

Abstract
Patients at risk for preterm delivery are frequently administered both antenatal steroids for fetal maturation and magnesium sulfate for neuroprotection. In this study, we investigate whether steroids coadministered with magnesium sulfate preserve blood–brain barrier integrity in neuroinflammation. Human umbilical vein endothelial cells were grown in astroglial conditioned media in a 2-chamber cell culture apparatus. Treatment with tumor necrosis factor-α (TNF-α) or catalytically active recombinant matrix metalloproteinase 9 (MMP-9) simulated neuroinflammation. Membrane integrity was assessed by zona occludens 1 (ZO-1) immunoreactivity, permeability to fluorescently conjugated dextran, and transendothelial electrical resistance (TEER). The TNF-α and MMP-9 treatment increased the rate of dextran transit, decreased TEER, and decreased ZO-1 immunoreactivity at junctional interfaces. Dexamethasone pretreatment alone or in combination with 0.5 mmol/L magnesium sulfate preserved monolayer integrity after inflammatory insult. Magnesium sulfate alone was not protective. This study supports a possible interaction between steroids and magnesium in neuroprotection.
Keywords: magnesium, neuroprotection, matrix metalloproteinase 9
Introduction
Preterm birth affects more than 500 000 pregnancies annually in the United States, and premature infants are at increased risk of multiple complications including intellectual disabilities, cerebral palsy, respiratory, vision, and digestive problems. Neurodevelopmental disorders associated with preterm birth include motor deficits such as cerebral palsy, and cognitive deficits that are related to neuronal and axonal disease affecting the brain.1 Periventricular leukomalacia is the neuropathologic finding associated with cerebral palsy and corresponds to cerebral white matter injury.1,2 Additionally, preterm birth and inflammation are known risk factors for cerebral palsy and subsequent cognitive disabilities. The risk of cerebral palsy increases as gestational age at delivery decreases2 and with active infection such as chorioamnionitis.3,4
Inflammation is one of the principal causes of preterm birth and is a risk factor for the development of cerebral palsy in both preterm and term infants. Inflammatory cytokines such as interleukin (IL)-1β, IL-6, and tumor necrosis factor-α (TNF-α) have been implicated in intrauterine inflammation leading to preterm birth and also result in a fetal inflammatory response that leads to white matter damage and neonatal morbidity.5,6 Elevated levels of amniotic fluid cytokines have been associated with preterm infants at risk of developing brain white matter lesions,6,7 and elevated IL-6 levels in the cord blood of neonates with periventricular leukomalacia support a role for intrauterine inflammation in white matter injury.8,9 Other studies have demonstrated high levels of TNF-α and IL-1β in the brains of neonates with periventricular leukomalacia compared to neonates without periventricular leukomalacia.10–12 Localized or systemic inflammation may result in disruption of the fetal blood–brain barrier, with increased cytokine access to the brain and central nervous system.13 These cytokines then damage oligodendrocytes, the cells responsible for myelination of neurons, leading to hypomyelination.1,8
Matrix metalloproteinases (MMPs) are proteases involved in development, tissue remodeling, and inflammation. Astrocytes, microglia, and neurons have all been shown to express MMPs, and their activity is markedly increased in the presence of trauma, which can lead to direct proteolytic breakdown of the blood–brain barrier.14 The MMPs may also play an active role in inflammation, with leukocytes and macrophages using MMP-9 to migrate across blood vessels and MMPs playing an active role in angiogenesis. The increased oxidative stressors seen in preterm infants may also lead to upregulation of MMP-9 with resultant disruption of the blood–brain barrier and cellular apoptosis.14 The MMPs have been associated with preterm labor, preterm premature rupture of the membranes, and fetal neurologic injury.15–17 The MMP-9 levels in the fetal membranes increase with labor,17 and patients with preterm premature rupture of the membranes have elevated levels of fetal plasma MMP-9 without an association with infection or inflammatory cytokines.15
Because of the detrimental effect of inflammatory cytokines and proteases, prevention of neonatal brain injury may involve preservation of the neonatal blood–brain barrier. Both antenatal corticosteroids and magnesium sulfate are given to patients at risk for preterm delivery and may preserve the integrity of the neonatal blood–brain barrier. Antenatal corticosteroids, given to promote fetal maturation,18 also decrease the incidence of periventricular leukomalacia in preterm infants and reduce cytokine production in both the mother and the fetus.19,20 Magnesium sulfate, administered to decrease the risk of cerebral palsy in cases of threatened premature delivery,21,22 may also decrease cytokine production.23 In the 3 largest trials of magnesium sulfate for neuroprotection, the majority of women also received antenatal corticosteroids for fetal lung maturation.24–26 It remains to be established whether steroids contribute mechanistically to magnesium sulfate’s role in neuroprotection in these trials.
Inflammatory cytokines and/or MMPs may contribute to white matter damage either directly, by producing free radicals which damage cellular white matter, or indirectly, by proteolytically degrading the blood–brain barrier and allowing cytokines produced by maternal and fetal white blood cells to invade the fetal central nervous system.7,27,28 Elevated cytokines, including TNF-α, are present in the amniotic fluid and blood of preterm infants with brain white matter lesions6,29,30 and in the cord blood of term infants born to mothers with clinical or histologic chorioamnionitis.31,32
To differentiate among these mechanisms, we developed an in vitro model of the fetal microvasculature composing the blood–brain barrier to test the hypothesis that magnesium sulfate alone or in combination with dexamethasone can prevent cytokine-mediated blood–brain barrier degradation. The purpose of our study was 2-fold. The first objective was to study the effect of inflammation on blood–brain barrier integrity using the inflammatory mediators TNF-α and MMP-9. The second objective was to determine whether magnesium sulfate and/or dexamethasone preserved blood–brain barrier integrity after TNF-α or MMP-9.
Methods
Research data were derived from a protocol approved by the Madigan Army Medical Center institutional review board. A blood–brain barrier model was constructed using a 2-chamber cell culture apparatus separated by a Millicell polycarbonate (EMD-Millipore, Billerica, Massachusetts) permeable membrane with a 1-µm pore size (Figure 1A).33,34 The apical compartment was seeded with human umbilical vein endothelial cells (HUVECs; EMD-Millipore) with a density of 1000 cells/cm2 (Figure 1B). The HUVECs passage 3 to 4 were used and were grown to confluence over 3 to 4 days in EndoGRO complete medium (EMD-Millipore). In parallel, C6 rat astroglial cells (American Tissue Type Collection, Manassas, Virginia) were plated at 91 000 cells/cm2 and grown to confluence (approximately 2-3 days). The medium was changed to EndoGRO, and the cells were incubated 16 to 24 hours. Conditioned medium from these C6 astroglial cells was applied in 50% concentration to the HUVECs for an additional 16 to 24 hours (Figure 1D).35–37
Figure 1.

