

Abstract
Sym004 represents a novel EGFR targeting approach comprised of a mixture of two anti-EGFR antibodies directed against distinct epitopes of EGFR. In contrast to single anti-EGFR antibodies, Sym004 induces rapid and highly efficient degradation of EGFR. In the current study, we examine the capacity of Sym004 to augment radiation response in lung cancer and head and neck (H&N) cancer model systems. We first examined the anti-proliferative effect of Sym004 and confirmed 40∼60% growth inhibition by Sym004. Using clonogenic survival analysis, we identified that Sym004 potently increased cell kill by up to 10-fold following radiation exposure. A significant increase of γH2AX foci resulting from DNA double strand breaks was observed in Sym004-treated cells following exposure to radiation. Mechanistic studies further demonstrated that Sym004 enhanced radiation response via induction of cell cycle arrest followed by induction of apoptosis and cell death reflecting inhibitory effects on DNA damage repair. The expression of several critical molecules involved in radiation-induced DNA damage repair were significantly inhibited by Sym004, including DNAPK, NBS1, RAD50, and BRCA1. Using single and fractionated radiation in human tumor xenograft models, we confirmed that the combination of Sym004 and radiation resulted in significant tumor regrowth delay and superior anti-tumor effects compared to treatment with Sym004 or radiation alone. Taken together, these data reveal the strong capacity of Sym004 to augment radiation response in lung and H&N cancers. The unique action mechanism of Sym004 warrants further investigation as a promising EGFR targeting agent combined with radiotherapy in cancer therapy.
Keywords: Sym004, EGFR, Antibody, Radiation, Repair
Introduction
The epidermal growth factor receptor (EGFR) plays an important role in human tumorigenesis. Targeting EGFR with molecular inhibitors has been intensely pursued as a cancer treatment strategy following landmark studies by Mendelsohn and colleagues who demonstrated the anti-tumor effect of a monoclonal antibody (mAb) to EGFR (1). To date, four EGFR targeting agents (cetuximab, panitumumab, gefitinib and erlotinib) from two distinct drug classes have gained FDA approval (2). These include mAbs directed against the extracellular ligand-binding domain of EGFR and small molecules tyrosine kinase inhibitors (TKIs) directed against the cytosolic catalytic domain of the EGFR. Among these, cetuximab (Erbitux®) and panitumumab (Vectibix®) are approved mAbs for the treatment of metastatic colorectal cancer and cetuximab for the treatment of H&N cancer.
A series of preclinical studies provide proof-of-principle that cetuximab and paniumumab can enhance radiation response in a variety of tumor model systems (3-5). Although selected patients receive clear benefit from anti-EGFR mAbs, overall single agent response rates are on the order of 10% (6). Favorable response rates and improved survival is achieved when cetuximab is combined with radiotherapy as in the landmark phase III clinical trial (7, 8), although a substantial proportion of patients do not respond to anti-EGFR therapy reflecting primary or acquired resistance. Moreover, many patients who initially respond well to treatment still manifest subsequent tumor recurrence (9, 10). Hence, the advancement of next generation EGFR inhibitors and further refinement of complementary therapeutic approaches are valuable to further enhance anti-EGFR treatment efficacy.
When two mAbs against distinct receptor epitopes are combined, a rapid and more efficient receptor internalization is observed and followed by EGFR degradation (11). The mixture antibody treatment is also more effective than single Abs in inhibiting signaling and tumor growth in tissue culture and animal models (12, 13). Furthermore, antibody mixtures have been shown to activate complement–dependent cytotoxicity that may contribute to a more effective anti-tumor capacity than single mAbs (14). Sym004 was developed following screening of >400 different mAb combinations based on the highest capacity to inhibit cell growth (15). Sym004 is a mixture of anti-EGFR mAb 992 and 1024 that targets non-overlapping epitopes (epitope 992 vs 1024) in EGFR extracellular domain III. Interestingly, mAb 992 and 1024 work highly synergistically when combined. These two epitopes are different from the epitopes of cetuximab and panitumumab. Similar to cetuximab, Sym004 can inhibit tumor growth by blocking ligand-induced receptor activation and signaling. However, Sym004 induces a highly efficient degradation of EGFR which is not observed with cetuximab (16). It is postulated that the rapid and efficient EGFR internalization results from clustering of receptor-antibody complexes at the cell membrane. Preliminary studies demonstrate that Sym004 exhibits superior anti-tumor capacity in comparison to cetuximab or panitumumab in both in vitro and in vivo models (16). Furthermore, Sym004 inhibits growth of cancer cells with acquired resistance to cetuximab resulting from increased EGFR ligand production. These findings highlight Sym004 as a promising strategy to maximize EGFR inhibition that may induce more potent tumor suppression than current clinically used EGFR targeting mAbs.
Materials and Methods
Reagents and antibodies
Sym004 was provided by Symphogen A/S (Lyngby, Denmark). Antibodies against EGFR, p-EGFR (Y1173), BAD, Importinβ1 and Histone 3 were obtained from Santa Cruz Biotechnology Inc. (Santa Cruz, CA) and anti-DNAPK was obtained from Thermal Scientific Lab Vision (Kalamazoo, MI). Anti-α-tubulin was obtained from Calbiochem (San Diego, CA) and anti-Sec61β was obtained from Upstate (Lake Placid, NY). All other antibodies were obtained from Cell Signaling Technology (Beverly, MA) and all other chemicals were purchased from Sigma (St. Louis, MO).
Cell lines
The primary human non-small cell lung carcinoma (NSCLC) H226 cell line was provided by Drs John Minna and Adi Gazdar (University of Texas Southwestern Medical School, Dallas, TX) 10 years ago and H292 cell line was obtained from the American Type Culture Collection in 2005. The human head and neck squamous cell carcinoma SCC1 (UM-SCC1) cell line was provided by Dr. Thomas E. Carey (University of Michigan, Ann Arbor, MI) and SCC1483 cell line was provided by Dr. Jennifer Grandis (University of Pittsburgh, Pittsburgh, PA) in 2004. NSCLC cells were maintained in RPMI medium with 10% FBS and HNSCC cells were cultured in DMEM supplemented with 10% FBS and 1 μg/ml hydrocortisone. The authenticity of these cell lines was regularly verified on the basis of cell morphology and genomic short tandem repeat (STR) profile of each cell line. All cell culture media and supplements were obtained from Life Technologies, Inc. (Gaithersburg, MD).
