

Abstract
MEKK2 is a serine/threonine kinase that functions as a MAPKinase kinase kinase (MAP3K) to regulate activation of MAP kinases. We recently have demonstrated that ablation of MEKK2 expression in invasive breast tumor cells dramatically inhibits xenograft metastasis, but the mechanism by which MEKK2 influences metastasis-related tumor cell function is unknown. In this study, we investigate MEKK2 function and demonstrate that silencing MEKK2 expression in breast tumor cell significantly enhances cell spread area and focal adhesion stability while reducing cell migration. We show that cell attachment to the matrix proteins fibronectin or Matrigel induces MEKK2 activation and localization to focal adhesions. Further, we reveal that MEKK2 ablation enhances focal adhesion size and frequency, thereby linking MEKK2 function to focal adhesion stability. Finally, we show that MEKK2 knockdown inhibits fibronectin-induced ERK5 signaling and FAK autophosphorylation. Taken together, our results strongly support a role for MEKK2 as a regulator of signaling that modulates breast tumor cell spread area and migration through control of focal adhesion stability.
Keywords: MEKK2, focal adhesion, fibronectin, kinase
1. Introduction
Breast cancer progression from primary tumor to metastatic carcinoma requires that tumor cells attain the ability to detach from the primary tumor mass and move through surrounding tissue stroma to gain access to distant tissues via the circulatory or lymphatic systems [1]. Therefore, tumor cell migration is absolutely essential for metastasis to occur, and therapeutic intervention that blocks tumor cell migration would be predicted to inhibit solid tumor metastasis. As with other adherent cells, breast tumor cell migration requires the coordinated initiation and release of adhesion to substrata [2]. Regulation of cell adhesion to extracellular matrix proteins such as fibronectin is accomplished through membrane-proximal protein complexes associated with integrin receptors [3]. Integrins are dimeric cell membrane receptors with extracellular domains that selectively bind to a range of matrix protein ligands to elicit cellular responses [4]. The small integrin intracellular domain functions as an anchor for the formation of multi-protein complexes called focal adhesions that include the cytoskeletal proteins actin, talin and vinculin along with signaling proteins in a complex network estimated to include more than 180 protein-protein interactions [5]. Focal adhesions link signaling proteins such as paxillin and focal adhesion kinase (FAK) to integrin cytoplasmic domains and thereby connect the intracellular tumor cell regulatory signaling to the extracellular environment [6]. Thus focal adhesion complexes regulate cell migration via control of adhesion, and factors that promote or inhibit focal adhesion stability or disassembly strongly influence cell migration [6]. While numerous proteins have been shown to associate with focal adhesion proteins in recent years [6, 7], the understanding of how signaling networks coordinate control of adhesion stability to promote cell movement is incomplete.
Mitogen-activated protein kinases (MAPK) are signaling regulators that influence multiple cellular functions, including (but not limited to) cell survival, differentiation, gene expression, and cell migration. MAPKs have been categorized into four main groups, including ERK1/2, JNK, p38 and ERK5, although multiple isoforms have been identified that expand the over number to at least twelve member proteins [9]. MAPK are the terminal kinases of a three kinase activation cascade system in which MAPK are phosphorylated and consequently activated by a MAPK kinase (MAP2K), which is itself phosphorylated and activated by a MAPK kinase kinase (MAP3K). The MAP3K class of MAPK regulators include over twenty member proteins; more than the MAPK or MAP2K protein groups combined. As the stimuli that activate each MAP3K can vary widely, the relative abundance of distinct MAP3K proteins represents an important regulatory mechanism that contributes to the specificity of MAPK activation. While the ERK1/2, JNK and p38 MAPK all are responsive to activating signals originating from several different MAP3K, ERK5 activity has been shown to be predominantly under the control of only the MEKK2 and MEKK3 members of the MAP3K class [9].
While similar in sequence (55% protein sequence identity) and predicted domain structure, gene knockout studies indicate that MEKK2 and MEKK3 are have functionally distinct developmental roles in mice [9]. MEKK3 knockout is associated with inadequate angiogenesis, abnormal cardiac development and consequent embryo lethality, whereas MEKK2 knockout mice are viable and fertile. MEKK2 has been demonstrated to phosphorylate the MAP2K proteins MKK4, and MKK7 that, in turn, phosphorylate and activate JNK. In addition to JNK regulation, MEKK2 can phosphorlylate and activate the MAP2K protein MEK5, that then phosphorylates and activates ERK5 [9]. In addition to established regulatory roles in gene expression and cell survival [10, 11], JNK also has been linked to control of cell migration [12]. While multiple MAP3K proteins can influence JNK activation [9], suggesting that significant functional redundancy exists within this protein family, our group and others have found that individual MAP3K signaling modules respond to different environmental inputs to specifically control JNK activation. For example, c-Kit-induced JNK activation in stem cell-derived mast cells requires MEKK2, but not related kinase MEKK1 [13], whereas FGF-2 induces JNK-dependent uPA expression in fibroblasts that is blocked by ablation of MEKK1, but not MEKK2 [14]. How MEKK2 specifically regulates JNK activity to influence tumor cell biology has not been investigated, and the tumor cell functions controlled by MEKK2 have not been defined. While JNK regulation and function has been extensively examined, the current understanding of ERK5 is far more limited. Knockout studies have linked ERK5 to vascular and cardiac development [15, 16], and thus functional analysis of ERK5 in tumor cells have focused on the importance of ERK5-dependent gene expression in tumor vascularity and angiogenesis. Interestingly, Sawhney and colleagues found that ERK5 co-immunoprecipitates with integrins and promotes FAK autophosphorylation and consequent activation in invasive tumor cells [17], linking ERK5 to cell adhesion and migration. However, the signaling mechanisms that mediate ERK5 activity in these complexes was not defined.
Some of the cellular functions under the control of MAPK signaling, including cell migration influence the course of solid tumor progression and metastasis. We have recently conducted an shRNA screen of MAP3K to identify potential metastasis regulators, and we reported that silencing MEKK2 expression in breast tumor cells dramatically inhibits metastasis arising from breast tumor xenografts, whereas silencing MEKK3 did not reduce metastasis [8]. To understand the role of MEKK2 in solid tumor progression, our subsequent studies have focused on defining the mechanisms by which MEKK2 breast tumor cell biology. In this report, we reveal that MEKK2 regulates breast tumor cell migration, at least in part, through control of focal adhesion stability. Furthermore, we demonstrate that MEKK2 is recruited to focal adhesions and activated downstream of attachment to extracellular matrix. Finally, we examine the impact of MEKK2 expression on signaling to propose a mechanism for MEKK2-dependent tumor cell migration.