Diagram of the in vitro blood–brain barrier model. A, HUVEC cells were seeded in a monolayer on the apical chamber of a 2-chamber cell culture apparatus B, C6 cell astroglial conditioned medium was applied to the top chamber, and membrane integrity was assessed by measuring the rate of fluorescent dye transit or TEER (C). D, Schematic of experimental design, including time and dose schedule for inhibitors. The HUVEC cells were plated onto the apical membrane of the 2-chamber apparatus, and C6 cells were plated in 24-well format. Both cells were grown to confluence (48-96 hours for HUVECs and 24-48 hours for C6 cells), then C6 cells were conditioned by changing the medium to HUVEC Endogro for 24 hours. The resulting conditioned medium was applied to confluent HUVEC monolayers (50%) for 16 to 24 hours. Dexamethasone (or vehicle control) was applied for 16 to 24 hours, followed by either an overnight incubation with TNF-α for 16 hours or MMP-9 for 2 to 3 hours in M199 (low magnesium) media. Then TEER and FITC-dextran permeability assays were performed. FITC-dextran indicates fluorescein isothiocyanate-conjugated dextran; HUVEC, human umbilical vein endothelial cell; TEER, transendothelial electrical resistance.
Transendothelial electrical resistance (TEER) across the apical and basal compartments was measured by volt-ohmmeter ([v-Ωm] Millicell-Electrical Resistance System; EMD-Millipore; Figure 1C). Resistances were reported in ohm-centimeter squared (Ω cm2). Membrane permeability was assessed by measuring the diffusion of fluorescein isothiocyanate-conjugated dextran (FITC-dextran) across the membrane. A multiwell spectrophotometer was used to measure fluorescence in the basal and apical chambers at an excitation wavelength of 485 nm and an emission wavelength of 530 nm, as recommended by the manufacturer (EMD-Millipore).38 Permeability scores were calculated by dividing basal chamber fluorescence intensities by apical chamber intensities and multiplying by 1000. Scores were calculated at 0, 20, 60, 120, 180, and 240 minutes after FITC-dextran addition.
The TNF-α (10 µg/mL)39 was added to the monolayers for 16 to 24 hours during C6 astroglial media conditioning.39 The dose of TNF-α was chosen based on Burke-Gaffney et al39 who used a dose of 10 µg/mL in a study on the effects of cytokines (including TNF-α) on HUVEC monolayers. To assess changes in monolayer integrity after MMP-9, monolayers were conditioned 16 to 24 hours in C6 astroglial media. Dexamethasone (100 nmol/L)40 was added to the monolayers for 16 to 24 hours.33 The dose of dexamethasone was chosen based on Forster et al40 who demonstrated inhibition of MMP-9 production by cEND cells (mouse brain capillary endothelial cells) after treatment with 100 nmol/L dexamethasone. In humans, the average maternal plasma dexamethasone level 2 hours after a 5-mg injection, dosed every 12 hours, is reported to be 37 ng/mL.41 Dexamethasone 100 nmol/L is equivalent to 39.2 ng/mL and is similar to the levels seen in humans 2 hours after injection. The culture medium was exchanged with fresh magnesium-free Dulbecco modified Eagle medium containing 1.25 μg recombinant active MMP-9 (Calbiochem, San Diego, California) and incubated for 2 hours.34,35 Magnesium sulfate treatment (0.5 or 5 mmol/L; Sigma Chemicals, St Louis, Missouri) was concomitant with MMP-9 incubation (2 hours).29,36
Low-dose concentrations of magnesium sulfate (0.5 mmol/L) were comparable to second-quartile levels of magnesium measured in the cord blood of infants born to mothers who received a 6 g loading dose, then 2 g/h infusion for up to 12 hours.42 High doses (5 mmol/L) were approximately 1.5- to 2-fold higher than the highest concentrations measured in the cord blood.42,43 Two hours after MMP-9 application, monolayer integrity was measured by TEER or FITC-dextran transit from the apical to basal chambers as described previously. The conditioning and treatment strategies are described in Figure 1D.
Immunocytochemistry was conducted by seeding HUVECs at a density of 75 000 cells per chamber of an 8-chamber glass slide (Nunc, Fisher Scientific, Pittsburgh, Pennsylvania). After growing to confluence, the cells were treated overnight with 10 µg/mL TNF-α or 1.25 μg MMP-9, fixed in paraformaldehyde, permeabilized in 0.2% triton-X 100, blocked 1 hour with 10% normal goat serum, and incubated with primary antibodies immunoreactive to human zona occludens 1 (ZO-1; raised in rabbit) as recommended by the supplier (Life Technologies, Carlsbad, California). Chamber slides were washed and incubated with a goat-antirabbit secondary antibody conjugated to Alexa-fluor 568 (Life Technologies) diluted in 10% bovine serum albumin (Sigma). Slides were mounted with Prolong Gold containing the nuclear-specific conjugate 4′,6-diamidino-2-phenylindole (DAPI; Life Technologies).
Wide field fluorescent images were digitally captured by an epifluorescent Nikon inverted microscope coupled to a Clara interline CCD camera (Andor Technology, South Windsor, Connecticut) and controlled by the NIS-Elements software Version 3.22.1 (Nikon Instruments, Melville, New York). Image z-stacks were obtained using a Nikon Plan Fluor 40× (1.3 NA, oil; Nikon Instruments) objective and deconvolved with the AutoQuant Blind Deconvolution module within the NIS-Elements software. Whole image level adjustments were made in the acquisition software, converted to TIFF format, and figures assembled using PhotoShop Elements 7.0 (Adobe Systems Inc, Seattle, Washington).
Quantification of linear ZO-1 staining was accomplished. Images of HUVECs cultured in 96-well plates and processed for immunofluorescence were analyzed for total ZO-1 length using a modified procedure described by Liu et al.44 Images from 6 random fields were processed with a band-pass filter using program default settings, a binary mask was generated, and images were despeckled using NIH ImageJ (ver 1.47t).45 Linear ZO-1 length was automatically determined using the AnalyzeSkeleton ImageJ plugin,46 and only line segments greater than 0.5 µm were included for analysis in Microsoft Excel.
Statistical significance was determined by 1-way analysis of variance (ANOVA) with Bonferroni correction using Prism 4 Graphpad software (La Jolla, California). An α of P < .01 was considered statistically significant. The F test confirmed normal distribution of the data.
