drug, regulate tumor resistance mechanisms to immune effector cells. These results may provide rational molecular basis for further investigations to combine these therapies clinically.
Keywords: Vemurafenib, MAPK, BRAFV600E kinase inhibitor, apoptosis, immunotherapy, signal transduction, adoptive cell transfer, HDACi, SAHA, gene expression, sensitization, melanoma, NK cells, TCR transgenic CTL
Introduction
Activating somatic BRAFV600E mutation is found in nearly half of human melanomas and results in sequential phosphorylation and activation of MEK1/2 and ERK1/2 leading to apoptosis resistance, increased survival, and proliferation of melanomas [1,2]. The BRAFV600E-inhibitor (BRAFi), vemurafenib, selectively binds to BRAFV600E in its active conformation, effectively inhibiting its kinase activity [3,4], blocking constitutively active ERK1/2 pathway, and inducing apoptosis in melanomas. It induces cell cycle arrest and apoptosis of BRAFV600E harboring cells, inhibits tumor growth and increases survival of experimental animals in melanoma xenograft models [5]. In clinical trials, treatment with optimal concentration of vemurafenib results in complete or partial tumor regression in >80% of BRAFV600E harboring melanoma patients [6]. However, progression free survival is limited due to the development of resistance to vemurafenib over a 6-8 months period [7].
The principal BRAFi escape mechanisms include recovery of ERK1/2 phosphorylation through activating NRAS mutations, paradoxical MAPK activation, COT kinase reactivation, PTEN loss, AKT amplification/mutation, CRAF dimerization, BRAF genomic amplification, BIM suppression, cyclin D1 induction, and overexpression of various receptor tyrosine kinases (RTK) [8-18]. These bypass mechanisms provide melanomas with the advantage to resist apoptosis and proliferate [19]. Thus, combining vemurafenib with strategies that impede these compensatory/resistance mechanisms is an area of active basic and clinical research [20,21].
Remarkable clinical responses have also been seen in patients with metastatic melanoma with modern immune-based approaches CTLA-4 and PD-1/PD-L-1 checkpoints blockade, adoptive cell transfer (ACT), as well as earlier experiences with high-dose IL-2 [21]. The proximal mediators of these therapies are tumor-reactive cytotoxic T lymphocytes and natural killer (NK) cells. Various resistance mechanisms to immune-mediated apoptotic death signals have been described including phenotypic changes and effector cell exhaustion [22], functional tolerance, deficiencies in antigen processing and presentation, and mutation or down-regulation of antigenic epitopes. Consi-dering that the immune system eradicates tumors via apoptosis, a more fundamental property of tumors that may limit the efficacy of immunotherapy-resistance to apoptosis- may also be a determining factor [23,24].
Vorinostat (suberoylanilide hydroxamic acid, SAHA) is the first Food and Drug Administration (FDA) approved histone deacetylase inhibitor (HDACi) with well-established clinical efficacy in cutaneous T-cell lymphoma (CTCL) patients [25], and other cancer cell types. As single agent or combined with other agents, HDACi has some anti-melanoma activity in vitro and in vivo [26]. Given the aberrant expression profile of apoptosis-associated genes in drug- and immune-resistant tumors, and the ability of SAHA to negatively regulate these resistance mechanisms [26], the efficacy of SAHA in conjunction with vemurafenib and/or immunotherapy in melanoma treatment warrants further investigation.
The availability of these two clinically effective melanoma therapies, which deliver death signals through different mechanisms, presents the obvious opportunity for their use in combination. We developed an in vitro model to understand the mechanisms of tumor-acquired resistance to BRAFi and to immune effector cells and whether these resistant pathways intersect. Our in vitro model consists of a melanoma cell line M249 which harbors BRAFV600E and expresses the melanoma antigenic epitope MART-127-35 in the context of HLA A*0201; this cell line is sensitive to both vemurafenib and to MART-specific CTL (F5 CTL). A second melanoma cell line M238, lacking MART-127-35 and HLA A*0201 yet sensitive to both vemurafenib and NK cells, was also used. Serial exposure of M249 and M238 cells to vemurafenib yielded vemurafenib-resistant [M249(VemR), M238 (VemR)] sublines, which were completely resistant to MART CTL despite having unaltered MART-1/A*0201 expression, and NK cell killing, respectively. This cross-resistance incriminates disruption of common intracellular apoptotic machinery by both modalities. Pretreatment of M249(VemR) and M238(VemR) sublines with SAHA restored sensitivity to MART CTL and NK killing, respectively. These results imply that a common apoptosis machinery regulates resistance, which can be restored by an epigenetic modifier, thus, sensitizing melanomas to immune effector cells.
Materials and methods
Cell lines and clones
Human melanoma lines were established from surgical specimens as described [23,24]. For the generation of CTLR sublines, parental cells were grown in the presence of step-wise increasing numbers of F5 CTLs (E:T 20:1, 40:1, 60:1) for a total of 8 weeks (2-3 weeks for each E:T). Thirty percent to 50% of melanoma cells survived the first cycle of selection (20:1, 2 weeks), percentage of which drastically reduced during subsequent selection cycles until no further killing was observed. Remaining viable melanoma cells were then subjected to two consecutive rounds of limiting dilution analysis. Single cells were propagated and maintained in RPMI-1640 supplemented with 10% (v/v) heat-inactivated fetal bovine serum (FBS). After immunoselection, cells were maintained in medium containing excess (10:1) F5 CTLs, but were grown in F5 CTL-free medium at least 1 week prior to analysis. Vemurafenib-resistant sublines were established by the growth of vemurafenib-sensitive parental lines in the presence of step-wise increasing concentrations of vemurafenib (0.1-10 μM for 3 month). Cells were grown in Vemurafenib-free medium at least one week prior to analysis. Cultures were incubated in controlled atmosphere incubator at 37°C with saturated humidity at 0.3-0.5 × 106 cells/mL and were used at 50% to 70% growth confluency for each experiment. Cultures were routinely (once/month) checked for mycoplasma contamination (Lonza).
Reagents
Vemurafenib (PLX4032) was purchased from Selleck (Houston, TX). Stock vemurafenib was stored at –80°C at 10 mM in DMSO prior to being used in experiments. MART-1, actin and tubulin antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA), Millipore (Temecula, CA) and Sigma. Antibodies to various receptor tyrosine kinases (RTK) were purchased from Cell Signaling Technology (Beverly, MA).
ELISA
The amount of cytokine release was measured using ELISA assay kits (eBiosciences) according to manufacturer’s instructions.
