

Abstract
Ideal cell-free expression systems can theoretically emulate an in vivo cellular environment in a controlled in vitro platform.1 This is useful for expressing proteins and genetic circuits in a controlled manner as well as for providing a prototyping environment for synthetic biology.2,3 To achieve the latter goal, cell-free expression systems that preserve endogenous Escherichia coli transcription-translation mechanisms are able to more accurately reflect in vivo cellular dynamics than those based on T7 RNA polymerase transcription. We describe the preparation and execution of an efficient endogenous E. coli based transcription-translation (TX-TL) cell-free expression system that can produce equivalent amounts of protein as T7-based systems at a 98% cost reduction to similar commercial systems.4,5 The preparation of buffers and crude cell extract are described, as well as the execution of a three tube TX-TL reaction. The entire protocol takes five days to prepare and yields enough material for up to 3000 single reactions in one preparation. Once prepared, each reaction takes under 8 hr from setup to data collection and analysis. Mechanisms of regulation and transcription exogenous to E. coli, such as lac/tet repressors and T7 RNA polymerase, can be supplemented.6 Endogenous properties, such as mRNA and DNA degradation rates, can also be adjusted.7 The TX-TL cell-free expression system has been demonstrated for large-scale circuit assembly, exploring biological phenomena, and expression of proteins under both T7- and endogenous promoters.6,8 Accompanying mathematical models are available.9,10 The resulting system has unique applications in synthetic biology as a prototyping environment, or "TX-TL biomolecular breadboard."
Keywords: Cellular Biology, Issue 79, Bioengineering, Synthetic Biology, Chemistry Techniques, Synthetic, Molecular Biology, control theory, TX-TL, cell-free expression, in vitro, transcription-translation, cell-free protein synthesis, synthetic biology, systems biology, Escherichia coli cell extract, biological circuits, biomolecular breadboard
Introduction
Cell-free expression technology began in the 1950s as purely translational, advancing years later to encompass coupled transcription-translation mechanisms using T7 bacteriophage DNA.11,12 Since then, numerous efforts have been made to optimize the creation of crude cell extract (or E. coli S30 extract).13,14 These optimizations include prolonging cell-free protein synthesis through ATP regeneration or strain modifications, and reducing protocol time and cost.15-17 Alternative cell-free expression systems exist that use reconstituted components in lieu of crude cell extract for expression.5 Both crude cell extract and reconstitution methods have been developed for commercial use.
With the advent of synthetic biology, there is an increased need for a well-characterized platform to test and express engineered biological modules and circuits.18,19 This platform must be versatile, well-characterized, simple to manipulate, and focused on user-supplied components. Despite being developed half a century earlier, cell-free systems based on E. coli intrinsically share these requirements, as they are a simplified in vitro representation of cellular processes without the complexity of growth and metabolism. Additionally, all of the foundational knowledge from in vivo work on E. coli applies readily to E. coli cell-free systems.
Although cell-free expression systems can have applications in synthetic biology, to date the goal of most cell-free expression systems has been the maximization of protein and metabolite yield. This is accomplished by using T7 bacteriophage transcription of sequences driven by T7 promoters.20 Although expression is efficient and robust, these systems serve a highly specialized purpose. Cell regulation methods are limited, target DNA templates must be reengineered to include T7 promoters, and certain sequences such as ribosomal complexes cannot be transcribed and assembled.21,22 Existing cell-free expression systems are unable to maintain high yields while preserving endogenous regulatory mechanisms, a versatility necessary for synthetic biology.
We have developed an endogenous E. coli cell-free expression system that preserves the efficiency of protein expression demonstrated by previous systems but adds additional versatility by allowing expression and regulation based on both endogenous and exogenous (T7 or other) mechanisms. The protocol described here is originally based on Kigawa et al. (2004) and Liu et al. (2005), but has significant modifications. It utilizes Mg- and K- glutamate over Mg- and K- acetate for increased efficiency, removes 2-mercaptoethanol, and lyses cells using a bead-beater.17,23,24 Bead-beating is chosen over homogenization, pressure-based methods, or sonication due to its lower cost and comparable yields to competing systems.23 3-phosphoglyceric acid (3-PGA) is used as the energy source as it was found to give superior protein yields when compared to creatine phosphate and phosphoenolpyruvate.4,25 Our system can produce up to 0.75 mg/ml of reporter protein using either a sigma70-based promoter with lambda-phage operators or a T7-driven promoter, similar to yields from other commercial systems.4,6 Five days are required to produce all necessary reagents (Figure 1). Furthermore, it provides a 98% cost reduction compared to comparable commercial cell-free systems - material costs are $0.11 per 10 μl reaction, which rises to $0.26 with labor included (Figure 2).
Protocol
1. Crude Cell Extract Preparation
Preparing crude cell extract over three days requires two people to conduct efficiently. The protocol functionally consists of three parts: culture growth (step 1.1 to step 1.11), cell lysis (step 1.12 to step 1.37), and extract clarification (step 1.38 to step 1.52). It is presented divided into days for convenience. Ideal extract can produce 0.75 mg/ml of deGFP from plasmid pBEST-OR2-OR1-Pr-UTR1-deGFP-T500 (Addgene #40019), and has a crude cell extract concentration between 27-30 mg/ml of protein.4 However, extract characteristics vary from batch to batch. The following recipe supplies enough for approximately 3,000 single reactions (6 ml crude cell extract). If scaling down, it is recommended to use no less than 1/6 of values given here. Due to time constraints, scaling up is not recommended.
Day 1
Prepare bacterial culture media, culture plate, and media supplements as described in Table 1. See Supplemental Material 1 for recipes.
Streak BL21-Rosetta2 strain from -80 °C onto a 2xYT+P+Cm agar plate and incubate for at least 15 hr at 37 °C or until colonies are readily visible. Note: Chloramphenicol (Cm) is used to select for a plasmid encoding rare tRNAs in the BL21-Rosetta2 strain.
Day 2
Prepare buffers and supplements as described in Table 2. See Supplemental Material 1 for recipes.
Prepare and sterilize materials required for day 3, including: 6 x 4 L Erlenmeyer flasks with aluminum foil cover (autoclaved), 4 x 1 L sterile centrifuge bottles, funnel (autoclaved), 100 g of 0.1 mm glass beads (autoclaved), 2 stir-bars (autoclaved), 1 L and 500 ml graduated cylinder (autoclaved), 2 x 1 L beakers (autoclaved), 3 ml syringe with 18 G needles (sterile), 2-3 float buoys, 2-3 10k MWCO dialysis cassettes (sterile), cuvettes.
Prepare mini-culture 1. Add 4 ml of 2xYT+P media and 4 μl of Cm to a 12 ml sterile culture tube and pre-warm to 37 °C for 30 min.
Inoculate mini-culture 1 with a colony from the 2xYT+P+Cm agar plate. Incubate at 220 rpm, 37 °C for 8 hr.
7 hr and 30 min later, prepare mini-culture 2. Add 50 ml of 2xYT+P media and 50 μl of Cm to a sterile 250 ml Erlenmeyer flask and pre-warm to 37 °C for 30 min.
Inoculate mini-culture 2 with 100 μl of mini-culture 1 and incubate at 220 rpm, 37 °C for 8 hr.
Day 3
Weigh four empty sterile 50 ml Falcon tubes and record mass in Table 3. Chill Falcon tubes on ice; these will be subsequently used in step 1.18.
7 hr and 30 min after step 1.8, prepare final bacterial culture media. Using a sterile 1 L graduated cylinder, transfer 660 ml of 2xYT+P media into each of six 4 L Erlenmeyer flasks and pre-warm to 37 °C for 30 min. Note: 4 L or larger Erlenmeyer flasks are recommended for proper aeration.