Cell proliferation assay
Viable growing cells was determined by crystal violet staining as described previously (17).
Quantification of EGFR mRNA expression
EGFR mRNA level was quantified with real-time PCR (RT-qPCR) using a Bio-Rad iQ®5 RT-qPCR Detection System and SsoFast EvaGreen® Supermix reagent as recommended by manufacturer (Bio-Rad Laboratories, Hercules, CA). Detailed information is provided in the Supplementary Materials and Methods.
Cellular fractionation and immunoblotting analyses
Following harvesting, cells were lysed in a NP-40 lysis buffer (20 mM HEPES, pH 7.0, 10 mM KCl, 2 mM MgCl2, 0.5% NP-40, 1 mM Na3VO4, 10 mM NaF, 1 mM PMSF, 2 μg/ml aprotinin). Thereafter, the cells were homogenized by a tightly fitting Dounce homogenizer followed by centrifugation at 1,500 xg for 5 min to sediment the nuclei. The supernatant was then centrifuged at 16,100 xg for 20 min, and the resulting supernatant formed the non-nuclear fraction. To extract nuclear proteins, the isolated nuclei were resuspended in NETN buffer (20 mM Tris-Cl, pH 8.0, 150mM NaCl, 1mM EDTA, 0.5% NP-40, 1 mM Na3VO4, 10 mM NaF, 1 mM PMSF, and 2 mg/ml aprotinin) followed by sonication. Nuclear lysates were then collected after centrifugation at 16,100 xg for 20 min. To obtain whole cell lysates for western blot analysis, cells were lysed with Tween-20 lysis buffer and sonicated. Following quantification by Bradford analysis, equal protein amounts were loaded and analyzed by SDS-PAGE as described previously (17).
Radiation survival
Survival following radiation exposure was defined as the ability of the cells to maintain their clonogenic capacity and to form colonies as described previously (17).
Immunofluorescent staining of γH2AX Foci
Cells were plated on chamber slides and exposed to 10 μg/ml of drugs for 1.5 hrs before irradiation. Twenty-four hrs following 2 Gy radiation, cells were fixed in 2% paraformaldehyde and permeabilized in 0.2% Triton X-100. The cells were then probed with anti-γH2AX antibody (Upstate, Billerica, MA) followed by Alexa Fluor 488-conjugated secondary antibody (Invitrogen, Carlsbad, CA). Fluorescent γH2AX foci were then captured using a Zeiss Axioplan fluorescent microscope. To quantitate γH2AX foci, visual scoring of foci in 200 randomly chosen intact nuclei from irradiated samples was determined after subtracting the background numbers of foci from un-irradiated samples.
Cell cycle analysis
Cell cycle phase of tumor cell was determined by flow cytometry using popidium iodide (PI) staining as described previously (17).
Apoptosis assessment
Apoptosis was detected by flow cytometry using an Annexin V/Propidium iodide (PI) dual staining kit from BD Biosciences Pharmingen (San Diego, CA) as described previously (17).
Determination of radiation response in human tumor xenografts
Athymic nude mice were obtained from Harlan Bioproducts for science (Indianapolis, IN) and maintained in a laminar air-flow cabinet under aseptic conditions. The care and treatment of experimental animals was in accordance with institutional guidelines. H226 or SCC1483 cells were injected subcutaneously into the dorsal flank area of the mice. Tumor volume was monitored by direct measurement with calipers and calculated by the formula; π/6 × (large diameter) × (small diameter)2. Following the establishment of tumor, mice were randomly selected to receive control human IgG, radiation alone, Sym004 alone, or radiation in combination with Sym004. Sym004 was administered via i.p. injection twice per week at the specified doses and intervals. Radiation treatment was delivered by a cabinet X-ray biological irradiator X-RAD 320 from Precision X-Ray, Inc. (North Branford, CT). Mice were immobilized using custom-designed jigs that exposed the dorsal flank (harboring tumor xenografts) to irradiation without exposing non-tumor bearing normal tissues.
Statistical analysis
Student's t-test was used to evaluate the significance of difference between Sym004 and control group. Differences between treatments were considered statistically significant when p < 0.05.
Results
Sym004 induces EGFR degradation and inhibits proliferation
Sym004 is a mixture of two anti-EGFR antibodies that has been shown to induce a rapid and efficient degradation of EGFR (16). To confirm this observation, we first examined the level of EGFR by immunoblotting following Sym004 treatment for 0.5, 2 and 24 hrs in NSCLC and HNSCC cells. As shown in Fig 1A, treatment with 10 μg/ml of Sym004 significantly reduces the total EGFR level within 24 hrs in all tested cell lines. The reduction in EGFR after Sym004 treatment appears to reflect post-translational down-regulation of EGFR since no significant change of EGFR mRNA level was observed in cells treated with Sym004 up to 48 hrs (Fig 1B). Notably, we found that Sym004 inhibited phosphorylation of EGFR at tyrosine 1173 in a bi-phasic pattern that has been shown in several HNSCC cell lines following cetuximab treatment in a previous report (18). Although the precise mechanism for this bi-phasic inhibition of Sym004 is not clear, a pronounced inhibition of cell proliferation was observed after treatment with Sym004 (Fig. 1C). Sym004 significantly inhibits the proliferation of all tested cells in a dose-dependent manner.
Fig. 1. Effect of Sym004 on EGFR expression and cell proliferation.