Transduction of CD8 CTLs with F5 MART-1 TCRα/β retrovirus
Nonadherent population of healthy donor human peripheral blood mononuclear cells (PBMC) was isolated after obtaining informed consent form and Institutional Review Board (IRB) approval and cultured in AIM-V media supplemented with 5% human AB serum, OKT3 (50 ng/mL), and IL-2 (300 IU/mL) for 48 hours. CD16+CD56+ NK cells and CD3+CD8+ CTLs were both isolated by EasyStep Negative Selection enrichment kits (Stem Cell Technologies) according to manufacturer’s instructions. CTLs were transduced with MSCV MART-1 TCR as described [35,36]. CD8+ CTLs with more than 95% MART-1 TCRα/β expression and purified NK cells were used in all experiments.
Drug sensitivity and proliferation assay (XTT)
Inhibition of proliferation was assessed using the standard XTT assay kit (Roche, Indianapolis, IN) that measures the metabolic activity of viable cells. The percentage of proliferation (viability) was calculated using the background-corrected reading as follows: % of Control = [(OD of sample wells/OD of untreated cells)] × 100 [37].
Expression analysis of apoptotic genes by quantitative real-time PCR (qPCR)
Samples were analyzed with iQ SYBR Green Supermix using iCycler Sequence Detection System (BioRad). Gene expression analysis was performed using RT2 profiler apoptosis PCR arrays. Total RNA was extracted from 107 cells for each condition with RNeasy mini kit (Qiagen) and quantified by 3.1.2 NanoDrop ND-1000 spectrophotometer. Three micrograms of total RNA was reverse transcribed to first-strand cDNA for 1 hour at 42°C with 200 units SuperScript II RT and 20 μM random hexamer primers. Amplification of 2.5 μL of cDNAs was performed using gene-specific primers.
Cell-mediated cytotoxicity assay
Melanoma cultures were trypsinized for 5 minutes, washed once in cold PBS and labeled with 100 μCi of Na2 51CrO4 for 1 hour (37°C/5% CO2). After 3X washes, 104 cells were added to V-bottom 96-well plates and used immediately as described [23,24]. Percentage of specific 51Cr-release was measured as: % cytotoxicity = (experimental release - spontaneous release)/(total release - spontaneous release) × 100.
Flow cytometric analysis for evaluation of active caspase-3 levels (apoptosis)
Levels of active caspase-3 were evaluated for the measurement of apoptosis [37]. Melanoma cells were treated under the conditions routinely used for PI staining and cell cycle analysis. At the end of the incubation period, the cells were washed once with ice-cold 1 × PBS/0.1% BSA and were resuspended in 100 μl ice-cold 1 × PBS/0.1% BSA. Fifty microliters of cell suspension (containing 2 × 106 cells) were aliquoted to each sample and fixed with the perm/fix solution (PharMingen) for 20 min. Thereafter, the cells were washed twice with 1 × perm/wash (PharMingen) solution and stained with the FITC-labeled anti-active caspase-3 antibody for 30 min (light protected). Thereafter, the samples were washed once with 1 × perm/wash solution followed by flow cytometric analysis (Coulter Electronics, Miami, FL). As negative control, the cells were stained with isotype control (pure IgG1) under the same conditions described above.
Immunoblot analysis
A total of 107 cells were grown in complete medium (± inhibitors), lysed at 4°C in RIPA buffer [50 mmol/L Tris-HCl (pH 7.4), 1% NP-40, 0.25% sodium deoxycholate, 150 μmol/L NaCl] supplemented with protease inhibitor cocktail (Complete Mini; Roche) and subjected to immunoblot analysis as described [23,24].
BRAF and NRAS mutational analysis
Genomic DNA extracted from melanoma cell lines was subjected to polymerase chain reaction (PCR) using primer sets that were designed to amplify specific regions of exon 15 (activation domain) of BRAF, and exon 1 and 2 of NRAS. PCR products were first analyzed on agarose gels to determine single band amplicons, then purified and submitted for genotyping analysis (UCLA, Sequencing Core Facility) of both strands by Sanger sequencing. Final sequences were analyzed for novel and known mutations within genomic hot-spots.
Statistical analysis
Assays were set up in duplicates or triplicates and results were expressed as mean ± standard error of the mean (SEM). Statistical analysis and P values were calculated by two-tailed paired t test with a confidence interval (CI) of 95% for determination of significance of differences between treatment groups (P < 0.05: significant). ANOVA was used to test significance among the groups using InStat 2.01 software.
Results
Cytotoxic effects of BRAFi vemurafenib (PLX4032) and MART TCR-transduced CTL on human melanoma cell lines
The M249 melanoma cell line harbors the BRAFV600E oncogenic mutation, expresses the MART-1 melanoma antigen, is stably transfected with HLA A*0201 allowing M249 melanomas to present the MART-1 epitope in the context of this class-I restricting element and be recognized and killed by MART-127-35 TCR-engineered human T cells (F5 CTL). The BRAFV600E/MART-1 negative/A*0201 negative melanoma line M238 cannot be recognized and killed by F5 CTL (Figure 1A). M249 and M238 melanomas were exposed to various concentrations of the BRAFi vemurafenib (0.0001-10 μmol/L, 72 hr) and an XTT proliferation assay was used to determine inhibition of proliferation and survival rate; both cell lines showed dramatic sensitivity to the cytostatic effect of vemurafenib with an IC50 value between 0.1 and 1.0 μmol/L at 72 hr post treatment (Figure 1B). Vemurafenib treatment (0.01-2.5 μmol/L, 8-72 hr) induced apoptosis in both cell lines in a dose- and time-dependent manner as measured by active caspase-3 levels, albeit with different kinetics (Figure 1C).
Figure 1.
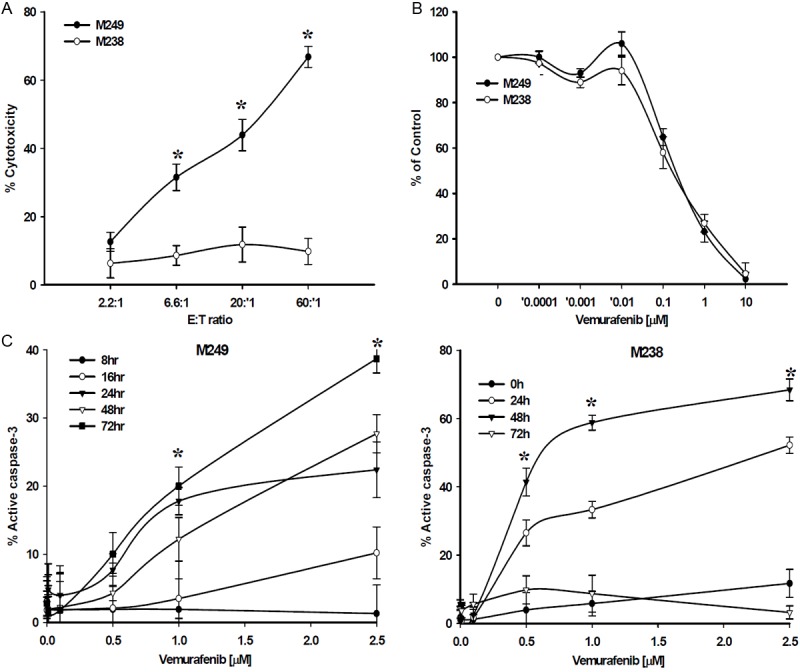
Cytotoxic and cytostatic effects of Vemurafenib and F5 CTL on M249. (A) Cytotoxic activity of F5 CTLs against M249 and M238. 51Cr-labeld tumor cells were co-incubated with F5 CTLs at various E:T ratios in a 6 hr standard 51Cr-release assay. Cytostatic (B) and Apoptotic (C) effects of vemurafenib on M249 and M238. Cells were treated with (B) Vemurafenib (0.0001-10 μmol/L) for 72 hours, or (C) vemurafenib (0.1-2.5 μmol/L) for 8-72 hours and percent proliferation (% of control) and apoptosis were assessed by XTT proliferation assay (IC50 0.1-1.0 μmol/L) and active caspase-3 levels by FACS analysis, respectively. Samples were set up in triplicates and the results are represented as mean ± SEM of three independent experiments. *P values < 0.05.