Results
The HUVECs reached 85% to 90% confluence in 48 hours, and monolayer confluence was confirmed by an increase in TEER. The TEER readings increased from approximately 192 ± 6.5 Ω cm2 immediately after plating to 276.7 ± 4.4 Ω cm2 after 48 to 72 hours.
Preconditioning HUVEC monolayers with C6 astroglial media increased ZO-1 immunoreactivity at the junctional interfaces (Figure 2A and B). Treatment of conditioned HUVEC monolayers with either TNF-α or MMP-9 disrupted monolayer integrity, resulting in punctual staining at the junctional interfaces (Figure 2C and D).
Figure 2.
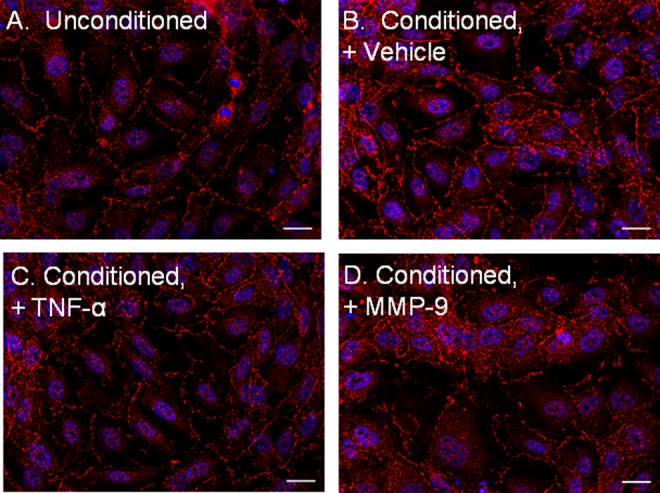
The ZO-1 immunocytochemistry is consistent with tight junction formation after treating HUVECs with astroglial conditioned media. The HUVEC monolayers were treated with unconditioned media (A) or C6 astroglial conditioned media with (B) vehicle, (C) C6 astroglial media with TNF-α (0.1 µg/mL), or (D) C6 astroglial media with MMP-9 (2.5 µg) and probed for ZO-1 by immunocytochemistry (red, ZO-1; blue, DAPI nuclear probe; scale bar = 20 µm). DAPI indicates 4′,6-diamidino-2-phenylindole; HUVECs, human umbilical vein endothelial cells; MMP-9, matrix metalloproteinase 9; TNF-α, tumor necrosis factor-α; ZO-1, zona occludens 1.
Conditioning HUVECs with C6 astroglial media also decreased the amount of FITC-dextran transit across the HUVEC monolayer. Permeability scores decreased from 10 ± 0.7 in vehicle-treated monolayers to 4.5 ± 1.1 in C6 astroglial-conditioned monolayers at 240 minutes. Treatment with TNF-α and the protease MMP-9 was used to study the effects of these agents on endothelial cell tight junctions and endothelial cell permeability. The TNF-α increased the permeability of the monolayer to FITC-dextran: permeability scores increased from 10 ± 0.7 in vehicle controls to 31 ± 8 in TNF-α-treated monolayers at 240 minutes (Figure 3). The MMP-9 treatment increased FITC-dextran transit to permeability scores of 27 ± 6 at 240 minutes (Figure 3). Dexamethasone decreased the amount of FITC-dextran transit in MMP-9-treated monolayers at 240 minutes (from permeability scores of 27 ± 6 to 8 ± 1), but only marginally decreased TNF-α-induced permeability at 240 minutes (from permeability scores of 31 ± 8 to 22 ± 3; Figure 3, right panel).
Figure 3.

Dexamethasone pretreatment reduces MMP-9-induced HUVEC monolayer permeability to FITC-dextran. The HUVEC monolayers were pretreated with dexamethasone (100 nmol/L, 1 hour) prior to 1.25 µg MMP-9 or (10 µg/mL) TNF-α application, and FITC-dextran transit was measured from the apical to the basal chambers at 240 minutes (n = 4-18 replicates per group). *P < .01 relative to vehicle by 1-way ANOVA, post hoc multiple comparisons test with Bonferroni correction. Permeability score = basal chamber fluorescence/apical chamber fluorescence × 1000, read over 20 ms. ANOVA indicates analysis of variance; FITC-dextran, fluorescein isothiocyanate-conjugated dextran; HUVEC, human umbilical vein endothelial cell; MMP-9, matrix metalloproteinase 9; TNF-α, tumor necrosis factor-α.
The MMP-9 and TNF-α treatments also resulted in TEER measurements consistent with a disruption in monolayer integrity. The TNF-α decreased TEER across the HUVEC monolayer from 268 ± 1.8 to 243 ± 2.1 Ω cm2 (Figure 4). The MMP-9 treatment decreased TEER from 268 ± 1.8 to 222 ± 6 Ω cm2 (Figure 4). Dexamethasone pretreatment resulted in higher TEER measurements in MMP-9-treated monolayers compared to monolayers treated with MMP-9 alone (from 259 ± 5 to 222 ± 6 Ω cm2, respectively; Figure 4). Dexamethasone had less positive effect on TEER when cotreated with TNF-α (slight increase from 243 ± 2.1 in vehicle to 259 ± 5 Ω cm2 with dexamethasone; Figure 4).
Figure 4.
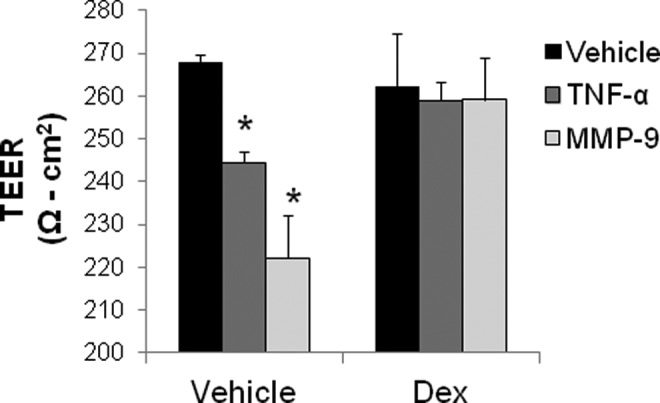
Dexamethasone pretreatment (100 nmol/L) for 1 hour before MMP-9 restores HUVEC monolayer transendothelial electrical resistance (n = 8-20 replicates per group). *P < .01 relative to vehicle by 1-way ANOVA, post hoc multiple comparisons test with Bonferroni correction. ANOVA indicates analysis of variance; HUVEC, human umbilical vein endothelial cell; MMP-9, matrix metalloproteinase 9.