Add 6.6 ml of mini-culture 2 into each 4 L Erlenmeyer flask. Incubate at 220 rpm, 37 °C until the culture reaches an OD of 1.5-2.0 at 600 nm (corresponding to mid-log growth phase). Check OD periodically with a 1:10 culture dilution for accuracy. This step should take no more than 3 hr - 3 hr 45 min; rapid growth and collection during mid-log phase is critical for extract quality.
Immediately after growth, transfer all cultures evenly into four 1 L centrifuge bottles and centrifuge at 5000 x g for 12 min at 4 °C to pellet bacterial cells.
While centrifuging, complete S30A buffer preparation by adding 4 ml of 1 M DTT to 2 L of previously prepared S30A. Mix and maintain buffer on ice.
When centrifuging is finished, completely remove supernatant from step 1.12 by decanting and blotting the centrifuge bottles on a sterile paper towel.
Add 200 ml of S30A buffer at 4 °C to each of the four centrifuge bottles, and shake the bottles vigorously until pellet is completely solubilized with no remaining clumps. Centrifuge the four bottles at 5,000 g for 12 min at 4 °C.
Completely remove supernatant from previous step by decanting and blotting the centrifuge bottles on a sterile paper towel.
Repeat steps 1.15 and 1.16.
Add 40 ml S30A buffer at 4 °C to each centrifuge bottle. Transfer each pellet and S30A combination into a chilled Falcon tube from 1.9). Note: This step is to transfer the pellets into a smaller container.
Centrifuge the Falcon tubes at 2,000 g for 8 min at 4 °C. Remove supernatant by decanting.
Re-centrifuge the Falcon tubes at 2,000 g for 2 min at 4 °C. Completely remove residual supernatant by pipette. Keep on ice.
Weigh the four Falcon tubes with pellet and record mass in Table 3. Calculate pellet mass, S30A buffer volume needed, and mass of beads needed based on the specific formulas in Table 3.
Correct loading and bead beating of the pellet is critical to making quality extract, and is the most challenging step. It is recommended to review the video before attempting. Failure to avoid air bubbles and distribute beads evenly will result in inefficient extract.
Add the amount of S30A buffer calculated in Table 3 to each Falcon tube, vortex until homogenous, and return to ice.
While keeping the other Falcon tubes on ice, add beads intermittently to a single Falcon tube in three aliquots, each using 1/3 of the total beads. After addition of each aliquot of beads, vortex for 30 sec. Place Falcon tube on ice between vortex steps and after final vortex. After last aliquot is added, ensure beads are uniformly distributed. A thick paste should be formed.
Prepare a 5 ml (volume) pipette tip by cutting off the end using a sterile razor blade to create a 3-4 mm opening. Dial pipette to 2 ml. Note: Different pipette sets and tips provide different amounts of suction that may not be sufficient to pull and release thick bead-cell solution; a 1 ml pipette tip with end removed can be used in lieu.
Place 20 bead-beating tubes on ice.
Verify high viscosity of cell-bead solution using modified pipet. It should be viscous to the point of barely exiting the pipette tip during ejection. If too viscous, re-adjust pipette tip according to step 1.25. If not viscous enough, beads can be added in increments of (pellet mass * 0.05), to a maximum mass of (pellet mass * 5.1). After each addition of beads, vortex for 30 sec and return to ice. See Figure 3a for a demonstration of viscosity.
Remove bead-cell solution from Falcon tube using modified pipet, and transfer into a sterile bead-beating tube, filling it three-quarters full with bead-cell solution. Spin extremely briefly (1s) on a counter mini-centrifuge to remove air bubbles without redistributing beads. See Figures 3b-d for still images of bead-beating tube loading.
Finish adding bead-cell solution to form a concave meniscus.
Add a very small drop of bead-cell solution onto the inside of a bead-beating tube cap, being careful to not impede the outside lip of the cap; otherwise, the bead-beating tube will not close sufficiently. Tap the cap on a flat surface and verify that there are no air bubbles on the bottom of the cap.
Cap the bead-beating tube with the bead-beating cap from the previous step. Hand to assistant for bead beating. If done correctly, the cap should be tightly sealed, no air bubbles should be visible, and little (if any) bead-cell solution should overflow. Redo the loading process if air bubbles are visible or the cap does not fully close.
Vortex Falcon tube from step 1.24 with the remaining bead-cell solution to ensure even distribution of beads. Repeat steps 1.28 to 1.31 until Falcon tube is empty; then repeat steps 1.24 to 1.31 for each additional Falcon tube.
Conduct steps 1.33 to 1.38 simultaneously. Have assistant take filled bead-beating tubes from 1.31 and place on ice. Once two filled bead-beating tubes have been collected and have been on ice for at least one minute, begin bead beating.
Beat one tube for 30 sec at 46 rpm. Place upside down on ice for 30 sec while beating the other tube.
Repeat previous step such that each filled bead-beating tube has been beat for 1 min total.
Repeat steps 1.33 to 1.35 until 8 filled bead-beating tubes (or the maximum amount the centrifuge can hold) have been processed. Then, construct filter apparatus from 15 ml Falcon (Figure 3e). Add a new bead-beating cap, flat-part face up, to the bottom of a 15 ml Falcon. Then, remove cap from processed bead-beating tube and press micro-chromatography column firmly onto end of processed bead-beating tube until completely sealed. Snap off elution end of micro-chromatography column, and place micro-chromatography column, elution end down, into empty bead-beading tube. Place this complex into 15 ml Falcon. Repeat for all 8 filled bead-beating tubes; keep on ice when complete.
Centrifuge 8 filter apparatuses, Falcon tube uncapped, at 6,000 g for 5 min at 4 °C to separate extract and pellet from beads.
Verify each bead-beating tube has produced viable extract. Properly beat extract will not be turbid, and the pellet will have two distinct layers. Discard all turbid tubes, and transfer the supernatant from non-turbid tubes into individual 1.75 ml micro-centrifuge tubes, taking as little pellet as possible. Keep on ice until all bead-beating tubes have been processed. See Figure 3f comparing a correctly vs. incorrectly processed bead-beating tube.
Centrifuge micro-centrifuge tubes from previous step at 12,000 g for 10 min at 4 °C.
Transfer pellet-free supernatant into empty bead-beating tubes using a pipet, consolidating 500 μl into a new bead-beating tube.
Incubate previous step, with bead-beating caps removed, at 220 rpm, 37 °C for 80 min. This step digests remaining nucleic acids using endogenous exonucleases released during the bead-beading process, and can be done by standing the bead-beating tube up in a tissue culture tube.
Prepare dialysis materials. Complete S30B buffer preparation by adding 2 ml of 1 M DTT to 2 L of previously prepared S30B. Mix and add 900 ml into each of two sterile 1 L beakers. Add sterile magnetic stirrer into each beaker; keep at 4 °C.
After step 1.41, extract should look turbid. Consolidate extract into 1.5 ml aliquots in 1.75 ml micro-centrifuge tubes, and centrifuge at 12000 g for 10 min at 4 °C.
Using a pipet, consolidate pellet-free supernatant into 15 ml Falcon tubes on ice, and mix well by capping the tube and inverting. Save 10 μl of supernatant on ice for step 1.47.
Determine total amount of extract produced, and hydrate the necessary number of 10k MWCO dialysis cassettes by submersing in S30B for 2 min, assuming 2.5 ml of extract per cassette.
Load cassettes with 2.5 ml of extract. Each beaker can take up to 2 cassettes; dialyze, stirring, at 4 °C for 3 hr. Note: partial loading of cassettes is acceptable. Dialyzing increases protein production yield.
During the previous step, characterize extract protein concentration with a Bradford assay, using extract saved in step 1.44. See Supplemental Material 2 for details.