Generation of vemurafenib-resistant (VemR) melanomas
Serial exposure of M249 and M238 cells to increasing concentrations of vemurafenib (0.1-10 μmol/L) over a three month period yielded multiple tumor sublines resistant to the cytostatic (Figure 2A) and apoptotic (Figure 2B, 2C) effects of vemurafenib, sublines which we term VemR. Sequence analysis showed that M249 (VemR) and M238(VemR) sublines retained BRAFV600E mutation and acquired no secondary NRAS mutations (Figure 2D). Protein levels of EGFR, c-KIT, Met, and PDGFRβ were upregulated, and IGFRβ was downregulated in M249 (VemR) sublines. However, in M238(VemR) sublines c-Kit levels were undetectable, EGFR levels were significantly upregulated whereas PDGFRβ levels were reduced. Protein levels of Met and IGFRβ remained unchanged (Figure 2E).
Figure 2.
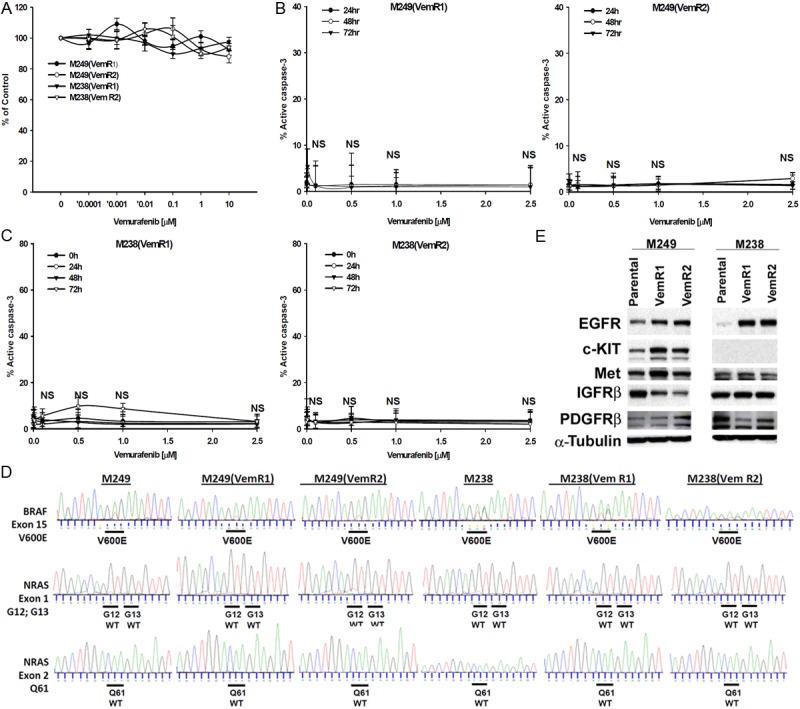
Generation of VemR sublines. M249 and M238 cells were grown in the presence of increasing concentrations of Vemurafenib (0.1-10 μmol/L) for 3 months and subjected to LDA analysis. (A) Cytostatic effects of Vemurafenib on M249(VemR) and M238(VemR) sublines. Cells were grown in the presence of vemurafenib (0.0001-10 μmol/L, 72 hr) and percentage of proliferation was assessed by XTT assay. Samples were set up in triplicates and results are presented as mean ± SEM of three independent experiments. Sensitivity of (B) M249(VemR) and (C) M238(VemR) sublines to vemurafenib-mediated apoptosis. M249(VemR1, VemR2) and M238(VemR1, VemR2) sublines were grown in the presence of Vemurafenib (0.1-2.5 μmol/L) for various time points (24-72 hr) and percent apoptosis was assessed by measuring the levels of active caspase-3. Experiment was repeated three times with similar results. Higher vemurafenib concentrations (up to 10 μmol/L) had no apoptotic effect (not shown). NS: not significant. (D) Mutational Analysis of Vemurafenib-resistant sublines. Genomic amplification and sequence analysis of VemR sublines compared to their parental counterparts. VemR sublines harbor BRAFV600E mutation in exon 15, do not harbor NRAS mutations (wild type NRAS) and no secondary NRAS genomic hot-spot mutations at exon 1 [G12, G13] and exon 2 [Q61] were identified. (E) Expression of various receptor tyrosine kinases (RTK) in VemR sublines by western blot analysis. Results are representative of two independent experiments.
Recognition and killing of VemR sublines by immune effector cells
The M249(VemR) sublines could be recognized (as measured by IFN-γ and IL-2 release) (Figure 3A) but not killed (using 51Cr-release assay) by F5 CTL (Figure 3B), whereas, M238(VemR) sublines were neither recognized nor killed by F5 CTLs. While the M238 cells were sensitive to NK cell-mediated killing, the VemR sublines were resistant (Figure 3C). Slight induction in the expression of MART-1 protein levels in the M249(VemR), but not M238(VemR), sublines were observed (Figure 3D). These results indicate that the recognition machinery (peptide/MHC complex) of M249(VemR) cells was intact, but these cells have developed cross-resistance to apoptotic death signals delivered by F5 CTL. Similarly, while the MART-1 negative, A*0201 negative M238 cells were sensitive to NK cells, its VemR derivatives were NK-resistant.
Figure 3.