2. Materials and methods
2.1. Antibodies and Reagents
Anti-MEKK2, anti-MEKK3, and anti-ERK2 antibodies were purchased from Santa Cruz Biotechnology, anti-vinculin antibodies was purchased from Sigma. Growth factor reduced Matrigel was purchased from BD Biosciences. Antibodies specific to phosphorylated or total p38, JNK, ERK1/2, ERK5, FAK, and MKK4 were purchased from Cell Signaling Technology, and anti-MEKK3 antibodies were purchased from Epitomics. Protein A and agarose beads were purchased from Roche Applied Science. Alexa Fluor®-conjugated phalloidin and Alexa Fluor®-conjugated secondary antibodies were purchased from Invitrogen. Fibronectin and collagen were purchased from Sigma. Horseradish peroxidase conjugated secondary donkey anti-rabbit was purchased from Jackson ImmunoResearch. Horseradish peroxidase conjugated secondary sheep anti-mouse was purchased from Amersham-GE Health. BIX02189 was purchased from Selleck Chemicals, and XMD 8–92 from TOCRIS Bioscience.
2.2. Plasmid vectors
The cDNA vectors encoding wild-type HA-tagged MEKK2 and kinase-inactive mutant MEKK2 (K385M) cloned into pCMV5 were previously described [18]. FLAG-tagged MEKK2 kinase domain (amino acids 357-619) was produced by PCR and cloned into pCDNA3. The vector encoding inactive GST-MKK4 was a generous gift of Dr. Ajay Rana (Loyola University Chicago). Lentiviral vectors encoding MEKK2 shRNA were purchased from Open Biosystems (ThermoScientific)
2.3. Cell culture and transfection
MDA-MB 231 breast tumor cells stably expressing MEKK2 shRNA cells were described previously [19]. Briefly, stable lines were created by infecting cells with lentivirus encoding either of two distinct MEKK2 shRNA sequences (OpenBiosystems clone ID TRCN0000002043 and TRCN0000002045), followed by selection with puromycin-containing growth media (2 μg/ml) where indicated. HEK-293T cells were purchased from A.T.C.C. All cells were cultured in DMEM (Dulbecco's modified Eagle's medium) (Invitrogen) containing 10% (v/v) fetal bovine serum (Atlanta Biologicals) at 37°C and maintained in 5% CO2 in a humidified atmosphere. All transfections were conducted using Lipofectamine Plus (Invitrogen) as per the manufacturer's recommendations.
2.4. Immunofluorescence
MDA-MB 231 cells expressing either empty vector or MEKK2-specific shRNA were seeded onto matrix protein-coated glass coverslips as follows: sterile washed coverslips were incubated in phosphate-buffered saline (PBS) containing matrix proteins (20 μg/ml of fibronectin, 100 μg/ml of Matrigel and 20 μg/ml collagen) for 1 hour at 37°C, then were removed and allowed to dry. Cultured cells were dispersed with enzyme-free cell dissociation buffer (Life technologies), washed and resuspended in serum-free growth media before seeding into culture dishes containing the coated coverslips and allowed to adhere for the indicated times. Cells were then fixed in methanol-free 4% (w/v) formaldehyde (Thermo Scientific) in PBS for 15 minutes. Following three PBS washes, the cells were permeabilized for 15 minutes with 0.1% Triton X-100 in PBS. After washing, the coverslips were blocked in 5% (v/v) goat serum/PBS for 1 hour at room temperature (23°C), then incubated with either anti-MEKK2 or anti-vinculin antibodies as indicated overnight at 4°C. After three washes, the coverslips were incubated with Alexa Fluor® 488-conjugated anti-rabbit, Alexa Fluor® 594-conjugated anti-mouse and Alexa Fluor® 647-conjugated phalloidin (Invitrogen) in blocking solution overnight at 4°C. Following washing, cells were mounted in Fluoro-Gel (Electron Microscopy Services). Images were acquired using 60× objective on an Olympus IX81 microscope.
2.5. Quantification of protein colocalization
MDA-MB 231 cells seeded on glass coverslips coated with Matrigel were fixed and incubated with anti-vinculin and anti-MEKK2 antibodies as described above. A minimum of 12 sequential Z-stack images were taken at 0.5 μm intervals on a DeltaVision microscope and deconvolved using softWoRx software (Applied Precision). The deconvolved images were rendered in three-dimensions using Imaris software (Bitplane Inc.). Surfaces were then constructed around regions of high vinculin concentration using a constant fluorescence threshold for vinculin and a minimum volume of 0.4 μm3 using the Surface Finder function of the software and was globally applied to all images. Total fluorescence intensity of MEKK2 within the defined focal adhesion surfaces was calculated by the program and reported as intensity units/μm3. Statistical correlation was calculated using the Pearson Correlation Coefficient test.
2.6. Focal adhesion quantification
The total number and associated area of focal adhesions detected within fixed MDA-MB 231 cells seeded on FN (20 μg/ml) and stained with anti-vinculin antibodies was quantified with ImageJ software using a procedure adapted from a previously described approach [20, 21]. Briefly, center region of cells containing the nucleus and majority of the cell body was manually traced and masked. Total cells analyzed is n = 48 control MDA-MB 231 cells, and n= 46 MDAMB 231 cells with stable MEKK2 knockdown. The detection threshold for pixel intensity was initially set by the program to distinguish focal adhesions from background fluorescence and was globally applied to all images thereafter. The resulting pixels were then converted to binary images and the number and area of focal adhesions were determined using the “Analyze Particles” function in ImageJ. The data were analyzed and graphed using Prism 4 from GraphPad Software, Inc. (La Jolla, CA). The statistical significance was calculated using unpaired t-test.
2.7. Attachment Assay
Cells were seeded on FN-coated 96-well plates (20 μg/ml) and incubated undisturbed at 37° C for 30 minutes. The plates were then vortexed at 2000 RPM for 30 seconds, after which the media and unattached cells were removed. Attached cells were fixed in methanol and Images were acquired with an inverted microscope equipped with a camera and Micron imaging software. Images of at least four wells taken prior and post vortexing were counted using ImageJ v1.46h (NIH). The data were analyzed and graphed using Prism 4 from GraphPad Software, Inc. (La Jolla, CA) and statistical significance was calculated using unpaired t-test.
2.8. Spreading Assay
Cells were seeded on FN-coated 96-well plates (20 μg/ml) and incubated undisturbed at 37° C for 2 hours to allow for adherence and spreading. Media was then removed and cells were fixed in methanol. Images were acquired using an inverted microscope equipped with a camera and Micron imaging software. The perimeter of each cell was traced using ImageJ v1.46h (NIH) to calculate the surface area; cells sharing an edge or more were excluded. Approximately 600 individual cells were measured in images from at least four wells. The data were analyzed and graphed using Prism 4 from GraphPad Software, Inc. (La Jolla, CA) and statistical significance was calculated using Mann-Whitney U-test.