After dialysis is complete, aliquot extract by 1.5 ml in 1.75 ml micro-centrifuge tubes. Centrifuge at 12,000 g for 10 min at 4 °C. A pellet will form at the bottom of the tube.
Consolidate clear supernatant from previous step by pipetting into a 15 ml Falcon tube on ice. Homogenize by inverting 5-10x.
Based on concentration determined by Bradford in step 1.47, determine amount of extract to aliquot into individual 1.75 ml tubes. Each individual tube should have a volume with 810-900 mg of total protein. Extract should have a total protein concentration greater than 27 mg/ml. This step requires assistance to conduct expediently. Note: Aliquot extract below 30 mg/ml into 30 μl aliquots, and scale if concentration is higher; for example, aliquot extract at 28 mg/ml by 30 μl, and aliquot extract at 32 mg/ml by 28.1 μl.
Aliquot extract following step 1.50, taking care to avoid bubbles. Flash-freeze extract in liquid nitrogen. Note: Aliquots with bubbles can be removed by centrifuging at 10,000 x g for 30 sec at 4 °C.
Remove tubes from liquid nitrogen using a strainer and immediately store at -80 °C. Safety: Wear protective eyewear; the caps of extract tubes may come off due to the temperature difference between liquid nitrogen and room temperature.
2. Amino Acid Solution Preparation
Amino Acid Solution should be prepared in bulk. The following recipe utilizes one full kit of RTS Amino Acid Sampler, supplying enough for approximately 11,000 single reactions. If scaling down, it is recommended to use no less than half a kit. Each amino acid in the stock is supplied at 1.5 ml, 168 mM, except for leucine at 140 mM. The final composition of Amino Acid Solution is: leucine, 5 mM, all other amino acids, 6 mM. This is 4x working concentration.
Remove all 20 amino acids from -20 °C and thaw at room temperature. Once thawed, vortex until amino acids dissolve, incubating at 37 °C if necessary. After amino acids are dissolved, put all amino acids on ice except for Asn, Phe, and Cys, which are kept at room temperature. Cys may not fully dissolve.
On ice, add 12 ml of sterile water to a sterile 50 ml Falcon tube.
Add 1.5 ml of each amino acid in the following order, taking care to vortex the Falcon tube after each addition and to keep the solution on ice: Ala, Arg, Asn, Asp, Gln, Glu, Gly, His, Ile, Lys, Met, Phe, Pro, Ser, Thr, Val, Trp, Tyr, Leu, Cys. Cys can be added as a suspension. After addition, vortex until solution is relatively clear, incubating at 37 °C if necessary. Cys may not fully dissolve.
Aliquot Amino Acid Solution into 50 tubes at 26 μl each on ice. Aliquot the rest at 500 μl per tube on ice. The 26 μl aliquots will be used for calibrating extract, while the 500 μl aliquots will be used for preparing buffer. While aliquoting, vortex the main stock frequently to avoid unequal distribution of suspension.
Flash freeze aliquots in liquid nitrogen and store at -80 °C. Safety: Wear protective eyewear; the caps of extract tubes may come off due to the temperature difference between liquid nitrogen and room temperature.
Optional: Conduct an activity assay of newly-made Amino Acid Solution against previously made Amino Acid Solutions.
3. Energy Solution Preparation
Energy Solution is used both for calibrating crude cell extract and for creating buffer, and should be prepared in bulk. The following recipe supplies enough for approximately 10000 single reactions. If scaling down, it is recommended to use no less than 1/24 of values given here. As the Energy Solution is a significant monetary cost, first time users may want to prepare at 1/24 scale. The final composition of Energy Solution is: HEPES pH 8 700 mM, ATP 21 mM, GTP 21 mM, CTP 12.6 mM, UTP 12.6 mM, tRNA 2.8 mg/ml, CoA 3.64 mM, NAD 4.62 mM, cAMP 10.5 mM, folinic Acid 0.95 mM, Spermidine 14 mM, 3-PGA 420 mM. This is 14x working concentration. If desired, each individual item in Table 4 can be stored at -80 °C for later use.
Remove all chemicals in Table 4 from -80 °C, -20 °C, or 4 °C to room temperature for 30 min.
Prepare stock solutions as described in Table 4. See Supplemental Material 1 for recipes. Place all solutions on ice after preparation.
In a 15 ml Falcon tube, add in the following order, taking care to vortex the Falcon tube after each addition and to keep the solutions on ice: 3.6 ml 2 M HEPES, 144 μl water, 1.39 ml nucleotide mix, 576 μl 50 mg/ml tRNA, 576 μl 65 mM CoA, 276 μl 175 mM NAD, 170 μl 650 mM cAMP, 288 μl 33.9 mM Folinic acid, 144 μl 1 M spermidine, and 3.09 ml 1.4 M 3-PGA.
Aliquot Energy Solution into 50 tubes at 7 μl each on ice. Aliquot the rest at 150 μl per tube on ice. The 7 μl aliquots will be used for calibrating extract, while the 150 μl aliquots will be used for preparing buffer. While aliquoting, vortex the main stock frequently.
Flash freeze aliquots in liquid nitrogen and store at -80 °C. Safety: Wear protective eyewear; the caps of extract tubes may come off due to the temperature difference between liquid nitrogen and room temperature.
Optional: Conduct an activity assay of newly made Energy Solution against previously made Energy Solutions.
4. Buffer Preparation
Buffer Preparation requires the completion of Crude Cell Extract Preparation, Amino Acid Solution Preparation, and Energy Solution Preparation. Each buffer is unique to a batch of crude cell extract. Mg-glutamate, K-glutamate, and DTT (in that order) are optimized in this section to produce reactions with maximum levels of expression. The following protocol utilizes a pre-written template, TXTL_e(template)_calibration_JoVE.xlsx (Supplemental Material 3), to calibrate pre-prepared crude cell extract and prepare buffer. However, one can also calibrate crude cell extract and prepare buffer without the template by optimizing Mg-glutamate, K-glutamate, and DTT manually and setting up buffer such that along with extract, it is 75% of a total reaction volume. If calibrating manually, final reaction conditions can be found in step 5.
Complete the "General Data" form.
Thaw on ice 100 mM Mg-glutamate (4 °C), 3 M K-glutamate (4 °C), 6 mM Amino Acid Solution (26 μl, -80 °C), Energy Solution (7 μl, -80 °C), 100 mM DTT (-20 °C), positive control DNA (-20 °C), 40% PEG-8000 (4 °C), crude cell extract (-80 °C), and water (4 °C). Note: Use 1 nM working concentration pBEST-OR2-OR1-Pr-UTR1-deGFP-T500 (Addgene plasmid 40019) for the positive control (excitation 485 nm, emission 525 nm), or another reference which produces high signal intensity.4
Prepare seven 10.5 μl reactions, testing a range of 4-10 mM additional Mg-glutamate, by aliquoting set amounts of stock Mg-glutamate into individual micro-centrifuge tubes. Note: Although 10.5 μl reactions are initially prepared, the final reaction is 10 μl.
Prepare master mix as indicated in the template under "Mg-glutamate calibration," adding an extra 80 mM of K-glutamate. Keep on ice and vortex after the addition of each item. Note: The values given here and in the template are in addition to the amounts of Mg-glutamate, K-glutamate, and DTT present in the S30B buffer used to make crude cell extract.
Add master mix to samples containing Mg-glutamate and prepare reactions. See steps 5.10 to 5.13 for detailed instructions.
Run reaction at 29 °C, either in an incubator or a plate reader.
Determine optimum Mg-glutamate concentration by end-expression level and maximal rate of protein expression (Figure 4a). Note: Runtimes vary depending on experiment but typically last under 8 hr.
