

Abstract
Early onset familial Alzheimer’s disease (FAD) is caused by mutations of Presenilin 1, Presenilin 2, and amyloid precursor protein. Beyond the effects of PS1 mutations on proteolytic functions of the gamma-secretase complex, mutant or deficient PS1 disrupts lysosomal function and calcium homeostasis, both of which are considered strong pathogenic factors in FAD. Loss of PS1 function compromises assembly and proton-pumping activity of the vacuolar-ATPase on lysosomes, leading to defective lysosomal acidification and marked impairment of autophagy. Additional dysregulation of cellular calcium by mutant PS1 in FAD has been ascribed to altered ion channels in the endoplasmic reticulum; however, rich stores of calcium in lysosomes are also abnormally released in PS1-deficient cells secondary to the lysosomal acidification defect. The resultant rise in cytosolic calcium activates calcium-dependent enzymes, contributing substantially to calpain over-activation that is a final common pathway leading to neurofibrillary degeneration in all forms of AD. Here we discuss the close inter-relationships among deficits of lysosomal function, autophagy, and calcium homeostasis as a pathogenic process in PS1-related FAD and their relevance to sporadic AD.
Keywords: Alzheimer’s disease, lysosomes, calcium regulation, calpains
Introduction
Autosomal dominant mutations of Presenilin 1 (PS1), Presenilin 2 (PS2), and amyloid precursor protein (APP) cause an early-onset form of Alzheimer’s disease (EOAD). Although EOAD accounts for fewer than 5% of all AD cases, investigations of the three responsible genes have thus far provided many of the available clues to suspected pathogenic mechanisms in AD. Mutations of PS1, which are responsible for the vast majority of early-onset AD cases, can accelerate disease onset to ages as early as the late 20’s. In most cases of EOAD, the defining lesions of AD – neurofibrillary tangles (NFT) and neuritic plaques-- as well as characteristic autophagic –lysosomal pathology, resemble the features of later onset “sporadic” AD (sAD), although these abnormalities are usually more severe. Notable clinical and neuropathological heterogeneity, however, is sometimes seen among families carrying one of the >120 known PS1 mutations [1, 2] [3].
Presenilin 1 (PS1), a ubiquitous protein with 9 transmembrane domains, exists as a 65Kda holoprotein in the endoplasmic reticulum (ER). The molecule is cleaved by a furin-like “presenilinase” to generate a two-chain form [4, 5], which constitutes the catalytic subunit of the gamma (γ)-secretase enzyme complex composed of four additional subunits. Delivered from the ER to diverse vesicular destinations in the cell, gamma-secretase mediates the intramembranous cleavage of well over 25 different substrates, which are mainly type 1 membrane proteins [6, 7], including most notably APP. The gamma-secretase generates aneurotoxic amyloid-β peptide (Aβ) from a membrane bound carboxyl-terminal fragment of APP generated by β-APP cleaving enzyme (BACE-1)[8]. The pathogenic effects of PS1 mutations in AD are commonly ascribed to a small increase in the generation of a toxic 42 amino acid peptide (Aβ42) relative to a less toxic 40 amino acid form (Aβ40) [7, 9, 10]. Recent findings, however, indicate that AD-causing PS1 mutations often confer loss of function of the secretase, so that even if the Aβ42/Aβ40 ratio is modestly higher, as is often, though not invariably the case [11], the absolute levels of these peptides may be markedly lowered [12, 13] and not always a higher Aβ42 to Aβ40 ratio [11]. Thus, additional or alternative explanations for pathogenicity of PS1 mutations have been sought [14][15, 16].
PS1 may contribute to AD pathogenesis through loss of its other functions [17]while serving as a component of gamma-secretase, which may include cell adhesion, neurite outgrowth, and synaptic plasticity [18, 19]or alternatively, by acting via its secretase-independent roles as PS1 holoprotein, which include lysosomal acidification essential for autophagic protein degradation [20], wnt signaling [21], and cellular calcium regulation[22–24]. In this brief review, we will focus specifically on the multiple roles of the PS1 holoprotein and how loss of its function in the ER may link ER and lysosome pathogenic mechanisms in EOAD which accelerate neurodegeneration.
Mutations or deletion of PS1 cause autophagy defects by disrupting v-ATPase assembly and lysosome acidification
Autophagy is a lysosomal degradative pathway for recycling diverse cellular constituents [25, 26], particularly under conditions of metabolic stress (Figure 1). Essential for survival of neurons, autophagy is solely responsible for the cellular turnover of damaged or obsolete organelles and is vital to eliminating misfolded and aggregated proteins, which are poorly degraded by the ubiquitin-proteasome system. Autophagy has been reported to be altered in different neurological disorders, and is considered to be a pathogenic factor in several of these diseases, particularly AD and Parkinson’s disease [27]. In AD, disruption of autophagy results in a particularly profuse accumulation of autophagic vacuoles within grossly swollen dystrophic neurites of affected neurons [28]which stems from defects in the lysosomal clearance of autophagy substrates by lysosomes [29, 30]. Growing evidence has linked lysosome system failure during autophagy to multiple pathological outcomes in AD, including accelerated amyloidogenesis, neuritic dystrophy, apoptosis, and possibly, tauopathy [31–34].
Figure 1.
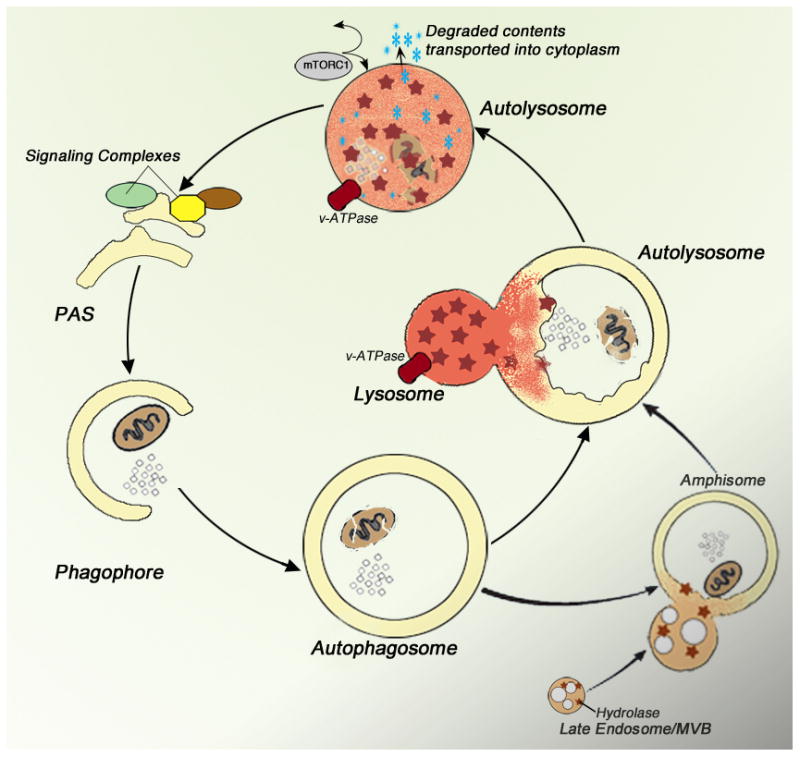
An overview of macroautophagy (autophagy). Multiple signaling pathways and protein modification assemblies initiate formation of an isolation membrane which is then elongated around the selected substrate or region of cytoplasm (phagophore). The inner and outer bi-layers of the isolation membrane close to form a double membrane-limited autophagosome. Lysosomes fuse with either an autophagosome or an amphisome created by autophagosome– endosome fusion, yielding autolysosomes. The contents of the autolysosome are degraded by hydrolytic enzymes activated within a highly acidic environment by the proton pump, v-ATPase. Digestion yields basic metabolites which are released into the cytoplasm to be used for new synthesis or as sources of energy and can also modulate TORC1 activity and other aspects of autophagy induction [26, 102]. MVB, multivesicular body; PAS, preautophagosomal structure (adapted from Nixon, 2013).