Dexamethasone did not affect the TNF-α-induced increase in endothelial cell permeability at 240 minutes (Figure 5A; permeability scores were 11 ± 0.4 without TNF-α, 28 ± 4 with TNF-α, and 29 ± 3 with TNF-α after pretreatment with dexamethasone). However, dexamethasone prevented the MMP-9-mediated increase in permeability at 240 minutes (Figure 5B, left panel; permeability scores were 12 ± 1 without MMP-9, 34 ± 8 with MMP-9, and 12 ± 3 with MMP-9 after pretreatment with dexamethasone). Combination pretreatment with dexamethasone and low-dose magnesium sulfate (0.5 mmol/L) preserved monolayer integrity after MMP-9 treatment. This did not occur with dexamethasone and high-dose magnesium (5 mmol/L; Figure 5B, middle and right panels). Neither magnesium dose alone affected permeability when treated with MMP-9 (Figure 5B, middle and right panels). Permeability scores at 240 minutes decreased from 34 ± 1 in monolayers treated with MMP-9 and 0.5 mmol/L magnesium to 23 ± 5 after dexamethasone pretreatment followed by MMP-9 and 0.5 mmol/L magnesium (Figure 5B, middle panel). Treatment with 5.0 mmol/L magnesium and TNF-α resulted in an increase in permeability scores at 240 minutes from 11 ± 4 with 5.0 mmol/L magnesium alone to 29 ± 4 with TNF-α alone, and 30 ± 5 with dexamethasone and TNF-α.
Figure 5.
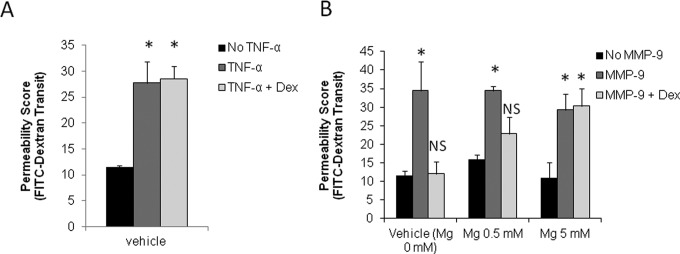
Differential effect of dexamethasone and/or magnesium sulfate on TNF-α and MMP-9-induced permeability increase. A, The TNF-α increases permeability in the presence of magnesium sulfate at 240 minutes. B, Dexamethasone and 0.5 mmol/L magnesium sulfate preserve monolayer membrane integrity after MMP-9 at 240 minutes (n = 2-4 replicates per group in 2 independent experiments). *P < .01 relative to vehicle by 1-way ANOVA, post hoc multiple comparisons test with Bonferroni correction. Permeability score = basal chamber fluorescence/apical chamber fluorescence × 1000, read over 20 ms. ANOVA indicates analysis of variance; MMP-9, matrix metalloproteinase 9; TNF-α, tumor necrosis factor-α.
The ZO-1 immunocytochemistry was quantified by measuring ZO-1 length. A 1-way ANOVA was used to test for ZO-1 length per wide field microscopy view of treated HUVEC cells stained for ZO-1. Length of ZO-1 per µm2 differed significantly across treatment groups, F 3, 20 = 9.04, P = .0005. The pair wise comparison with Bonferroni adjustment between vehicle and MMP-9 showed that differences in ZO-1 lengths approached significance (P = .02). There was no significant difference between vehicle and low-dose magnesium (0.5 mmol/L) + MMP-9 groups (P = 1.0) as shown in Figure 6.
Figure 6.
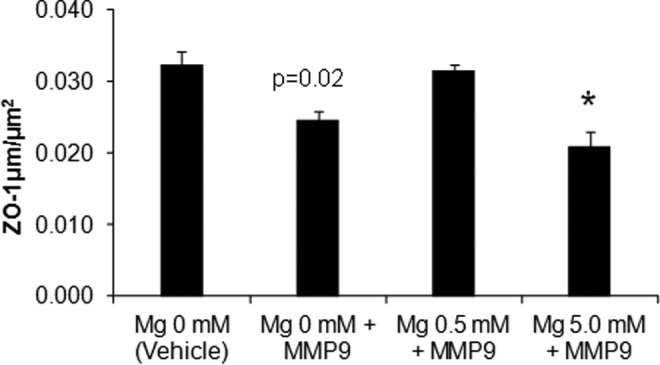
Combination dexamethasone, low-dose magnesium (0.5 mmol/L) preserve tight junctions after MMP-9 treatment. Dexamethasone and 0.5 mmol/L magnesium sulfate prevent MMP-9-induced degradation of ZO-1 immunoreactivity (n = 6-8 replicates per group). *P < .01 relative to vehicle by 1-way ANOVA, post hoc multiple comparison test with Bonferroni correction. ANOVA indicates analysis of variance; MMP-9, matrix metalloproteinase 9; ZO-1, zona occludens 1.