Repeat steps 4.2 to 4.7 for K-glutamate under "K-glutamate calibration," setting Mg-glutamate levels to those found in step 4.7.
Repeat steps 4.2 to 4.7 for DTT under "DTT calibration," setting Mg-glutamate levels to those found in step 4.7 and K-glutamate levels to those found in step 4.8. Note: We have found that added DTT does not significantly affect end-expression levels.
Use values found in calibration under "Buffer composition" to determine the composition of buffer to be prepared. Based on the amount of crude cell extract produced, a master mix recipe is produced for a set amount of buffers.
Thaw aliquots as listed in master mix recipe on ice. Once thawed, prepare master mix, keeping on ice and vortexing after the addition of each item.
Aliquot by amount stated under "Buffer composition." Flash-freeze buffer tubes in liquid nitrogen. While aliquoting, vortex the main stock frequently.
Remove tubes from liquid nitrogen using a strainer and immediately store at -80 °C. Safety: Wear protective eyewear; the caps of extract tubes may come off due to the temperature difference between liquid nitrogen and room temperature.
5. Experimental Execution of a TX-TL Reaction
Final reaction conditions are: 8.9-9.9 mg/ml protein (from crude extract), 4.5 mM-10.5 mM Mg-glutamate, 40-160 mM K-glutamate, 0.33-3.33 mM DTT, 1.5 mM each amino acid except leucine, 1.25 mM leucine, 50 mM HEPES, 1.5 mM ATP and GTP, 0.9 mM CTP and UTP, 0.2 mg/ml tRNA, 0.26 mM CoA, 0.33 mM NAD, 0.75 mM cAMP, 0.068 mM folinic acid, 1 mM spermidine, 30 mM 3-PGA, 2% PEG-8000. A basic TX-TL reaction has 3 parts (tubes): crude cell extract, buffer, and DNA. The ratio is: 75% buffer and extract, 25% DNA. Reactions can vary in volume, and we use 10 μl by convention to minimize reaction volume and enable running in a 384-well plate. Larger volumes require agitation for proper oxygenation. The following protocol utilizes a pre-written template, TXTL_JoVE.xlsx (Supplemental Material 4), to conduct a 10 μl reaction. Items in purple indicate user-input values, and items in blue indicate additional reagents to add to the reaction. However, one can also conduct a reaction without the template by following reaction conditions outlined above.
Complete the "General Data" form.
Under "Master Mix Preparation," insert the extract percentage value from step 4.1 into the purple box.
Design your experiment in silico using the "Master Mix Preparation" (rows 10-17) and "DNA Preparation" (rows 19-50) sections. Generally, constants can be put into the "Master Mix Preparation" section, while variables can be put into the "DNA Preparation" section. Minimize samples per experiment to avoid sample evaporation and experimental start time bias. See Figure 6 for a sample setup.
Under "Master Mix Preparation," add reagents such as inducers or proteins, which will go in all samples at a constant concentration. Starting with row 14, fill out the blue shaded areas, keeping one reagent to each line. Units are relative ratios.
Under "DNA Preparation," add DNA which will be sample specific. Sample IDs #1 and #2 correspond to positive and negative controls, respectively. Sample IDs #3 and above are user-modifiable for DNA, stock concentration in ng/μl, length in base pairs, desired final concentration in nM, and repeats (of 10 μl reactions). The amount of stock DNA to reach desired final concentration is automatically calculated. The total across the row sums to 10.5*n, where n is the number of repeats. Note: Although the final reaction volume is 10 μl, the calculations assume a total volume of 10.5 μl per reaction, to account for volume lost during pipetting.
Under "DNA Preparation," add reagents or additional DNA which will be sample specific to blue columns. Stock DNA concentrations in nM can be calculated under "DNA Preparation," while sample specific reagents require manual calculation based on a total reaction volume of 10.5*n. The entered volumes are subtracted out of the water volume of the same row.
Remove needed number of tubes of buffer, crude cell extract, and positive control under "Tubes to thaw," from -20 °C or -80 °C and thaw on ice.
Prepare DNA samples. For each sample ID, aliquot out the indicated DNA, water, and user-supplied items per the "DNA Preparation" section into a micro-centrifuge tube, at room temperature. Note: to avoid sample loss, recently calibrated pipets and low-stick pipette tips and micro-centrifuge tubes are recommended.
When tubes from step 5.7 are thawed, prepare the master mix consisting of buffer, extract, and any global user-supplied items based on the orange-shaded boxes, keeping on ice and vortexing after the addition of each item. Note: Extract is extremely viscous. Aliquots with bubbles can be removed by centrifuging at 10,000 x g for 30 sec at 4 °C.
Add the amount of master mix indicated in the orange cells under "DNA Preparation" (column O) to each DNA sample, and keep at room temperature. Treat this as the reaction start time.
Vortex each sample, and centrifuge at 10,000 x g for 30 sec at room temperature to bring down any residual sample and to reduce bubbles.
If conducting reaction in micro-centrifuge tubes, incubate directly at 29 °C. Otherwise, pipette 10 μl of sample into a 384-well plate. Note: Reactions in volumes greater than 10 μl may require agitation for oxygenation.
Centrifuge plate at 4,000 x g for 30 sec at room temperature to bring down any residual sample and to reduce bubbles. Seal plate afterwards to prevent evaporation.
Run reaction at 29 °C. Note: Runtimes vary depending on experiment but typically last under 8 hr.
Representative Results
We have presented a five day protocol for the preparation of an endogenous Escherichia coli based TX-TL cell-free expression system. A sample timeline for creating the reagents - crude cell extract and buffer - can be found in Figure 1. Once created, these can be stored at -80 °C for up to one year. After reagents are created, experimental setup and execution can be done in less than 8 hr.
In addition, we optimized the expression conditions of the TX-TL cell-free expression system. Other user-supplied additions, such as buffers or DNA solutions, should be calibrated for toxicity beforehand. For example, different methods of processing plasmids result in different expression due to salt content. We also tested the effect of Tris-Cl elution buffer on reaction efficiency (Figure 5).
An example of crude cell extract calibration, referring step 4.1 to 4.9, is shown in Figure 4a. In general, our experiments show that the crude cell extract is most sensitive to Mg-glutamate levels, followed by K-glutamate levels. To demonstrate the cell-free expression system, we constructed and tested a negative feedback loop based on tet repression.26 (Figure 6). In the cell-free expression system, the same circuit run with and without aTc shows a 7-fold end-point expression change of deGFP reporter after eight hours of expression. Although this experiment does not require global inducers or repressors, if necessary they can be added under "Master Mix Preparation."
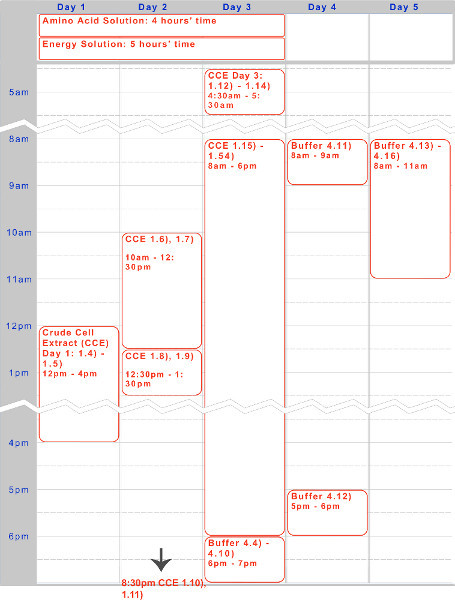 Figure 1. Timeline for crude cell extract, amino acid solution, and energy solution preparation. A five-day timeline for a typical execution of the protocol is given above, optimized for overnight incubations and daytime working steps.
Figure 1. Timeline for crude cell extract, amino acid solution, and energy solution preparation. A five-day timeline for a typical execution of the protocol is given above, optimized for overnight incubations and daytime working steps.