Mutations of PS1 considerably exacerbate autophagy pathology in EOAD and mouse models of AD [35] and similar pathology can be induced in neurons and non-neuronal cells by deleting PS1 [20, 36, 37]. Exploring the basis for these effects, Lee and colleagues [20] demonstrated that PS1 holoprotein serves as a chaperone in the ER for the vATPase V0a1 subunit, a 6-pass transmembrane protein constituent of the proton pump responsible for full acidification of lysosomes. Binding to the PS1 holoprotein, as this subunit is being translated in the ER, stabilizes the correct folded state and facilitates its glycosylation, believed to be necessary for transit from the ER [20, 38–40]. In cells lacking PS1, the V0a1 subunit of the v-ATPase is poorly glycosylated and unstable, which prevents adequate delivery to lysosomes and assembly of the multi-subunit v-ATPase, leading to greatly diminished levels and activity of the proton pump [41] resulting in failure to fully acidify lysosomes, which is a requirement for lysosomal protease activation and autophagy[20, 30]. Similar lysosomal acidification defects have now been observed in PS1 KO blastocysts, PS1 KO and PS1/2 KO MEFS, PS1-FAD human fibroblast lines, PS1 KO neurons, and PS1/APP mice [30, 41]. The roles of the V0a1 subunit and of PS1 in lysosome acidification have been confirmed in recent studies of PS1 ablated cells or mutant PS1 mouse models and other systems [30, 38–40, 42] though Coen et al. has proposed that neither of these components is involved in lysosomal pH regulation [43]. The importance of this PS1 chaperone function in AD pathogenesis is underscored by the ability of pharmacological inhibitors of vATPase to induce identical AD-related autophagy dysfunction in normal cells and by the complete reversal this phenotype in PS1 deficient cells by pharmacologically normalizing lysosomal acidification[41]. The connection of PS1 loss of function to wnt signaling was recently demonstrated in which the loss of PS1 led to an increase in nuclear β-catenin and phosphorylated LRP6 due to increased wnt activity, all of which was attributed to defective acidification of the multivesicular body [38].
Defective lysosome acidification – an emerging factor in pathogenesis of degenerative diseases
Interestingly, disruptions of normal v-ATPase activity appear to be important for the progression of other diseases. Mutations within the a3 subunit of the v-ATPase cause misfolding and ER retention of this subunit, leading to osteopetrosis, a bone disease that may be accompanied by neurodegeneration [44]. Mice lacking the a3 subunit also have impaired acidification of lysosomes in macrophages[45]. In a rare familial form of Parkinson’s disease, the loss of function of ATPase13A2 (Park9), believed to be involved in lysosome acidification, leads to impaired autophagy and neurodegeneration[46]. Mutations in leucine-rich repeat kinase 2 (LRRK2) increase the activity of the protein and lead to late onset familial Parkinson’s disease. Among other pathological effects that have been attributed to this protein kinase, increase in LRRK2 activity leads to pathologically increased autophagy induction and defective lysosomal acidification [47]. Reduced v-ATPase activity and abnormally elevated lysosomal pH due to a deficiency of VMA21, an integral membrane protein required for the assembly of the vATPaseV0 complex in the ER, leads to x-linked myopathy, a form of muscle fiber degeneration [48].
Although lysosomal pH is predominantly maintained through the activity of the vacuolar-ATPase (v-ATPase), other ion channels localized to the lysosomal membrane are suspected to be important in maintaining or regulating pH shifts during lysosomal proteolysis and may be particularly important when the intralumenal lysosomal environment is disturbed in disease states. The chloride channel, CLC7, has been implicated in fine-tuning lysosomal pH in conjunction with v-ATPase [49] and other evidence has suggested an involvement of calcium channels, mucolipin (TRPML1) and TPC2, in lysosomal pH regulation [50–52]. Lysosomal storage disorders exhibit elevated lysosomal pH as a part of the progression of the disease. Elevated lysosomal pH has been detected in several forms of neuronal ceroid lipofuscinosis, including juvenile NCL (Batten disease) and infantile NCL leading to altered lysosomal enzyme activity [53].
Calcium dysregulation in EOAD – the important contribution of defective lysosomal acidification
PS1 holoprotein has also been implicated in mediating the gamma-secretase-independent effects of AD-causing PS1 mutations (or PS1 deletion) in dysregulating calcium homeostasis at several different subcellular levels, including the ER [54–57], the mitochondrial-associated membranes (MAM)[58] and lysosomes[43, 59] (Figure 2). Calcium homeostasis in the ER is normally maintained by regulating calcium efflux through ryanodine receptors (RyR) and inositol 1,4,5-triphosphate receptors (IP3R), and by regulating the import of calcium through the sarco-/endoplasmic reticulum Ca2+ ATPase ( SERCA) channel [60]. Although the mechanism(s)are unresolved, loss of function of PS1 leads to ER calcium dysregulation. Exaggerated calcium release by IP3R in response to IP3 was initially reported [61] and confirmed by other groups in different cells types [57, 62, 63]and recent studies into the underlying mechanism of the IP3R mediated calcium release linked the exaggerated release to a gain-of-function mechanism in which mutations in PS1 stimulate IP3R gating [64]. The expression of ryanodine receptors is also upregulated in the brains of transgenic mice harboring PS1 mutations [65]as well as in PC12 cells expressing PS1 mutations and in primary cells from PS1 KI mice[63], This increase in RyRchannels mediates calcium release [63, 65]. Additionally, PS1 may act as a passive calcium leak channel within the ER: mutations within the PS1 holoprotein are proposed to disrupt the leak function leading ER stores to overfill, thereby over-activating IP3R and increasing calcium release [22, 66]. Another group of investigators, however, was unable to observe changes in ER calcium fill or leak rates in cells lacking PS or expressing FAD linking PS mutations [24] and the basis for the observed differences is not yet resolved. The removal of calcium from the cytosol back to the ER is controlled by the SERCA pump, but in cells lacking PS1, PS2, or PS1/2, SERCA activity is diminished leading to increased cytosolic calcium. A similar effect has been seen in cells expressing either PS2 or FAD-PS2 [67], although others have observed increased SERCA function leading to the overfilling of ER calcium stores in cells overexpressing PS2 or AD- linked mutant PS1[68]. Despite these uncertainties about the precise mechanisms, there is consensus in the field that ER calcium is dysregulated in response to loss of PS1 function [56, 60].