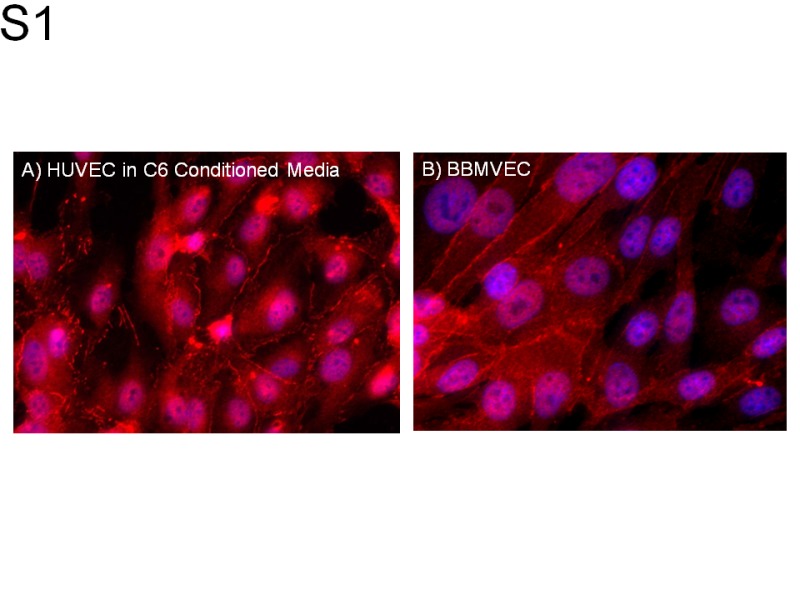
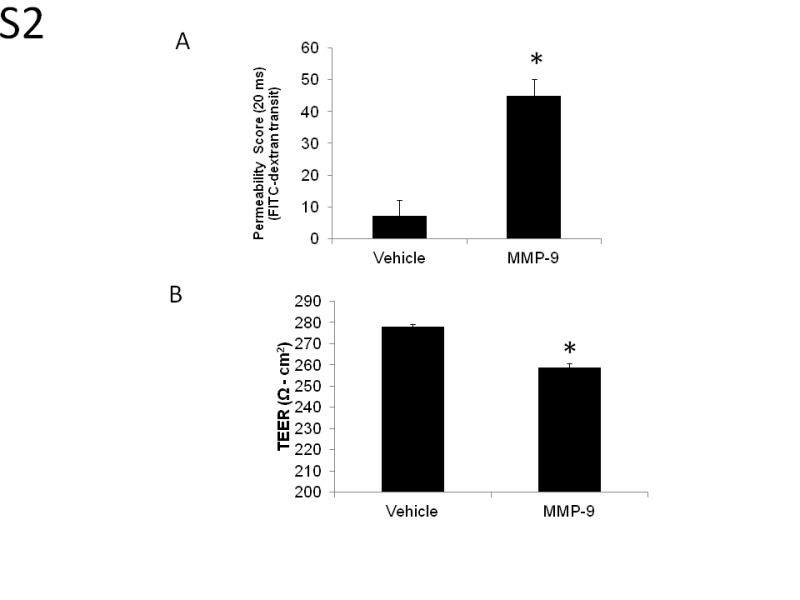
To determine whether measures of plasma membrane integrity in C6-conditioned HUVECs were applicable to endothelial cells of the brain microvasculature, we compared ZO-1 immunocytochemistry in C6-conditioned HUVECs with bovine brain microvascular endothelial cells (BBMVECs; Supplemental Figure S1). The ZO-1 immunocytochemistry in unconditioned BBMVECs was qualitatively comparable to immunocytochemistry in C6-conditioned HUVEC monolayers. Permeability and TEER measurements in BBMVECs were comparable to HUVEC results (Supplemental Figures S2A and 2B). Treatment with MMP-9 increased the permeability from 7.1 ± 6.2 in vehicle controls to 45 ± 10.2 in MMP-9-treated cells at 240 minutes (Supplemental Figure S2A). Treatment with MMP-9 decreased TEER from 278.0 ± 1.1 in vehicle controls to 258.8 ± 2.0 Ω cm2 in MMP-9-treated cells (P = .02; Supplemental Figure S2B).
Discussion
The MMP-9 and TNF-α disrupted vascular integrity, increased vascular endothelial cell permeability, and decreased TEER in an in vitro model of the blood–brain barrier. Dexamethasone preserved tight junction protein expression and reduced permeability between the simulated “blood” and “brain” compartments after MMP-9 but not TNF-α. Monolayer integrity and tight junctions (as measured by ZO-1 immunoreactivity) were preserved only when magnesium was administered at low doses and in the presence of dexamethasone: neither 0.5 mmol/L nor 5 mmol/L magnesium sulfate in isolation prevented monolayer degradation in the presence of MMP-9.
Antenatal corticosteroids are routinely administered to women in preterm labor to promote fetal maturation. In the trials studying neuroprotection, 97% of the patients received at least 1 dose of steroids for fetal maturity in the Maternal Fetal Medicine Network trial (D. J. Rouse, MD, written communication, May 2012).23 Almost all women in the PREMAG trial22 received steroids in addition to magnesium, and all patients in the ACTOMgS04 trial24 received steroids (C. A. Crowther, MD, written communication, February 2013). Earlier studies support a role for antenatal steroids in reducing cerebral white matter lesions in very-low-birth-weight infants,3,46 however, a connection between concomitant steroid administration and magnesium use has not been investigated.
The preterm fetal brain is exquisitely sensitive to hypoxic/ischemic and excitotoxic insults, which result in the white matter damage and axonal injury seen in cerebral palsy.6 Both corticosteroids and magnesium reduce cytotoxic damage to white matter by multiple cellular mechanisms. Corticosteroids counter neuroinflammation primarily by reducing cytokine release from leukocytes and/or glial cells,7,13,28 or by upregulating tissue inhibitors of metalloproteinases (TIMPs), the cellular inhibitor of MMPs.40 In contrast, 2 principal mechanisms have been proposed for neuroprotection by magnesium sulfate: (1) preventing damage to preoligodendrocytes1 by inhibiting ionotropic glutamate receptors, activating N-methyl-d-aspartic acid receptors, and competitively inhibiting calcium influx2 and (2) inhibiting proteolytic and cytotoxic degradation of the blood–brain barrier.36,47
Recent work from our laboratory supports direct inhibition of MMP-9 proteolytic activity by magnesium sulfate.47 It is interesting that in our model, magnesium failed to prevent MMP-9-mediated blood–brain barrier degradation in the absence of dexamethasone. The dose and timing of magnesium may have affected these results, and the timing of steroid and magnesium administration may also affect neuroprotection in the setting of inflammation-induced preterm labor. Steroids and magnesium sulfate may need to be administered soon after infection or inflammatory insult to be effective, and lower doses of magnesium may be more effective with less potential fetal risk.48 Our study also provides a cellular mechanism supporting aggressive reduction in inflammation with multiple agents for maximum clinical benefit.28,49
We simulated neuroinflammation by applying inflammatory mediators with discrete cellular mechanisms of action at the level of the microvasculature comprising the blood–brain barrier: TNF-α, which acts via a surface receptor to induce cellular apoptosis50 and MMP-9, which directly degrades the cellular junctions by intrinsic proteolytic activity.51 In this model, dexamethasone and/or magnesium prevented inhibition of MMP-9-induced blood–brain barrier degradation but did not affect TNF-α induced degradation. This discrepancy may relate to the discrete mechanisms of action of the 2 inflammatory mediators, including a dexamethasone-induced upregulation of TIMPs.40
Although dexamethasone may not be sufficient to inhibit TNF-α receptor-mediated apoptosis in our system, it does inhibit cytokine release from leukocytes and glial cells in vivo.10 Thus, dexamethasone may act in vivo by indirectly inhibiting TNF-α-induced blood–brain barrier degradation, while directly interfering with MMP-9 activity. We plan to conduct future studies incorporating the major producers of inflammatory cytokines such as astrocytes, monocytes, and neutrophils into this model to investigate whether corticosteroids and magnesium affect blood–brain barrier integrity by inhibiting cytokine production in these cells.
The blood–brain barrier is a specialized layer of endothelial cells separating the blood from the brain parenchyma. Endothelial cell tight junctions maintain the low permeability and the high electrical resistance of the blood–brain barrier.33 Neuroinflammatory substances (such as MMPs) can undermine the integrity of tight junctions and functionality of efflux transporters, resulting in an influx of white blood cells and subsequent secretion of proinflammatory cytokines in the central nervous system.52 It is possible that MMP-9-mediated breakdown of the fetal blood–brain barrier in inflammation-mediated spontaneous preterm birth allows increased access of inflammatory cytokines, resulting in damage to the fetal central nervous system.