2.9. In vitro wound healing assay
The procedure was adapted and modified from one previously described [22]. Briefly, cells were seeded and incubated until confluent. The cell layer was gently wounded using a pipet tip, washed and incubated in complete media at 37° C for 20 hours or less. Images were captured with an inverted microscope equipped with a camera and Micron imaging software then processed using ImageJ v1.46h (NIH) to measure the distance traveled by the cell front and the rate of wound closure. The cell front velocity (v) was calculated as v = d / t, where d is the distance traveled by the cell front at time t. The percent wound healing rate (Wr) was calculated as Wr = (1-(At/A0))/t, where A0 and At are the area of the wound at the start of the experiment and at time t respectively. The data were analyzed and graphed using Prism 4 from GraphPad Software, Inc. (La Jolla, CA) and statistical significance was calculated using unpaired t-test.
2.10. Immunoblotting
Proteins were separated by SDS-PAGE and transferred on to Protran nitrocellulose membranes (Whatman). Membranes were blocked in 5% (w/v) non-fat dried skimmed milk powder diluted in TBST (20 mM Tris, 137 mM NaCl and 0.1% Tween-20, adjusted to pH 7.6) or 5% globulin-free BSA (Sigma) in TBST and incubated in the appropriate antibody at 4°C overnight. After extensive washing, the membranes were then incubated with HRP (horseradish peroxidase)–conjugated donkey anti-rabbit IgG (Jackson ImmunoResearch) or HRP–sheep anti-mouse IgG (Amersham-GE Healthcare) secondary antibodies for one hour at room temperature. After extensive washing, the targeted proteins were detected by ECL (enhanced chemiluminescence, Thermo Scientific). Where indicated, blots were stripped by treatment with 2% (w/v) SDS and 100 mM 2-mercaptoethanol in TBS, and then re-probed with the desired antibodies.
2.11. MEKK2 kinase assay
MDA-MB 231 cells were dispersed, washed in serum-free medium and seeded on fibronectin-coated plates or mixed in suspension end-over-end for 20 minutes. Cells were then lysed in kinase lysis buffer (20 mM Tris, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 1 mM PMSF, 1 mM sodium vanadate, 0.05 mM DTT, and 1μg/ml leupeptin) and cleared by centrifugation at 13,000 rpm for 10 min at 4°C and cytosolic cell lysate were collected. 500 μg of total protein from cell lysates was brought to a volume of 350 μl with lysis buffer and rotated end-over-end at 4 °C with anti-MEKK2 antibodies overnight. Protein A beads (15 μl packed volume) were then added to the lysates and were again rotated end-over-end at 4 °C. After 2–4 hours mixing, the beads were washed twice in lysis buffer and once in kinase buffer (20 mM HEPES, pH 7.5, 10 mM MgCl2, 5 mM p-nitrophenyl phosphate). The beads were then incubated for 20 min at 30 °C in 50 μl of kinase buffer supplemented with and 1 μg of purified MKK4. The mixture was separated by SDS-PAGE and transferred to nitrocellulose membranes and then immunoblotted with anti-phospho-MKK4 antibodies.
2.12. Transwell migration assay
MDA-MB 231 cells were suspended in serum-free medium containing either MEK5 inhibitor BIX021089 (10 μM), ERK5 inhibitor XMD 8–92 (5 μM) or DMSO. Chemotaxis was assessed by Transwell (Corning) assay, wherein 105 cells were allowed to migrate through a filter (8 μm pore) toward 4% serum for 5 hours, and then migrating cells were stained with Wright's stain and counted as previously described [22].
3. Results
3.1. Attachment to fibronectin regulates MEKK2 localization in breast tumor cells
To define the location of MEKK2 in breast tumor cells, and thereby gain insight into possible mechanisms by which MEKK2 promotes tumor metastasis, we conducted immunofluorescence analysis on fixed MDA-MB 231 breast tumor cells using antibodies specific for MEKK2. In these experiments, we seeded tumor cells onto non-coated glass coverslips, fixed the cells in formaldehyde and utilized anti-MEKK2 antibodies to detect endogenous MEKK2 protein. In addition, we also seeded cells onto fibronectin-coated coverslips to enhance attachment and cell spreading prior to fixation. Interestingly, we observed that MEKK2 localization changed dramatically when cells were attached to fibronectin compared to MEKK2 in cells adhered to uncoated coverslips. While MEKK2 was observed in the cytoplasm of cells attached to uncoated coverslips, we discovered that attachment to fibronectin induced a marked MEKK2 enrichment in focused areas proximal to the membrane area attached to fibronectin (Fig. 1). As some of these MEKK2 enriched areas resembled focal adhesion complexes, we compared MEKK2 localization with that of the focal adhesion protein vinculin (Fig. 1). When fibronectin-adhered cells were treated with both anti-vinculin and anti-MEKK2 antibodies, we observed that some of the MEKK2-rich areas co-localized with vinculin in focal adhesions (Fig. 1). We then examined whether cellular attachment to other matrix proteins would similarly induce redistribution of MEKK2 to focal adhesions. We discovered that cell attachment to Matrigel, but not collagen, also induced MEKK2 to co-localize with vinculin (Fig. 1). These findings strongly suggest that MEKK2 redistribution is induced by ligation of a subset of integrin receptors. Furthermore, as Matrigel consists predominantly of laminin, collagen IV, and entactin matrix proteins and not fibronectin, these results indicate that at binding of at least two distinct extracellular matrix proteins induces MEKK2 translocalization to focal adhesions. Importantly, we detected a similar MEKK2 localization pattern in a second breast cancer cell line (BT474), indicating that focal adhesion-associated MEKK2 was not a cell line-specific characteristic (data not shown). We concluded that MEKK2 localization in invasive breast tumor cells is consistent with proteins known to regulate cell adhesion and migration, and therefore we elected to investigate the role of MEKK2 in these functions.
Fig. 1.

Cell attachment to matrix protein induces MEKK2 re-distribution. MDA-MB 231 cells seeded on glass coverslips that were either uncoated (first row), or coated with collagen (second row), fibronectin (third row), or Matrigel (fourth row) for 6 hours, fixed and stained for immunofluorescence analysis with anti-MEKK2 (green) or anti-vinculin (red) antibodies. Arrowheads indicate areas of MEKK2 localization to focal adhesions. Scale bar represents 20 μm. Images are representative of at least three independent experiments.