Figure 2.
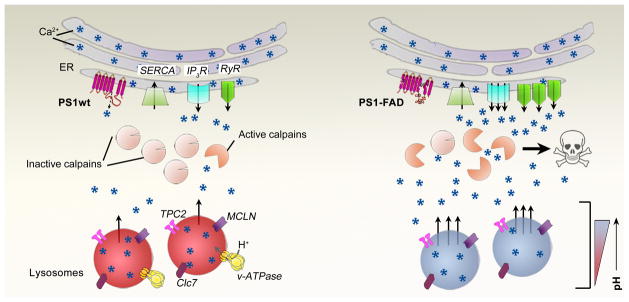
Diagram of calcium dysregulation in PS1wt (left) versus PS1-FAD conditions (right). In the ER, PS1-FAD induces an upregulation of RyR expression, induces a gain of function in IP3R, enhances SERCA function, and inhibits passive leak function of PS1. At the lysosomes, PS1-FAD elevates lysosomal pH by blocking vATPase maturation in the ER and delivery to lysosomes leading to increased calcium efflux though TPC2 and/or MCLN. Consequently, cytosolic calcium levels are increased leading to calpain activation. All of which contributes to impairments of cellular function and neurodegeneration (depicted by skull and crossbones). Black asterisks are symbolic of PS1-FAD mutations. Arrows depict movement of calcium. TPC2, two-pore calcium channel; MCLN, mucolipin1; CLC7, chloride channel protein 7
Although the ER contains the largest stores of calcium, with concentrations in the low millimolar range [69]lysosomes are not far behind with calcium concentrations in the range of 500–600 μM[70]. Recently, efflux of calcium from lysosomes in PS1/2KO MEF was reported, although the underlying mechanism was not investigated (Coen et al. 2012). A subsequent study confirming this phenomenon also revealed reduced levels of the lysosome –associated NAADP-dependent calcium channel, TPC2 [59], although it remains unclear how diminished amounts of a calcium efflux channel would explain elevated lysosomal calcium efflux. An earlier study using the lysosomal pH modulators chloroquine and bafilomycin, however, provides an attractive possible basis for calcium dysregulation in PS1 loss of function conditions by linking lysosomal calcium efflux to changes in lysosomal pH[71]. The possibility that decreased lysosomal calcium is a direct consequence of elevated lysosomal pH is indeed consistent with our recent findings [41]that raising lysosomal pH in wild-type blastocysts not only reproduces the autophagy defects seen in PS1 mutant or deficient cells but also induces the abnormal lysosomal calcium efflux. Importantly, reversing lysosomal pH deficits in PS1 deleted cells rapidly rescues both the lysosomal calcium abnormalities and the autophagy phenotype. By contrast, reversing the calcium efflux abnormalities alone neither corrects the acidification defect nor the autophagy deficits in PS1 deleted cells, indicating that lysosomal calcium dysregulation is a secondary consequence of pH elevation. These studies therefore link two γ-secretase independent effects of PS1 loss of function. They imply that vATPase deficiency is the common basis for these deficits and suggest that calcium efflux from lysosomes may be a significant contributor to the overall defect in cytosolic calcium elevation seen in PS1-deficient cells.
Altered ER calcium homeostasis reflected in a depletion of ER stores leading to elevated cytosolic calcium is common among several lysosomal storage disorders and it is possible that the mechanisms are similar to those proposed in the case of PS1 loss of function. Enhanced release of calcium by the ryanodine receptor has been observed in models of Gaucher’s disease due to the accumulation of glycosylceramide[72–74]. The ER calcium stores are depleted in GM1 gangliosidosis due to the interaction of GM1 with IP3R [75] and downregulation of SERCA pumps has been observed in Sandhoff disease [74] and in Neimann-Pick type A [76]due to interactions with ganglioside GM2 resulting in elevation of cytosolic calcium [77]. Lowered amounts of lysosomal calcium have also been observed in Neimann-Pick type C due to accumulation of sphingosine[78].
PS1 mediated calcium dysregulation–- a pathway to accelerated calpain – dependent neurofibrillary degeneration in FAD
Altered ER and lysosomal calcium homeostasis leads to an increase in cytosolic calcium levels [54], one consequence of which is the activation of calpain 1 (μ-calpain) and calpain 2 (m-calpain), proteases that are highly expressed in the central nervous system [79] (Figure 2). Targets of calpain cleavage include cytoskeletal proteins, signal transduction proteins, synaptic proteins, and transcription factors [80]. Abnormally increased activities of both calpain 1 and calpain 2 have been linked to AD [81–83]with calpain 1 actions possibly concentrated at synapses [82] and calpain 2 bound to neurofibrillary tangles (NFTs) and more activated within the neurites of neurons at risk for developing NFTs [83]. Calpain over-activation is well-established to have many deleterious effects, among them being the pathological activation of CDK5 (through the cleavage of p35 to p25) [84], ERK1/2 [85], and GSK3beta [86] and consequent hyperphosphorylation of tau leading to NFT formation. Calpains can also cleave IP3R making it constituently active which may further disrupt ER calcium homeostasis in PS1- related EOAD[87]. The pathogenic role of calpains in AD is further indicated by the marked depletion in AD brain of calpastatin[88], the specific endogenous inhibitor of calpains[89, 90]. Moreover, overexpressing calpastatin reduced calpain activity, tau phosphorylation, ERK activation, and amyloid plaque load in either of two different mouse models of AD [91, 92]. A synthetic calpain inhibitor, BDA-410, improved spatial memory, fear conditioning, and synaptic plasticity associated with increased CREB phosphorylation in APP/PS1 transgenic mice [93]. Finally, we have recently observed that calpastatin overexpression in the JNPL3 tauopathy mouse model leads to decreased tau phosphorylation, increased life span, and loss of motor neurons (McBrayer, MK unpublished results). Because ER and lysosomal calcium dysregulation is altered in PS1 FAD and PS1 KO cell lines [43, 59], it is not surprising that we have also found calpains to be robustly activated in PS1 KO blastocysts and PS1-FAD fibroblasts as reflected in marked increase in calpain-cleaved spectrin and significantly decreased calpastatin levels (McBrayer, MK unpublished results).
