

Abstract
In various animal and human studies, early administration of 17β-estradiol, a strong antioxidant, anti-inflammatory, and anti-apoptotic agent, significantly decreases the severity of injury in the brain associated with cell death. Estrone, the predominant estrogen in postmenopausal women, has been shown to be a promising neuroprotective agent. The overall goal of this project was to determine if estrone mitigates secondary injury following traumatic brain injury (TBI) in rats. Male rats were given either placebo (corn oil) or estrone (0.5 mg/kg) at 30 min after severe TBI. Using a controlled cortical impact device in rats that underwent a craniotomy, the right parietal cortex was injured using the impactor tip. Non-injured control and sham animals were also included. At 72 h following injury, the animals were perfused intracardially with 0.9% saline followed by 10% phosphate-buffered formalin. The whole brain was removed, sliced, and stained for TUNEL-positive cells. Estrone decreased cortical lesion volume (p<0.01) and neuronal injury (p<0.001), and it reduced cerebral cortical levels of TUNEL-positive staining (p<0.0001), and decreased numbers of TUNEL-positive cells in the corpus callosum (p<0.03). We assessed the levels of β-amyloid in the injured animals and found that estrone significantly decreased the cortical levels of β-amyloid after brain injury. Cortical levels of phospho-ERK1/2 were significantly (p<0.01) increased by estrone. This increase was associated with an increase in phospho-CREB levels (p<0.021), and brain-derived neurotrophic factor (BDNF) expression (p<0.0006). In conclusion, estrone given acutely after injury increases the signaling of protective pathways such as the ERK1/2 and BDNF pathways, decreases ischemic secondary injury, and decreases apoptotic-mediated cell death. These results suggest that estrone may afford protection to those suffering from TBI.
Key words: brain-derived neurotrophic factor, estrone, extracellular signal-regulated kinase (ERK), traumatic brain injury
Introduction
Each year in the United States alone, a third of a million persons are hospitalized for traumatic brain injury (TBI), of which approximately 50,000 die. Despite advances in neurosurgical interventions and intensive care monitoring, many of the survivors do not fully recover and are left with permanent disability (Adamides et al., 2006; Alderson and Roberts, 2005; Sahuquillo and Arikan, 2006; Wakai et al., 2005). Secondary brain damage, which is associated with oxidant injury, inflammation, and apoptosis, may begin almost immediately after the primary injury. Brain regions such as the cerebral cortex and hippocampus are affected by secondary injury after the initial impact, resulting in diminished memory and cognition. For example, in mice after TBI, neuronal cell loss is commonly found in the hippocampus, primarily within the CA3 and dentate gyrus regions. In addition, behavioral deficits correlate with neuronal loss in these regions (Conti et al., 1998; Nathoo et al., 2004; Ng et al., 2000; Rink et al., 1995; Tomasevic et al., 2010; Yakovlev et al., 1997).
In various animal and human studies, early administration of estrogen, a strong antioxidant, anti-inflammatory, and anti-apoptotic agent, significantly decreases the severity of injury in the brain caused by early cell death (Kondo et al., 1997; McCullough et al., 2001; Merchenthaler et al., 2003; Simpkins et al., 1997; Sudo et al., 1997; Suzuki et al., 2007; Yang et al., 2000). Steroid hormones were first postulated as a potential resuscitative therapy based on epidemiologic studies revealing that women have better outcomes following a variety of traumatic injuries compared to their male counterparts. Subsequently, early treatment with 17β-estradiol was tested in numerous animal models of resuscitation, ranging from TBI and stroke to spinal cord injury (Chen et al., 2009; Emerson et al., 1993; McCullough et al., 2001; Nakamura et al., 2005; O'Connor et al., 2005; Roof and Hall, 2000; Simpkins et al., 1997; Soustiel et al., 2005; Sribnick et al., 2003,2005; Yang et al., 2003; Yune et al., 2004; Zhang et al., 1998), trauma-related hemorrhage, and sepsis (Knoferl et al., 2000; Mizushima et al., 2000; Palacios and Pang, 1998; Sener et al., 2005; Szalay et al., 2005; Yu et al., 2006a,2006b). Another estrogen, estrone, has been shown to be neuroprotective after injuries such as excitotoxic cell death and stroke (Bae et al., 2000; Perez et al., 2005; Regan and Guo, 1997), but the neuroprotective capacity of this estrogen is not very well described. Estrone is the most abundant estrogen in menopausal women (Blair, 2010; Cirigliano, 2007), and is hypothesized to be neuroprotective in this population.
In this study, we sought to determine if estrone is neuroprotective in combating secondary brain injury in the rat brain after TBI. We hypothesized that estrone decreases apoptosis and promotes neural repair by increasing the production of neurotrophins such as brain-derived neurotrophic factor (BDNF).
Methods
Animals
The experimental animals were housed and cared for by the Animal Resource Center (ARC) at the University of Texas Southwestern Medical Center (UTSWMC), which is certified by the Association for Assessment and Accreditation of Laboratory Animal Care. All procedures listed in this article were approved by the Institutional Animal Care and Use Committee at UTSWMC.
Rat TBI model and treatment
The Benchmark™ Stereotaxic Impactor (Leica Micro-Systems, Wetzlar, Germany) was used to administer severe TBI in rats. In brief, adult Sprague-Dawley male rats (weighing 300–325 g) were anesthetized with isoflurane (5%) and placed in an adapted nose-cone device. To maintain the animal's body temperature during the TBI procedure a heating pad was placed under the animal, and the body temperature was monitored with a rectal thermometer. An incision was made to expose the skull, and the craniotomy procedure was performed. Following the craniotomy procedure, a cortical contusion was delivered to the right hemisphere via a vertically-directed pneumatic cylinder. The impact device is well described (Whalen et al., 1999), and consists of a 4-mm flat-tip impounder that delivers a velocity of 3 m/sec to a depth of 2.5 mm. The skin was closed with surgical wound clips. At 30 min following brain injury, the animals were treated subcutaneously with estrone (0.5 mg/kg) or vehicle (corn oil). Control and sham (craniotomy only) animals were also included as controls. The animals were monitored for pain every 6 h for the first 24 h, and once a day thereafter. To maintain the animal's temperature following surgery, a heating pad was placed under the cage for 24 h.
Tissue preparation and quantification of labeled cells
At 72 h following TBI, the rats received a transcardiac perfusion with 4% formaldehyde in PBS, followed by immersion fixation. The brains were removed and paraffin-embedded. The brains were sectioned (5-μm) using the Thermo Scientific Rotation Microtome (Thermo Scientific, Pittsburgh, PA). To quantify positive staining, the Zeiss Imager A.2 and Axiovision software was used (Carl Zeiss, Oberkochen, Germany). In this study, we devised a cell-counting method based on the stereology counting methods that have been previously reported (Kuhn et al., 1996; Yu et al., 2008; Zhang et al., 2001). Coronal brain slices were collected from a rostral to caudal direction starting at the epicenter of the injury zone. The brain sections (5 μm) spanned a total of 12 microscope slides. A total of 36 slices (3 slices per slide) were collected per brain. Staining of 1/12 from each series of sections were counted.