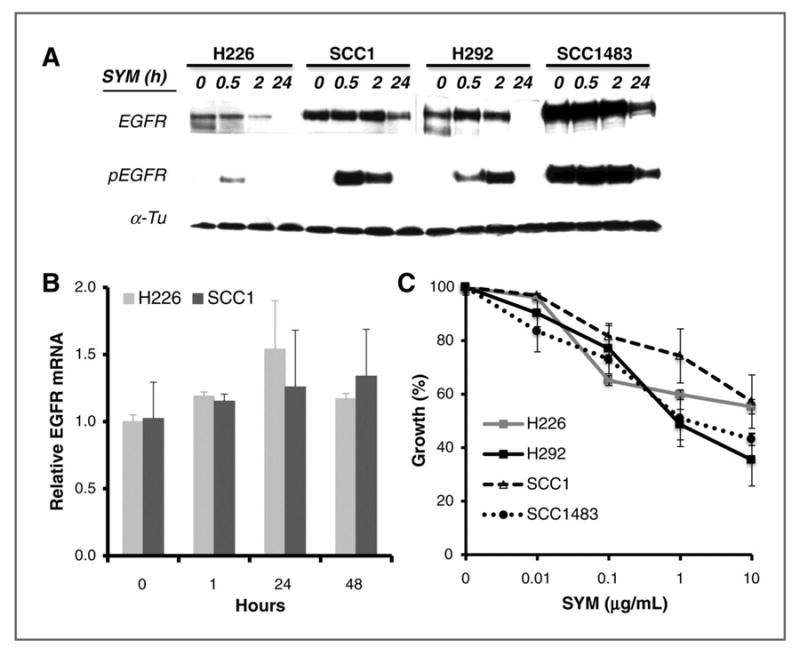
(A) Cells were treated with 10 μg/ml Sym004 for 0, 0.5, 2 or 24 hrs. Thereafter, total and phosphorylated EGFR level was determined by immunoblotting analysis. α-Tubulin (α-Tu) serves as a loading control. (B) Effect of Sym004 on EGFR gene expression was examined by RT-qPCR as described in “Materials and Methods”. (C) Cells were exposed to serial concentrations of Sym004 for 72 hrs. Thereafter, growth of tumor cells was determined by cell proliferation analysis.
Sym004 enhances radiosensitivity
We next investigated whether the superior down-regulation of EGFR induced by Sym004 could be translated into augmentation of radiation response in tumor cells. We examined radiation response of tumor cells following Sym004 treatment using clonogenic survival analysis. As shown in Fig 2, treatment with Sym004 prior to radiation significantly reduced clonogenic survival when compared with controls in all cell lines tested. The radiation dose enhancement ratios of survival at 10% (ER10) induced by Sym004 are 1.45 (H226), 1.21 (H292), 1.12 (SCC1) and 1.21 (SCC1483) respectively.
Fig. 2. Effect of Sym004 on radiosensitivity.
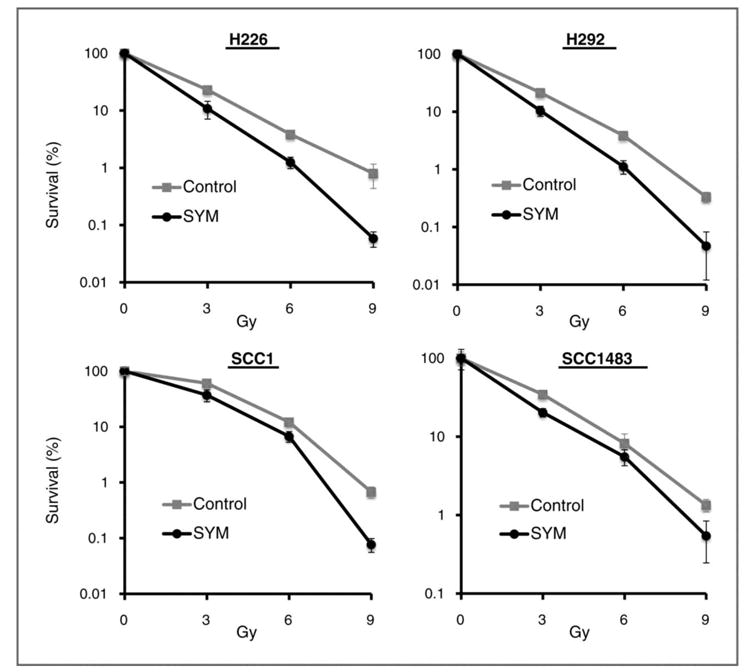
Radiosensitivity of control or Sym004 (10 μg/ml, 72 hrs) pre-treated cells was examined by clonogenic survival analysis as described in “Materials and Methods”. Results were expressed as the percentage of colony formation relative to cells without radiation in each group. Data points are represented as mean ± SD
Sym004 inhibits radiation-induced EGFR signaling and cell cycle progression
To further investigate underlying mechanisms for the effect of Sym004 on radiation response, immunoblotting analysis was conducted to examine the effect of Sym004 on radiation-induced EGFR signaling and cell cycle progression. Consistent with previous findings (19), we observed radiation-induced activation of EGFR (T1173) and downstream MAPK signaling in the control H226 cells 1∼4 hours after 6 Gy radiation as shown in Fig 3A. This radiation-induced EGFR signaling correlated well with an increase of p-RB that serves as a key regulatory factor to stimulate G1-S progression. However, pretreatment with Sym004 inhibited radiation-induced EGFR survival signaling and the expression level of p-RB. A significant increase of cleaved PARP (ΔPARP) and BAD that resulted from the activation of apoptosis was also found in Sym004-treated cells 24 hrs after radiation.
Fig. 3. Effect of Sym004 on radiation-induced signaling and cell cycle progression.

(A) H226 cells were either non-treated (control) or pre-treated with 10 μg/ml of Sym004 for 24 hrs followed by 6 Gy radiation. Thereafter, cells were harvested at 0, 1, 4 or 24 hrs after radiation and lysed for immunoblotting analysis. Δ represents cleaved fragment of PARP. (B) Following pretreatment with 10 μg/ml Sym004 for 24 hrs, H226 cells were fixed and analyzed by flow cytometry. Figure depicts cell populations in G0/G1, S and G2/M phase at 0 (D0), 1 (D1) or 2 (D2) days following radiation.
To validate the effect of Sym004 on radiation-induced cell cycle progression, we examined the cell cycle phase distribution 1 and 2 days following exposure to radiation treatment. As shown in Fig 3B, the S phase cell cycle populations were enhanced 1 day (D1) after radiation in the control H226 cells. This result correlates well with previous observation of an increase of EGFR signaling and p-Rb following radiation treatment in Fig 3A. However, treatment with Sym004 significantly inhibited radiation-induced cell cycle G1-S progression and resulted in cell cycle arrest in G0/G1 phase. In addition, augmentation of radiation-induced G2/M arrest was observed on Sym004-treated cells when compared to controls. Similar results were found in 3 additional tumor cell lines (data not shown).