3.2. MEKK2 is recruited to focal adhesions
Our results suggested that MEKK2 is specifically recruited to focal adhesions in response to cellular attachment to Matrigel or fibronectin. To define the temporal regulation of MEKK2 localization, we first identified the time required for focal complex formation, then asked when MEKK2 co-localized in these complexes. We determined by immunofluorescence analysis that focal complexes were detectable in fixed cells as early as 15 minutes after attachment to Matrigel. We developed a new Imaris software-based approach to quantify protein co-localization, then utilized our technique to quantify MEKK2 co-localization with vinculin in focal adhesion complexes over a one-hour time course. Using vinculin fluorescence as a constant to define focal adhesion boundaries, three-dimensional surfaces were built around vinculin enriched regions using the Surface Finder function of the Imaris program revealing conical focal complexes extending from the center out to the periphery of cells (Fig. 2A-1,2,3). We discovered that MEKK2 concentration in these focal complexes increased over time (Fig. 2B) and is statistically correlated with incubation time on Matrigel when subjected to a Pearson Correlation Coefficient test (r (3) = 0.9937, p <0.005). Furthermore, vinculin localization in the focal adhesion complexes precedes that of MEKK2, suggesting that MEKK2 localizes to formed adhesion complexes and is not required for the formation of focal adhesions. Altogether, our results strongly suggest that MEKK2 is recruited to established focal adhesions in response to breast tumor cell attachment to fibronectin or Matrigel, and suggest that ligation of specific integrin receptors are required for matrix-induced MEKK2 translocation.
Fig. 2.
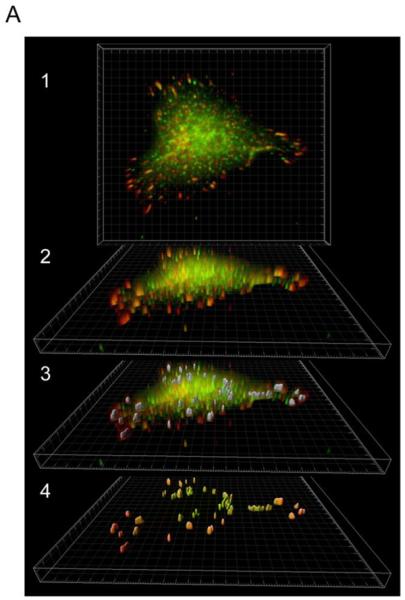
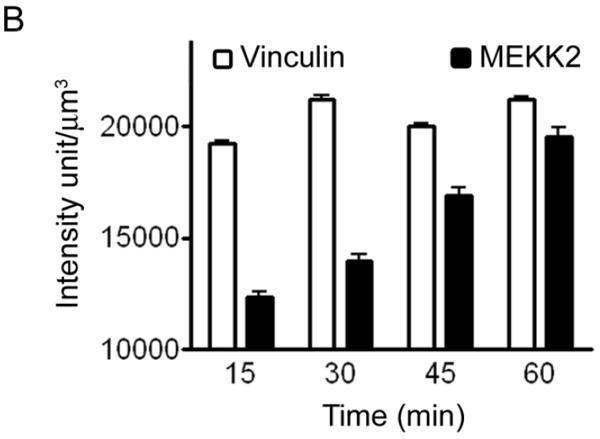
Quantification of MEKK2 colocalization with vinculin in three-dimensional focal adhesions. (A) Three dimensional rendering of Z-stack images of MDA-MB 231 cells seeded on Matrigel in the XY plane revealing colocalization of MEKK2 (green) and vinculin (red) (A1) or rotated to show focal adhesions volume (A2). Three-dimensional surfaces (in gray) were constructed around regions of high vinculin fluorescence intensity (A3). The cell body was subtracted from the image and the colocalized fluorescence signal was over imposed on the three-dimensional surfaces of focal adhesions. Image is representative of >140 images taken at four time points. (B) Bar-graph representation of vinculin and MEKK2 quantification showing a significant linear correlation of MEKK2 recruitment in focal adhesion with incubation time (r (3) = 0.9937, **p <0.005).
3.3. MEKK2 regulates cell spread area and focal adhesion stability but not attachment
Cell spreading is dependent upon the dynamics of focal adhesion formation and disassembly, therefore we asked whether MEKK2 regulates focal adhesion formation and stability. To determine whether MEKK2 influences these parameters, we stably knocked down MEKK2 expression utilizing the shRNA vectors we had used previously to block xenograft metastasis (Fig. 3A) [19]. MEKK2 shows high protein sequence similarity to another MAP3K called MEKK3, so we confirmed the specificity of our MEKK2 shRNA vectors by performing anti-MEKK3 immunoblot analysis using lysates from cells with stable MEKK2 knockdown. Although MEKK3 protein shares 55% sequence identity with MEKK2, the MEKK2 sequences targeted by either shRNA vector used in this study are not conserved in MEKK3, and as predicted MEKK2 shRNA did not affect MEKK3 expression (Fig. 3A). These results strongly suggest that our MEKK2 shRNA are both very effective at silencing MEKK2 expression and very specific for knocking down only MEKK2. Utilizing immunofluorescence microscopy to detect endogenous vinculin as a marker of focal adhesions in cells attached to fibronectin (Fig. 3B), we discovered that both the incidence and size of focal adhesions are strongly influenced by MEKK2 expression. MEKK2 knockdown significantly enhanced the number (Figs. 3C) and area (Fig. 3D) of focal adhesions in breast tumor cells. We next examined the effect of MEKK2 knockdown on the cell adhesion parameters of cell surface spread area and attachment. We compared attachment and spreading on fibronectin of cells with stable MEKK2 knockdown to that of control cells. We found that cell spread area is enhanced in cells with stable MEKK2 knockdown (Fig. 4A), and that cell area was rescued to control levels by expression of shRNA-resistant MEKK2 (add-back). In contrast, MEKK2 knockdown did not alter the ability of cells to attach to fibronectin-coated plates (Fig. 4B) indicating that the enhanced spreading of surface area in cells with MEKK2 knockdown was not due to increased or accelerated cell attachment. Furthermore, when we knocked down MEKK2 expression in SKBR3 breast tumor cells, we observed similar effects of MEKK2 knockdown on cell area (data not shown), indicating that MEKK2 knockdown-associated cell spreading is not limited to MDA-MB 231 cells, and therefore is not a cell line-specific effect of MEKK2 shRNA. Altogether, these results suggest that MEKK2 activity promotes focal adhesion turnover in invasive breast tumor cells.
Fig. 3.

MEKK2 knockdown stabilizes focal adhesions. (A) Immunoblot of MDA-MB 231 lysates displaying expression of MEKK2 (upper panel), MEKK3 (middle panel), and total ERK2 as a loading control (lower panel) from control cells, cells treated with MEKK2 shRNA #'s 1 and 2, or MEKK2 shRNA with stably expressed shRNA-resistant MEKK2 (Add-back). (B) Fluorescence microscopy with anti-vinculin antibodies was used to detect focal adhesions in MDA-MB 231 cells attached to fibronectin. Representative images after 16 hours of attachment showing vinculin-associated focal adhesions in control cells (upper panel, left) or cells with MEKK2 shRNA (upper panel, right). Original images were converted to binary images (lower panel) for analysis. (C) Quantification of number of focal adhesions per cell, and (D) mean area of individual focal adhesions after attachment to fibronectin for 5 hours and 16 hours in MDAMB 231 cells. The data represented in the graphs was derived from at least three independent experiments. Total cells analyzed is n = 48 control MDA-MB 231 cells, and n= 46 MDA-MB 231 cells with stable MEKK2 knockdown (D). The data represented in the graphs was derived from at least three independent experiments. *p <0.05, **p <0.01, ***p <0.001. Scale bar represents 10 μm.