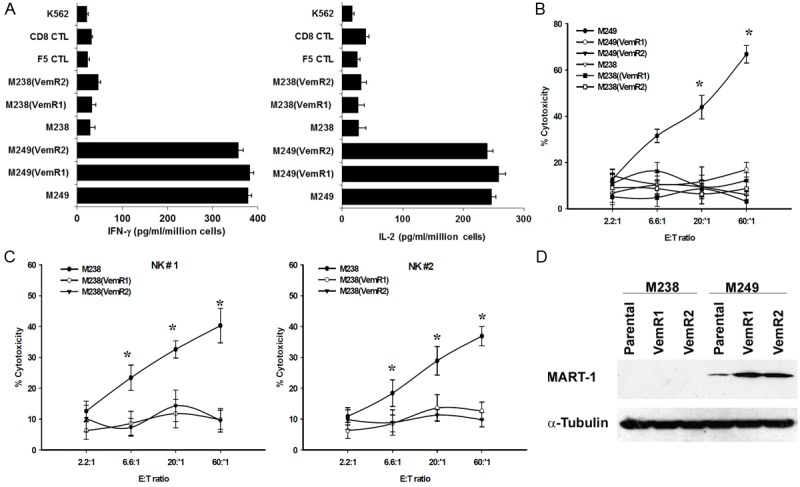
Recognition and killing of M249(VemR) and M238(VemR) sublines by F5 CTLs and NK cells. (A) M249 parental and M249(VemR) sublines express comparable levels of surface MART-1/ HLA A*0201 complex. 106 tumors were co-incubated overnight with F5 CTLs at 1:1 E:T ratio. IL-2 and IFN-γ released was measured using ELISA. Samples were set up in quadruplicate; results are presented as mean ± SEM of two independent experiments. F5 CTL, CD8 CTL and K562 cells were used as control. (B) M249(VemR) and (C) M238(VemR) cells exhibit resistance to F5 CTL- and NK-mediated killing. Cells were used in a 6 hr 51Cr-release assay. Samples were set up in duplicate, results presented as mean ± SEM of two independent experiments. *P values < 0.05. (D) MART-1 protein expression. Whole cell extract (40 μg) were subjected to immunoblotting. Levels of α-tubulin were used for equal loading (n = 3).
F5 CTL-resistant M249 cells are not sensitive to vemurafenib
We have recently described the generation of several F5 CTL-resistant melanoma sublines and have characterized their mechanism of resistance, which includes constitutive activation of survival pathways and over expression of anti-apoptotic factors [23,24]. Two sublines of M249 cells completely resistant to F5 CTL [M249(CTLR1, R2)] were generated by serial exposure to these cytotoxic TCR-transgenic CD8+ T cells. F5 CTL resistant cells [M249(CTLR1, R2)] were exposed to vemurafenib over a range of concentrations (0.1-2.5 μmol/L) for various time points (24-72 hr). These CTL-resistant tumor cells were resistant to BRAFi as measured by active caspase-3 levels (Figure 4A, 4B). Reinforcing this observation of cross-resistance to apoptotic death signals, CTL-resistant melanoma targets were resistant to F5 CTL and vemurafenib in combination (Figure 4C).
Figure 4.
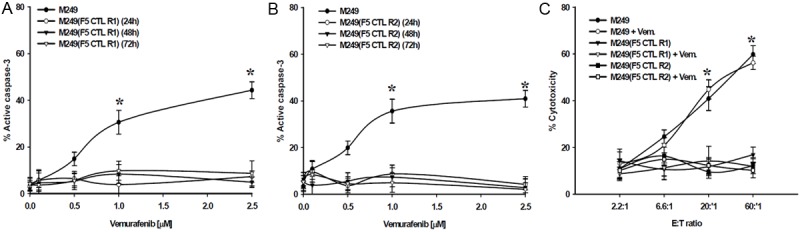
Effects of Vemurafenib on (A) M249(CTLR1) and (B) M249(CTLR2) sublines. Cells were grown in the presence of vemurafenib (0.1-2.5 μmol/L) for various time points (24-72 hr). Percentage of apoptosis was assessed by measuring the levels of active caspase-3. (C) Inability of vemurafenib to immunosensitize. M249(F5 CTLR) sublines were treated with 1 μmol/L vemurafenib (24, 48 hr), washed twice, labeled, and co-incubated with F5 CTLs at various E:T ratios in a 6 hr standard 51Cr-release assay. Only results of 24 hr Vemurafenib pretreatment are shown. Similar results were obtained at 48 hr vemurafenib pretreatment. Samples were set up in duplicates; results are presented as mean ± SEM of two independent experiments. Parental M249 cells (± vemurafenib) were used as control. *P values < 0.05.
The HDACi SAHA reverses resistance to immune effector cells
HDACi regulate the expression pattern of apoptotic genes rendering tumors more susceptible to apoptotic stimuli [26] and overcome BRAFi-resistance [27]. Incubation of M249(VemR) and M249(CTLR) sublines with 1 μmol/L SAHA for 48 hr largely reversed their resistance to F5 CTL killing (Figure 5A). We confirmed these findings with two patient-derived MART-specific CTLs (Figure 5B). Similarly, pretreatment of M238(VemR) sublines with SAHA reversed their resistance to NK killing (Figure 5C). Pretreatment of either of these categories of resistant sublines (VemR and CTLR) with SAHA, however, did not restore their sensitivity to vemurafenib (Figure 5D).
Figure 5.
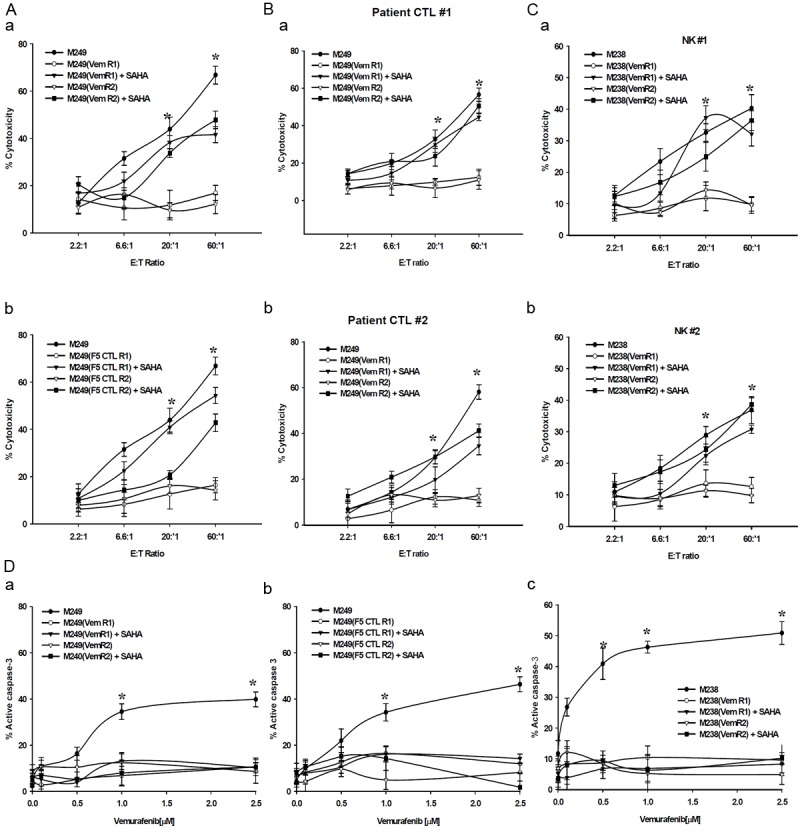
Immunosensitization of M249 VemR, F5 CTLR and M238(VemR) sublines by SAHA. Cells (106) were left either untreated or pretreated with SAHA (1 μmol/L-48 hr) and used in 51Cr-release assay using various effectors: (A) MART TCR transgenic CTL, (B) two patient derived MART-1 specific CTLs, (C) two freshly-isolated NK cells. (D) Inability of SAHA to sensitize: (a) M249(VemR), (b) M249(F5 CTLR), (c) M238(VemR) sublines to vemurafenib. SAHA pretreated cells (1 μmol/L-48 hr) were washed, grown in the presence of vemurafenib (0.1-2.5 μmol/L), and apoptosis was assessed by measuring levels of active caspase-3. Untreated M238 and M249 cells were used as control. Samples were set up in duplicates and results are presented as mean ± SEM (n = 4). *P values < 0.05.