H&E and Nissl staining
The sectioned brains were stained using hematoxylin and eosin (H&E) and cresyl violet (Nissl) staining. The sections were visualized using the Zeiss Imager A.2. To quantify the amount of Nissl staining, the numbers of intact and non-intact (pyknotic nuclei) cells were counted. The averages of nine different fields of view were calculated for each animal (counts from three fields of view/coronal slice).
Measurement of lesion volume
Image J software was used to assess differences in lesion volume between the control, sham, placebo, and estrone H&E-stained sections. In brief, within the injury zone, the cerebral cortex of the ipsilateral and contralateral hemispheres were traced, and the area from the ipsilateral hemisphere was normalized to the contralateral hemisphere. The density values were subtracted from 1 to determine the lesion volume percentage (1 – ipsilateral area/contralateral area).
Immunohistochemistry
Following the fixation steps, paraffin sections were cut at 5 μm and fixed to the microscope slides. The sections were treated with hydrogen peroxide to inactivate endogenous peroxidase. The primary antibody was added to the sections and the sections were treated overnight at 4°C. The sections were treated with rabbit anti-amyloid-β primary antibody (7.5 μg/mL; Invitrogen, Carlsbad, CA). Single antigen detection was performed using an Alexa fluorophore-conjugated secondary antibody. Detection of the fluorophore was achieved using the Zeiss Imager A.2 microscope. The averages of nine different fields of view were calculated for each animal (counts from three fields of view/coronal slice). The graphs depict the average numbers of positively-stained cells for each group.
TUNEL assay
To determine if estrone protects from apoptosis, a separate set of sections were used in the DeadEnd Fluorometric Apoptosis Detection System, as recommended by the manufacturer (Promega, Fitchburg, WI).
Pretreatment of paraffin-embedded tissues
Briefly, after fixation and paraffin-embedding of the brain, the brain tissue was sectioned. The brain tissue was deparaffinized by immersing the slides in fresh xylene in a Coplin jar for 5 min at room temperature. This step was repeated once for a total of two xylene washes. The slides were washed by immersing the slides in 100% ethanol for 5 min at room temperature in a Coplin jar. The slides were rehydrated by sequentially immersing the slides through graded ethanol washes (100%, 95%, 85%, 70%, and 50%) for 3 min each at room temperature. The slides were then washed by immersion in 0.85% NaCl for 5 min at room temperature, and also washed by immersing the slides in PBS for 5 min at room temperature. The tissue sections were fixed by immersing the slides in 4% methanol-free formaldehyde solution in PBS for 15 min at room temperature, followed by a washing step in PBS for 5 min at room temperature. A total of two PBS washes were conducted. About 100 μL of 20 μg/mL proteinase K was added to each slide to permeabilize the tissue sections, and the slides were incubated for 8–10 min at room temperature. The tissue sections were washed by immersing the slides in PBS for 5 min at room temperature in a Coplin jar. In addition, the tissue sections were fixed and washed in PBS for 5 min at room temperature.
Detection of apoptosis
The tissue sections were covered with 100 μL of equilibration buffer, then the equilibration buffer was removed and 50 μL of rTdT incubation buffer was added to the tissue sections. The treated sections were then incubated at 37°C for 60 min. Following the incubation period, the sections were treated with 2×SSC in a Coplin jar for 15 min at room temperature. The sections were washed by immersing the slides in fresh PBS for 5 min at room temperature. This step was repeated two additional times. The sections were mounted in anti-fade solution (cat. no. S7461; Molecular Probes, Eugene, OR) and covered with glass cover-slips. The edges were sealed using clear nail polish and allowed to dry for 5–10 min. The samples were immediately analyzed using a fluorescence microscope. Positive and negative controls were also prepared as recommended by the manufacturer. The standard fluorescein filter set was used to view the green fluorescence of fluorescein at 520±20 nm, and blue fluorescence of 4,6-diamino-2-phenylindole (DAPI) at 460 nm. The slides were stored at −20°Celsius under dark conditions. The averages of nine different fields of view were calculated for each animal (counts from three fields of view/coronal slice) using the Zeiss Imager A.2 microscope. The graphs depicts the average numbers of TUNEL-positive cells for each group.
Western blot analysis
The harvested rat brains were placed in 0.5 mL of lysis buffer (50 mM Tris (pH 7.4); 150 mM NaCl; 10% glycerol; 1 mM EGTA; 1 mM Na3VO4; 5 mM ZnCl2; 100 mM NaF; 1% Triton X-100; 10 mg/mL aprotinin; 1 mg/mL leupeptin; and 1 mM phenylmethylsulfonyl fluoride), then homogenized and centrifuged for 10 min. Following centrifugation, the supernatant was collected and analyzed for protein concentration. Protein concentrations were determined using the Bio-Rad DC protein assay kit (based on the method of Lowry; Lowry et al., 1951). Total protein (100 μg) was loaded onto a sodium dodecyl sulfate 10% polyacrylamide gel and run at 100 V for 1 h. After electrophoresis, the protein was transferred to a polyvinylidene difluoride membrane (0.22 mm pore size; Bio-Rad Inc., Hercules, CA), and blocked for 3 h in a solution of 3% bovine serum albumin and 0.2% Tween-containing TBS. The membrane was subsequently probed with phospho-p44/42 MAP kinase (1:1000; Cell Signaling Technology, Inc., Beverly, MA), phospho-CREB (1:1000; Cell Signaling), total CREB (1:1000; Cell Signaling), and BDNF (1:1000; Abcam, Cambridge, MA) antibodies. Antibody binding to the membrane was detected using a secondary antibody (goat anti-rabbit) conjugated to horseradish peroxidase (1:20,000; Pierce, Rockford, IL), and visualized with the aid of an imaging system, using enzyme-linked chemiluminescence (ECL; Amersham, Arlington Heights, IL). All blots were stained using ponceau S stain (Sigma-Aldrich, Inc., St. Louis, MO) to ensure equal loading. Densitometric analyses of the protein levels were performed to conduct statistical analyses. In brief, the density from each band from the sham (n=8), placebo (n=8), and estrone (n=8) groups were normalized to the control group (n=8).
Statistical analysis
Data obtained from no fewer than three independent experiments were analyzed using analysis of variance (ANOVA), followed by Tukey's post-hoc test. Groups were considered significantly different if p<0.05. The data are presented as graphs depicting the mean±standard error of the mean (SEM), created with GraphPad software (GraphPad, San Diego, CA).
Results
Estrone treatment after brain injury reduces cortical injury in rats
After TBI, we stained the brain sections using H&E stain and found that the rats treated with placebo had extensive damage to the parietal cortex and hippocampus compared to the control, sham, and estrone groups (Fig. 1A). Using the light microscope, it was determined that the injury to the parietal cortical region led to increased cell death (as indicated by the observed cortical lesions; Fig. 1B). No lesions were observed in the control and sham groups. After TBI, there was a significant (*p<0.006) increase in lesion volume compared to the control and sham groups. The estrone group had a significant (#p<0.01) decrease in the amount of cortical lesions compared to the placebo-treated group (Fig. 1C).
FIG. 1.