Several cell line models have been reported in the literature for studying properties of the blood–brain barrier in vitro. Both primary cultures and cell lines have been proposed. Although primary cultures from brain have higher TEER values (exceeding 1200 ohm-cm2), these cells are more difficult to obtain and culture. Cell lines represent a viable alternative to using primary cultures. Both brain microvascular endothelial cells and astroglial-conditioned endothelial cell lines are well established in vitro models of the blood–brain barrier. Both lines have been shown to develop a distinct network of functional endothelial cell tight junctions, low permeability to small molecules, and high TEER.33,37,53–58 Astroglial cells grown in coculture or astroglial conditioned media are both well documented to increase tight junction formation between endothelial cells in blood–brain barrier models.35,37,53–55 Our TEER results for BBMVEC and HUVEC are comparable to those reported in the literature for HUVEC-derived ECV lines (200 Ω cm2))33,37,54 However, the BBMVEC and HUVEC cell lines are distinct cell lines, and results from our study using an HUVEC cell line cannot be directly extrapolated to the BBMVEC cell line.
Directly studying the human fetal blood–brain barrier presents obvious clinical and ethical challenges. In vitro models are often a necessary and useful first step to assert proof of principle and guide development of in vivo studies by enabling the isolation and investigation of specific cellular mechanisms.26,27,54 Such models isolate features of the blood–brain barrier to investigate specific pharmacological mechanisms.26 Such information is an important first step toward additional study of the specific mechanisms in an in vivo animal model.
Our study had several strengths and weaknesses. The strengths of our experimental approach include the use of an in vitro blood–brain barrier model to study in isolation the effects of specific inflammatory mediators and proteases on endothelial tight junctions. This allowed us to investigate a specific mechanism behind the effect of dexamethasone and magnesium both alone and in combination on blood–brain barrier integrity in the context of neuroinflammation.
The weaknesses of our study include the fact that our in vitro model of the neonatal blood–brain barrier is not as complex as the actual blood–brain barrier. The pathophysiology behind inflammation-mediated fetal brain injury may involve excitotoxicity, hypoxia/ischemia, reactive oxygen species, and other mechanisms not studied in our model.1,13 Additionally, the dose of TNF-α used in our model (10 µg/mL) was chosen based on prior work studying the effect of cytokines on HUVEC monolayers.39 Limited human data suggest that elevated levels of TNF-α in the amniotic fluid and blood of preterm infants with brain white matter lesions and in the cord blood of term infants with chorioamnionitis are much lower, with TNF-α levels reported between 0.4 and 247 pg/mL.30–32 The higher doses used in our model could have impacted the results of our study. It is however interesting that even with high levels of TNF-α, there was preservation of monolayer integrity and tight junctions with a combination of low-dose magnesium sulfate and dexamethasone. Our initial data suggest that dexamethasone in combination with low-dose magnesium (0.5 mmol/L) does not affect TNF-α-induced membrane degradation, but a larger number is necessary before definitive conclusions can be reached. Logistically, we were unable to collect both TEER and permeability for some combinations of treatments due to the number of experimental conditions.
Conclusion
Our study of an in vitro model of the blood–brain barrier supports a role of antenatal steroids in the neuroprotective effect of magnesium in the context of neuroinflammation. We recognize that steroids were given to a high percentage of women in the studies of magnesium for neuroprotection. In our model, we saw an effect with steroids and low-dose magnesium; therefore, it seems that timing and dosage of magnesium play critical roles in neuroprotection. These results support further investigation into a therapeutic window and the timing of combination steroid and magnesium treatment to optimize neuroprotection in preterm infants.
Supplementary Material
Supplementary Material
Acknowledgments
We thank Rick Burney, MD, MS, for expert consultation on experimental design, and Dan Fong, PhD (Nikon Instruments), for technical assistance in Nikon deconvolution microscopy. We gratefully acknowledge Ms Cindy Kirker for library support.
Authors’ Note: MAL and DLI contributed equally to this work. This paper was originally presented at the 60th Annual Meeting for the Society for Gynecologic Investigation, Orlando, FL; March 20-23, 2013. The views expressed in this article are those of the author(s) and do not necessarily reflect the official policy or position of the Department of the Navy, Department of the Army, Department of Defense, or the United States Government. Several of the authors are military service members. This work was prepared as part of their official duties. Title 17 U.S.C. 105 provides that “Copyright protection under this title is not available for any work of the United States Government”. Title 17 U.S.C. 101 defines a United States Government work as a work prepared by a military service member or employee of the United States Government as part of that person’s official duties.
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was internally funded by the Graduate Medical Education program at Madigan Army Medical Center .
Supplemental Material: The online supplemental figures are available at http://rs.sagepub.com/supplemental.