Expression of apoptotic genes in M249 sensitive and resistant sublines
Focused apoptosis array qPCR analysis showed that the expression of several groups of apoptosis-associated genes was altered in M249(VemR) and M249(CTLR) sublines compared to their parental cells. The expression levels of positive regulators of apoptosis (e.g., Apaf-1, BAD, CIDE-A, -B), various caspases (e.g., caspases-2, -3, -5, -6, -8), TNF/TNFR superfamily and death domain proteins (e.g., TNFSF10, TNFRSF10B, 11B), DNA damage (GADD45) were reduced, while the expression levels of several negative regulators of apoptosis (e.g., Bcl-2 members) were increased, consistent with a resistant phenotype (Table 1A). Treatment of these resistant sublines (VemR and CTLR) with SAHA increased the expression levels of positive regulators of apoptosis, caspases, TNF/TNFR family and death domain proteins and decreased the expression levels of several negative regulators of apoptosis (Table 1B), consistent with a proapoptotic gene program. In comparison, a larger number of apoptotic genes were modified in M238(VemR) (± SAHA) sublines, however, a trend very similar to M249(VemR) was observed (Supplemental Table 1).
Table 1.
Differential expression of apoptotic genes in (A) M249(VemR) and (B) M249(CTLR) and their regulation by SAHA as measured by real time qPCR analysis using apoptosis arrays (representative of two independent experiments, genes with > 2.5 fold mRNA changes were considered significant)
| A. M249(VemR) | |||
|
| |||
| M249(VemR) vs M249 | M249(VemR) + SAHA vs M249(VemR) | ||
|
|
|||
| Gene name | Fold mRNA change | Fold mRNA change | |
|
| |||
| Positive apoptosis regulators | TBP53BP2 | -4.3 | 2.5 |
| APAF-1 | -2.6 | 3.9 | |
| BAD | -3.2 | 4 | |
| CIDEA | -7.2 | 9.8 | |
| CIDEB | -3.5 | 2.4 | |
| FADD | -2.7 | 3.5 | |
| AKT1 | 2.6 | -1.9 | |
| Negative apoptosis regulators | BAG1 | 8.9 | -6 |
| BCLAF1 | 3.8 | -2.8 | |
| BCL2L1 | 3.6 | -4.2 | |
| BCL2 | 2.8 | -3 | |
| NAIP(BIRC1) | -2.8 | 2.1 | |
| Caspases and caspase activators/regulator | CASP2 | 3.7 | -1.3 |
| CASP3 | -4.1 | 2.5 | |
| CASP5 | 5.5 | -4.82 | |
| CASP6 | -3.2 | 3.14 | |
| CASP8 | -4.8 | 2.8 | |
| TP73 | -3.7 | 4.6 | |
| CARD6 | -6.5 | 9.8 | |
| CARD8 | -10.3 | 3.8 | |
| Death domain and TNF/TNFR family | MGC:45012/TRAP/TRAF2 | -3.3 | 2.1 |
| CART-1/MLN62/TRAF4 | -2.8 | 3.4 | |
| TNFRSF5/CD40 | -1.3 | 2.6 | |
| TNFSF10/TRAIL | -3.4 | 7.3 | |
| TNFRSF10B/CD262 | -7.2 | 11.3 | |
| TNFRSF11B/OPG | -5 | 2.3 | |
|
| |||
| B. M249(CTLR) | |||
|
| |||
| M249(CTLR) vs M249 | M249(CTLR) + SAHA vs M249(CTLR) | ||
|
|
|||
| Gene name | Fold mRNA change | Fold mRNA change | |
|
| |||
| Positive apoptosis regulators | AKT1/PKB | -2 | 5.3 |
| BBC2/BCL2L8 | -5.1 | 8.2 | |
| BAK/BAK Like | -3 | 5.3 | |
| BCL2L4/BAX | -5.3 | 5 | |
| BCLXs | -3.3 | 5 | |
| BCL-B/BOO | -2.7 | 6.6 | |
| CIDE-A | -1.5 | 2.5 | |
| CIDE-B | -1.6 | 6.3 | |
| TRP53/LFS-1 | -6.4 | 8.7 | |
| APO-1/ALPS1A | -4.4 | 3.5 | |
| Death domain proteins | FADD/GIG3/MORT-1 | -2.8 | 6.9 |
| TNFRSF10a/CD261 | -2.15 | 10 | |
| TNFRSF10B/CD262 | -4.5 | 7.6 | |
| TNFRSF11B/OPG | -52.5 | 57.3 | |
| TNFRSF21/DR6 | -1.7 | 4.5 | |
| TNF/TNFR superfamily | APO2L/TNFSF10 | -23.5 | 7.3 |
| TNFRSF1A/Hs.89862 | -2.15 | 3.5 | |
| MGC:45012/TRAP/TRAF2 | -3.1 | 4 | |
| CAP-1/CD40BP/TRAF3 | -1.5 | 2.5 | |
| CART-1/MLN62/TRAF4 | -3 | 4.5 | |
| Caspases and activators | CASP1/ICE/IL1BC | -4.6 | 13.1 |
| CASP2/ICH-1L | -2 | 7.3 | |
| CASP4/ICE(rel)III | -3 | 2.5 | |
| CASP7(CMH-1/LAP3) | -1.5 | 2.8 | |
| APAF1 | -2.1 | 4 | |
| FADD/GIG3/MORT-1 | -2.8 | 6.9 | |
| Negative apoptosis regulators | DAPK/DKFZP78 | 3.6 | -4.5 |
| DFF45/DFF1 | 2.5 | -4.4 | |
| DP5/HARAKIRI | 6.5 | -6.7 | |
| BNIP-2/NIP2 | 2.8 | -2.8 | |
| P73 | -3 | 3.1 | |
| ACC-1/ACC-2 A1 | -4.4 | 3.1 | |
| BCL2L2/BCL-W | 4.1 | -6.22 | |
| BCL2L3/EAT/MCL1 | 1.7 | -2.5 | |
| LT/TNFB | 6.1 | -10.1 | |
| DNA damage | GADD45/DDIT1 | -2.2 | 3.3 |
Discussion
Remarkable clinical responses in patients with metastatic melanoma can be achieved with BRAFi vemurafenib as well as immune-based approaches such as CTLA-4 and PD-1/PD-L1 checkpoints blockade, and adoptive cell transfer (ACT) [21,28,29]. Having two effective therapeutic modalities - small molecule BRAFi and immunotherapy - raises the question whether there is a biological rationale to use them in combination.