Sym004 inhibits DNA damage repair
We then characterized the DNA damage profile following radiation by examining the numbers of phosphorylated histone 2AX (γH2AX) foci in the nucleus which resulted from radiation-induced DNA double strand breaks (DSB). As shown in Fig 4A, a significant increase of γH2AX foci was observed in Sym004-pretreated H226 and SCC1 cells by a factor of 1.4∼1.6 at two hrs after radiation when compared to control cells. Since radiation response is initiated with the recognition of DNA damage and often results in cell cycle arrest for repair, we next examined the effect of Sym004 on several check point proteins for initiation of radiation-induced DNA damage repair, such as BRCA1, ATM and CHK1. As shown in Fig 4B, all these check point molecules were significantly activated in in a dose-dependent response following radiation in control cells without Sym004 pretreatment. Following 2 or 24 hrs of pretreatment with Sym004, the activation of these molecules was significantly inhibited, especially in the 10 Gy-treated cells. Notably, the inhibitory effect of Sym004 was more profound in cells exposed to a high dose of radiation at 10 Gy than 2 Gy. This finding also validates the more profound cell killing observed in Sym004-treated cells following a high dose 9 Gy radiation in Fig 2.
Fig. 4. Effect of Sym004 on DNA damage repair.
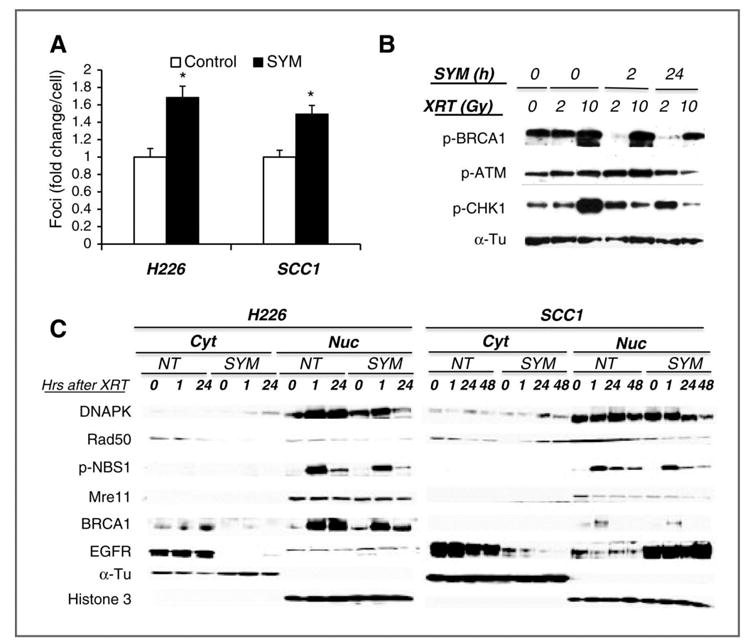
(A) Radiation-induced DNA damage was determined by examining γH2AX foci in the nucleus by immunofluorescent staining as described in “Materials and Methods”. Results were expressed as the fold-change of foci relative to controls. Data points are represented as mean ± SD with *p<0.05 (B) H292 cells were either non-treated (control) or pre-treated with 10 μg/ml of Sym004 for 24 hrs followed by 6 Gy radiation for 0, 2 or 24 hrs followed by 0, 2 or 10 Gy of radiation. Twenty-four hrs after radiation, whole cell lysates were analyzed by immunoblotting analysis. (C) depicts the effect of Sym004 on the expression of nuclear DNA damage repair proteins 0∼48 hrs following 6 Gy of radiation. Histone 3 and α-Tu serve as loading and purity controls of nuclear and non-nuclear fractions respectively. Figure is representative of either 2 or 3 independent experiments with similar results.
We further examined the capacity of Sym004 to inhibit DNA damage repair by examining several key proteins involved in the repair of lethal DSB in the nucleus, such as DNAPK, NBS1, and Mre11. Among them, the block of nuclear DNAPK translocation is known to play an important role in regulating cetuximab-induced radiosensitivity (20, 21). Consistently, we found a significant inhibition of nuclear translocation of DNAPK following 6 Gy radiation in both Sym004-treated H226 and SCC1 cells as shown in Fig 4C. In addition, the levels of Mre11, Rad50 and pNBS1 that form a complex in DSB sites to promote repair processes in the nucleus were reduced by Sym004 treatment. Consistent with the inhibition of pBRCA1 in Fig 4B, the level of nuclear BRCA1 was also decreased in Sym004-treated cells. Since inhibition of all these repair molecules is known to sensitize tumor cells to radiation, these results suggest that Sym004 appears to augment the radiation effect via inhibiting DNA repair machinery in tumor cells arrested in G0/G1 and G2/M. Notably, we found that a higher level of EGFR was present in the nuclear fraction of Sym004-pretreated SCC1 cells, but not in H226 cells as shown in Fig 4C. Since previous studies have shown a negative impact of nuclear EGFR on the sensitivity and survival following radiation treatment (22, 23), the finding of high nuclear EGFR in SCC1 cells compared to H226 cells correlates well with the reduced radiation response in SCC1 cells than that of H226 cells following Sym004 treatment at a dose ≤ 6 Gy.
Sym004 enhances radiation-induced apoptosis
Since un-repaired tumor cells are prone to apoptotic cell death, we examined the capacity of Sym004 to induce apoptosis following radiation using Annexin V/PI flow cytometry analysis. Fig. 5 depicts representative FACS cytograms of dual stained cells from control or Sym004-pretreated cells in all cell lines tested. Following 6 Gy radiation exposure, the percentage of early apoptotic cells in the lower right quadrants (Annexin V-positive/PI-negative) was slightly increased in the control cells. The fold increase of the early apoptotic population was 2.6, 1.5, 1.3 and 1.3 for H226, H292, SCC1 and SCC1483, respectively. However, treatment with Sym004 prior to radiation induced a more profound induction of apoptosis with fold increases of 6.7(H226), 2.7(SCC1), 2.5(SCC1) or 2.4(SCC1483) respectively. Interestingly, the highest enhancement of apoptosis was observed in H226 cells. This result also correlated with a more profound cell killing of H226 cells in the clonogenic survival analysis. In addition, a more profound increase of cells in the late apoptotic/necrotic phase (italic number) in the upper right quadrants of Fig 5, was found in most of Sym004-treated cells when compared to control. These results strongly suggest that Sym004 hinders the EGFR-mediated radiation damage repair response and results in a stronger induction of apoptosis.