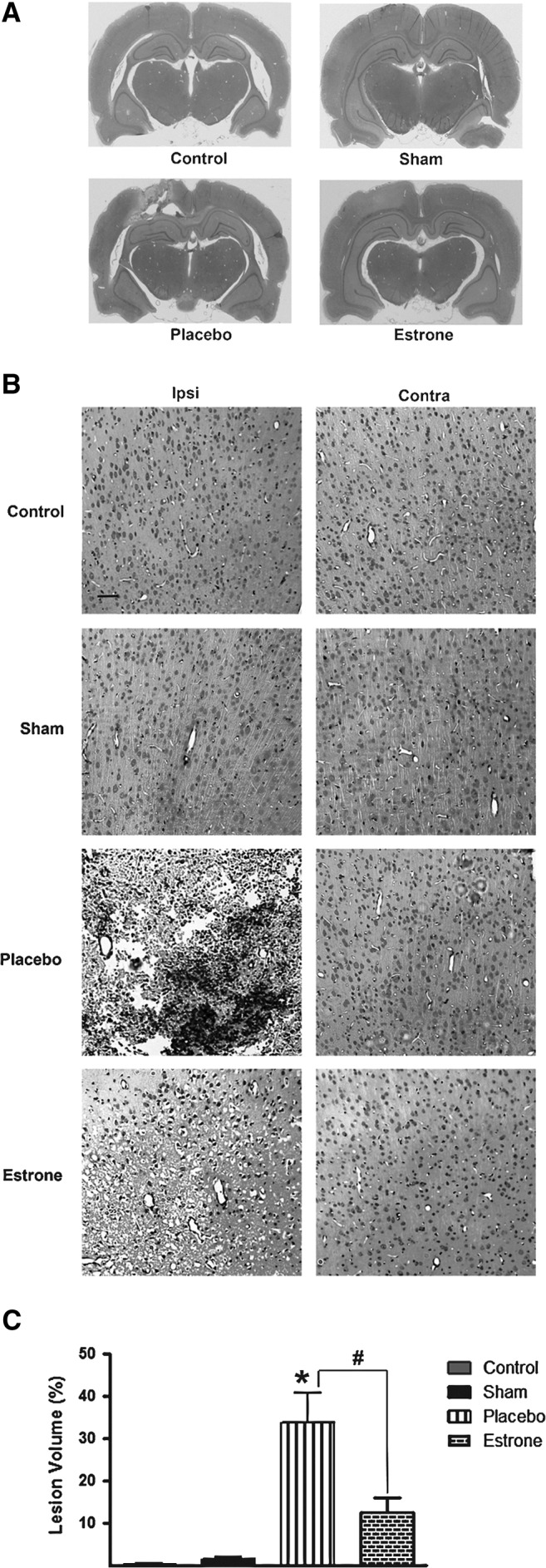
Estrone treatment post-TBI reduced cortical lesion size. At 72 h after injury, the brains were collected, fixed, and stained using H&E stain. In the placebo-treated group, there was an increase in cortical and hippocampal injury (A). Also, there was a significant (*p<0.006) increase in lesion volume within the cerebral cortex of the placebo-treated animals. Estrone treatment resulted in a significant (#p<0.01) decrease in lesion volume (B and C; scale bar=50 μm; n=6 for non-injured controls, 8 for shams, 10 for placebo, and 10 for estrone; TBI, traumatic brain injury; H&E, hematoxylin and eosin; Ipsi, ipsilateral; Contra, contralateral).
After TBI, estrone maintains the integrity of cortical neurons
To elucidate whether cortical neurons are depleted/injured after injury to the parietal cortex, we stained the brain sections with cresyl violet (Nissl stain) to determine the number of intact versus non-intact neurons in the various treatment groups. Compared to the control groups and the estrone-treatment group, we found an increase in the number of pyknotic nuclei in the placebo group, indicating that these neurons were undergoing apoptosis (non-intact morphology) (Fig. 2A). At 72 h after injury, in the placebo group we found a significant decrease in the number of intact neurons compared to the control (p<0.0001), sham (p<0.0001), and estrone-treated (p<0.001) groups. The increase in non-intact cells was significantly different in the placebo group compared to the control (p<0.05) and sham groups (p<0.03), but not the estrone group (Fig. 2B).
FIG. 2.
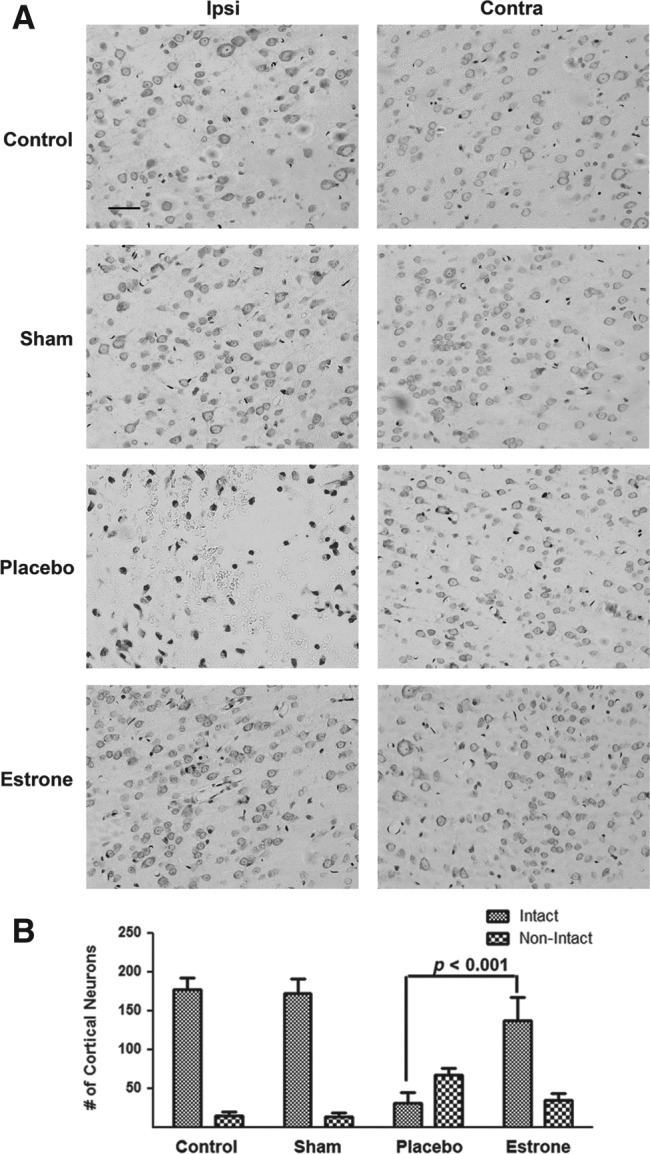
In the TBI rats treated with placebo only, there was an increase in neural degeneration. In the placebo group there was an increase in the number of condensed nuclei (A). Also, there was a decrease in the numbers of cortical neurons in the ipsilateral cortex, and an increase in non-intact neurons (B). One-way analysis of variance with Tukey's post-hoc test was used to determine if the observations were significantly different (p<0.001; scale bar=50 μm; n=6 for non-injured controls, 8 for shams, 12 for placebo, and 11 for estrone; TBI, traumatic brain injury; Ipsi, ipsilateral; Contra, contralateral).