Here we report that in the course of acquisition of resistance to BRAFi, VemR sublines were recognized, but not killed, by highly avid and specific melanoma-reactive CTLs. Similarly, CTL-resistant sublines developed cross-resistance to vemurafenib indicating the inversion of a common apoptotic machinery. Exposure to SAHA largely restored sensitivity of both M249(VemR) and M249(CTLR) sublines to TCR engineered and to naturally occurring patient-derived MART-1 CTLs by increasing the expression profile of proapoptotic genes. The observation of cross-resistance was further reinforced by using a second melanoma line, M238, and its vemurafenib-resistant derivatives. The MA-RT-127-35/A*0201 negative M238(VemR) sublines had altered expression profile of apoptotic genes consistent with a resistant phenotype, and developed cross-resistance to NK killing; a phenomenon that was largely reversed by SAHA. These in vitro data support the requirement of intact apoptotic machinery in tumors for the full execution of apoptotic death signals delivered by otherwise robust immune effector cells. However, other factors such as tumor heterogeneity and the effects of tumor microenvironment and immune suppressive cytokines might be important contributing factors influencing the outcome of immunotherapy in vivo.
The effectiveness of both BRAFi and immune therapies raises the obvious question whether these can be combined. The ability of BRAFi to upregulate MART-1 and gp-100, resulting in increased in vitro recognition by TCR transgenic antigen-specific human CTL without negatively affecting the viability and function of T cells is shown [30-32]. However, similar to our results, these reports found vemurafenib unable of sensitizing BRAFV600E melanomas to CTL killing. In contrast, improved efficacy of ACT by vemurafenib in animal models of melanoma, however, has been recently reported [33]. The discrepancy might be explained by use of different cell lines and experimental settings (in vitro versus in vivo), and contributions of the tumor microenvironment.
Upregulation of the immune checkpoint molecule CTLA-4 on activated T cells and its interaction with CD80/86 blocks T cell activation. The fully humanized mAb ipilimumab blocks this interaction resulting in sustained T cell stimulation [21]. Likewise, the programmed death receptor 1 (PD-1) is another member of the B7:CD28 family of costimulatory molecules that regulate T cell activation, whose ligand (PD-L1) is expressed on melanomas. The human mAb, MDX-1106 (BMS-936558), directed against PD-1 plays a role in breaking tolerance [34]. Similarly, ACT uses antigen-specific autologous T cells in eradicating melanomas [35,36]. These modern immune therapies have shown remarkable improvement in the treatment of metastatic melanoma [21,28]. The efficacy of these immune therapies is dependent on the cytotoxic potential of CTL and NK cells [21], and would be predicted to have limited efficacy in patients whose melanomas developed resistance to vemurafenib. In fact, combination of vemurafenib and CTL was ineffective to overcome CTL-resistance [30-32].
Upregulation of various RTKs has been implicated in vemurafenib-resistance [16-20]. Protein expression analysis of various RTKs revealed upregulation of EGFR, c-KIT, Met, and PDGFRβ, and downregulation of IGFRβ in M249(VemR) sublines. In contrast, in M238 (VemR) sublines c-Kit levels were undetectable, levels of EGFR were significantly upregulated and PDGFRβ were reduced, while IGFRβ and Met remained unchanged. These data suggest that the two melanoma lines used in our studies utilize different means to develop resistance to vemurafenib. The activation status and contribution of these changes in vemurafenib-resistance, however, requires further scrutiny. Nonetheless, irrespective of the aberrant upstream signaling operative in VemR sublines, activation of any of the above pathways eventually leads to “resistance to apoptosis”. In fact, the expression of several gene categories including positive regulators of apoptosis, inducers of apoptosis, death domain and TNF/TNFR family, caspases and regulators, were decreased while anti-apoptosis genes were upregulated in M249(VemR) and M249(CTLR) sublines, which might account for their resistance phenotype. Although a larger number of apoptotic genes were modified in M238(VemR) sublines, however, a trend similar to M249 (VemR) was observed.
Pretreatment of both CTL- and vemurafenib-resistant sublines with clinically achievable low micromolar concentration of SAHA largely restored their CTL-sensitivity. TCR transgenic MART CTL was used as a reliable and reproducible source of specific CTL [36,37]. Cross-resistance of VemR and CTLR sublines and immune sensitization by SAHA were also confirmed using naturally occurring MART CTL derived from metastatic melanoma patients. The observed cross-resistance was not limited to M249(VemR) sublines. In fact, VemR derivatives of the MART-127-35 /A*0201 negative M238 line, which cannot be targeted by F5 CTLs, developed cross-resistant to NK cell killing. SAHA regulated the expression pattern of several groups of apoptotic genes simultaneously: there was a general trend that anti-apoptotic genes were reduced while positive apoptosis regulators were upregulated indicating that SAHA sensitizes through cooperative collaboration among multiple groups of apoptotic genes. Although, the precise nature of the resistant factor(s) remains to be identified, by lowering the threshold of apoptosis and generating a pro-apoptotic intracellular setting, SAHA dooms the resistant cells to undergo apoptosis upon immune effector (CTL and NK) encounter.
Upon adoption of alternative resistant mechanisms, VemR sublines lose their survival/growth dependency on BRAFV600E, which may explain the inability of SAHA to restore vemurafenib sensitivity. CTL and NK killing is independent of BRAFV600E, therefore, SAHA-mediated modulation of apoptotic machinery sensitizes both VemR and CTLR sublines to CTL-mediated, but not vemurafenib, killing.
These in vitro results suggest that modulation of aberrant apoptotic machinery via inclusion of SAHA to BRAFV600E targeted therapy as adjuvant will overcome the acquired dual resistance to vemurafenib and CTL and will immunosensitize BRAFV600E-harboring melanomas to CTL and NK killing. These results may provide a rationale molecular basis for future investigations to combine these therapies.