Conclusions ----- lysosomal and calcium dysfunction in sporadic AD
While calcium dysregulation may be initially driven by presenilin mutations in FAD, additional cellular calcium derangements are also seen in the more prevalent late-onset forms of AD [94]and likely have a multifactorial origin, including contributions from excitotoxicity, ischemia, and Abeta neurotoxicity [95, 96]. It is tempting to speculate that the notable disruption of lysosomal function in AD [97, 98] may give rise to additional lysosomal-related calcium dysregulation. Interestingly, a further connection has been suggested by Yamashima between calpain activation and lysosomal dysfunction leading to lysosomal membrane rupture, which involves increased calpain-mediated cleavage of carbonylated HSP70.1. HSP70.1 has roles as both a protein chaperone and stabilizer of lysosomal membrane integrity. Calpain-mediated loss of HSP70.1, coupled with other factors impairing autophagic clearance [98], are proposed to lead to lysosomal membrane disruption and likely to diverse downstream pathological events [95]. Calpain activation is observed in patients with sAD and recent research indicates that calcium homeostasis is also disrupted in sAD[99–101]. EOAD and sAD likely have different initiation points leading to dysregulated lysosome dysfunction and calcium dysregulation but ultimately share pathological outcomes, including calpain over-activation, that should prove to be useful drug targets for AD therapy.
Acknowledgments
We gratefully acknowledge the expert assistance of Nicole Piorkowski in manuscript preparation and Corrinne Peterhoff in figure preparation and thank Devin Wolfe for helpful discussions. Studies from this laboratory are supported by the National Institute on Aging (P01AG17617and R01AG5604).
References
- 1.St George-Hyslop P, Haines J, Rogaev E, Mortilla M, Vaula G, Pericak-Vance M, Foncin JF, Montesi M, Bruni A, Sorbi S, et al. Genetic evidence for a novel familial Alzheimer’s disease locus on chromosome 14. Nat Genet. 1992;2:330–334. doi: 10.1038/ng1292-330. [DOI] [PubMed] [Google Scholar]
- 2.Campion D, Flaman J-M, Brice A, Hannequin D, Dubois B, Martin C, Moreau V, Charbonnier F, Didierjean O, Tardieu S, Penet C, Puel M, Pasquier F, Le Doze F, Bellis G, Calenda A, Heilig R, Martinez M, Mallet J, Bellis M, Clerget-Darpoux F, Agid Y, Frebourg T. Mutations of the presenilin I gene in families with early-onset Alzheimer’s disease. Hum Mol Genet. 1995;4:2373–2377. doi: 10.1093/hmg/4.12.2373. [DOI] [PubMed] [Google Scholar]
- 3.Ryan NS, Rossor MN. Correlating familial Alzheimer’s disease gene mutations with clinical phenotype. Biomark Med. 2010;4:99–112. doi: 10.2217/bmm.09.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang J, Kang DE, Xia W, Okochi M, Mori H, Selkoe DJ, Koo EH. Subcellular distribution and turnover of presenilins in transfected cells. J Biol Chem. 1998;273:12436–12442. doi: 10.1074/jbc.273.20.12436. [DOI] [PubMed] [Google Scholar]
- 5.Yu G, Chen F, Levesque G, Nishimura M, Zhang DM, Levesque L, Rogaeva E, Xu D, Liang Y, Duthie M, George-Hyslop PH, Fraser PE. The presenilin 1 protein is a component of a high molecular weight intracellular complex that contains beta-catenin 2686. J Biol Chem. 1998;273:16470–16475. doi: 10.1074/jbc.273.26.16470. [DOI] [PubMed] [Google Scholar]
- 6.Citron M, Westaway D, Xia W, Carlson G, Diehl T, Levesque G, Johnson-Wood K, Lee M, Seubert P, Davis A, Kholodenko D, Motter R, Sherrington R, Perry B, Yao H, Strome R, Lieberburg I, Rommens J, Kim S, Schenk D, Fraser P, St George Hyslop P, Selkoe DJ. Mutant presenilins of Alzheimer’s disease increase production of 42- residue amyloid beta-protein in both transfected cells and transgenic mice. Nat Med. 1997;3:67–72. doi: 10.1038/nm0197-67. [DOI] [PubMed] [Google Scholar]
- 7.De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, Guhde G, Annaert W, von Figura K, Van Leuven F. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature. 1998;391:387–390. doi: 10.1038/34910. [DOI] [PubMed] [Google Scholar]
- 8.Cai H, Wang Y, McCarthy D, Wen H, Borchelt DR, Price DL, Wong PC. BACE1 is the major beta-secretase for generation of Abeta peptides by neurons. Nat Neurosci. 2001;4:233–234. doi: 10.1038/85064. [DOI] [PubMed] [Google Scholar]
- 9.Selkoe DJ. The cell biology of beta-amyloid precursor protein and presenilin in Alzheimer’s disease. Trends Cell Biol. 1998;8:447–453. doi: 10.1016/s0962-8924(98)01363-4. [DOI] [PubMed] [Google Scholar]
- 10.Steiner H, Duff K, Capell A, Romig H, Grim MG, Lincoln S, Hardy J, Yu X, Picciano M, Fechteler K, Citron M, Kopan R, Pesold B, Keck S, Baader M, Tomita T, Iwatsubo T, Baumeister R, Haass C. A Loss of Function Mutation of Presenilin-2 Interferes with Amyloid beta -Peptide Production and Notch Signaling. J Biol Chem. 1999;274:28669–28673. doi: 10.1074/jbc.274.40.28669. [DOI] [PubMed] [Google Scholar]
- 11.Shioi J, Georgakopoulos A, Mehta P, Kouchi Z, Litterst CM, Baki L, Robakis NK. FAD mutants unable to increase neurotoxic Abeta 42 suggest that mutation effects on neurodegeneration may be independent of effects on Abeta. J Neurochem. 2007;101:674–681. doi: 10.1111/j.1471-4159.2006.04391.x. [DOI] [PubMed] [Google Scholar]
- 12.Bentahir M, Nyabi O, Verhamme J, Tolia A, Horre K, Wiltfang J, Esselmann H, Strooper B. Presenilin clinical mutations can affect γ-secretase activity by different mechanisms. Journal of Neurochemistry. 2006;96:732–742. doi: 10.1111/j.1471-4159.2005.03578.x. [DOI] [PubMed] [Google Scholar]
- 13.Kumar-Singh S, Theuns J, Van Broeck B, Pirici D, Vennekens K, Corsmit E, Cruts M, Dermaut B, Wang R, Van Broeckhoven C. Mean age-of-onset of familial alzheimer disease caused by presenilin mutations correlates with both increased Abeta42 and decreased Abeta40. Human mutation. 2006;27:686–695. doi: 10.1002/humu.20336. [DOI] [PubMed] [Google Scholar]