Estrone reduces cortical β-amyloid immunoreactivity after TBI
Increases in β-amyloid in neurons is a reliable neural marker of injury after TBI (Blasko et al., 2004; Bramlett et al., 1997; Lewen et al., 1995,1996; Otsuka et al., 1991). Here, after TBI in rats, β-amyloid was detected and there was an increase in β-amyloid staining in the placebo-treated group compared to the estrone-treated group. There was very little β-amyloid in the control and sham groups. The β-amyloid staining was determined to be localized within cortical neurons (Fig. 3A). In the estrone-treated group, there were significantly lower amounts of amyloid in the cerebral cortex. No difference was observed between the control, sham, and estrone groups (Fig. 3B).
FIG. 3.

Estrone decreases β-amyloid levels in the cortex after TBI. The levels of β-amyloid (a neural marker of injury) were greatly increased in the cerebral cortex in the placebo group. This increase was not observed on the contralateral side of the cerebral cortex (A). Estrone treatment significantly (p<0.01) reduced β-amyloid levels in the cortex (B). One-way analysis of variance with Tukey's post-hoc test was used to determine if the observations were significantly different (n=6 for on-injured controls, 8 for shams, 12 for placebo, and 11 for estrone; DAPI, 4,6-diamino-2-phenylindole; TBI, traumatic brain injury).
Apoptosis in the cortex is significantly decreased in rats treated with estrone after TBI
In the injured rats treated with estrone, we found a decrease in TUNEL-positive cells in the parietal cortex. In the animals treated with placebo, injury to the cortex led to more TUNEL-positive cells. The control and sham groups had very low levels of TUNEL-positive cells (Fig. 4A). We found no difference between groups in the TUNEL staining in the contralateral brain sections (data not shown). The numbers of positive cells were counted in the placebo group and it was determined that there was a significant increase (p<0.01) in the number of TUNEL-positive cells compared to the control, sham, and estrone groups. The estrone group had an approximately 1.5-fold increase in TUNEL-positive cells compared to the control and sham groups (Fig. 4B).
FIG. 4.

Estrone treatment after TBI decreases the number of apoptotic bodies in the cortex. The placebo group had greater DNA damage compared to the control and sham groups (A). Compared to the placebo group, estrone significantly (p<0.0001) lowered DNA damage (B). One-way analysis of variance with Tukey's post-hoc test was used to determine if the observations were significantly different (n=8 for non-injured controls, 8 for shams, 16 for placebo, and 14 for estrone; TBI, traumatic brain injury; DAPI, 4,6-diamino-2-phenylindole).
TUNEL-positive staining is decreased in the corpus callosum
In addition to the parietal cortex, we found an increase in the levels of TUNEL-positive cells in the corpus callosum compared to the estrone-treated groups. The control and sham groups exhibited a low level of TUNEL staining (Fig. 5A). There was no difference between groups in TUNEL staining in the contralateral brain sections (data not shown). This increase was found to be significant (p<0.03), with an approximately 2.5-fold increase in TUNEL-positive cells in the rats treated with placebo versus estrone after TBI (Fig. 5B).
FIG. 5.

The levels of TUNEL-positive cells are decreased in the corpus callosum after TBI in the presence of estrone. In the estrone-treated group there was a decrease in TUNEL staining compared to the placebo group (A). This decrease was significant (B; p<0.03), and only observed in the ipsilateral cerebral cortex. One-way analysis of variance with Tukey's post-hoc test was used to determine if the observations were significantly different (n=8 for non-injured controls, 8 for shams, 16 for placebo, and 14 for estrone; TBI, traumatic brain injury; DAPI, 4,6-diamino-2-phenylindole).
Increased ERK1/2 activity results in increased cortical BDNF expression in TBI rats
To determine if the neuroprotective effects of estrone were involved in pathways of repair such as the MAP kinase pathway we harvested brain tissue, and analyzed total protein lysate using Western blot analysis. We found that compared to the placebo-treated group, estrone treatment resulted in the activation of extracellular signal-regulated kinase (ERK)1/2 (p<0.01; Fig. 6A), and cyclic adenosine monophosphate-responsive element-binding (CREB; p<0.021; Fig. 6B). The activation of these signaling factors was associated with a significant (p<0.0006) increase in the production of BDNF (Fig. 6C). These results suggest that estrone is also involved in growth and regeneration after TBI.
FIG. 6.
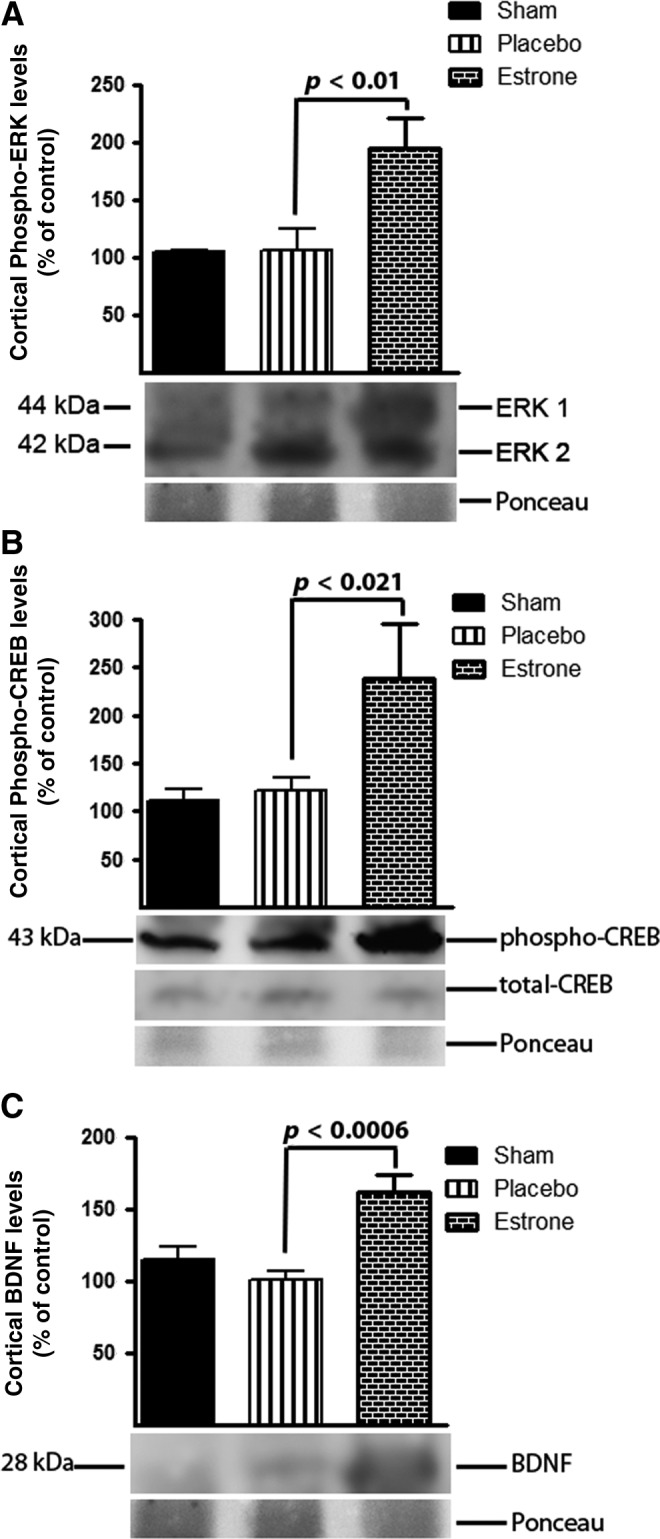
The neuroprotective effects of estrone are mediated by the ERK signaling pathway. We found that after injury, estrone significantly increased the levels of phospho-ERK (A; p<0.01), phospho-CREB (B; p<0.021), and subsequent BDNF expression (p<0.0006) in the cerebral cortex. One-way analysis of variance with Tukey's post-hoc test was used to determine if the observations were significantly different (n=6 for non-injured controls, 8 for shams, 10 for placebo, and 9 for estrone; ERK, extracellular signal-regulated kinase; CREB, cyclic adenosine monophosphate-responsive element-binding; BDNF, brain-derived neurotrophic factor).