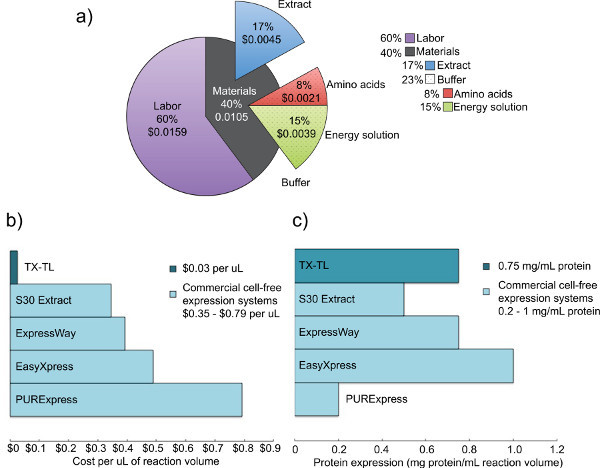 Figure 2. Cost and expression analysis of competing crude cell extracts. a) Breakdown of the costs of labor and materials of the TX-TL cell-free expression system. Based on costs of reagents as of December 2012, and labor costs of $14 per hour. b) Comparison of TX-TL cell-free expression system costs vs. other commercial systems. Costs are broken down per μl, although reaction volumes may vary per kit. c) Comparison of TX-TL cell-free expression system yield vs. other commercial systems. Protein expression yield determined by manufacturer standards. Click here to view larger figure.
Figure 2. Cost and expression analysis of competing crude cell extracts. a) Breakdown of the costs of labor and materials of the TX-TL cell-free expression system. Based on costs of reagents as of December 2012, and labor costs of $14 per hour. b) Comparison of TX-TL cell-free expression system costs vs. other commercial systems. Costs are broken down per μl, although reaction volumes may vary per kit. c) Comparison of TX-TL cell-free expression system yield vs. other commercial systems. Protein expression yield determined by manufacturer standards. Click here to view larger figure.
 Figure 3. Loading and processing of a bead-beating tube. a) Demonstration of correct viscosity of cell-bead solution. Cell-bead solution will have a viscosity dependent on many factors, including amount of S30A buffer added, amount of beads added, and time spent on ice. b) Loading of bead-beating tube before quick tabletop centrifugation. The centrifugation removes bubbles accumulated during loading. c) Bubbles surfacing after tabletop centrifugation. The size of the bubbles will vary; they can be popped or removed using a pipette tip. d) Completely filled bead-beating tube before capping. A meniscus is formed in the bead-beating tube, and the cap has enough to cover and cause small amounts to overfill. e) Correctly loaded filter apparatus. These can be reused. f) Comparison of correctly vs. incorrectly processed bead-beating tube. The tube on the left is a well-beat tube - it features a small and well-delineated top layer, and very clear supernatant. The tube on the right is suboptimal, based on the larger, hazy second layer and the hazy supernatant. Tubes that are suboptimal should not undergo additional processing.
Figure 3. Loading and processing of a bead-beating tube. a) Demonstration of correct viscosity of cell-bead solution. Cell-bead solution will have a viscosity dependent on many factors, including amount of S30A buffer added, amount of beads added, and time spent on ice. b) Loading of bead-beating tube before quick tabletop centrifugation. The centrifugation removes bubbles accumulated during loading. c) Bubbles surfacing after tabletop centrifugation. The size of the bubbles will vary; they can be popped or removed using a pipette tip. d) Completely filled bead-beating tube before capping. A meniscus is formed in the bead-beating tube, and the cap has enough to cover and cause small amounts to overfill. e) Correctly loaded filter apparatus. These can be reused. f) Comparison of correctly vs. incorrectly processed bead-beating tube. The tube on the left is a well-beat tube - it features a small and well-delineated top layer, and very clear supernatant. The tube on the right is suboptimal, based on the larger, hazy second layer and the hazy supernatant. Tubes that are suboptimal should not undergo additional processing.
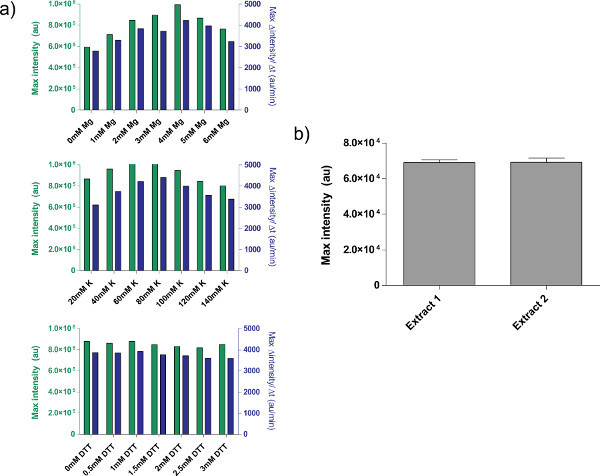 Figure 4. Properties of crude extract preparations. a) Typical calibration plots for crude cell extract. Crude extract is calibrated for additional Mg-glutamate, K-glutamate, and DTT levels, in that order. Shown is endpoint fluorescence after 8 hr, as well as maximal rate of protein production based on a 12-minute moving average. Based on these plots, an acceptable range of additional Mg-glutamate is 4 mM, K-glutamate is 60-80 mM, and DTT is 0-3 mM. Note that every crude extract needs to be calibrated independently for these three variables. b) Variation from extract preparations. Endpoint fluorescence of two crude extracts prepared on different dates is shown; error bars are 1 standard deviation from three independent runs on different days. Click here to view larger figure.
Figure 4. Properties of crude extract preparations. a) Typical calibration plots for crude cell extract. Crude extract is calibrated for additional Mg-glutamate, K-glutamate, and DTT levels, in that order. Shown is endpoint fluorescence after 8 hr, as well as maximal rate of protein production based on a 12-minute moving average. Based on these plots, an acceptable range of additional Mg-glutamate is 4 mM, K-glutamate is 60-80 mM, and DTT is 0-3 mM. Note that every crude extract needs to be calibrated independently for these three variables. b) Variation from extract preparations. Endpoint fluorescence of two crude extracts prepared on different dates is shown; error bars are 1 standard deviation from three independent runs on different days. Click here to view larger figure.
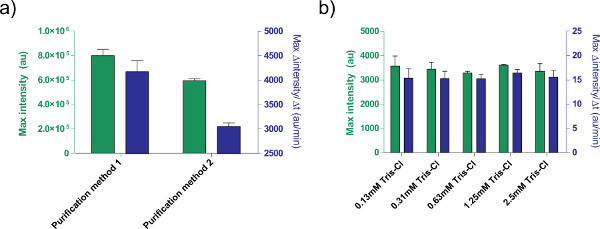 Figure 5. Effects of DNA solution on expression efficiency. a) Comparison of two different purification methods for processing plasmids. 1 nM of pBEST-OR2-OR1-Pr-UTR1-deGFP-T500 is prepared using only a QiaPrep Spin Miniprep Kit (Purification method 1) or post-processed with a QiaQuick PCR purification kit (Purification method 2). Shown is endpoint fluorescence after 8 hr, as well as maximal rate of protein production based on a 12-minute moving average. Error bars are 1 standard deviation from four independent runs on different days. b) Effect of elution buffer (Tris-Cl). Different concentrations of Tris-Cl are compared in a cell-free expression reaction based on the expression of 1 nM of pBEST-OR2-OR1-Pr-UTR1-deGFP-T500. Concentrations given are final concentrations of Tris-Cl in the reaction; elution buffer used is 10 mM Tris-Cl. Error bars are 1 standard deviation from three independent runs on different days. Click here to view larger figure.