Acknowledgements
The authors wish to acknowledge Dr. Steven Rosenberg (NCI, Surgery Branch) for the kind gift of MCV-MART-1 F5 TCR vector, Dr. Bijay Mukherji (University of Connecticut, Medical Center) for providing MART-1 specific CTLs, and Dr. Antoni Ribas (UCLA, Hematology/Oncology) for the review of the manuscript. The authors wish to thank the UCLA Sequencing and Flow Cytometry Core Facilities for assistance with analysis. This work was supported by the National Center for Research Resources and the National Cancer Institute (NCI) of the National Institutes of Health through Grants Number NIH R21CA 149938 (ARJ), RO1 CA129816 (JSE) and PO1 1088934 (JSE), The Stacy and Evelyn Kesselman Research Fund and The Joy and Jerry Monkarsh Fund.
Disclosure of conflict of interest
The authors claim no conflicts of interest.
Supporting Information
References
- 1.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, Floyd Y, Gray K, Hall S, Hawes R, Hughes J, Kosmidou V, Menzies A, Mould C, Parker A, Stevens C, Watt S, Hooper S, Wilson R, Jayatilake H, Gusterson BA, Cooper C, Shipley J, Hargrave D, Pritchard-Jones K, Maitland N, Chenevix-Trench G, Riggins GJ, Bigner DD, Palmieri G, Cossu A, Flanagan A, Nicholson A, Ho JW, Leung SY, Yuen ST, Weber BL, Seigler HF, Darrow TL, Paterson H, Marais R, Marshall CJ, Wooster R, Stratton MR, Futreal PA. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–54. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 2.Sumimoto H, Imabayashi F, Iwata T, Kawakami Y. The BRAF-MAPK signaling pathway is essential for cancer-immune evasion in human melanoma cells. J Exp Med. 2006;203:1651–6. doi: 10.1084/jem.20051848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tsai J, Lee JT, Wang W, Zhang J, Cho H, Mamo S, Bremer R, Gillette S, Kong J, Haass NK, Sproesser K, Li L, Smalley KS, Fong D, Zhu YL, Marimuthu A, Nguyen H, Lam B, Liu J, Cheung I, Rice J, Suzuki Y, Luu C, Settachatgul C, Shellooe R, Cantwell J, Kim SH, Schlessinger J, Zhang KY, West BL, Powell B, Habets G, Zhang C, Ibrahim PN, Hirth P, Artis DR, Herlyn M, Bollag G. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc Natl Acad Sci U S A. 2008;105:3041–6. doi: 10.1073/pnas.0711741105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bollag G, Hirth P, Tsai J, Zhang J, Ibrahim PN, Cho H, Spevak W, Zhang C, Zhang Y, Habets G, Burton EA, Wong B, Tsang G, West BL, Powell B, Shellooe R, Marimuthu A, Nguyen H, Zhang KY, Artis DR, Schlessinger J, Su F, Higgins B, Iyer R, D’Andrea K, Koehler A, Stumm M, Lin PS, Lee RJ, Grippo J, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, Chapman PB, Flaherty KT, Xu X, Nathanson KL, Nolop K. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature. 2010;467:596–9. doi: 10.1038/nature09454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang H, Higgins B, Kolinsky K, Packman K, Go Z, Iyer R, Kolis S, Zhao S, Lee R, Grippo JF, Schostack K, Simcox ME, Heimbrook D, Bollag G, Su F. RG7204 (PLX4032), a selective BRAFV600E inhibitor, displays potent antitumor activity in preclinical melanoma models. Cancer Res. 2010;70:5518–27. doi: 10.1158/0008-5472.CAN-10-0646. [DOI] [PubMed] [Google Scholar]
- 6.Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, O’Dwyer PJ, Lee RJ, Grippo JF, Nolop K, Chapman PB. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809–19. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Solit DB, Rosen N. Resistance to BRAF inhibition in melanomas. N Engl J Med. 2011;364:772–4. doi: 10.1056/NEJMcibr1013704. [DOI] [PubMed] [Google Scholar]
- 8.Dummer R, Flaherty KT. Resistance patterns with tyrosine kinase inhibitors in melanoma: new insights. Curr Opin Oncol. 2012;24:150–4. doi: 10.1097/CCO.0b013e32834fca92. [DOI] [PubMed] [Google Scholar]
- 9.Johannessen CM, Boehm JS, Kim SY, Thomas SR, Wardwell L, Johnson LA, Emery CM, Stransky N, Cogdill AP, Barretina J, Caponigro G, Hieronymus H, Murray RR, Salehi-Ashtiani K, Hill DE, Vidal M, Zhao JJ, Yang X, Alkan O, Kim S, Harris JL, Wilson CJ, Myer VE, Finan PM, Root DE, Roberts TM, Golub T, Flaherty KT, Dummer R, Weber BL, Sellers WR, Schlegel R, Wargo JA, Hahn WC, Garraway LA. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010;468:968–72. doi: 10.1038/nature09627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Atefi M, von Euw E, Attar N, Ng C, Chu C, Guo D, Nazarian R, Chmielowski B, Glaspy JA, Comin-Anduix B, Mischel PS, Lo RS, Ribas A. Reversing melanoma cross-resistance to BRAF and MEK inhibitors by co-targeting the AKT/mTOR pathway. PLoS One. 2011;6:e28973. doi: 10.1371/journal.pone.0028973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Montagut C, Sharma SV, Shioda T, McDermott U, Ulman M, Ulkus LE, Dias-Santagata D, Stubbs H, Lee DY, Singh A, Drew L, Haber DA, Settleman J. Elevated CRAF as a potential mechanism of acquired resistance to BRAF inhibition in melanoma. Cancer Res. 2008;68:4853–61. doi: 10.1158/0008-5472.CAN-07-6787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jiang CC, Lai F, Thorne RF, Yang F, Liu H, Hersey P, Zhang XD. MEK-independent survival of B-RAFV600E melanoma cells selected for resistance to apoptosis induced by the RAF inhibitor PLX4720. Clin Cancer Res. 2011;17:721–30. doi: 10.1158/1078-0432.CCR-10-2225. [DOI] [PubMed] [Google Scholar]
- 13.Bhatt KV, Spofford LS, Aram G, McMullen M, Pumiglia K, Aplin AE. Adhesion control of cyclin D1 and p27Kip1 levels is deregulated in melanoma cells through BRAF-MEK-ERK signaling. Oncogene. 2005;24:3459–71. doi: 10.1038/sj.onc.1208544. [DOI] [PubMed] [Google Scholar]