Discussion
In this study, we found that estrone is neuroprotective in severely brain-injured rats. Administration of estrone (0.5 mg/kg) at 30 min post-injury to the right parietal cortex resulted in a decrease in lesion size and in the numbers of cells undergoing apoptosis in the cortex and corpus callosum. This neuroprotection was associated with an increase in the levels of phospho-ERK1/2 and CREB. ERK1/2 signaling mediates cellular processes such as mitosis, cellular differentiation, cell survival, and the expression of growth factors (Marshall, 1995; Stanciu and DeFranco, 2002). CREB is a transcription factor and increased activation results in the transcription of BDNF (Carlezon et al., 2005). BDNF is vital for the growth of neurons and establishment of synapses. This growth factor is also neuroprotective and stimulates neurogenesis following injury (Acheson et al., 1995; Bekinschtein et al., 2008; Ferrer et al., 2001; Huang and Reichardt, 2001; Su et al., 2011; Yamada and Nabeshima, 2003). Here, BDNF protein levels were significantly increased by estrone in the cerebral cortex at 72 h after injury. These results suggest that estrone is neuroprotective after TBI, and may stimulate neurogenesis, since BDNF levels increased in the treatment group.
Another interesting finding from this study is that β-amyloid levels were significantly increased in the cerebral cortex in our controlled cortical impact animal model. To our surprise, we found a decrease in β-amyloid immunoreactivity in the estrone-treated group. As demonstrated previously, β-amyloid is a biomarker of neurodegeneration and may predict long-term cognitive deficits. Amyloid precursor protein (APP) is normally found in neurons and is involved in axonal transport. This protein has proven to be a reliable marker for axonal injury after TBI (Blasko et al., 2004; Bramlett et al., 1997; Otsuka et al., 1991, Lewen et al., 1995,1996). Post-translational modification of APP yields toxic amyloid products such as β-amyloid 40/42. Following TBI, an increase in APP and β-amyloid has been observed within the brain following a severe head injury in both animals and humans. For example, in rats that experienced a significant brain injury, a marked increase in APP immunoreactivity was detected in axons (axonal swelling) and cell bodies in the cortex, subcortical region, hippocampus, and thalamus (Blasko et al., 2004; Bramlett et al., 1997; Lewen et al., 1995,1996; Otsuka et al., 1991). In addition to neurons, a significant increase in APP was observed in glial cells (Otsuka et al., 1991). This increase in APP results in significant axonal injury, disruption of axonal transport, and neurodegeneration (Blasko et al., 2004; Bramlett et al., 1997; Ciallella et al., 2002; Lewen et al., 1995,1996; Otsuka et al., 1991; Pierce et al., 1996). Since β-amyloid is known to be neurotoxic, we hypothesize that decreases in β-amyloid levels in the cerebral cortex is one mechanism by which estrone protects after TBI.
This study stems from previous findings from our laboratory that increased pressure in the brain after injury results in the local production of estrogens such as estradiol and estrone. Previously we determined that clinically-relevant increases in pressure results in a substantial increase in the production of aromatase and subsequent estrone and/or estradiol production. The production of estrone was blocked by an aromatase inhibitor, suggesting that estrogen is produced locally after injury and may protect from secondary injury (Gatson et al., 2011).
In various animal and human studies, early administration of estrogen, a strong antioxidant, anti-inflammatory, and anti-apoptotic agent, significantly decreased the severity of injury in the brain caused by early, devastating cell death (Kondo et al., 1997; McCullough et al., 2001; Merchenthaler et al., 2003; Simpkins et al., 1997; Sudo et al., 1997; Suzuki et al., 2007; Yang et al., 2003). The efficacy of 17β-estradiol was tested in numerous animal models of resuscitation ranging from TBI and stroke to spinal cord injury (Khaksari et al., 2011; McCullough et al., 2001; Nakamura et al., 2005; O'Connor et al., 2005; Roof and Hall, 2000; Shahrokhi et al., 2010; Simpkins et al., 1997; Soustiel et al., 2005; Sribnick et al., 2003,2005; Yang et al., 2003; Yune et al., 2004; Zhang et al., 1998), trauma-related hemorrhage, and sepsis (Knoferl et al., 2000; Mizushima et al., 2000; Palacios and Pang, 1998; Sener et al., 2005; Szalay et al., 2005; Yu et al., 2006a,2006b). In these scenarios, studies using old and young, and male and female animals found estrogen to be a strikingly effective therapy. In addition to estradiol, estrone has shown promise as a neuroprotective agent in the brain (Bae et al., 2000; Perez et al., 2005; Regan and Guo, 1997). For example, after stroke and reperfusion injury, estrone reduced apoptotic signaling and decreased lesion size. Also, estrone decreases excitotoxic cell death of neurons after injury. Treatment with estrone was also found to improve long-term cognition in injured animals (Bae et al., 2000; Bhavnani et al., 2003; Kajta et al., 2002,2004,2005; Perez et al., 2005; Regan and Guo, 1997; Shughrue and Merchenthaler, 2003). These studies support the use of estrone as a therapeutic agent to combat secondary brain injury after TBI.
Since estrone is the predominant form of estrogen in post-menopausal women, we posit that physiological levels of estrone (∼ 20 pg/mL) may protect from increased inflammation and oxidative stress after stroke and TBI in older women. In post-menopausal women, even though estradiol levels decrease, androstenedione and testosterone can be converted to estrone and estradiol, respectively (Blair, 2010; Cirigliano, 2007). In some instances the local production of these estrogens may be adequate to afford protection after trauma in certain tissues.
In conclusion, as demonstrated previously in stroke and reperfusion injury studies, estrone also protects the brain from heightened secondary brain injury after TBI. After TBI, estrone decreases β-amyloid levels in the cerebral cortex, decreases apoptosis, and promotes the expression of growth factors such as BDNF. In the clinical setting, estrone may be an effective therapeutic agent to treat brain injury in patients suffering from severe head injuries.
Acknowledgments
The authors would like to thank Martha Romero for technical assistance with the animal perfusion protocol and use of the fluorescence microscope. We would also like to acknowledge Dr. John Shelton for assisting with the slicing of the brains for immunohistochemical analysis and acquiring photomicrographs of the coronal brain slices. This work was supported by the Department of Surgery, Division of Burn/Trauma/Critical Care, University of Texas Southwestern Medical Center, Dallas, Texas.
Author Disclosure Statement
No competing financial interests exist.