- 14.Pimplikar SW, Nixon RA, Robakis NK, Shen J, Tsai LH. Amyloid-independent mechanisms in Alzheimer’s disease pathogenesis. J Neurosci. 2010;30:14946–14954. doi: 10.1523/JNEUROSCI.4305-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harris H, Rubinsztein DC. Control of autophagy as a therapy for neurodegenerative disease. Nat Rev Neurol. 2011;8:108–117. doi: 10.1038/nrneurol.2011.200. [DOI] [PubMed] [Google Scholar]
- 16.Selkoe DJ. Resolving controversies on the path to Alzheimer’s therapeutics. Nat Med. 2011;17:1060–1065. doi: 10.1038/nm.2460. [DOI] [PubMed] [Google Scholar]
- 17.Naruse S, Thinakaran G, Luo JJ, Kusiak JW, Tomita T, Iwatsubo T, Qian X, Ginty DD, Price DL, Borchelt DR, Wong PC, Sisodia SS. Effects of PS1 deficiency on membrane protein trafficking in neurons. Neuron. 1998;21:1213–1221. doi: 10.1016/s0896-6273(00)80637-6. [DOI] [PubMed] [Google Scholar]
- 18.Kim TW, Tanzi RE. Presenilins and Alzheimer’s disease. Curr Opin Neurobiol. 1997;7:683–688. doi: 10.1016/s0959-4388(97)80089-x. [DOI] [PubMed] [Google Scholar]
- 19.Shen J, Kelleher RJ., 3rd The presenilin hypothesis of Alzheimer’s disease: evidence for a loss-of-function pathogenic mechanism. Proc Natl Acad Sci U S A. 2007;104:403–409. doi: 10.1073/pnas.0608332104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee JH, Yu WH, Kumar A, Lee S, Mohan PS, Peterhoff CM, Marinez-Vicente M, Massey AG, Sovak G, Uchiyama Y, Cuervo AM, Nixon RA. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell. 2010;141:1146–1158. doi: 10.1016/j.cell.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kang DE, Soriano S, Frosch MP, Collins T, Naruse S, Sisodia SS, Leibowitz G, Levine F, Koo EH. Presenilin 1 facilitates the constitutive turnover of beta-catenin: differential activity of Alzheimer’s disease-linked PS1 mutants in the beta-catenin-signaling pathway. J Neurosci. 1999;19:4229–4237. doi: 10.1523/JNEUROSCI.19-11-04229.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tu H, Nelson O, Bezprozvanny A, Wang Z, Lee SF, Hao YH, Serneels L, De Strooper B, Yu G, Bezprozvanny I. Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer’s disease-linked mutations. Cell. 2006;126:981–993. doi: 10.1016/j.cell.2006.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zampese E, Fasolato C, Kipanyula MJ, Bortolozzi M, Pozzan T, Pizzo P. Presenilin 2 modulates endoplasmic reticulum (ER)-mitochondria interactions and Ca2+ cross-talk. Proc Natl Acad Sci U S A. 2011;108:2777–2782. doi: 10.1073/pnas.1100735108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shilling D, Mak DO, Kang DE, Foskett JK. Lack of evidence for presenilins as endoplasmic reticulum Ca2+ leak channels. J Biol Chem. 2012;287:10933–10944. doi: 10.1074/jbc.M111.300491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant α-synuclein by chaperone-mediated autophagy. Science. 2004;305:1292–1295. doi: 10.1126/science.1101738. [DOI] [PubMed] [Google Scholar]
- 26.Klionsky DJ, Codogno P, Cuervo AM, Deretic V, Elazar Z, Fueyo-Margareto J, Gewirtz DA, Kroemer G, Levine B, Mizushima N, Rubinsztein DC, Thumm M, Tooze SA. A comprehensive glossary of autophagy-related molecules and processes. Autophagy. 2010;6:438–448. doi: 10.4161/auto.6.4.12244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med. 2013;19:983–997. doi: 10.1038/nm.3232. [DOI] [PubMed] [Google Scholar]
- 28.Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A, Cuervo AM. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol. 2005;64:113–122. doi: 10.1093/jnen/64.2.113. [DOI] [PubMed] [Google Scholar]
- 29.Nixon RA, Yang DS. Autophagy failure in Alzheimer’s disease-locating the primary defect. Neurobiol Dis. 2011;43:38–45. doi: 10.1016/j.nbd.2011.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wolfe DM, Lee J-H, Kumar A, Lee S, Orenstein S, Nixon RA. Autophagy failure in Alzheimer’s disease and the role of defective lysosomal acidification. Euro J Neurosci. 2013;37:1949–1961. doi: 10.1111/ejn.12169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boland B, Kumar A, Lee S, Platt FM, Wegiel J, Yu WH, Nixon RA. Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer’s disease. J Neurosci. 2008;28:6926–6937. doi: 10.1523/JNEUROSCI.0800-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu WH, Cuervo AM, Kumar A, Peterhoff CM, Schmidt SD, Lee J-H, Mohan PS, Mercken M, Farmery MR, Tjernberg LO, Jiang Y, Duff K, Uchiyama Y, Naslund J, Mathews PM, Cataldo AM, Nixon RA. Macroautophagy--a novel β-amyloid peptide-generating pathway activated in Alzheimer’s disease. J Cell Biol. 2005;171:87–98. doi: 10.1083/jcb.200505082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee S, Sato Y, Nixon RA. Lysosomal Proteolysis Inhibition Selectively Disrupts Axonal Transport of Degradative Organelles and Causes an Alzheimer’s-Like Axonal Dystrophy. J Neurosci. 2011;31:7817–7830. doi: 10.1523/JNEUROSCI.6412-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang DS, Kumar A, Stavrides P, Peterson J, Peterhoff CM, Pawlik M, Levy E, Cataldo AM, Nixon RA. Neuronal apoptosis and autophagy cross talk in aging PS/APP mice, a model of Alzheimer’s disease. Am J Pathol. 2008;173:665–681. doi: 10.2353/ajpath.2008.071176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cataldo AM, Peterhoff CM, Schmidt SD, Terio NB, Duff K, Beard M, Mathews PM, Nixon RA. Presenilin mutations in familial Alzheimer disease and transgenic mouse models accelerate neuronal lysosomal pathology. J Neuropathol Exp Neurol. 2004;63:821–830. doi: 10.1093/jnen/63.8.821. [DOI] [PubMed] [Google Scholar]