Fig. 5. Effect of Sym004 on radiation-induced apoptosis.
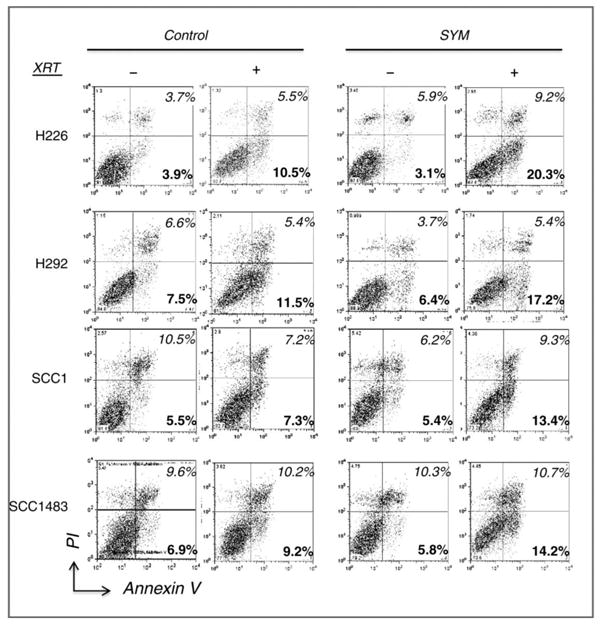
Tumor cells were either non-treated (control) or pre-treated with 10 μg/ml of Sym004 for 24 hrs followed by 6 Gy radiation. Cells were then harvested 2 days following radiation and processed for Annexin V/PI apoptosis analysis as described in “Materials and Methods”. Numbers indicated in black and grey in the lower and upper right quadrants represent percentage of cells in early (Annexin V-positive/PI-negative) or late (double negative) apoptotic phase. Figure is representative of either 2 or 3 independent experiments with similar results.
Sym004 augments radiation response in human tumor xenografts
To expand upon the in vitro findings, we investigated the capacity of Sym004 to augment radiation in both NSCLC and HNSCC human xenografts using a variety of treatment dose and schedule schemes. We first examined the response of NSCLC xenografts (H226) following combination treatment of radiation with two different doses of Sym004 at 1.6 (S1.6) or 4.8 (S4.8) mg/kg. As expected, Sym004 alone inhibited tumor growth in a dose dependent manner as shown in Fig 6A. Interestingly, the combination of radiation (1.5 Gy) and either dose of Sym004 induced a dramatic inhibition of tumor growth while single treatments only caused a modest effect when compared to the control group. The combined radiation and high dose Sym004 (S4.8) achieved a more pronounced antitumor effect than low dose Sym004 S1.6 reflecting its capacity to delay tumor regrowth.
Fig. 6. Effect of Sym004 on radiation response of human tumor xenografts.
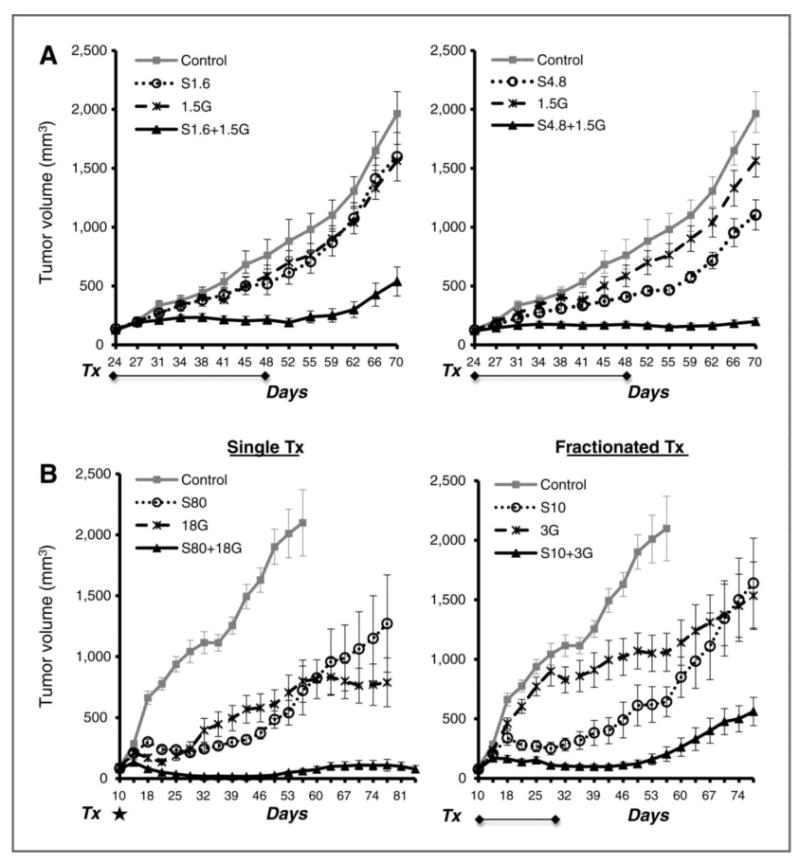
(A) Mice with H226 tumor xenografts were treated with Sym004 at 1.6 (left panel) or 4.8 (right panel) mg/kg/dose twice weekly from day 24-48. Radiation was delivered simultaneously with Sym004 at a dose of 1.5 Gy/fraction. (B) Depicts growth profile of SCC1483 tumor xenografts following single (left panel) or fractionated (right) treatment with radiation and/or Sym004. For single treatment, mice were treated with a single dose of Sym004 (80 mg/kg) and radiation (18Gy) on day 10 as highlighted by *. For fractionated treatment, mice received 6 fractions of radiation at 3 Gy and 4 doses of Sym004 at 10 mg/kg/dose. Tumor volume was monitored and values represent mean tumor size (mm3) ± SEM (n=8∼10 per group).