References
- Acheson A. Conover J.C. Fandl J.P. DeChiara T.M. Russell M. Thadani A. Squinto S.P. Yancopoulos G.D. Lindsay R.M. A BDNF autocrine loop in adult sensory neurons prevents cell death. Nature. 1995;374:450–453. doi: 10.1038/374450a0. [DOI] [PubMed] [Google Scholar]
- Adamides A.A. Winter C.D. Lewis P.M. Cooper D.J. Kossmann T. Rosenfeld J.V. Current controversies in the management of patients with severe traumatic brain injury. ANZ J. Surg. 2006;76:163–174. doi: 10.1111/j.1445-2197.2006.03674.x. [DOI] [PubMed] [Google Scholar]
- Alderson P. Roberts I. Corticosteroids for acute traumatic brain injury. Cochrane Database Syst Rev. 2005:CD000196. doi: 10.1002/14651858.CD000196.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae Y.H. Hwang J.Y. Kim Y.H. Koh J.Y. Anti-oxidative neuroprotection by estrogens in mouse cortical cultures. J. Korean Med. Sci. 2000;15:327–336. doi: 10.3346/jkms.2000.15.3.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekinschtein P. Cammarota M. Katche C. Slipczuk L. Rossato J.I. Goldin A. Izquierdo I. Medina J.H. BDNF is essential to promote persistence of long-term memory storage. Proc. Natl. Acad. Sci. USA. 2008;105:2711–2716. doi: 10.1073/pnas.0711863105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhavnani B.R. Berco M. Binkley J. Equine estrogens differentially prevent neuronal cell death induced by glutamate. J. Soc. Gynecol. Investig. 2003;10:302–308. doi: 10.1016/s1071-5576(03)00087-x. [DOI] [PubMed] [Google Scholar]
- Blair I.A. Analysis of estrogens in serum and plasma from postmenopausal women: past present, and future. Steroids. 2010;75:297–306. doi: 10.1016/j.steroids.2010.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasko I. Beer R. Bigl M. Apelt J. Franz G. Rudzki D. Ransmayr G. Kampfl A. Schliebs R. Experimental traumatic brain injury in rats stimulates the expression, production and activity of Alzheimer's disease beta-secretase (BACE-1) J. Neural. Transm. 2004;111:523–536. doi: 10.1007/s00702-003-0095-6. [DOI] [PubMed] [Google Scholar]
- Bramlett H.M. Kraydieh S. Green E.J. Dietrich W.D. Temporal and regional patterns of axonal damage following traumatic brain injury: a beta-amyloid precursor protein immunocytochemical study in rats. J. Neuropathol. Exp. Neurol. 1997;56:1132–1141. doi: 10.1097/00005072-199710000-00007. [DOI] [PubMed] [Google Scholar]
- Carlezon W.A., Jr. Duman R.S. Nestler E.J. The many faces of CREB. Trends Neurosci. 2005;28:436–445. doi: 10.1016/j.tins.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Chen S.H. Chang C.Y. Chang H.K. Chen W.C. Lin M.T. Wang J.J. Chen J.C. Chang F.M. Premarin stimulates estrogen receptor-alpha to protect against traumatic brain injury in male rats. Crit. Care Med. 2009;37:3097–3106. doi: 10.1097/CCM.0b013e3181bc7986. [DOI] [PubMed] [Google Scholar]
- Ciallella J.R. Ikonomovic M.D. Paljug W.R. Wilbur Y.I. Dixon C.E. Kochanek P.M. Marion D.W. DeKosky S.T. Changes in expression of amyloid precursor protein and interleukin-1beta after experimental traumatic brain injury in rats. J. Neurotrauma. 2002;19:1555–1567. doi: 10.1089/089771502762300229. [DOI] [PubMed] [Google Scholar]
- Cirigliano M. Bioidentical hormone therapy: a review of the evidence. J. Womens Health (Larchmt.) 2007;16:600–631. doi: 10.1089/jwh.2006.0311. [DOI] [PubMed] [Google Scholar]
- Conti A.C. Raghupathi R. Trojanowski J.Q. McIntosh T.K. Experimental brain injury induces regionally distinct apoptosis during the acute and delayed post-traumatic period. J. Neurosci. 1998;18:5663–5672. doi: 10.1523/JNEUROSCI.18-15-05663.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerson C.S. Headrick J.P. Vink R. Estrogen improves biochemical and neurologic outcome following traumatic brain injury in male rats, but not in females. Brain Res. 1993;608:95–100. doi: 10.1016/0006-8993(93)90778-l. [DOI] [PubMed] [Google Scholar]
- Ferrer I. Krupinski J. Goutan E. Marti E. Ambrosio S. Arenas E. Brain-derived neurotrophic factor reduces cortical cell death by ischemia after middle cerebral artery occlusion in the rat. Acta Neuropathol. 2001;101:229–238. doi: 10.1007/s004010000268. [DOI] [PubMed] [Google Scholar]
- Gatson J.W. Simpkins J.W. Yi K.D. Idris A.H. Minei J.P. Wigginton J.G. Aromatase is increased in astrocytes in the presence of elevated pressure. Endocrinology. 2011;152:207–213. doi: 10.1210/en.2010-0724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang E.J. Reichardt L.F. Neurotrophins: roles in neuronal development and function. Annu. Rev. Neurosci. 2001;24:677–736. doi: 10.1146/annurev.neuro.24.1.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajta M. Lason W. Kupiec T. Effects of estrone on N-methyl-D-aspartic acid- and staurosporine-induced changes in caspase-3-like protease activity and lactate dehydrogenase-release: time- and tissue-dependent effects in neuronal primary cultures. Neuroscience. 2004;123:515–526. doi: 10.1016/j.neuroscience.2003.09.005. [DOI] [PubMed] [Google Scholar]
- Kajta M. Lason W. Bien E. Marszal M. Neuroprotective effects of estrone on NMDA-induced toxicity in primary cultures of rat cortical neurons are independent of estrogen receptors. Pol. J. Pharmacol. 2002;54:727–729. [PubMed] [Google Scholar]
- Kajta M. Marszal M. Kubera M. Lason W. Effects of estrone on quisqualate-induced toxicity in primary cultures of rat cortical neurons. J. Physiol. Pharmacol. 2005;56:233–245. [PubMed] [Google Scholar]
- Khaksari M. Soltani Z. Shahrokhi N. Moshtaghi G. Asadikaram G. The role of estrogen and progesterone, administered alone and in combination, in modulating cytokine concentration following traumatic brain injury. Can. J. Physiol. Pharmacol. 2011;89:31–40. doi: 10.1139/y10-103. [DOI] [PubMed] [Google Scholar]
- Knoferl M.W. Diodato M.D. Angele M.K. Ayala A. Cioffi W.G. Bland K.I. Chaudry I.H. Do female sex steroids adversely or beneficially affect the depressed immune responses in males after trauma-hemorrhage? Arch. Surg. 2000;135:425–433. doi: 10.1001/archsurg.135.4.425. [DOI] [PubMed] [Google Scholar]