Figure 5. Effects of DNA solution on expression efficiency. a) Comparison of two different purification methods for processing plasmids. 1 nM of pBEST-OR2-OR1-Pr-UTR1-deGFP-T500 is prepared using only a QiaPrep Spin Miniprep Kit (Purification method 1) or post-processed with a QiaQuick PCR purification kit (Purification method 2). Shown is endpoint fluorescence after 8 hr, as well as maximal rate of protein production based on a 12-minute moving average. Error bars are 1 standard deviation from four independent runs on different days. b) Effect of elution buffer (Tris-Cl). Different concentrations of Tris-Cl are compared in a cell-free expression reaction based on the expression of 1 nM of pBEST-OR2-OR1-Pr-UTR1-deGFP-T500. Concentrations given are final concentrations of Tris-Cl in the reaction; elution buffer used is 10 mM Tris-Cl. Error bars are 1 standard deviation from three independent runs on different days. Click here to view larger figure.
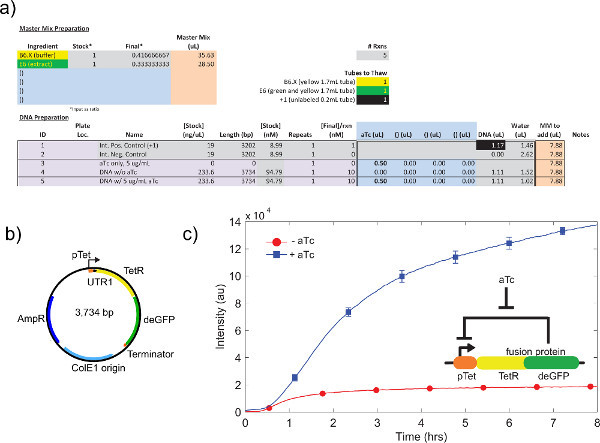 Figure 6. Sample TX-TL run of a negative feedback loop. a) Sample setup of a cell-free execution reaction. Tests "on" vs. "off" state of the negative feedback loop, with positive and negative controls. b) Plasmid map of negative feedback loop. c) Representative results. Data reflects experiment in a) and b), with negative control subtracted from signal. Genetic circuit shown in insert. Error bars are 1 standard deviation from three independent runs on different days. Click here to view larger figure.
Figure 6. Sample TX-TL run of a negative feedback loop. a) Sample setup of a cell-free execution reaction. Tests "on" vs. "off" state of the negative feedback loop, with positive and negative controls. b) Plasmid map of negative feedback loop. c) Representative results. Data reflects experiment in a) and b), with negative control subtracted from signal. Genetic circuit shown in insert. Error bars are 1 standard deviation from three independent runs on different days. Click here to view larger figure.
| Name | Concentration | Amount | Sterilization | Notes |
| Chloramphenicol (Cm) | 34 mg/ml in ethanol | 1 ml | Filter sterilize (0.22 μM) | Can be made in larger volumes stored at -20 °C for later use. |
| 2xYT+P+Cm agar plate | 31 g/L 2xYT, 40 mM potassium phosphate dibasic, 22 mM potassium phosphate monobasic, 34 μg/ml chloramphenicol | 1 plate | Autoclave | |
| 2xYT+P media | 31 g/L 2xYT, 40 mM potassium phosphate dibasic, 22 mM potassium phosphate monobasic | 4 L | Autoclave |
Table 1. Reagents for day 1 of Crude Cell Extract protocol.
| Name | Concentration | Amount | Sterilization | Notes |
| Tris base | 2 M | 250 ml | Filter sterilize (0.22 μM) or autoclave | Can be stored at room temperature. |
| DTT | 1 M | 6 ml | Filter sterilize (0.22 μM) | Can be made in larger volumes and stored at -20 °C for later use. |
| S30A buffer | 14 mM Mg-glutamate, 60 mM K-glutamate, 50 mM Tris, pH 7.7 | 2 L | Autoclave | To reach pH 7.7, titrate with acetic acid. Add DTT to 2 mM final concentration just before use. Store at 4 °C. |
| S30B buffer | 14 mM Mg-glutamate, 60 mM K-glutamate, ~5 mM Tris, pH 8.2 | 2 L | Autoclave | To reach pH 8.2, titrate with 2M Tris. Add DTT to 1 mM final concentration just before use. Store at 4 °C. |
Table 2. Reagents for day 2 of Crude Cell Extract protocol.
| Falcon | ||||
| 1 | 2 | 3 | 4 | |
| Empty 50 ml Falcon (g) | ||||
| 50 ml Falcon with pellet (g) | ||||
| Pellet mass (50 ml Falcon with pellet - empty 50 ml Falcon) (g) | ||||
| S30A buffer volume to add (pellet mass* 0.9) (ml) | ||||
| Total mass of beads to add (pellet mass * 5.0) (g) |
Table 3. S30A buffer and bead mass calculator, for day 3 of Crude Cell Extract protocol.
| Name | Concentration | Amount | Sterilization | Notes |
| HEPES | 2 M, pH 8 | 4 ml | None | To reach pH 8, titrate with KOH. |
| Nucleotide Mix | 156 mM ATP and GTP, 94 mM CTP and UTP, pH 7.5 | 1.5 ml | None | To reach pH 7.5, titrate with KOH. |
| tRNA | 50 mg/ml | 600 μl | None | |
| CoA | 65 mM | 600 μl | None | |
| NAD | 175 mM, pH 7.5-8 | 300 μl | None | To reach pH 7.5-8, titrate with Tris at 2 M. |
| cAMP | 650 mM, pH 8 | 200 μl | None | To reach pH 8, titrate with Tris at 2 M. |
| Folinic Acid | 33.9 mM | 300 μl | None | Although only 300 μl is needed, recipe in supplemental is for 1.15 ml. |
| Spermidine | 1 M | 150 μl | None | Store at 4 °C, heat to 37 °C to melt. |
| 3-PGA | 1.4 M, pH 7.5 | 3.2 ml | None | To reach pH 7.5, titrate with Tris at 2 M. |
Table 4. Reagents to prepare for Energy Solution protocol.
Supplemental Material 1. Recipes for Items.
Chloramphenicol, 34 mg/ml: Prepare 0.51 g chloramphenicol and add ethanol to 15 ml. Filter sterilize (0.22 μM), aliquot to 1 ml tubes, store at -20 °C for later use.
2xYT+P+Cm agar plate: Prepare 1.24 g 2xYT, 1.6 ml potassium phosphate dibasic solution @ 1 M, 0.88 ml potassium phosphate monobasic solution @ 1 M, 0.6 g agar, and water to 40 ml. Autoclave. Let cool to 50 °C and add 40 μl Cm. Aliquot 25 ml into a 100 x 15 mm Petri dish, and let cool for an hour.
2xYT+P media: Prepare 124 g 2xYT, 160 ml potassium phosphate dibasic solution @1 M, 88 ml potassium phosphate monobasic solution @ 1 M, and water to 4 L. Aliquot out into 2 x 1.88 L and 0.24 L. Autoclave.
Tris base, 2 M: Prepare 60.57 g Tris base and water to 250 ml. Sterilize, store at RT for later use.
DTT, 1 M: Prepare 2.31 g DTT and water to 15 ml. Filter sterilize (0.22 μM), aliquot to 1 ml tubes, store at -20 °C for later use.
S30A buffer: Prepare 10.88 g Mg-glutamate and 24.39 g K-glutamate, 50 ml Tris at 2M, acetic acid (to pH 7.7), and water to 2 L. Autoclave, store at 4 °C, add 4 ml 1 M DTT before use.
S30B buffer: Prepare 10.88 g Mg-glutamate and 24.39 g K-glutamate, Tris at 2 M (to pH 8.2), and water to 2 L. Autoclave, store at 4 °C, add 2 ml 1 M DTT before use.