- 36.Esselens C, Oorschot V, Baert V, Raemaekers T, Spittaels K, Serneels L, Zheng H, Saftig P, De Strooper B, Klumperman J, Annaert W. Presenilin 1 mediates the turnover of telencephalin in hippocampal neurons via an autophagic degradative pathway. J Cell Biol. 2004;166:1041–1054. doi: 10.1083/jcb.200406060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wilson CA, Murphy DD, Giasson BI, Zhang B, Trojanowski JQ, Lee VM. Degradative organelles containing mislocalized α- and β-synuclein proliferate in presenilin-1 null neurons. J Cell Biol. 2004;165:335–346. doi: 10.1083/jcb.200403061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dobrowolski R, Vick P, Ploper D, Gumper I, Snitkin H, Sabatini DD, De Robertis EM. Presenilin Deficiency or Lysosomal Inhibition Enhances Wnt Signaling through Relocalization of GSK3 to the Late-Endosomal Compartment. Cell Rep. 2012 doi: 10.1016/j.celrep.2012.09.026. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Torres M, Jimenez S, Sanchez-Varo R, Navarro V, Trujillo-Estrada L, Sanchez-Mejias E, Carmona I, Davila JC, Vizuete M, Gutierrez A, Vitorica J. Defective lysosomal proteolysis and axonal transport are early pathogenic events that worsen with age leading to increased APP metabolism and synaptic Abeta in transgenic APP/PS1 hippocampus. Mol Neurodegener. 2012;7:59. doi: 10.1186/1750-1326-7-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Avrahami L, Farfara D, Shaham-Kol M, Vassar R, Frenkel D, Eldar-Finkelman H. Inhibition of GSK-3 ameliorates beta-amyloid(A-beta) pathology and restores lysosomal acidification and mTOR activity in the Alzheimer’s disease mouse model: in vivo and in vitro studies. J Biol Chem. 2013;288:1295–1306. doi: 10.1074/jbc.M112.409250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee J-H, Wolfe D, McBrayer M, Haslett L, Kumar A, Sato Y, Mohan P, Coffey EE, Mitchell C, Lloyd-Evans E, Nixon RA. Society for Neuroscience, editor. Presenilin maintains lysosomal calcium homeostasis by regulating v-ATPase-mediated lysosomal acidification. San Diego, CA, USA: 2013. [Google Scholar]
- 42.Sawa A. Neuronal cell death in Down’s syndrome. J Neural Transm Suppl. 1999;57:87–97. doi: 10.1007/978-3-7091-6380-1_6. [DOI] [PubMed] [Google Scholar]
- 43.Coen K, Flannagan R, Baron S, Carraro-Lacroix L, Wang D, Vermeire W, Michiels C, Munck S, Baert V, Sugita S, Wuytack F, Hiesinger P, Grinstein S, Annaert W. Lysosomal calcium homeostasis defects, not proton pump defects, cause endo-lysosomal dysfunction in PSEN-deficient cells. J Cell Biol. 2012;198:23–35. doi: 10.1083/jcb.201201076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bhargava A, Voronov I, Wang Y, Glogauer M, Kartner N, Manolson MF. Osteopetrosis mutation R444L causes endoplasmic reticulum retention and misprocessing of vacuolar H+-ATPase a3 subunit. J Biol Chem. 2012;287:26829–26839. doi: 10.1074/jbc.M112.345702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sun-Wada GH, Tabata H, Kawamura N, Aoyama M, Wada Y. Direct recruitment of H+-ATPase from lysosomes for phagosomal acidification. J Cell Sci. 2009;122:2504–2513. doi: 10.1242/jcs.050443. [DOI] [PubMed] [Google Scholar]
- 46.Dehay B, Ramirez A, Martinez-Vicente M, Perier C, Canron MH, Doudnikoff E, Vital A, Vila M, Klien C, Bezard E. Loss of P-type ATPase ATP13A2/PARK9 function induces general lysosomal deficiency and leads to Parkinson disease neurodegeneration. Proceedings of the National Academy of Sciences. 2012;109:9611–9616. doi: 10.1073/pnas.1112368109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gomez-Suaga P, Luzon-Toro B, Churamani D, Zhang L, Bloor-Young D, Patel S, Woodman PG, Churchill GC, Hilfiker S. Leucine-rich repeat kinase 2 regulates autophagy through a calcium-dependent pathway involving NAADP. Hum Mol Genet. 2012;21:511–525. doi: 10.1093/hmg/ddr481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ramachandran N, Munteanu I, Wang P, Ruggieri A, Rilstone JJ, Israelian N, Naranian T, Paroutis P, Guo R, Ren ZP, Nishino I, Chabrol B, Pellissier JF, Minetti C, Udd B, Fardeau M, Tailor CS, Mahuran DJ, Kissel JT, Kalimo H, Levy N, Manolson MF, Ackerley CA, Minassian BA. VMA21 deficiency prevents vacuolar ATPase assembly and causes autophagic vacuolar myopathy. Acta Neuropathol. 2013;125:439–457. doi: 10.1007/s00401-012-1073-6. [DOI] [PubMed] [Google Scholar]
- 49.Mindell JA. Lysosomal acidification mechanisms. Annu Rev Physiol. 2012;74:69–86. doi: 10.1146/annurev-physiol-012110-142317. [DOI] [PubMed] [Google Scholar]
- 50.Bach G, Chen CS, Pagano RE. Elevated lysosomal pH in Mucolipidosis type IV cells. Clin Chim Acta. 1999;280:173–179. doi: 10.1016/s0009-8981(98)00183-1. [DOI] [PubMed] [Google Scholar]
- 51.Soyombo AA, Tjon-Kon-Sang S, Rbaibi Y, Bashllari E, Bisceglia J, Muallem S, Kiselyov K. TRP-ML1 regulates lysosomal pH and acidic lysosomal lipid hydrolytic activity. J Biol Chem. 2006;281:7294–7301. doi: 10.1074/jbc.M508211200. [DOI] [PubMed] [Google Scholar]
- 52.Lu YY, Hao BX, Graeff R, Wong CW, Wu WT, Yue J. TPC2 Signaling Inhibits Autophagosomal-Lysosomal Fusion by Alkalizing Lysosomal pH. J Biol Chem. 2013 doi: 10.1074/jbc.M113.484253. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 53.Holopainen JM, Saarikoski J, Kinnunen PK, Jarvela I. Elevated lysosomal pH in neuronal ceroid lipofuscinoses (NCLs) Eur J Biochem. 2001;268:5851–5856. doi: 10.1046/j.0014-2956.2001.02530.x. [DOI] [PubMed] [Google Scholar]
- 54.LaFerla FM. Calcium dyshomeostasis and intracellular signalling in Alzheimer’s disease. Nat Rev Neurosci. 2002;3:862–872. doi: 10.1038/nrn960. [DOI] [PubMed] [Google Scholar]
- 55.Bezprozvanny I. Presenilins: a novel link between intracellular calcium signaling and lysosomal function? J Cell Biol. 2012;198:7–10. doi: 10.1083/jcb.201206003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mattson MP. ER calcium and Alzheimer’s disease: in a state of flux. Sci Signal. 2010;3:pe10. doi: 10.1126/scisignal.3114pe10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stutzmann GE, Caccamo A, LaFerla FM, Parker I. Dysregulated IP3 signaling in cortical neurons of knock-in mice expressing an Alzheimer’s-linked mutation in presenilin1 results in exaggerated Ca2+ signals and altered membrane excitability. J Neurosci. 2004;24:508–513. doi: 10.1523/JNEUROSCI.4386-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Area-Gomez E, Del Carmen Lara Castillo M, Tambini MD, Guardia-Laguarta C, de Groof AJ, Madra M, Ikenouchi J, Umeda M, Bird TD, Sturley SL, Schon EA. Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. EMBO J. 2012;31:4106–4123. doi: 10.1038/emboj.2012.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Neely Kayala KM, Dickinson GD, Minassian A, Walls KC, Green KN, Laferla FM. Presenilin-null cells have altered two-pore calcium channel expression and lysosomal calcium: implications for lysosomal function. Brain Res. 2012;1489:8–16. doi: 10.1016/j.brainres.2012.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Honarnejad K, Herms J. Presenilins: role in calcium homeostasis. Int J Biochem Cell Biol. 2012;44:1983–1986. doi: 10.1016/j.biocel.2012.07.019. [DOI] [PubMed] [Google Scholar]