References
- 1. Volpe JJ. Brain injury in premature infants: a complex amalgam of destructive and developmental disturbances. Lancet Neurol. 2009;8(1):110–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Conde-Agudelo A, Romero R. Antenatal magnesium sulfate for the prevention of cerebral palsy in preterm infants less than 34 weeks' gestation: a systematic review and metaanalysis. Am J Obstet Gynecol. 2009;200(6):595–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Murata Y, Itakura A, Matsuzawa K, Okumura A, Wakai K, Mizutani S. Possible antenatal and perinatal related factors in development of cystic periventricular leukomalacia. Brain Dev. 2005;27(1):17–21. [DOI] [PubMed] [Google Scholar]
- 4. Wu YW, Colford JM., Jr Chorioamnionitis as a risk factor for cerebral palsy: A meta-analysis. JAMA. 2000;284(11):1417–1424. [DOI] [PubMed] [Google Scholar]
- 5. Gomez R, Romero R, Ghezzi F, Yoon BH, Mazor M, Berry SM. The fetal inflammatory response syndrome. Am J Obstet Gynecol. 1998;179(1):194–202. [DOI] [PubMed] [Google Scholar]
- 6. Yoon BH, Jun JK, Romero R, et al. Amniotic fluid inflammatory cytokines (interleukin-6, interleukin-1beta, and tumor necrosis factor-alpha), neonatal brain white matter lesions, and cerebral palsy. Am J Obstet Gynecol. 1997;177(1):19–26. [DOI] [PubMed] [Google Scholar]
- 7. Yoon BH, Romero R, Park JS, et al. Fetal exposure to an intra-amniotic inflammation and the development of cerebral palsy at the age of three years. Am J Obstet Gynecol. 2000;182(3):675–681. [DOI] [PubMed] [Google Scholar]
- 8. Yoon BH, Park CW, Chaiworapongsa T. Intrauterine infection and the development of cerebral palsy. BJOG. 2003;110(suppl 20):124–127. [DOI] [PubMed] [Google Scholar]
- 9. Yoon BH, Romero R, Yang SH, et al. Interleukin-6 concentrations in umbilical cord plasma are elevated in neonates with white matter lesions associated with periventricular leukomalacia. Am J Obstet Gynecol. 1996;174(5):1433–1440. [DOI] [PubMed] [Google Scholar]
- 10. Deguchi K, Mizuguchi M, Takashima S. Immunohistochemical expression of tumor necrosis factor alpha in neonatal leukomalacia. Pediatr Neurol. 1996;14(1):13–16. [DOI] [PubMed] [Google Scholar]
- 11. Kadhim H, Tabarki B, De Prez C, Sebire G. Cytokine immunoreactivity in cortical and subcortical neurons in periventricular leukomalacia: are cytokines implicated in neuronal dysfunction in cerebral palsy? Acta Neuropathol. 2003;105(3):209–216. [DOI] [PubMed] [Google Scholar]
- 12. Yoon BH, Romero R, Kim CJ, et al. High expression of tumor necrosis factor-alpha and interleukin-6 in periventricular leukomalacia. Am J Obstet Gynecol. 1997;177(2):406–411. [DOI] [PubMed] [Google Scholar]
- 13. McAdams RM, Juul SE. The role of cytokines and inflammatory cells in perinatal brain injury. Neurol Res Int. 2012;. 2012:561494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang H, Adwanikar H, Werb Z, Noble-Haeusslein LJ. Matrix metalloproteinases and neurotrauma: evolving roles in injury and reparative processes. Neuroscientist. 2010;16(2):156–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Romero R, Chaiworapongsa T, Espinoza J, et al. Fetal plasma MMP-9 concentrations are elevated in preterm premature rupture of the membranes. Am J Obstet Gynecol. 2002;187(5):1125–1130. [DOI] [PubMed] [Google Scholar]
- 16. Vadillo-Ortega F, Hernandez A, Gonzalez-Avila G, Bermejo L, Iwata K, Strauss JF, 3r. Increased matrix metalloproteinase activity and reduced tissue inhibitor of metalloproteinases-1 levels in amniotic fluids from pregnancies complicated by premature rupture of membranes. Am J Obstet Gynecol. 1996;174(4):1371–1376. [DOI] [PubMed] [Google Scholar]
- 17. Weiss A, Goldman S, Shalev E. The matrix metalloproteinases (MMPS) in the decidua and fetal membranes. Front Biosci. 2007;12:649–659. [DOI] [PubMed] [Google Scholar]
- 18. Effect of corticosteroids for fetal maturation on perinatal outcomes. NIH Consensus Development Panel on the Effect of Corticosteroids for Fetal Maturation on Perinatal Outcomes. JAMA. 1995;273(5):413–418. [DOI] [PubMed] [Google Scholar]
- 19. Baud O, Foix-L'Helias L, Kaminski M, et al. Antenatal glucocorticoid treatment and cystic periventricular leukomalacia in very premature infants. N Engl J Med. 1999;341(16):1190–1196. [DOI] [PubMed] [Google Scholar]
- 20. Kent A, Lomas F, Hurrion E, Dahlstrom JE. Antenatal steroids may reduce adverse neurological outcome following chorioamnionitis: neurodevelopmental outcome and chorioamnionitis in premature infants. J Paediatr Child Health. 2005;41(4):186–190. [DOI] [PubMed] [Google Scholar]
- 21. American College of Obstetricians and Gynecologists Committee on Obstetric Practice; Society for Maternal-Fetal Medicine.Committee Opinion No. 455: Magnesium sulfate before anticipated preterm birth for neuroprotection. Obstet Gynecol. 2010;115(3):669–671. [DOI] [PubMed] [Google Scholar]
- 22. Doyle LW, Crowther CA, Middleton P, Marret S, Rouse D. Magnesium sulphate for women at risk of preterm birth for neuroprotection of the fetus. Cochrane Database Syst Rev. 2009;(1):CD004661. [DOI] [PubMed] [Google Scholar]
- 23. Sugimoto J, Romani AM, Valentin-Torres AM, et al. Magnesium decreases inflammatory cytokine production: a novel innate immunomodulatory mechanism. J Immunol. 2012;188(12):6338–6346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Crowther CA, Hiller JE, Doyle LW, Haslam RR. Effect of magnesium sulfate given for neuroprotection before preterm birth: a randomized controlled trial. JAMA. 2003;290(20):2669–2676. [DOI] [PubMed] [Google Scholar]
- 25. Marret S, Marpeau L, Zupan-Simunek V, et al. Magnesium sulphate given before very-preterm birth to protect infant brain: the randomised controlled PREMAG trial*. BJOG. 2007;114(3):310–318. [DOI] [PubMed] [Google Scholar]
- 26. Rouse DJ, Hirtz DG, Thom E, et al. A randomized, controlled trial of magnesium sulfate for the prevention of cerebral palsy. N Engl J Med. 2008;359(9):895–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Berger I, Peleg O, Ofek-Shlomai N. Inflammation and early brain injury in term and preterm infants. Isr Med Assoc J. 2012;14(5):318–323. [PubMed] [Google Scholar]
- 28. Dammann O, Leviton A. Maternal intrauterine infection, cytokines, and brain damage in the preterm newborn. Pediatr Res. 1997;42(1):1–8. [DOI] [PubMed] [Google Scholar]
- 29. Baud O, Emilie D, Pelletier E, et al. Amniotic fluid concentrations of interleukin-1beta, interleukin-6 and TNF-alpha in chorioamnionitis before 32 weeks of gestation: histological associations and neonatal outcome. Br J Obstet Gynaecol. 1999;106(1):72–77. [DOI] [PubMed] [Google Scholar]