Fig. 4.
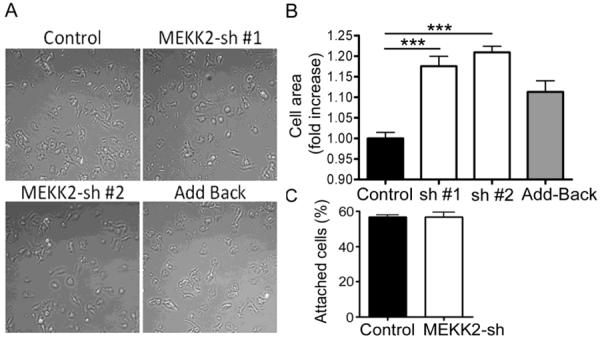
MEKK2 regulates the surface area of spreading cells. (A) MDA-MB 231 control cells, cells treated with MEKK2 shRNAs, or MEKK2 shRNA with stably expressed YFP-MEKK2 (Add-back) were plated on fibronectin-coated cell culture plates for 2 hr. (B) Quantification of the impact of MEKK2 knockdown on cell spread area. (C) MEKK2 knockdown did not have an effect on MDA-MB231 cells in attachment assays on fibronectin after 30 min. The results were normalized to control and compiled from at least three independent experiments. ***p <0.001.
3.4. MEKK2 knockdown inhibits breast tumor cell migration
Adhesion is an integral component of cell migration, therefore to determine whether MEKK2 influences cell migration, we compared the migration of cells with stable MEKK2 knockdown to that of control cells. We utilized in vitro wound-healing assays to assess cell migration as a function of the time required for a confluent culture of cells to fill the area cleared of cells by “wounding” with a pipet tip (wound-healing rate). Strikingly, we observed that while cells stably transfected with empty vector (control) had nearly filled the wound by twenty hours, cells stably expressing either of two distinct MEKK2 shRNAs showed a markedly reduced wound healing rate (Fig 5A, B). We discovered that cells with stable MEKK2 knockdown consistently displayed prolonged wound-healing time compared to control cells, indicating a clear reduction in directed cell migration of MEKK2-deficient cells (Fig. 5B). Since cell surface area can influence scratch assay results independently of migration rate, we confirmed our findings by quantifying the velocity of cell movement along the wound edge (cell front), and found that silencing MEKK2 expression was associated with reduced cell front velocity (Fig. 5C). Furthermore, re-expression of MEKK2 in cells with stable knockdown (add-back cells) rescued cell migration to a level similar to controls, thereby confirming that the reduced migration was due to specific MEKK2 knockdown and not an off-target shRNA effect (Fig. 5B). Consistent with these findings, cell-front velocity was also partially rescued when MEKK2 expression was restored (Fig. 5C). Taken together, these data strongly suggest that MEKK2 regulates cell migration in MDA-MB 231 cells.
Fig. 5.

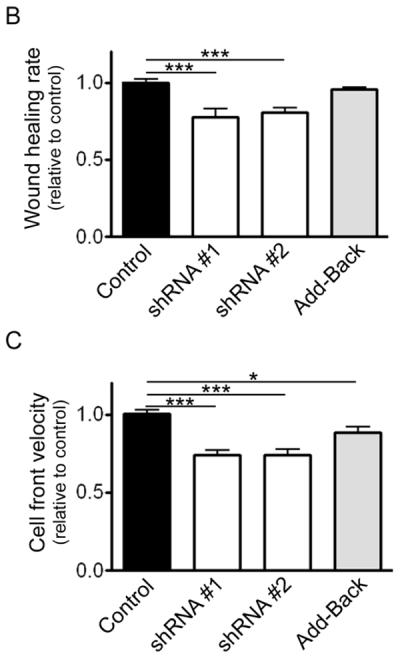
Silencing MEKK2 expression inhibits tumor cell migration. (A) In vitro wound healing assay showing effect on migration parameters of stable MEKK2 expression knockdown. The blue lines indicate the cell front at the beginning of the assay (0 hours) and the red line indicates the cell front at the end of the assay (20 hours). ImageJ was used to measure the wound healing rate (B) and cell-front velocity of cells migrating into the wound (C) on non-coated cell culture plastic plates. The results were normalized to control, and were compiled from at least three independent experiments. *p <0.05, ***p <0.001.
3.5. Attachment to fibronectin activates MEKK2
Our data strongly suggest that MEKK2 activity is subject to spatial and temporal regulation by fibronectin. We asked whether attachment to fibronectin was sufficient to activate MEKK2 by performing semi-quantitative kinase assays with MEKK2 purified from cells seeded on fibronectin. Using recombinant inactive MKK4 as a substrate, we detected phosphorylation of the MKK4 activation loop residues by immunoprecipitated MEKK2 with phosphor-specific MKK4 antibodies (phospho-MKK4 S257/T261). We discovered that endogenous MEKK2 was activated by cellular attachment to fibronectin, compared to MEKK2 purified from cells in suspension (Fig. 6). This result indicate that attachment to fibronectin and so the activation of its receptor was sufficient to activate MEKK2.
Fig. 6.
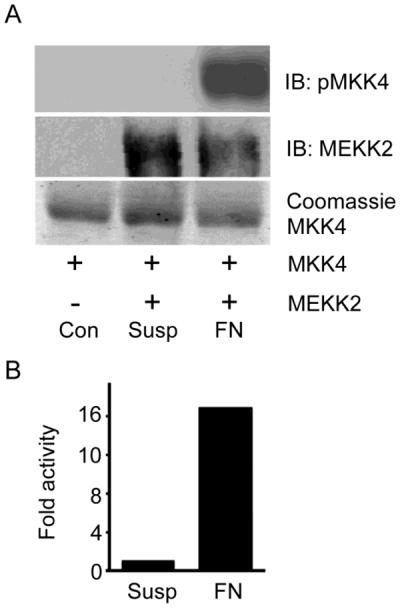
Adhesion to fibronectin activates MEKK2 in MDA-MB 231 cells. (A) In vitro kinase assay using anti-phospho-MKK4 (S257/T261) immunoblot to reveal phosphorylation of recombinant MKK4 by endogenous MEKK2 immunoprecipitated from MDA-MB 231 cells (upper panel). Cells were either adhered to fibronectin-coated plastic cell culture plate (FN) for 20 minutes or maintained in suspension (Susp) for 20 minutes prior to lysis. The substrate alone without immunoprecipitated MEKK2 (cont) is included for comparison. MEKK2 was immunoprecipitated as shown by immunoblot (middle panel), and recombinant MKK4 substrate loading is detected by coomassie stain (bottom panel). (B) Bar-graph showing relative MEKK2 kinase activity as determined by immunoblot densitometry of pMKK4 induced by MEKK2 from fibronectin-attached cells or cells in suspension of the data shown in A. The results are representative of at least three independent experiments, and comparison of the densitometry from these experiments by paired T test analysis yields a p<0.05.