- Kondo Y. Suzuki K. Sakuma Y. Estrogen alleviates cognitive dysfunction following transient brain ischemia in ovariectomized gerbils. Neurosci. Lett. 1997;238:45–48. doi: 10.1016/s0304-3940(97)00847-1. [DOI] [PubMed] [Google Scholar]
- Kuhn H.G. Dickinson-Anson H. Gage F.H. Neurogenesis in the dentate gyrus of the adult rat: age-related decrease of neuronal progenitor proliferation. J. Neurosci. 1996;16:2027–2033. doi: 10.1523/JNEUROSCI.16-06-02027.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewen A. Li G.L. Nilsson P. Olsson Y. Hillered L. Traumatic brain injury in rat produces changes of beta-amyloid precursor protein immunoreactivity. Neuroreport. 1995;6:357–360. doi: 10.1097/00001756-199501000-00032. [DOI] [PubMed] [Google Scholar]
- Lewen A. Li G.L. Olsson Y. Hillered L. Changes in microtubule-associated protein 2 and amyloid precursor protein immunoreactivity following traumatic brain injury in rat: influence of MK-801 treatment. Brain Res. 1996;719:161–171. doi: 10.1016/0006-8993(96)00081-9. [DOI] [PubMed] [Google Scholar]
- Lowry O.H. Rosebrough N.J. Farr A.L. Randall R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- Marshall C.J. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell. 1995;80:179–185. doi: 10.1016/0092-8674(95)90401-8. [DOI] [PubMed] [Google Scholar]
- McCullough L.D. Alkayed N.J. Traystman R.J. Williams M.J. Hurn P.D. Postischemic estrogen reduces hypoperfusion and secondary ischemia after experimental stroke. Stroke. 2001;32:796–802. doi: 10.1161/01.str.32.3.796. [DOI] [PubMed] [Google Scholar]
- Merchenthaler I. Dellovade T.L. Shughrue P.J. Neuroprotection by estrogen in animal models of global and focal ischemia. Ann. NY Acad. Sci. 2003;1007:89–100. doi: 10.1196/annals.1286.009. [DOI] [PubMed] [Google Scholar]
- Mizushima Y. Wang P. Jarrar D. Cioffi W.G. Bland K.I. Chaudry I.H. Estradiol administration after trauma-hemorrhage improves cardiovascular and hepatocellular functions in male animals. Ann. Surg. 2000;232:673–679. doi: 10.1097/00000658-200011000-00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T. Hua Y. Keep R.F. Park J.W. Xi G. Hoff J.T. Estrogen therapy for experimental intracerebral hemorrhage in rats. J. Neurosurg. 2005;103:97–103. doi: 10.3171/jns.2005.103.1.0097. [DOI] [PubMed] [Google Scholar]
- Nathoo N. Narotam P.K. Agrawal D.K. Connolly C.A. van Dellen J.R. Barnett G.H. Chetty R. Influence of apoptosis on neurological outcome following traumatic cerebral contusion. J. Neurosurg. 2004;101:233–240. doi: 10.3171/jns.2004.101.2.0233. [DOI] [PubMed] [Google Scholar]
- Ng I. Yeo T.T. Tang W.Y. Soong R. Ng P.Y. Smith D.R. Apoptosis occurs after cerebral contusions in humans. Neurosurgery. 2000;46:949–956. doi: 10.1097/00006123-200004000-00034. [DOI] [PubMed] [Google Scholar]
- O'Connor C.A. Cernak I. Vink R. Both estrogen and progesterone attenuate edema formation following diffuse traumatic brain injury in rats. Brain Res. 2005;1062:171–174. doi: 10.1016/j.brainres.2005.09.011. [DOI] [PubMed] [Google Scholar]
- Otsuka N. Tomonaga M. Ikeda K. Rapid appearance of beta-amyloid precursor protein immunoreactivity in damaged axons and reactive glial cells in rat brain following needle stab injury. Brain Res. 1991;568:335–338. doi: 10.1016/0006-8993(91)91422-w. [DOI] [PubMed] [Google Scholar]
- Palacios B. Pang C.C. Protective effects of ethynylestradiol on the hemodynamic changes induced by lipopolysaccharide in anesthetized rats. J. Cardiovasc. Pharmacol. 1998;31:479–483. doi: 10.1097/00005344-199804000-00001. [DOI] [PubMed] [Google Scholar]
- Perez E. Liu R. Yang S.H. Cai Z.Y. Covey D.F. Simpkins J.W. Neuroprotective effects of an estratriene analog are estrogen receptor independent in vitro and in vivo. Brain Res. 2005;1038:216–222. doi: 10.1016/j.brainres.2005.01.026. [DOI] [PubMed] [Google Scholar]
- Pierce J.E. Trojanowski J.Q. Graham D.I. Smith D.H. McIntosh T.K. Immunohistochemical characterization of alterations in the distribution of amyloid precursor proteins and beta-amyloid peptide after experimental brain injury in the rat. J. Neurosci. 1996;16:1083–1090. doi: 10.1523/JNEUROSCI.16-03-01083.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regan R.F. Guo Y. Estrogens attenuate neuronal injury due to hemoglobin, chemical hypoxia, and excitatory amino acids in murine cortical cultures. Brain Res. 1997;764:133–140. doi: 10.1016/s0006-8993(97)00437-x. [DOI] [PubMed] [Google Scholar]
- Rink A. Fung K.M. Trojanowski J.Q. Lee V.M. Neugebauer E. McIntosh T.K. Evidence of apoptotic cell death after experimental traumatic brain injury in the rat. Am. J. Pathol. 1995;147:1575–1583. [PMC free article] [PubMed] [Google Scholar]
- Roof R.L. Hall E.D. Estrogen-related gender difference in survival rate and cortical blood flow after impact-acceleration head injury in rats. J. Neurotrauma. 2000;17:1155–1169. doi: 10.1089/neu.2000.17.1155. [DOI] [PubMed] [Google Scholar]
- Sahuquillo J. Arikan F. Decompressive craniectomy for the treatment of refractory high intracranial pressure in traumatic brain injury. Cochrane Database Syst Rev. 2006:CD003983. doi: 10.1002/14651858.CD003983.pub2. [DOI] [PubMed] [Google Scholar]
- Sener G. Arbak S. Kurtaran P. Gedik N. Yegen B.C. Estrogen protects the liver and intestines against sepsis-induced injury in rats. J. Surg. Res. 2005;128:70–78. doi: 10.1016/j.jss.2005.02.019. [DOI] [PubMed] [Google Scholar]
- Shahrokhi N. Khaksari M. Soltani Z. Mahmoodi M. Nakhaee N. Effect of sex steroid hormones on brain edema, intracranial pressure, and neurologic outcomes after traumatic brain injury. Can. J. Physiol. Pharmacol. 2010;88:414–421. doi: 10.1139/y09-126. [DOI] [PubMed] [Google Scholar]
- Shughrue P.J. Merchenthaler I. Estrogen prevents the loss of CA1 hippocampal neurons in gerbils after ischemic injury. Neuroscience. 2003;116:851–861. doi: 10.1016/s0306-4522(02)00790-x. [DOI] [PubMed] [Google Scholar]