To further evaluate the impact of combined Sym004 and radiation treatment, we examined radiation response in a HNSCC xenograft (SCC1483) following 2 different treatment schemes as shown in Fig 6B. The left panel shows the tumor response following a single treatment of high dose Sym004 (80 mg/kg, S80) and/or radiation (18 Gy) at day 10. As expected, high dose radiation resulted in significant tumor shrinkage during the first few weeks following treatment. However, surviving tumor cells commenced regrowth within two weeks after radiation. The combination of radiation and Sym004 was very potent to regress established tumors and thereafter induced significant tumor regrowth delay over 9-10 weeks after treatment. Similarly, the combination treatment of fractionated radiation and Sym004 also caused a significant growth delay as shown in the right panel of Fig 6B, but the capacity of the fractionated treatment to inhibit tumor regrowth was more modest than that of the single treatment. This likely reflects more modest cell killing in the fractionated treatments with low dose radiation and drug exposure. Although there are slightly different response profiles between the single and fractionated treatment regimens, these results confirm and extend the previous in vitro findings and indicate a profound capacity of Sym004 to augment radiation response in vivo.
Discussion
Sym004 is a recently developed novel EGFR targeting approach comprising a mixture of two distinct anti-EGFR mAbs. With a potent capacity to inhibit EGFR via the induction of rapid and efficient internalization and degradation of EGFR, Sym004 exhibits a more pronounced growth inhibition than current EGFR targeting agents across a variety of tumor cells (16). In the present study, we provide evidence that the powerful downregulation of EGFR induced by Sym004 can be translated into a profound augmentation of radiation response in tumor cells. In tumor xenografts, we observed a superior anti-tumor capacity of Sym004 when combined with either single or fractionated radiation (Fig 6). In addition, we found potent cell killing in Sym004-treated cells, especially when cells were exposed to high dose radiation in clonogenic survival analysis (Fig 2). These results are consistent with the more profound inhibition of check point proteins for DNA damage by Sym004 after 10 Gy radiation when compared to 2 Gy as shown in Fig 4B. These data suggest that the effect of Sym004-induced EGFR downregulation can be amplified when cells are exposed to high dose radiation.
The potent enhancement of radiosensitivity we observed with Sym004 was seldom observed with other EGFR targeting agents including cetuximab from our previous studies over the last 14 years (5, 20, 24). To establish a direct comparison, we performed side-by-side analysis of clonogenic survival in Sym004- and cetuximab-treated cells following a 9-Gy radiation exposure. As shown in Supplemental Fig 1A, we observed a 2.5∼7.2 fold greater reduction in clonogenic survival in Sym004-treated cells when compared to cetuximab-treated cells in all of the cell lines tested. Furthermore, western blot analysis demonstrated that Sym004 is more potent than cetuximb to inhibit radiation-induced phosphorylation of EGFR, AKT and CDC2 24 hrs after radiation that regulate survival signaling and cell cycle progression (Supplemental Fig 1B). Most importantly, Sym004 demonstrated a stronger impact than cetuximab to inhibit DNA damage repair signaling and prompt the induction of apoptosis. A significant inhibition of radiation-induced p-NBS1 and p-p53 as well as induction of γH2AX was observed in Sym004-treated cells (Supplemental Fig 1B). With the observed loss of EGFR in Sym004-treated cells, these results suggest that Sym004 is more potent than cetuximab to enhance radiosensitivity likely reflecting its profound capacity to down-regulate EGFR.
Consistently, we observed a correlation between Sym004-induced radiation response and EGFR downregulation across several lines of study in the current report. As shown in Fig 1A, EGFR and pEGFR levels were robustly decreased at 24 hrs. In turn, a more significant inhibition of radiation-induced pBRCA1, pATM and pCHK1 was observed in cells exposed to Sym004 for 24 hrs than at 2 h hrs as shown in Fig 4B. In addition, Sym004 was more potent at enhancing radiosensitivity in H226 and H292 cells than in SCC1 and SCC1483 cells, especially at a dose ≤ 6 Gy (Fig 2). The enhanced radiosensitivity in the H226 and H292 cell lines correlated with a more rapid and efficient EGFR down-regulation in H226 and H292 compared to SCC1 and SCC1483 cell lines (Fig 1A). These consistent results suggest EGFR down-regulation as a key contributor to Sym004-induced radiosensitivity. However, a more detailed comparison to include additional cell lines and radiation doses will be valuable to expand these observations from the currently tested 4 cell lines.
Surprisingly, we found that a significant proportion of EGFR was present in the nuclear, but not cytosolic fraction of SCC1 following 24 hrs of Sym004 pretreatment while EGFR was present in the cytosolic fraction of untreated control cells as shown in Fig 4C. Further studies showed that Sym004 caused a significant nuclear accumulation within 30 min in all cell lines tested (Supplemental Fig 2A). Although nuclear EGFR was down regulated in all cell lines at 24 hrs, SCC1 and SCC1483 demonstrated a lower rate and intensity of EGFR down regulation when compared to H226 and H292 cells. This finding is consistent with a lower radiation response in SCC1 cells than that of H226 cells following 24-hrs of Sym004 treatment. Using immunoprecipitation, we further identified that the association of EGFR and Importin β1 or Sce61β that are involved in EGFR nuclear import (25, 26) was higher in SCC1 than those in H226 cells 24 hrs after Sym004 treatment (Supplemental Fig 2B). These findings suggest that Sym004 can induce EGFR nuclear translocation and that nuclear EGFR may be involved in regulating radiation response.
In addition to Sym004, cetuximab has been previously shown to induce nuclear translocation of EGFR in tumor cells, including SCC1 and SCC1483 (27, 28). However, different response profiles of nuclear EGFR translocation were observed when cetuximab-treated and Sym004-treated SCC1 and SCC1483 cells were compared. Cetuximab-induced nuclear EGFR was maintained up to 50∼96 hrs in both SCC cells (28). In contrast, Sym004-induced nuclear EGFR was maintained less than 24 hrs as shown in Supplemental Fig 2A. The rapid reduction of nuclear EGFR translocation could result from the degradation of EGFR induced by Sym004. The lower level of nuclear EGFR correlated with a higher radiation response in Sym004-treated SCC cells when compared to cetuximab-treated cells as shown in Supplemental Fig 1
A more detailed investigation of Sym004- and cetuximab-induced EGFR nuclear translocation and the resultant impact on radiosensitivity is highly desired. Specifically, anti-EGFR mAb can induce nuclear EGFR accumulation that may enhance resistance to radiation (28, 29). Several molecular image-tracking experiments are underway to examine EGFR trafficking following Sym004 or cetuximab treatment. Surprisingly, our preliminary results from 3D imaging suggest that EGFR is located on the nuclear surface, as opposed to inside the nucleus, following treatment with both anti-EGFR agents (data not shown). Side views of the intact nucleus show the accumulation of EGFR on the external (surface) of nucleus. Since most studies of EGFR trafficking have been performed using cellular fractionation and 2D imaging, the nuclear translocation of EGFR induced by cetuximab or Sym004 may reflect limitations of cellular fractionation and 2D imaging. More detailed molecular imaging studies are in progress to further clarify the effect of anti-EGFR agents on EGFR nuclear trafficking.