- 61.Ito E, Oka K, Etcheberrigaray R, Nelson TJ, McPhie DL, Tofel-Grehl B, Gibson GE, Alkon DL. Internal Ca2+ mobilization is altered in fibroblasts from patients with Alzheimer disease. Proc Natl Acad Sci U S A. 1994;91:534–538. doi: 10.1073/pnas.91.2.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Guo Q, Furukawa K, Sopher BL, Pham DG, Xie J, Robinson N, Martin GM, Mattson MP. Alzheimer’s PS-1 mutation perturbs calcium homeostasis and sensitizes PC12 cells to death induced by amyloid beta-peptide 791. Neuroreport. 1996;8:379–383. doi: 10.1097/00001756-199612200-00074. [DOI] [PubMed] [Google Scholar]
- 63.Chan SL, Mayne M, Holden CP, Geiger JD, Mattson MP. Presenilin-1 mutations increase levels of ryanodine receptors and calcium release in PC12 cells and cortical neurons. J Biol Chem. 2000;275:18195–18200. doi: 10.1074/jbc.M000040200. [DOI] [PubMed] [Google Scholar]
- 64.Cheung KH, Mei L, Mak DO, Hayashi I, Iwatsubo T, Kang DE, Foskett JK. Gain-of-function enhancement of IP3 receptor modal gating by familial Alzheimer’s disease-linked presenilin mutants in human cells and mouse neurons. Sci Signal. 2010;3:ra22. doi: 10.1126/scisignal.2000818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stutzmann GE, Smith I, Caccamo A, Oddo S, Laferla FM, Parker I. Enhanced ryanodine receptor recruitment contributes to Ca2+ disruptions in young, adult, and aged Alzheimer’s disease mice. J Neurosci. 2006;26:5180–5189. doi: 10.1523/JNEUROSCI.0739-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Supnet C, Bezprozvanny I. Presenilins function in ER calcium leak and Alzheimer’s disease pathogenesis. Cell Calcium. 2011;50:303–309. doi: 10.1016/j.ceca.2011.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Brunello L, Zampese E, Florean C, Pozzan T, Pizzo P, Fasolato C. Presenilin-2 dampens intracellular Ca2+ stores by increasing Ca2+ leakage and reducing Ca2+ uptake. J Cell Mol Med. 2009;13:3358–3369. doi: 10.1111/j.1582-4934.2009.00755.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Green KN, Demuro A, Akbari Y, Hitt BD, Smith IF, Parker I, LaFerla FM. SERCA pump activity is physiologically regulated by presenilin and regulates amyloid beta production. J Cell Biol. 2008;181:1107–1116. doi: 10.1083/jcb.200706171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Alvarez J, Montero M, Garcia-Sancho J. Subcellular Ca(2+) Dynamics. News Physiol Sci. 1999;14:161–168. [PubMed] [Google Scholar]
- 70.Lloyd-Evans E, Platt FM. Lysosomal Ca(2+) homeostasis: role in pathogenesis of lysosomal storage diseases. Cell Calcium. 2011;50:200–205. doi: 10.1016/j.ceca.2011.03.010. [DOI] [PubMed] [Google Scholar]
- 71.Christensen KA, Myers JT, Swanson JA. pH-dependent regulation of lysosomal calcium in macrophages. J Cell Sci. 2002;115:599–607. doi: 10.1242/jcs.115.3.599. [DOI] [PubMed] [Google Scholar]
- 72.Lloyd-Evans E, Pelled D, Riebeling C, Bodennec J, de-Morgan A, Waller H, Schiffmann R, Futerman AH. Glucosylceramide and glucosylsphingosine modulate calcium mobilization from brain microsomes via different mechanisms. J Biol Chem. 2003;278:23594–23599. doi: 10.1074/jbc.M300212200. [DOI] [PubMed] [Google Scholar]
- 73.Korkotian E, Schwarz A, Pelled D, Schwarzmann G, Segal M, Futerman AH. Elevation of intracellular glucosylceramide levels results in an increase in endoplasmic reticulum density and in functional calcium stores in cultured neurons. J Biol Chem. 1999;274:21673–21678. doi: 10.1074/jbc.274.31.21673. [DOI] [PubMed] [Google Scholar]
- 74.Pelled D, Trajkovic-Bodennec S, Lloyd-Evans E, Sidransky E, Schiffmann R, Futerman AH. Enhanced calcium release in the acute neuronopathic form of Gaucher disease. Neurobiol Dis. 2005;18:83–88. doi: 10.1016/j.nbd.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 75.Sano R, Annunziata I, Patterson A, Moshiach S, Gomero E, Opferman J, Forte M, d’Azzo A. GM1-ganglioside accumulation at the mitochondria-associated ER membranes links ER stress to Ca(2+)-dependent mitochondrial apoptosis. Mol Cell. 2009;36:500–511. doi: 10.1016/j.molcel.2009.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ginzburg L, Futerman AH. Defective calcium homeostasis in the cerebellum in a mouse model of Niemann-Pick A disease. J Neurochem. 2005;95:1619–1628. doi: 10.1111/j.1471-4159.2005.03534.x. [DOI] [PubMed] [Google Scholar]
- 77.Vitner EB, Platt FM, Futerman AH. Common and uncommon pathogenic cascades in lysosomal storage diseases. J Biol Chem. 2010;285:20423–20427. doi: 10.1074/jbc.R110.134452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lloyd-Evans E, Morgan AJ, He X, Smith DA, Elliot-Smith E, Sillence DJ, Churchill GC, Schuchman EH, Galione A, Platt FM. Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat Med. 2008;14:1247–1255. doi: 10.1038/nm.1876. [DOI] [PubMed] [Google Scholar]
- 79.Goll DE, Thompson VF, Li H, Wei W, Cong J. The calpain system. Physiol Rev. 2003;83:731–801. doi: 10.1152/physrev.00029.2002. [DOI] [PubMed] [Google Scholar]
- 80.Ferreira A. Calpain dysregulation in Alzheimer’s disease. ISRN Biochemistry. 2012;2012 doi: 10.5402/2012/728571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nixon RA. The calpains in aging and aging-related diseases. Ageing Res Rev. 2003;2:407–418. doi: 10.1016/s1568-1637(03)00029-1. [DOI] [PubMed] [Google Scholar]