3.7. MEKK2 regulates fibronectin-induced ERK5 and FAK phosphorylation
Next we examined the functional consequences of fibronectin attachment on signaling proteins downstream of MEKK2 by immunoblot analysis using phospho-specific MAPK antibodies as surrogate indicators of MAPK activation. Integrin ligation clearly activates signaling leading to MEKK2-dependent phosphorylation of ERK5, but surprisingly we did not detect fibronectin-induced JNK activity-associated phosphorylation (Fig. 7A). Similarly, we did not detect p38 phosphorylation in response to attachment to fibronectin. Activity-associated ERK1/2 phosphorylation was consistently detectable although variable, and pERK1/2 from integrin bound cells was usually similar to that of the controls. This consistent high pERK1/2 background level was likely due to the fact that MDA-MB 231 cells express mutated active KRAS. Regardless, pERK1/2 levels in either untreated or fibronectin-bound cells were independent of MEKK2 expression. Collectively, these data indicate that attachment to fibronectin induces signaling that leads to activation of MEKK2 and ERK5, but not JNK.
Fig. 7.
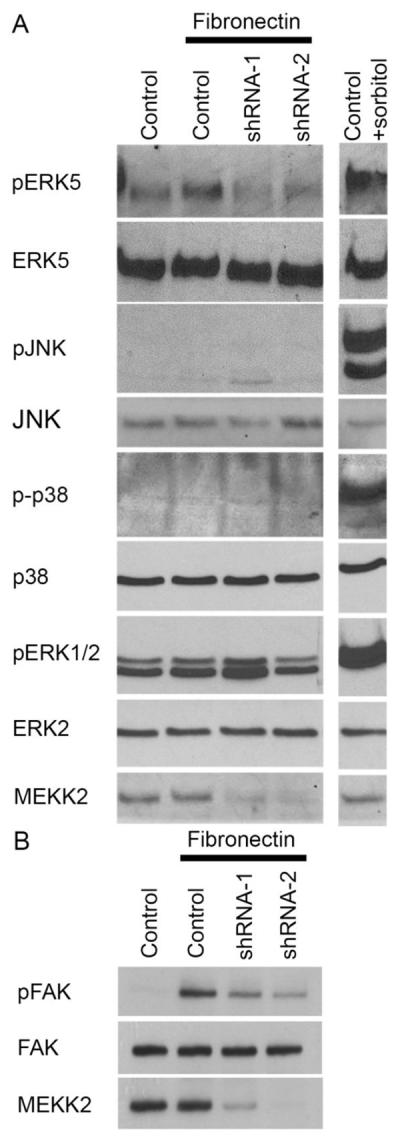

MEKK2 regulates FN-induced signaling. (A and B) MDA-MB 231 control cells and cells with stable MEKK2 knockdown were seeded on uncoated culture plates (not treated) or plates coated with fibronectin. Positive control cells for JNK, ERK5, p38 or ERK1/2 activation were exposed to 0.2M sorbitol for 15 minutes. (B) Immunoblots showing the level of fibronectin-induced FAK Y397 phosphorylation (upper panel) in MDA-MB 231 control cells and in cells with stable MEKK2 knockdown. Total FAK immunoblot (middle panel) shows equal protein content of test lanes and MEKK2 immunoblot (bottom panel) confirms MEKK2 knockdown. (C and D) Graphical representation of a transwell migration assays of MDA-MB 231 cells treated with vehicle (DMSO), BIX02189 MEK5 inhibitor (10 μM) or ERK5 inhibitor XMD 8–92 (5 μM) during the 5 hour migration period. (D) Serum-induced chemotaxis of MDA-MB 231 cells that stably express activated MEKK2 (357–619) is compared to that of MDA-MB 231 cells that express endogenous MEKK2 (control) or cells that express MEKK2 (357–619) but migrated in the presence of the inhibitors. Results are compiled from three independent experiments.
To gain additional insight into the mechanism by which MEKK2 expression influences cell migration, we examined the effect of MEKK2 knockdown on fibronectin-induced FAK autophosphorylation. FAK autophosphorylates tyrosine residue 397 (Y397), that leads to association with multiple proteins and can be used as a surrogate assay for FAK activation. As expected, our immunoblot analysis showed FAK autophosphorylation in response to fibronectin compared to FAK from cells kept in suspension. However, we observed that fibronectin-induced Y397 phosphorylation was reduced in cells with stable MEKK2 knockdown, indicating that FAK autophosphorylation is partly MEKK2-dependent (Fig. 7B). To test this hypothesis, we examined whether ERK5 activity was required for MDA-MB 231 cell migration. Using a modified Boyden chamber assay, we tested to determine the impact of ERK5 inhibition on chemotaxis induced by serum with specific ERK5 inhibitor XMD 8-92 (x). We observed that ERK5 inhibition with either XMD 8-92 or MEK5 inhibitor BIX02189 significantly reduced the ability of cells to migrate (Fig 7C, Supplemental Figure 1), inhibiting chemotaxis to an extent similar to MEKK2 knockdown (Fig. 5B). As MEKK2 phosphorylates and activates MEK5, which, in turn phosphorylates and activates ERK5, these data indicate that MEK5 regulates breast tumor cell chemotaxis, and suggest that the MEKK2 regulates MDA-MB 231 cell migration, at least in part, by control of ERK5 activity. To address this possibility, we expressed constitutively active MEKK2 (MEKK2 kinase domain, amino acids 357–619) [26] in cells with MEKK2 knockdown and repeated the chemotaxis assays in the presence of the inhibitors. We found that stable expression of the MEKK2 kinase domain was associated with an increase in serum-induced chemotaxis, and that this increase was partially blocked by treatment with either an ERK5 inhibitor (XMD 8-92) or a MEK5 inhibitor (BIX02189). These data suggest that MEKK2 activity promotes MDA-MB 231 cell chemotaxis via a mechanism that is partly dependent upon ERK5 activity. Taken together, these results indicate that both MEKK2 and ERK5 activity are important regulators of cell migration, and suggest that MEKK2 signaling contributes to the regulation of FAK phosphorylation.