In conclusion, Sym004 demonstrates the capacity to augment radiation response in NSCLC and HNSCC tumors both in vitro and in vivo. With a potent ability to induce rapid and efficient EGFR internalization and degradation, Sym004 may afford a more powerful anti-EGFR therapy approach than the currently used single EGFR mAbs. Preclinical pharmacokinetic studies in primates and early clinical trial safety and feasibility studies in humans indicate that Sym004 is well tolerated and does not induce unexpected toxicities (30). Several clinical trials are in progress to evaluate the clinical potential of Sym004 for patients with HNSCC and metastatic colorectal cancer (31, 32). With mature clinical trial data demonstrating a modest survival advantage (∼10%) for HNSCC patients treated with cetuximab with radiation (7), the more powerful impact of Sym004 compared to cetuximab in the current report provides a rationale to design clinical studies to test the impact of Sym004 with radiation for HNSCC patients in an effort to further improve overall outcome.
Supplementary Material
Acknowledgments
Grant Support: This work was supported in part by research funding from Symphogen A/S, Lyngby, Denmark (P.M. Harari).
Footnotes
Disclosure of Potential Conflicts of Interest: P.M. Harari holds a laboratory research agreement with Symphogen. M. Kragh and M.W. Pedersen are employees of Symphogen.
References
- 1.Masui H, Kawamoto T, Sato JD, Wolf B, Sato G, Mendelsohn J. Growth inhibition of human tumor cells in athymic mice by anti-epidermal growth factor receptor monoclonal antibodies. Cancer Res. 1984;44:1002–7. [PubMed] [Google Scholar]
- 2.Wheeler DL, Dunn EF, Harari PM. Understanding resistance to egfr inhibitors[mdash]impact on future treatment strategies. Nat Rev Clin Oncol. 2010;7:493–507. doi: 10.1038/nrclinonc.2010.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huang S, Bock JM, Harari PM. Epidermal growth factor receptor blockade with c225 modulates proliferation, apoptosis, and radiosensitivity in squamous cell carcinomas of the head and neck. Cancer Res. 1999;59:1935–40. [PubMed] [Google Scholar]
- 4.Milas L, Mason K, Hunter N, Petersen S, Yamakawa M, Ang K, et al. In vivo enhancement of tumor radioresponse by c225 antiepidermal growth factor receptor antibody. Clin Cancer Res. 2000;6:701–8. [PubMed] [Google Scholar]
- 5.Kruser TJ, Armstrong EA, Ghia AJ, Huang S, Wheeler DL, Radinsky R, et al. Augmentation of radiation response by panitumumab in models of upper aerodigestive tract cancer. International Journal of Radiation Oncology*Biology*Physics. 2008;72:534–42. doi: 10.1016/j.ijrobp.2008.06.1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mendelsohn J, Baselga J. Epidermal growth factor receptor targeting in cancer. Semin Oncol. 2006;33:369–85. doi: 10.1053/j.seminoncol.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 7.Bonner JA, Harari PM, Giralt J, Azarnia N, Shin DM, Cohen RB, et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N Engl J Med. 2006;354:567–78. doi: 10.1056/NEJMoa053422. [DOI] [PubMed] [Google Scholar]
- 8.Bonner JA, Harari PM, Giralt J, Cohen RB, Jones CU, Sur RK, et al. Radiotherapy plus cetuximab for locoregionally advanced head and neck cancer: 5-year survival data from a phase 3 randomised trial, and relation between cetuximab-induced rash and survival. The Lancet Oncol. 2010;11:21–28. doi: 10.1016/S1470-2045(09)70311-0. [DOI] [PubMed] [Google Scholar]
- 9.Jackman D, Pao W, Riely GJ, Engelman JA, Kris MG, Janne PA, et al. Clinical definition of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. J Clin Oncol. 2010;28:357–60. doi: 10.1200/JCO.2009.24.7049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, et al. Genotypic and histological evolution of lung cancers acquiring resistance to egfr inhibitors. Sci Transl Med. 2011;3:75ra26. doi: 10.1126/scitranslmed.3002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Friedman LM, Rinon A, Schechter B, Lyass L, Lavi S, Bacus SS, et al. Synergistic down-regulation of receptor tyrosine kinases by combinations of mabs: Implications for cancer immunotherapy. PNAS. 2005;102:1915–20. doi: 10.1073/pnas.0409610102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ben-Kasus T, Schechter B, Lavi S, Yarden Y, Sela M. Persistent elimination of erbb-2/her2-overexpressing tumors using combinations of monoclonal antibodies: Relevance of receptor endocytosis. PNAS. 2009;106:3294–99. doi: 10.1073/pnas.0812059106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Spangler JB, Neil JR, Abramovitch S, Yarden Y, White FM, Lauffenburger DA, et al. Combination antibody treatment down-regulates epidermal growth factor receptor by inhibiting endosomal recycling. PNAS. 2010;107:13252–57. doi: 10.1073/pnas.0913476107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dechant M, Weisner W, Berger S, Peipp M, Beyer T, Schneider-Merck T, et al. Complement-dependent tumor cell lysis triggered by combinations of epidermal growth factor receptor antibodies. Cancer Res. 2008;68:4998–5003. doi: 10.1158/0008-5472.CAN-07-6226. [DOI] [PubMed] [Google Scholar]