- 82.Saito K, Elce JS, Hamos JE, Nixon RA. Widespread activation of calcium-activated neutral proteinase (calpain) in the brain in Alzheimer disease: a potential molecular basis for neuronal degeneration. Proc Natl Acad Sci U S A. 1993;90:2628–2632. doi: 10.1073/pnas.90.7.2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Grynspan F, Griffin WR, Cataldo A, Katayama S, Nixon RA. Active site-directed antibodies identify calpain II as an early-appearing and pervasive component of neurofibrillary pathology in Alzheimer’s disease. Brain Res. 1997;763:145–158. doi: 10.1016/s0006-8993(97)00384-3. [DOI] [PubMed] [Google Scholar]
- 84.Kusakawa G, Saito T, Onuki R, Ishiguro K, Kishimoto T, Hisanaga S. Calpain-dependent proteolytic cleavage of the p35 cyclin-dependent kinase 5 activator to p25. J Biol Chem. 2000;275:17166–17172. doi: 10.1074/jbc.M907757199. [DOI] [PubMed] [Google Scholar]
- 85.Veeranna Kaji T, Boland B, Odrljin T, Mohan P, Basavarajappa BS, Peterhoff C, Cataldo A, Rudnicki A, Amin N, Li BS, Pant HC, Hungund BL, Arancio O, Nixon RA. Calpain mediates calcium-induced activation of the erk1,2 MAPK pathway and cytoskeletal phosphorylation in neurons: relevance to Alzheimer’s disease. Am J Pathol. 2004;165:795–805. doi: 10.1016/S0002-9440(10)63342-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Goni-Oliver P, Lucas JJ, Avila J, Hernandez F. N-terminal cleavage of GSK-3 by calpain: a new form of GSK-3 regulation. J Biol Chem. 2007;282:22406–22413. doi: 10.1074/jbc.M702793200. [DOI] [PubMed] [Google Scholar]
- 87.Kopil CM, Vais H, Cheung KH, Siebert AP, Mak DO, Foskett JK, Neumar RW. Calpain-cleaved type 1 inositol 1,4,5-trisphosphate receptor (InsP(3)R1) has InsP(3)-independent gating and disrupts intracellular Ca(2+) homeostasis. J Biol Chem. 2011;286:35998–36010. doi: 10.1074/jbc.M111.254177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rao MV, Mohan PS, Peterhoff CM, Yang DS, Schmidt SD, Stavrides PH, Campbell J, Chen Y, Jiang Y, Paskevich PA, Cataldo AM, Haroutunian V, Nixon RA. Marked calpastatin (CAST) depletion in Alzheimer’s disease accelerates cytoskeleton disruption and neurodegeneration: neuroprotection by CAST overexpression. J Neurosci. 2008;28:12241–12254. doi: 10.1523/JNEUROSCI.4119-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Emori Y, Kawasaki H, Imajoh S, Imahori K, Suzuki K. Endogenous inhibitor for calcium-dependent cysteine protease contains four internal repeats that could be responsible for its multiple reactive sites. Proc Natl Acad Sci U S A. 1987;84:3590–3594. doi: 10.1073/pnas.84.11.3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Maki M, Takano E, Mori H, Kannagi R, Murachi T, Hatanaka M. Repetitive region of calpastatin is a functional unit of the proteinase inhibitor. Biochem Biophys Res Commun. 1987;143:300–308. doi: 10.1016/0006-291x(87)90665-6. [DOI] [PubMed] [Google Scholar]
- 91.Liang B, Duan BY, Zhou XP, Gong JX, Luo ZG. Calpain activation promotes BACE1 expression, amyloid precursor protein processing, and amyloid plaque formation in a transgenic mouse model of Alzheimer disease. J Biol Chem. 2010;285:27737–27744. doi: 10.1074/jbc.M110.117960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Morales-Corraliza J, Berger JD, Mazzella MJ, Veeranna Neubert TA, Ghiso J, Rao MV, Staufenbiel M, Nixon RA, Mathews PM. Calpastatin modulates APP processing in the brains of beta-amyloid depositing but not wild-type mice. Neurobiol Aging. 2012;33:1125 e1129–1118. doi: 10.1016/j.neurobiolaging.2011.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Trinchese F, Fa M, Liu S, Zhang H, Hidalgo A, Schmidt SD, Yamaguchi H, Yoshii N, Mathews PM, Nixon RA, Arancio O. Inhibition of calpains improves memory and synaptic transmission in a mouse model of Alzheimer disease. J Clin Invest. 2008;118:2796–2807. doi: 10.1172/JCI34254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Green KN. Calcium in the initiation, progression and as an effector of Alzheimer’s disease pathology. J Cell Mol Med. 2009;13:2787–2799. doi: 10.1111/j.1582-4934.2009.00861.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yamashima T. Reconsider Alzheimer’s disease by the ‘calpain-cathepsin hypothesis’--a perspective review. Prog Neurobiol. 2013;105:1–23. doi: 10.1016/j.pneurobio.2013.02.004. [DOI] [PubMed] [Google Scholar]
- 96.Bartus RT, Dean RL., 3rd Pharmaceutical treatment for cognitive deficits in Alzheimer’s disease and other neurodegenerative conditions: exploring new territory using traditional tools and established maps. Psychopharmacology (Berl) 2009;202:15–36. doi: 10.1007/s00213-008-1365-7. [DOI] [PubMed] [Google Scholar]
- 97.Cataldo AM, Hamilton DJ, Barnett JL, Paskevich PA, Nixon RA. Properties of the endosomal-lysosomal system in the human central nervous system: disturbances mark most neurons in populations at risk to degenerate in Alzheimer’s disease. J Neurosci. 1996;16:186–199. doi: 10.1523/JNEUROSCI.16-01-00186.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Nixon RA, Yang D. Autophagy and neuronal cell death in neurological disorders. Cold Spring Harb Perspect Biol. 2012;4 doi: 10.1101/cshperspect.a008839. pii:a008839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.van Vliet P, Oleksik AM, Mooijaart SP, de Craen AJ, Westendorp RG. APOE genotype modulates the effect of serum calcium levels on cognitive function in old age. Neurology. 2009;72:821–828. doi: 10.1212/01.wnl.0000343852.10018.24. [DOI] [PubMed] [Google Scholar]
- 100.Veinbergs I, Everson A, Sagara Y, Masliah E. Neurotoxic effects of apolipoprotein E4 are mediated via dysregulation of calcium homeostasis. J Neurosci Res. 2002;67:379–387. doi: 10.1002/jnr.10138. [DOI] [PubMed] [Google Scholar]
- 101.Jaworska A, Dzbek J, Styczynska M, Kuznicki J. Analysis of calcium homeostasis in fresh lymphocytes from patients with sporadic Alzheimer’s disease or mild cognitive impairment. Biochim Biophys Acta. 2013;1833:1692–1699. doi: 10.1016/j.bbamcr.2013.01.012. [DOI] [PubMed] [Google Scholar]
- 102.Settembre C, Fraldi A, Medina DL, Ballabio A. Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat Rev Mol Cell Biol. 2013;14:283–296. doi: 10.1038/nrm3565. [DOI] [PMC free article] [PubMed] [Google Scholar]