- Simpkins J.W. Rajakumar G. Zhang Y.Q. Simpkins C.E. Greenwald D. Yu C.J. Bodor N. Day A.L. Estrogens may reduce mortality and ischemic damage caused by middle cerebral artery occlusion in the female rat. J. Neurosurg. 1997;87:724–730. doi: 10.3171/jns.1997.87.5.0724. [DOI] [PubMed] [Google Scholar]
- Soustiel J.F. Palzur E. Nevo O. Thaler I. Vlodavsky E. Neuroprotective anti-apoptosis effect of estrogens in traumatic brain injury. J. Neurotrauma. 2005;22:345–352. doi: 10.1089/neu.2005.22.345. [DOI] [PubMed] [Google Scholar]
- Sribnick E.A. Wingrave J.M. Matzelle D.D. Ray S.K. Banik N.L. Estrogen as a neuroprotective agent in the treatment of spinal cord injury. Ann. NY Acad. Sci. 2003;993:125–133. doi: 10.1111/j.1749-6632.2003.tb07521.x. discussion 159–160. [DOI] [PubMed] [Google Scholar]
- Sribnick E.A. Wingrave J.M. Matzelle D.D. Wilford G.G. Ray S.K. Banik N.L. Estrogen attenuated markers of inflammation and decreased lesion volume in acute spinal cord injury in rats. J. Neurosci. Res. 2005;82:283–293. doi: 10.1002/jnr.20622. [DOI] [PubMed] [Google Scholar]
- Stanciu M. DeFranco D.B. Prolonged nuclear retention of activated extracellular signal-regulated protein kinase promotes cell death generated by oxidative toxicity or proteasome inhibition in a neuronal cell line. J. Biol. Chem. 2002;277:4010–4017. doi: 10.1074/jbc.M104479200. [DOI] [PubMed] [Google Scholar]
- Su C. Underwood W. Rybalchenko N. Singh M. ERK1/2 and ERK5 have distinct roles in the regulation of brain-derived neurotrophic factor expression. J. Neurosci. Res. 2011;89:1542–1550. doi: 10.1002/jnr.22683. [DOI] [PubMed] [Google Scholar]
- Sudo S. Wen T.C. Desaki J. Matsuda S. Tanaka J. Arai T. Maeda N. Sakanaka M. Beta-estradiol protects hippocampal CA1 neurons against transient forebrain ischemia in gerbil. Neurosci. Res. 1997;29:345–354. doi: 10.1016/s0168-0102(97)00106-5. [DOI] [PubMed] [Google Scholar]
- Suzuki S. Gerhold L.M. Bottner M. Rau S.W. Dela Cruz C. Yang E. Zhu H. Yu J. Cashion A.B. Kindy M.S. Merchenthaler I. Gage F.H. Wise P.M. Estradiol enhances neurogenesis following ischemic stroke through estrogen receptors alpha and beta. J. Comp. Neurol. 2007;500:1064–1075. doi: 10.1002/cne.21240. [DOI] [PubMed] [Google Scholar]
- Szalay L. Shimizu T. Schwacha M.G. Choudhry M.A. Rue L.W., 3rd Bland K.I. Chaudry I.H. Mechanism of salutary effects of estradiol on organ function after trauma-hemorrhage: upregulation of heme oxygenase. Am. J. Physiol. Heart Circ. Physiol. 2005;289:H92–H98. doi: 10.1152/ajpheart.01247.2004. [DOI] [PubMed] [Google Scholar]
- Tomasevic G. Raghupathi R. Scherbel U. Wieloch T. McIntosh T.K. Deletion of the p53 tumor suppressor gene improves neuromotor function but does not attenuate regional neuronal cell loss following experimental brain trauma in mice. J. Neurosci. Res. 2010;88:3414–3423. doi: 10.1002/jnr.22491. [DOI] [PubMed] [Google Scholar]
- Wakai A. Roberts I. Schierhout G. Mannitol for acute traumatic brain injury. Cochrane Database Syst Rev. 2005:CD001049. doi: 10.1002/14651858.CD001049.pub2. [DOI] [PubMed] [Google Scholar]
- Whalen M.J. Carlos T.M. Dixon C.E. Schiding J.K. Clark R.S. Baum E. Yan H.Q. Marion D.W. Kochanek P.M. Effect of traumatic brain injury in mice deficient in intercellular adhesion molecule-1: assessment of histopathologic and functional outcome. J. Neurotrauma. 1999;16:299–309. doi: 10.1089/neu.1999.16.299. [DOI] [PubMed] [Google Scholar]
- Yakovlev A.G. Knoblach S.M. Fan L. Fox G.B. Goodnight R. Faden A.I. Activation of CPP32-like caspases contributes to neuronal apoptosis and neurological dysfunction after traumatic brain injury. J. Neurosci. 1997;17:7415–7424. doi: 10.1523/JNEUROSCI.17-19-07415.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K. Nabeshima T. Brain-derived neurotrophic factor/TrkB signaling in memory processes. J. Pharmacol. Sci. 2003;91:267–270. doi: 10.1254/jphs.91.267. [DOI] [PubMed] [Google Scholar]
- Yang S.H. Liu R. Wu S.S. Simpkins J.W. The use of estrogens and related compounds in the treatment of damage from cerebral ischemia. Ann. NY Acad. Sci. 2003;1007:101–107. doi: 10.1196/annals.1286.010. [DOI] [PubMed] [Google Scholar]
- Yang S.H. Shi J. Day A.L. Simpkins J.W. Estradiol exerts neuroprotective effects when administered after ischemic insult. Stroke. 2000:745–749. doi: 10.1161/01.str.31.3.745. discussion 749–750. [DOI] [PubMed] [Google Scholar]
- Yu H.P. Hsieh Y.C. Suzuki T. Shimizu T. Choudhry M.A. Schwacha M.G. Chaudry I.H. Salutary effects of estrogen receptor-beta agonist on lung injury after trauma-hemorrhage. Am. J. Physiol. Lung Cell Mol. Physiol. 2006a;290:L1004–L1009. doi: 10.1152/ajplung.00504.2005. [DOI] [PubMed] [Google Scholar]
- Yu H.P. Shimizu T. Choudhry M.A. Hsieh Y.C. Suzuki T. Bland K.I. Chaudry I.H. Mechanism of cardioprotection following trauma-hemorrhagic shock by a selective estrogen receptor-beta agonist: up-regulation of cardiac heat shock factor-1 and heat shock proteins. J. Mol. Cell Cardiol. 2006b;40:185–194. doi: 10.1016/j.yjmcc.2005.10.001. [DOI] [PubMed] [Google Scholar]
- Yune T.Y. Kim S.J. Lee S.M. Lee Y.K. Oh Y.J. Kim Y.C. Markelonis G.J. Oh T.H. Systemic administration of 17beta-estradiol reduces apoptotic cell death and improves functional recovery following traumatic spinal cord injury in rats. J. Neurotrauma. 2004;21:293–306. doi: 10.1089/089771504322972086. [DOI] [PubMed] [Google Scholar]
- Yu T.S. Zhang G. Liebl D.J. Kernie S.G. Traumatic brain injury-induced hippocampal neurogenesis requires activation of early nestin-expressing progenitors. J. Neurosci. 2008;28:12901–12912. doi: 10.1523/JNEUROSCI.4629-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R.L. Zhang Z.G. Zhang L. Chopp M. Proliferation and differentiation of progenitor cells in the cortex and the subventricular zone in the adult rat after focal cerebral ischemia. Neuroscience. 2001;105:33–41. doi: 10.1016/s0306-4522(01)00117-8. [DOI] [PubMed] [Google Scholar]
- Zhang Y.Q. Shi J. Rajakumar G. Day A.L. Simpkins J.W. Effects of gender and estradiol treatment on focal brain ischemia. Brain Res. 1998;784:321–324. doi: 10.1016/s0006-8993(97)00502-7. [DOI] [PubMed] [Google Scholar]