- 15.Koefoed K, Steinaa L, Soderberg JN, Kjaer I, Jacobsen HJ, Meijer PJ, et al. Rational identification of an optimal antibody mixture for targeting the epidermal growth factor receptor. MAbs. 2011;3:584–95. doi: 10.4161/mabs.3.6.17955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pedersen MW, Jacobsen HJ, Koefoed K, Hey A, Pyke C, Haurum JS, et al. Sym004: A novel synergistic anti-epidermal growth factor receptor antibody mixture with superior anticancer efficacy. Cancer Res. 2010;70:588–97. doi: 10.1158/0008-5472.CAN-09-1417. [DOI] [PubMed] [Google Scholar]
- 17.Benavente S, Huang S, Armstrong EA, Chi A, Hsu KT, Wheeler DL, et al. Establishment and characterization of a model of acquired resistance to epidermal growth factor receptor targeting agents in human cancer cells. Clin Cancer Res. 2009;15:1585–92. doi: 10.1158/1078-0432.CCR-08-2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mandic R, Rodgarkia-Dara CJ, Zhu L, Folz BJ, Bette M, Weihe E, et al. Treatment of hnscc cell lines with the egfr-specific inhibitor cetuximab (erbituxæ) results in paradox phosphorylation of tyrosine 1173 in the receptor. FEBS Letters. 2006;580:4793–800. doi: 10.1016/j.febslet.2006.07.064. [DOI] [PubMed] [Google Scholar]
- 19.Schmidt-Ullrich RK, Contessa JN, Lammering G, Amorino G, Lin PS. Erbb receptor tyrosine kinases and cellular radiation responses. Oncogene. 2003;22:5855–65. doi: 10.1038/sj.onc.1206698. [DOI] [PubMed] [Google Scholar]
- 20.Huang SM, Harari PM. Modulation of radiation response after epidermal growth factor receptor blockade in squamous cell carcinomas: Inhibition of damage repair, cell cycle kinetics, and tumor angiogenesis. Clin Cancer Res. 2000;6:2166–74. [PubMed] [Google Scholar]
- 21.Dittmann K, Mayer C, Rodemann H. Inhibition of radiation-induced egfr nuclear import by c225 (cetuximab) suppresses DNA-pk activity. Radiother Oncol. 2005;76:157–61. doi: 10.1016/j.radonc.2005.06.022. [DOI] [PubMed] [Google Scholar]
- 22.Ryu SH, Lee SW, Yang YJ, Song SY, Kim JH, Choi EK, et al. Intracytoplasmic epidermal growth factor receptor shows poor response to the cetuximab antitumor effect in irradiated non-small cell lung cancer cell lines. Lung Cancer. 2012;77:482–87. doi: 10.1016/j.lungcan.2012.05.104. [DOI] [PubMed] [Google Scholar]
- 23.Psyrri A, Egleston B, Pectasides E, Weinberger P, Yu Z, Kowalski D, et al. Correlates and determinants of nuclear epidermal growth factor receptor content in an oropharyngeal cancer tissue microarray. Cancer Epidemiology Biomarkers & Prevention. 2008;17:1486–92. doi: 10.1158/1055-9965.EPI-07-2684. [DOI] [PubMed] [Google Scholar]
- 24.Chinnaiyan P, Huang S, Vallabhaneni G, Armstrong E, Varambally S, Tomlins SA, et al. Mechanisms of enhanced radiation response following epidermal growth factor receptor signaling inhibition by erlotinib (tarceva) Cancer Res. 2005;65:3328–35. doi: 10.1158/0008-5472.CAN-04-3547. [DOI] [PubMed] [Google Scholar]
- 25.Wang YN, Yamaguchi H, Huo L, Du Y, Lee HJ, Lee HH, et al. The translocon sec61œ≤ localized in the inner nuclear membrane transports membrane-embedded egf receptor to the nucleus. J Biol Chem. 2010;285:38720–29. doi: 10.1074/jbc.M110.158659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lo HW, Ali-Seyed M, Wu Y, Bartholomeusz G, Hsu SC, Hung MC. Nuclear-cytoplasmic transport of egfr involves receptor endocytosis, importin beta1 and crm1. J Cell Biochem. 2006;98:1570–83. doi: 10.1002/jcb.20876. [DOI] [PubMed] [Google Scholar]
- 27.Liao HJ, Carpenter G. Cetuximab/c225-induced intracellular trafficking of epidermal growth factor receptor. Cancer Res. 2009;69:6179–83. doi: 10.1158/0008-5472.CAN-09-0049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li C, Iida M, Dunn EF, Wheeler DL. Dasatinib blocks cetuximab- and radiation-induced nuclear translocation of the epidermal growth factor receptor in head and neck squamous cell carcinoma. Radiother Oncol. 2010;97:330–37. doi: 10.1016/j.radonc.2010.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yu YL, Chou RH, Wu CH, Wang YN, Chang WJ, Tseng YJ, et al. Nuclear egfr suppresses ribonuclease activity of polynucleotide phosphorylase through dnapk-mediated phosphorylation at serine 776. J Biol Chem. 2012 doi: 10.1074/jbc.M112.358077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Skartved NJØ, Jacobsen HJ, Pedersen MW, Jensen PF, Sen JW, Jørgensen TK, et al. Preclinical pharmacokinetics and safety of sym004: A synergistic antibody mixture directed against epidermal growth factor receptor. Clin Cancer Res. 2011;17:5962–72. doi: 10.1158/1078-0432.CCR-11-1209. [DOI] [PubMed] [Google Scholar]
- 31.Machiels JPH, Specenier PM, Krauss J, Dietz A, Kaminsky MC, Lalami Y, et al. Sym004, a novel strategy to target egfr with an antibody mixture, in patients with advanced scchn progressing after anti-egfr monoclonal antibody: A proof of concept study. J Clin Oncol. 2013;31(suppl) abstr 6002. [Google Scholar]
- 32.Dienstmann R, Tabernero J, Cutsem EV, Cervantes-Ruiperez A, Keranen SR, Viñuales MB, et al. Proof-of-concept study of sym004, an anti-egfr monoclonal antibody (mab) mixture, in patients (pts) with anti-egfr mab-refractory kras wild-type (wt) metastatic colorectal cancer (mcrc) J Clin Oncol. 2013;31(suppl) abstr 3551. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.