HEPES: Prepare 1.91 g HEPES (MW 238.21), KOH (to pH 8), and water to 4 ml.
tRNA: Prepare 30 mg of tRNA and water to 600 μl.
CoA: Prepare 30 mg of CoA (MW 767.53) and water to 600 μl.
NAD: Add 34.83 mg of NAD (MW 663.43), Tris at 2 M (to pH 7.5-8), and water to 300 μl. (Add 27 μl of Tris at 2 M to bring the solution to pH 7.5-8).
cAMP: Add 42.80 mg of cAMP (MW 329.22), Tris at 2 M (to pH 8), and water to 200 μl. (Add 73 μl of Tris at 2 M to bring the solution to pH 8).
Folinic Acid (33.9 mM): To 20 mg of solid folinic acid calcium salt (MW 511.5), add 1.15 ml water.
Spermidine: Prepare 23.55 μl of spermidine (MW 145.25) and water to 150 μl. Prepare at room temperature after melting briefly at 37 °C. 3-PGA: Add 1.03 g of 3-PGA (MW 230.02), Tris at 2 M (to pH 7.5), and water to 3.2 ml. (Add 1.73 ml of Tris at 2 M to bring the solution to pH 7.5).
Nucleotide Mix: Add 145 mg of ATP dipotassium salt dihydrate (MW 619.4), 133 mg of GTP disodium salt (MW 567.14), 79.4 mg of CTP disodium salt dihydrate (MW 563.16), 82.6 mg of UTP trisodium salt dihydrate (MW 586.12), KOH at 15% dilution (to pH 7.5), and water to 1.5 ml. (Add 353 μl of KOH at 15% dilution to bring the solution to pH 7.5).
Supplemental Material 2. Bradford Assay.
Remove Bradford agent from 4 °C and set at room temperature.
Prepare 50 μl BSA Standard at 1 mg/ml and at 0.1 mg/ml.
Prepare 40 μl 20x dilution of extract from step 1.47.
Add 800 μl water to 7 cuvettes.
Prepare standard cuvettes for 0 mg/ml, 1 mg/ml (10 μl 0.1 mg/ml BSA), 2 mg/ml (20 μl 0.1 mg/ml BSA), 4 mg/ml (4 μl 1 mg/ml BSA), 6 mg/ml (6 μl 1 mg/ml BSA).
Prepare experimental cuvettes for 2 μl of sample and 4 μl of sample.
Add 200 μl of Bradford agent to each cuvette and mix well by pipetting. Incubate at room temperature for at least 10 min.
Produce standard curve at OD 595 nm using cuvettes from step 6.5. Reject standard curve if r2 < 0.95.
Determine extract concentration at OD 595 nm using cuvettes from step 6.6.
Supplemental Material 3. Buffer calibration spreadsheet.
Supplemental Material 4. Cell-free expression run spreadsheet.
Discussion
The endogenous Escherichia coli based TX-TL cell-free expression system described here is an easy-to-run three tube reaction that can take less than eight hours from set up to data collection. The process of creating all reagents requires five days' time total (with significant labor requirements on only one day), but produces crude extract for 3,000 reactions and buffer-making reagents for 10,000 reactions (Figure 1). Furthermore, crude extract and buffer-making reagents are stable for at least 1 year at -80 °C, allowing for multiple uses of one preparation.4 At $0.11 per 10 μl reaction ($0.26 including labor), costs are 98% lower than comparable commercial systems (Figure 2).
There are some unresolved limitations, however, to the system. The end efficiency of each crude cell extract preparation can vary based on user proficiency and on environmental conditions, although typical yield variation is between 5-10% (Figure 4b). As a result, batch-to-batch variability in both end-point expression and in expression dynamics are to be expected. These variations will likely remain until extract is fully characterized or until extract creation is fully automated. If the cell-free expression system is used to conduct sensitive quantitative experiments, it is advisable to run all experiments with the same batch of crude cell extract. The yield from a single crude cell extract batch, about 3000 reactions, should be sufficient for typical experimental courses. Although we suspect variation can be removed by scaling up and automating the procedure, such attempts would involve a substantial resource investment.
Additionally, although end-point expression levels are reasonably easy to determine, more work needs to be done in understanding dynamics intrinsic to the cell-free system. It is known that both resource competition and resource limitation can affect expression dynamics. For example, limited endogenous sigma 70 can result in a saturating regime with increased DNA template producing an expression profile analogous to that of nucleotide or amino acid depletion.9,27 However, dynamics do not have to be fully understood to utilize the system. For pure increases of yield, optimization can be done by machine-learning approaches.28 Questions of resource competition and limitation can be addressed by mathematical models verified using experimental data.
The protocol presented here is optimized for a BL21-Rosetta2 strain, but is generalizable to other E. coli strains. Modifications in BL21-Rosetta2, such as the removal of the gene encoding lon protease and the addition of genes encoding rare tRNAs, allow for maximal protein production. We have attempted the protocol with two other extract strains - BL21 only and a BL21 trxA knockout -and found 50% less protein yield. We hypothesize that yields similarly decrease when using other strains. Other changes in parameters, such as switching 2xYT growth medium for LB and other rich broths, have resulted in decreased protein yield.
Cell-free expression systems utilizing both endogenous and exogenous transcription-translation machinery and regulation mechanisms have wide applications in both protein and metabolite expression and in synthetic biology.3,29 Instead of being limited to T7-regulated circuits, one can envision producing complex biomolecules in a user-controllable setting using a mix of native E. coli promoters and exogenously supplied transcription and regulation mechanisms. Without limitations of cell division and metabolism, variability in synthetic circuits such as the repressilator or in metabolic engineered pathways such as those producing artemisinin can be reduced or better understood.30,31 We have used these advantages to implement genetic switches, as well as to understand sigma factor sequestration.9,32 Such technology can also form the backbone of "minimal" or "artificial" cells - small, well-characterized and self-sufficient embodied units of extract.33,34
Ultimately, we anticipate immediate uses of this endogenous cell-free expression system as a prototyping environment for synthetic biology. Nicknamed the "TX-TL biomolecular breadboard," the cell-free expression system provides a controllable environment where synthetic circuits ultimately destined for in vivo expression can undergo rounds of prototyping - cycles of testing on basic plasmid, linear, or chemically synthetized DNA, followed by analysis and rapid modification. Prototyping rounds can be aided by predictive mathematical models currently being developed. By removing cloning and in vivo manipulation for non-final circuits, we anticipate engineering cycle times to be reduced to 1-3 days instead of the current weeks' standard.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
We thank Jongmin Kim, Dan Siegal-Gaskins, Anu Thubagere, and Enoch Yeung for assistance streamlining the protocol, and Clare Chen and Barclay Lee for assistance in the early stages of the project. This material is based upon work supported in part by the Defense Advanced Research Projects Agency (DARPA/MTO) Living Foundries program, contract number HR0011-12-C-0065 (DARPA/CMO.Z.Z.S. is also supported by a UCLA/Caltech Medical Scientist Training Program fellowship and by a DoD, Air Force Office of Scientific Research, National Defense Science and Engineering Graduate (NDSEG) Fellowship, 32 CFR 168a. The views and conclusions contained in this document are those of the authors and should not be interpreted as representing officially policies, either expressly or implied, of the Defense Advanced Research Projects Agency or the U.S. Government.