4. Discussion
Timely initiation and release of adhesion is required for migration of both normal cells and cancerous cells. In our previous work, we demonstrated that shRNA-mediated silencing of MEKK2 expression in invasive breast tumor cells resulted in a dramatic reduction in xenograft metastasis. This metastasis inhibition was specific to MEKK2 knockdown, as blockade of six other MAP3K did not show a similar reduction in the incidence of metastasis [19]. Clearly MEKK2 plays a key role in the regulation of one or more tumor cell functions required for metastasis to occur, but in that study we did not define the functional role of MEKK2 in breast tumor cells. Indeed, prior to that report, very little evidence linking MEKK2 to metastasis was available in the literature. Unlike other MAP3K proteins associated with cancer, such as c-RAF and BRAF [9], MEKK2 has not been extensively studied and the mechanisms responsible for MEKK2 activation are largely unknown. Therefore our goal for this study was to identify MEKK2-dependent tumor cell function, and thereby gain insight into the mechanisms by which MEKK2 ablation effectively blocks xenograft metastasis. We found that MEKK2 expression knockdown was associated with a significant reduction in directed cell migration, and MEKK2-deficient cells displayed an increased number and size of visible focal adhesions as well as significantly enhanced cell spreading that was inversely related to cell front velocity and migration (Supplemental Figure 2). Tumor cell migration is absolutely required for distant metastasis formation, and our data strongly suggest that the observed inhibition of metastasis was due, at least in part, to reduced tumor cell migration in cells in which MEKK2 expression is silenced. It is important to note that, while regulated cell spreading is an essential component of cell migration, dysregulated, exaggerated cell spreading associated with enhanced cell adhesion has been observed to inhibit cell migration [23].
As a result of our investigations, we have uncovered new insights into the regulation of MEKK2 signaling. For example, we discovered that the predominantly cytoplasmic MEKK2 translocates to membrane-proximal areas near focal adhesions complexes in response to cell attachment to fibronectin or Matrigel, an observation that strongly suggests that integrin ligation initiates the formation of a signaling complex that recruits MEKK2. Furthermore, during the course of these studies we have developed a novel technique for quantifying co-localization of two proteins using fluorescent images captured from fixed cells. Utilizing the Surface Builder function of the Imaris software package to analyze Z-stack sets of immunofluorescence images, we were able to accurately quantify the colocalization of two proteins, MEKK2 and vinculin, in three-dimensional space within the cell. As a result, we further report, for the first time, that MEKK2 is recruited to focal adhesion complexes and that attachment to fibronectin induces MEKK2 activation. To our knowledge, this report is the first to link MEKK2 activation to a specific matrix protein. Hence our studies provide a basis for future studies during which we will define the composition of the membrane-proximal signaling complex necessary for MEKK2 recruitment and activation.
Our observation that fibronectin induces MEKK2 activation that is detectable from analysis of whole cell lysates was somewhat surprising result given that only a subset of MEKK2 was translocated to the membrane focal adhesion complexes, whereas some MEKK2 remained localized to the cytoplasm. We postulate that the membrane-associated MEKK2 is active, therefore it is possible that MEKK2 localized to focal adhesions are differently regulated that cytoplasmic MEKK2. Other kinases, including known MEKK2 regulator Src [8], localize to focal adhesions, and thus when it is recruited to focal adhesion protein complexes MEKK2 may be subject to activation by Src and other kinases. The location of potential regulatory phosphorylation sites within MEKK2 remains largely undefined, and aside from the MEKK2 carboxyl-terminal kinase domain and the amino-terminal PB1 domain, the function of most of the remaining MEKK2 sequence remains unknown. A recent report from Matitau and colleagues reveals that serine phosphorylation promotes an interaction between MEKK2 and 14-3-3 that negatively regulates MEKK2 activity [25], and our analysis of the MEKK2 peptide sequence reveals additional potential phosphorylation sites, as well as proline-rich sequences that may serve as SH3-binding proline-rich motifs. Thus much of MEKK2 regulation and function remains to be defined. Indeed, as PB1 domains serve as docking ports for other PB1 domain-containing proteins [18], it is possible that MEKK2 performs a scaffolding function [24] within focal adhesion protein complexes. As MEKK2 interaction with MEK5 is mediated by their respective PB1 domains, MEKK2 serves as both an activator of ERK5 signaling and a scaffold for ERK5 activation complexes. Based on our results, we would postulate that this scaffold function would promote ERK5-dependent phosphorylation of FAK and other ERK5 substrates. Our future work will investigate the interactions between MEKK2 and focal adhesion proteins that mediate MEKK2 localization, activation and function within focal adhesion complexes.
Based on our results, we conclude that integrin receptor ligation to fibronectin initiates signaling that leads to MEKK2 activation and recruitment to focal adhesions. Consistent with these conclusions, we report that MEKK2 knockdown is associated with focal adhesion stabilization and inhibited cell migration. However, the mechanism by which MEKK2 is recruited to focal adhesions was not explored in this project and is unclear at present. Our future studies will examine the nature of the MEKK2-associated protein complex at focal adhesions as an essential step in understanding how MEKK2 integrates into focal complexes and whether any of these components are targets of MEKK2 activity in vivo. As specific MEKK2 inhibitors are not available at present, characterizing these functional MEKK2 interactions may reveal potential therapeutic targets for treatment of pathologies associated with inappropriate cell migration, including, but not limited to, breast cancer metastasis.
In summary, our findings show that cellular attachment to extracellular proteins regulates MEKK2 activity and distribution, and link MEKK2 to control of breast tumor cell migration. Our study is the first to provide evidence that MEKK2 regulates breast tumor cell adhesion complex stability and migration, and controls fibronectin-induced FAK and ERK5 activation.
Supplementary Material
HIGHLIGHTS
Cellular attachment to matrix proteins recruits MEKK2 to focal adhesions.
MEKK2 regulates focal adhesion stability and migration in breast tumor cells
MEKK2 regulates fibronectin-induced ERK5 and FAK phosphorylation
Acknowledgments
FUNDING This work was supported by National Institutes of Health [Grant CA120881 (to B.C)] and the American Cancer Society, Illinois Div. [Grant 160485 (to B.C.)].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTION Ahmed Mirza, Michael Kahle and Magdalene Ameka developed experimental procedures, and designed and performed experiments producing the results described in the manuscript. Bruce Cuevas and Edward Campbell provided technical expertise required for the execution of the experimental procedures. Ahmed Mirza, Michael Kahle, Magdalene Ameka and Bruce Cuevas conceptualized and developed the study. Ahmed Mirza and Bruce Cuevas compiled and wrote the manuscript.