- 14.Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, Chen Z, Lee MK, Attar N, Sazegar H, Chodon T, Nelson SF, McArthur G, Sosman JA, Ribas A, Lo RS. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468:973–7. doi: 10.1038/nature09626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bardeesy N, Kim M, Xu J, Kim RS, Shen Q, Bosenberg MW, Wong WH, Chin L. Role of epidermal growth factor receptor signaling in RAS-driven melanoma. Mol Cell Biol. 2005;25:4176–88. doi: 10.1128/MCB.25.10.4176-4188.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaplan FM, Shao Y, Mayberry MM, Aplin AE. Hyperactivation of MEK-ERK1/2 signaling and resistance to apoptosis induced by the oncogenic B-RAF inhibitor, PLX4720, in mutant N-RAS melanoma cells. Oncogene. 2011;30:366–71. doi: 10.1038/onc.2010.408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Paraiso KH, Fedorenko IV, Cantini LP, Munko AC, Hall M, Sondak VK, Messina JL, Flaherty KT, Smalley KS. Recovery of phospho-ERK activity allows melanoma cells to escape from BRAF inhibitor therapy. Br J Cancer. 2010;102:1724–30. doi: 10.1038/sj.bjc.6605714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shao Y, Aplin AE. Akt3-mediated resistance to apoptosis in B-RAF-targeted melanoma cells. Cancer Res. 2010;70:6670–81. doi: 10.1158/0008-5472.CAN-09-4471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pratilas CA, Taylor BS, Ye Q, Viale A, Sander C, Solit DB, Rosen N. (V600E)BRAF is associated with disabled feedback inhibition of RAF-MEK signaling and elevated transcriptional output of the pathway. Proc Natl Acad Sci U S A. 2009;106:4519–24. doi: 10.1073/pnas.0900780106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fedorenko IV, Paraiso KH, Smalley KS. Acquired and intrinsic BRAF inhibitor resistance in BRAF V600E mutant melanoma. Biochem Pharmacol. 2011;82:201–9. doi: 10.1016/j.bcp.2011.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mackiewicz J. What is new in the treatment of advanced melanoma? State of the art. Contemp Oncol (Pozn) 2012;16:363–70. doi: 10.5114/wo.2012.31763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma C, Fan R, Ahmad H, Shi Q, Comin-Anduix B, Chodon T, Koya RC, Liu CC, Kwong GA, Radu CG, Ribas A, Heath JR. A clinical microchip for evaluation of single immune cells reveals high functional heterogeneity in phenotypically similar T cells. Nat Med. 2011;17:738–43. doi: 10.1038/nm.2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jazirehi AR, Baritaki S, Koya RC, Bonavida B, Economou JS. Molecular mechanism of MART-1+/A*0201+ human melanoma resistance to specific CTL-killing despite functional tumor-CTL interaction. Cancer Res. 2011;71:1406–17. doi: 10.1158/0008-5472.CAN-10-1296. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 24.Jazirehi AR, Economou JS. Proteasome inhibition blocks NF-kappaB and ERK1/2 pathways, restores antigen expression, and sensitizes resistant human melanoma to TCR-engineered CTLs. Mol Cancer Ther. 2012;11:1332–41. doi: 10.1158/1535-7163.MCT-11-0814. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 25.Mann BS, Johnson JR, Cohen MH, Justice R, Pazdur R. FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist. 2007;12:1247–52. doi: 10.1634/theoncologist.12-10-1247. [DOI] [PubMed] [Google Scholar]
- 26.Jazirehi AR. Regulation of apoptosis-associated genes by histone deacetylase inhibitors: implications in cancer therapy. Anticancer Drugs. 2010;21:805–13. doi: 10.1097/CAD.0b013e32833dad91. [DOI] [PubMed] [Google Scholar]
- 27.Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, McDermott U, Azizian N, Zou L, Fischbach MA, Wong KK, Brandstetter K, Wittner B, Ramaswamy S, Classon M, Settleman J. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. 2010;141:69–80. doi: 10.1016/j.cell.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johnson DB, Sosman JA. Update on the targeted therapy of melanoma. Curr Treat Options Oncol. 2013;14:280–92. doi: 10.1007/s11864-013-0226-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sullivan RJ, Flaherty KT. Resistance to BRAF-targeted therapy in melanoma. Eur J Cancer. 2013;49:1297–304. doi: 10.1016/j.ejca.2012.11.019. [DOI] [PubMed] [Google Scholar]
- 30.Frederick DT, Piris A, Cogdill AP, Cooper ZA, Lezcano C, Ferrone CR, Mitra D, Boni A, Newton LP, Liu C, Peng W, Sullivan RJ, Lawrence DP, Hodi FS, Overwijk WW, Lizée G, Murphy GF, Hwu P, Flaherty KT, Fisher DE, Wargo JA. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin Cancer Res. 2013;19:1225–31. doi: 10.1158/1078-0432.CCR-12-1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boni A, Cogdill AP, Dang P, Udayakumar D, Njauw CN, Sloss CM, Ferrone CR, Flaherty KT, Lawrence DP, Fisher DE, Tsao H, Wargo JA. Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer Res. 2010;70:5213–9. doi: 10.1158/0008-5472.CAN-10-0118. [DOI] [PubMed] [Google Scholar]
- 32.Comin-Anduix B, Chodon T, Sazegar H, Matsunaga D, Mock S, Jalil J, Escuin-Ordinas H, Chmielowski B, Koya RC, Ribas A. The oncogenic BRAF kinase inhibitor PLX4032/RG7204 does not affect the viability or function of human lymphocytes across a wide range of concentrations. Clin Cancer Res. 2010;16:6040–8. doi: 10.1158/1078-0432.CCR-10-1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koya RC, Mok S, Otte N, Blacketor KJ, Comin-Anduix B, Tumeh PC, Minasyan A, Graham NA, Graeber TG, Chodon T, Ribas A. BRAF inhibitor vemurafenib improves the antitumor activity of adoptive cell immunotherapy. Cancer Res. 2012;72:3928–37. doi: 10.1158/0008-5472.CAN-11-2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity. 2007;27:111–22. doi: 10.1016/j.immuni.2007.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, Royal RE, Topalian SL, Kammula US, Restifo NP, Zheng Z, Nahvi A, de Vries CR, Rogers-Freezer LJ, Mavroukakis SA, Rosenberg SA. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–9. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, Wunderlich JR, Nahvi AV, Helman LJ, Mackall CL, Kammula US, Hughes MS, Restifo NP, Raffeld M, Lee CC, Levy CL, Li YF, El-Gamil M, Schwarz SL, Laurencot C, Rosenberg SA. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J. Clin. Oncol. 2011;29:917–24. doi: 10.1200/JCO.2010.32.2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jazirehi AR, Bonavida B. Resveratrol modifies the expression of apoptotic regulatory proteins and sensitizes non-Hodgkin’s lymphoma and multiple myeloma cell lines to paclitaxel-induced apoptosis. Mol Cancer Ther. 2004;3:71–84. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.