References
- Noireaux V, Bar-Ziv R, Libchaber A. Principles of cell-free genetic circuit assembly. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:12672–12677. doi: 10.1073/pnas.2135496100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He MY, He YZ, Luo Q, Wang MR. From DNA to protein: No living cells required. Process Biochem. 2011;46:615–620. [Google Scholar]
- Forster AC, Church GM. Synthetic biology projects in vitro. Genome Res. 1101;17:1–6. doi: 10.1101/gr.5776007. [DOI] [PubMed] [Google Scholar]
- Shin J, Noireaux V. Efficient cell-free expression with the endogenous E. Coli RNA polymerase and sigma factor 70. Journal of biological engineering. 2010;4:8. doi: 10.1186/1754-1611-4-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu Y, et al. Cell-free translation reconstituted with purified components. Nat Biotechnol. 2001;19:751–755. doi: 10.1038/90802. [DOI] [PubMed] [Google Scholar]
- Shin J, Noireaux V. An E. coli Cell-Free Expression Toolbox: Application to Synthetic Gene Circuits and Artificial Cells. Acs Synth Biol. 2012;1:29–41. doi: 10.1021/sb200016s. [DOI] [PubMed] [Google Scholar]
- Shin J, Noireaux V. Study of messenger RNA inactivation and protein degradation in an Escherichia coli cell-free expression system. Journal of biological engineering. 2010;4:9. doi: 10.1186/1754-1611-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin J, Jardine P, Noireaux V. Genome Replication, Synthesis, and Assembly of the Bacteriophage T7 in a Single Cell-Free Reaction. Acs Synth Biol. 2012;1:408–413. doi: 10.1021/sb300049p. [DOI] [PubMed] [Google Scholar]
- Siegal-Gaskins D, Noireaux V, Murray RM. Biomolecular resource utilization in elementary cell-free gene circuits. In: Pao L, Abramovitch D, editors. American Control Conference; Jun 17-19; Washington, DC. AACC; 2013. pp. 1531–1536. [Google Scholar]
- Karzbrun E, Shin J, Bar-Ziv RH, Noireaux V. Coarse-grained dynamics of protein synthesis in a cell-free system. Physical review letters. 2011;106:048104. doi: 10.1103/PhysRevLett.106.048104. [DOI] [PubMed] [Google Scholar]
- Hoagland MB, Stephenson ML, Scott JF, Hecht LI, Zamecnik PC. Soluble Ribonucleic Acid Intermediate in Protein Synthesis. J Biol Chem. 1958;231:241–257. [PubMed] [Google Scholar]
- Wood WB, Berg P. Effect of Enzymatically Synthesized Ribonucleic Acid on Amino Acid Incorporation by a Soluble Protein-Ribosome System from Escherichia Coli. Proceedings of the National Academy of Sciences of the United States of America. 1962;48:94. doi: 10.1073/pnas.48.1.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zubay G. In-Vitro Synthesis of Protein in Microbial Systems. Annu Rev Genet. 1973;7:267–287. doi: 10.1146/annurev.ge.07.120173.001411. [DOI] [PubMed] [Google Scholar]
- Pratt JM. In: Transcription and Translation: A Practical Approach. Hames BD, Higgins SJ, editors. IRL Press; 1984. pp. 179–209. [Google Scholar]
- Kim HC, Kim DM. Methods for energizing cell-free protein synthesis. Journal of bioscience and bioengineering. 2009;108:1–4. doi: 10.1016/j.jbiosc.2009.02.007. [DOI] [PubMed] [Google Scholar]
- Michel-Reydellet N, Calhoun K, Swartz J. Amino acid stabilization for cell-free protein synthesis by modification of the Escherichia coli genome. Metabolic engineering. 2004;6:197–203. doi: 10.1016/j.ymben.2004.01.003. [DOI] [PubMed] [Google Scholar]
- Liu DV, Zawada JF, Swartz JR. Streamlining Escherichia coli S30 extract preparation for economical cell-free protein synthesis. Biotechnology progress. 2005;21:460–465. doi: 10.1021/bp049789y. [DOI] [PubMed] [Google Scholar]
- Andrianantoandro E, Basu S, Karig DK, Weiss R. Synthetic biology: new engineering rules for an emerging discipline. Molecular systems biology. 2006;2:2006.0028. doi: 10.1038/msb4100073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwok R. Five hard truths for synthetic biology. Nature. 2010;463:288–290. doi: 10.1038/463288a. [DOI] [PubMed] [Google Scholar]
- Tabor S, Richardson CC. A bacteriophage T7 RNA polymerase/promoter system for controlled exclusive expression of specific genes. Proceedings of the National Academy of Sciences of the United States of America. 1985;82:1074–1078. doi: 10.1073/pnas.82.4.1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewicki BT, Margus T, Remme J, Nierhaus KH. Coupling of rRNA transcription and ribosomal assembly in vivo. Formation of active ribosomal subunits in Escherichia coli requires transcription of rRNA genes by host RNA polymerase which cannot be replaced by bacteriophage T7 RNA polymerase. Journal of molecular biology. 1993;231:581–593. doi: 10.1006/jmbi.1993.1311. [DOI] [PubMed] [Google Scholar]
- Iskakova MB, Szaflarski W, Dreyfus M, Remme J, Nierhaus KH. Troubleshooting coupled in vitro transcription-translation system derived from Escherichia coli cells: synthesis of high-yield fully active proteins. Nucleic acids research. 2006;34:e135. doi: 10.1093/nar/gkl462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kigawa T, et al. Preparation of Escherichia coli cell extract for highly productive cell-free protein expression. Journal of structural and. 2004;5:63–68. doi: 10.1023/B:JSFG.0000029204.57846.7d. [DOI] [PubMed] [Google Scholar]
- Matsuda T, et al. Improving cell-free protein synthesis for stable-isotope labeling. Journal of biomolecular. NMR. 2007;37:225–229. doi: 10.1007/s10858-006-9127-5. [DOI] [PubMed] [Google Scholar]
- Sitaraman K, et al. A novel cell-free protein synthesis system. Journal of biotechnology. 2004;110:257–263. doi: 10.1016/j.jbiotec.2004.02.014. [DOI] [PubMed] [Google Scholar]
- Becskei A, Serrano L. Engineering stability in gene networks by autoregulation. Nature. 2000;405:590–593. doi: 10.1038/35014651. [DOI] [PubMed] [Google Scholar]
- Maeda H, Fujita N, Ishihama A. Competition among seven Escherichia coli sigma subunits: relative binding affinities to the core RNA polymerase. Nucleic acids research. 2000;28:3497–3503. doi: 10.1093/nar/28.18.3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caschera F, et al. Coping with complexity: machine learning optimization of cell-free protein synthesis. Biotechnology and bioengineering. 2011;108:2218–2228. doi: 10.1002/bit.23178. [DOI] [PubMed] [Google Scholar]
- Hodgman CE, Jewett MC. Cell-free synthetic biology: thinking outside the cell. Metabolic engineering. 2012;14:261–269. doi: 10.1016/j.ymben.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elowitz MB, Leibler S. A synthetic oscillatory network of transcriptional regulators. Nature. 2000;403:335–338. doi: 10.1038/35002125. [DOI] [PubMed] [Google Scholar]
- Tsuruta H, et al. High-level production of amorpha-4,11-diene, a precursor of the antimalarial agent artemisinin, in Escherichia coli. Plos One. 2009;4:e4489. doi: 10.1371/journal.pone.0004489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner TS, Cantor CR, Collins JJ. Construction of a genetic toggle switch in Escherichia coli. Nature. 2000;403:339–342. doi: 10.1038/35002131. [DOI] [PubMed] [Google Scholar]
- Jewett MC, Forster AC. Update on designing and building minimal cells. Current opinion in biotechnology. 2010;21:697–703. doi: 10.1016/j.copbio.2010.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noireaux V, Libchaber A. A vesicle bioreactor as a step toward an artificial cell assembly. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:17669–17674. doi: 10.1073/pnas.0408236101. [DOI] [PMC free article] [PubMed] [Google Scholar]