5. References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Petrie RJ, Doyle AD, Yamada KM. Random versus directionally persistent cell migration. Nat. Rev. Mol. Cell Biol. 2009;10:538–549. doi: 10.1038/nrm2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huttenlocher A, Horwitz AR. Integrins in cell migration. Cold Spring Harb. Perspect. Biol. 2011;3:a005074. doi: 10.1101/cshperspect.a005074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Humphries JD, Byron A, Humphries MJ. Integrin ligands at a glance. J. Cell Sci. 2006;119:3901–3903. doi: 10.1242/jcs.03098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zaidel-Bar R, Geiger B. The switchable integrin adhesome. J. Cell Sci. 2010;123:1385–1388. doi: 10.1242/jcs.066183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zaidel-Bar R, Itzkovitz S, Ma'ayan A, Iyengar R, Geiger B. Functional atlas of the integrin adhesome. Nat. Cell Biol. 2007;9:858–867. doi: 10.1038/ncb0807-858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu S, Calderwood DA, Ginsberg MH. Integrin cytoplasmic domain-binding proteins. J. Cell Sci. 2000;113(Pt 20):3563–3571. doi: 10.1242/jcs.113.20.3563. [DOI] [PubMed] [Google Scholar]
- 8.Cronan MR, Nakamura K, Johnson NL, Granger DA, Cuevas BD, Wang JG, Mackman N, Scott JE, Dohlman HG, Johnson GL. Defining MAP3 kinases required for MDA-MB-231 cell tumor growth and metastasis. Oncogene. 2012;31:3889–3900. doi: 10.1038/onc.2011.544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cuevas BD, Abell AN, Johnson GL. Role of mitogen-activated protein kinase kinase kinases in signal integration. Oncogene. 2007;26:3159–3171. doi: 10.1038/sj.onc.1210409. [DOI] [PubMed] [Google Scholar]
- 10.Kennedy NJ, Davis RJ. Role of JNK in tumor development. Cell Cycle. 2003;2:199–201. [PubMed] [Google Scholar]
- 11.Ventura JJ, Hubner A, Zhang C, Flavell RA, Shokat KM, Davis RJ. Chemical genetic analysis of the time course of signal transduction by JNK. Mol. Cell. 2006;21:701–710. doi: 10.1016/j.molcel.2006.01.018. [DOI] [PubMed] [Google Scholar]
- 12.Huang C, Jacobson K, Schaller MD. MAP kinases and cell migration. J. Cell Sci. 2004;117:4619–4628. doi: 10.1242/jcs.01481. [DOI] [PubMed] [Google Scholar]
- 13.Garrington TP, Ishizuka T, Papst PJ, Chayama K, Webb S, Yujiri T, Sun W, Sather S, Russell DM, Gibson SB, Keller G, Gelfand EW, Johnson GL. MEKK2 gene disruption causes loss of cytokine production in response to IgE and c-Kit ligand stimulation of ES cell-derived mast cells. EMBO J. 2000;19:5387–5395. doi: 10.1093/emboj/19.20.5387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Witowsky JA, Johnson GL. Ubiquitylation of MEKK1 inhibits its phosphorylation of MKK1 and MKK4 and activation of the ERK1/2 and JNK pathways. J. Biol. Chem. 2003;278:1403–1406. doi: 10.1074/jbc.C200616200. [DOI] [PubMed] [Google Scholar]
- 15.Yan L, Carr J, Ashby PR, Murry-Tait V, Thompson C, Arthur JS. Knockout of ERK5 causes multiple defects in placental and embryonic development. BMC Dev. Biol. 2003;3:11. doi: 10.1186/1471-213X-3-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Regan CP, Li W, Boucher DM, Spatz S, Su MS, Kuida K. Erk5 null mice display multiple extraembryonic vascular and embryonic cardiovascular defects. Proc. Natl. Acad. Sci. U. S. A. 2002;99:9248–9253. doi: 10.1073/pnas.142293999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sawhney RS, Liu W, Brattain MG. A novel role of ERK5 in integrin-mediated cell adhesion and motility in cancer cells via Fak signaling. J. Cell. Physiol. 2009;219:152–161. doi: 10.1002/jcp.21662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nakamura K, Johnson GL. PB1 domains of MEKK2 and MEKK3 interact with the MEK5 PB1 domain for activation of the ERK5 pathway. J. Biol. Chem. 2003;278:36989–36992. doi: 10.1074/jbc.C300313200. [DOI] [PubMed] [Google Scholar]
- 19.Cronan MR, Nakamura K, Johnson NL, Granger DA, Cuevas BD, Wang JG, Mackman N, Scott JE, Dohlman HG, Johnson GL. Defining MAP3 kinases required for MDA-MB-231 cell tumor growth and metastasis. Oncogene. 31:3889–3900. doi: 10.1038/onc.2011.544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lyle KS, Raaijmakers JH, Bruinsma W, Bos JL, de Rooij J. cAMP-induced Epac-Rap activation inhibits epithelial cell migration by modulating focal adhesion and leading edge dynamics. Cell. Signal. 2008;20:1104–1116. doi: 10.1016/j.cellsig.2008.01.018. [DOI] [PubMed] [Google Scholar]
- 21.Jaerve A, Schiwy N, Schmitz C, Mueller HW. Differential effect of aging on axon sprouting and regenerative growth in spinal cord injury. Exp. Neurol. 2011;231:284–294. doi: 10.1016/j.expneurol.2011.07.002. [DOI] [PubMed] [Google Scholar]
- 22.Cuevas BD, Abell AN, Witowsky JA, Yujiri T, Johnson NL, Kesavan K, Ware M, Jones PL, Weed SA, DeBiasi RL, Oka Y, Tyler KL, Johnson GL. MEKK1 regulates calpain-dependent proteolysis of focal adhesion proteins for rear-end detachment of migrating fibroblasts. EMBO J. 2003;22:3346–3355. doi: 10.1093/emboj/cdg322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Webb K, Hlady V, Tresco PA. Relationships among cell attachment, spreading, cytoskeletal organization, and migration rate for anchorage-dependent cells on model surfaces. J. Biomed. Mater. Res. 2000;49:362–368. doi: 10.1002/(sici)1097-4636(20000305)49:3<362::aid-jbm9>3.0.co;2-s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dhanasekaran DN, Kashef K, Lee CM, Xu H, Reddy EP. Scaffold proteins of MAP-kinase modules. Oncogene. 2007;26:3185–3202. doi: 10.1038/sj.onc.1210411. [DOI] [PubMed] [Google Scholar]
- 25.Matitau AE, Gabor TV, Gill RM, Scheid MP. MEKK2 Kinase Association with 14-3-3 Protein Regulates Activation of c-Jun N-terminal Kinase. J. Biol. Chem. 2013;288:28293–28302. doi: 10.1074/jbc.M113.511352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamashita M, Ying S, Zhang G, Li C, Cheng S, Deng C, Zhang Y. Ubiquitin ligase Smurf1 controls osteoblast activity and bone homeostasis by targeting MEKK2 for degradation. Cell. 2005;121:101–113. doi: 10.1016/j.cell.2005.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.