- 30. Procianoy RS, Silveira RC. Association between high cytokine levels with white matter injury in preterm infants with sepsis. Pediatr Crit Care Med. 2012;13(2):183–187. [DOI] [PubMed] [Google Scholar]
- 31. Dollner H, Vatten L, Halgunset J, Rahimipoor S, Austgulen R. Histologic chorioamnionitis and umbilical serum levels of pro-inflammatory cytokines and cytokine inhibitors. BJOG. 2002;109(5):534–539. [PubMed] [Google Scholar]
- 32. Shalak LF, Laptook AR, Jafri HS, Ramilo O, Perlman JM. Clinical chorioamnionitis, elevated cytokines, and brain injury in term infants. Pediatrics. 2002;110(4):673–680. [DOI] [PubMed] [Google Scholar]
- 33. Deli MA, Abraham CS, Kataoka Y, Niwa M. Permeability studies on in vitro blood-brain barrier models: physiology, pathology, and pharmacology. Cell Mol Neurobiol. 2005;25(1):59–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kacimi R, Giffard RG, Yenari MA. Endotoxin-activated microglia injure brain derived endothelial cells via NF-kappaB, JAK-STAT and JNK stress kinase pathways. J Inflamm (Lond). 2011;8:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hurst RD, Fritz IB. Properties of an immortalised vascular endothelial/glioma cell co-culture model of the blood-brain barrier. J Cell Physiol. 1996;167(1):81–88. [DOI] [PubMed] [Google Scholar]
- 36. Rochelson B, Dowling O, Schwartz N, Metz CN. Magnesium sulfate suppresses inflammatory responses by human umbilical vein endothelial cells (HuVECs) through the NFkappaB pathway. J Reprod Immunol. 2007;73(2):101–107. [DOI] [PubMed] [Google Scholar]
- 37. Tan KH, Dobbie MS, Felix RA, Barrand MA, Hurst RD. A comparison of the induction of immortalized endothelial cell impermeability by astrocytes. Neuroreport. 2001;12(7):1329–1334. [DOI] [PubMed] [Google Scholar]
- 38. Cecchelli R, Dehouck B, Descamps L, et al. In vitro model for evaluating drug transport across the blood-brain barrier. Adv Drug Deliv Rev. 1999;36(2-3):165–178. [DOI] [PubMed] [Google Scholar]
- 39. Burke-Gaffney A, Keenan AK. Modulation of human endothelial cell permeability by combinations of the cytokines interleukin-1 alpha/beta, tumor necrosis factor-alpha and interferon-gamma. Immunopharmacology. 1993;25(1):1–9. [DOI] [PubMed] [Google Scholar]
- 40. Forster C, Kahles T, Kietz S, Drenckhahn D. Dexamethasone induces the expression of metalloproteinase inhibitor TIMP-1 in the murine cerebral vascular endothelial cell line cEND. J Physiol. 2007;580(pt 3):937–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kream J, Mulay S, Fukushima DK, Solomon S. Determination of plasma dexamethasone in the mother and the newborn after administration of the hormone in a clinical trial. J Clin Endocrinol Metab. 1983;56(1):127–133. [DOI] [PubMed] [Google Scholar]
- 42. Rouse DJ. Magnesium sulfate (MgSO4) dose and timing, and umbilical cord Mg++ concentration: relationship to cerebral palsy (CP). Am J Obstet Gynecol. 2008;199(6):S46. [Google Scholar]
- 43. Johnson LH, Mapp DC, Rouse DJ, et al. Association of cord blood magnesium concentration and neonatal resuscitation. J Pediatr. 2012;160(4):573–577 e571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Liu KC, Jacobs DT, Dunn BD, Fanning AS, Cheney RE. Myosin-X functions in polarized epithelial cells. Mol Biol Cell. 2012;23(9):1675–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9(7):671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Arganda-Carreras I, Fernandez-Gonzalez R, Munoz-Barrutia A, Ortiz-De-Solorzano C. 3D reconstruction of histological sections: application to mammary gland tissue. Microsc Res Tech. 2010;73(11):1019–1029. [DOI] [PubMed] [Google Scholar]
- 47. Dolinsky BM Ippolito DL Tinnemore D Stallings JD Zelig CM, Napolitano PG. The effect of magnesium sulfate on the activity of matrix metalloproteinase-9 in fetal cord plasma and human umbilical vein endothelial cells. Am J Obstet Gynecol. 2010;203(4):371 e371–375. [DOI] [PubMed] [Google Scholar]
- 48. Pryde PG, Mittendorf R. Contemporary usage of obstetric magnesium sulfate: indication, contraindication, and relevance of dose. Obstet Gynecol. 2009;114(3):669–673. [DOI] [PubMed] [Google Scholar]
- 49. Agarwal R, Chiswick ML, Rimmer S, et al. Antenatal steroids are associated with a reduction in the incidence of cerebral white matter lesions in very low birthweight infants. Arch Dis Child Fetal Neonatal Ed. 2002;86(2):F96–F101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kaur C, Ling EA. Periventricular white matter damage in the hypoxic neonatal brain: role of microglial cells. Prog Neurobiol. 2009;87(4):264–280. [DOI] [PubMed] [Google Scholar]
- 51. Grossetete M, Phelps J, Arko L, Yonas H, Rosenberg GA. Elevation of matrix metalloproteinases 3 and 9 in cerebrospinal fluid and blood in patients with severe traumatic brain injury. Neurosurgery. 2009;65(4):702–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bazzoni G, Dejana E. Endothelial cell-to-cell junctions: molecular organization and role in vascular homeostasis. Physiol Rev. 2004;84(3):869–901. [DOI] [PubMed] [Google Scholar]
- 53. Grabb PA, Gilbert MR. Neoplastic and pharmacological influence on the permeability of an in vitro blood-brain barrier. J Neurosurg. 1995;82(6):1053–1058. [DOI] [PubMed] [Google Scholar]
- 54. Dittmar MS, Petermichl W, Schlachetzki F, Graf BM, Gruber M. Isoflurane induces endothelial apoptosis of the post-hypoxic blood-brain barrier in a transdifferentiated human umbilical vein endothelial cell model. PLoS One. 2012;7(6):e38260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Raub TJ, Kuentzel SL, Sawada GA. Permeability of bovine brain microvessel endothelial cells in vitro: barrier tightening by a factor released from astroglioma cells. Exp Cell Res. 1992;199(2):330–340. [DOI] [PubMed] [Google Scholar]
- 56. Brown J, Reading SJ, Jones S, et al. Critical evaluation of ECV304 as a human endothelial cell model defined by genetic analysis and functional responses: a comparison with the human bladder cancer derived epithelial cell line T24/83. Lab Invest. 2000;80(1):37–45. [DOI] [PubMed] [Google Scholar]
- 57. Kiessling F, Kartenbeck J, Haller C. Cell-cell contacts in the human cell line ECV304 exhibit both endothelial and epithelial characteristics. Cell Tissue Res. 1999;297(1):131–140. [DOI] [PubMed] [Google Scholar]
- 58. Suda K, Rothen-Rutishauser B, Gunthert M, Wunderli-Allenspach H. Phenotypic characterization of human umbilical vein endothelial (ECV304) and urinary carcinoma (T24) cells: endothelial versus epithelial features. In Vitro Cell Dev Biol Anim. 2001;37(8):505–514. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials