

Abstract
Precise regulation of nuclear factor κB (NF-κB) signaling is crucial for normal immune responses, and defective NF-κB activity underlies a range of immunodeficiencies. NF-κB is activated through two signaling cascades: the classical and noncanonical pathways. The classical pathway requires inhibitor of κB kinase β (IKKβ) and NF-κB essential modulator (NEMO), and hypomorphic mutations in the gene encoding NEMO (ikbkg) lead to inherited immunodeficiencies, collectively termed NEMO-ID. Noncanonical NF-κB activation requires NF-κB–inducing kinase (NIK) and IKKα, but not NEMO. We found that noncanonical NF-κB was basally active in peripheral blood mononuclear cells from NEMO-ID patients, and that noncanonical NF-κB signaling was similarly enhanced in cell lines lacking functional NEMO. NIK, which normally undergoes constitutive degradation, was aberrantly present in resting NEMO-deficient cells, and regulation of its abundance was rescued by reconstitution with full-length NEMO, but not a mutant NEMO protein unable to physically associate with IKKα or IKKβ. Binding of NEMO to IKKα was not required for ligand-dependent stabilization of NIK or noncanonical NF-κB signaling. Rather, an intact and functional IKK complex was essential to suppress basal NIK activity in unstimulated cells. Despite interacting with IKKα and IKKβ to form an IKK complex, NEMO mutants associated with immunodeficiency failed to rescue classical NF-κB signaling or reverse the accumulation of NIK. Together, these findings identify a crucial role for classical NF-κB activity in the suppression of basal noncanonical NF-κB signaling.
Introduction
Members of the nuclear factor κB (NF-κB) family of inducible transcription factors play key roles in normal immune system development and function, cell survival, and cellular homeostasis (1). Aberrant activation of NF-κB underlies the pathogenesis of many inflammatory diseases and cancers (2), and failure to activate NF-κB is associated with immunodeficiency (3). Two major mechanisms of NF-κB activation that regulate distinct subsets of genes have been described: the classical and non-canonical NF-κB pathways. A wide range of stimuli, including pro-inflammatory cytokines and the ligation of innate and adaptive immune receptors, activate the classical NF-κB pathway (1). Signals initiated by these diverse stimuli converge on the heterotrimeric inhibitor of κB kinase (IKK) complex, which contains IKKα, IKKβ, and the non-catalytic regulatory subunit NF-κB essential modulator (NEMO, also known as IKKγ) (4). Activation of the IKK complex leads to the phosphorylation, ubiquitylation, and proteasomal degradation of members of the inhibitor of κB (IκB) family of proteins that sequester NF-κB in the cytoplasm of resting cells. Degradation of IκB enables the nuclear translocation of NF-κB homo- or heterodimers to regulate the expression of genes involved in diverse cellular responses (1). Genetic studies have established that classical NF-κB signaling absolutely requires NEMO and predominantly the catalytic activity of IKKβ to phosphorylate IκBα (5–11). For most inducers of classical signaling, IKKα is not required for the phosphorylation of IκBα, but instead plays a less well-defined role in classical NF-κB–dependent gene transcription (12–15); IKKα is also important for the termination of classical NF-κB–dependent transcription of target genes in macrophages (16).
IKKα is required for the activation of non-canonical NF-κB signaling (17, 18) in response to the stimulation of receptors for the tumor necrosis factor (TNF) super-family of cytokines, including the lymphotoxin-β receptor (LTβR), CD40, B cell activating factor receptor (BAFF-R or TNFRSF13C), TNF-related weak inducer of apoptosis (TWEAK) receptor (also known as TNFRSF12A, Fn14, or CD266), and receptor activator of NF-κB (RANK, also known as TRANCE) (19). Activation of these receptors leads to the IKKα-mediated phosphorylation of the NF-κB precursor protein p100, which associates with and sequesters another NF-κB subunit RelB in the cytoplasm of resting cells. IKKα-mediated phosphorylation of p100 facilitates its proteasomal processing to p52 and the release of p52-RelB heterodimers to the nucleus. Noncanonical NF-κB signaling regulates the expression of a small panel of genes, including cxcl12, cxcl13, ccl19, ccl21, and tnfsf13b, whose products are crucial for lymph node organogenesis and B cell homeostasis (20). Analyses of NF-κB signaling in cells lacking individual IKKs have established that p100 processing can be induced in the absence of either NEMO or IKKβ (17, 18, 21). Thus, it is thought that IKKα alone is necessary for the regulation of non-canonical NF-κB signaling.
Activation of the non-canonical NF-κB signaling pathway also requires the catalytic activity of NF-κB–inducing kinase (NIK) (22). In resting cells, newly synthesized NIK protein is rapidly degraded by a complex consisting of TNF receptor–associated factor 2 (TRAF2), TRAF3, cellular inhibitor of apoptosis 1 (cIAP1), and cIAP2 (23–26), and upon activation of an appropriate receptor, for example LTβR, NIK is freed from this complex and accumulates in the cytoplasm (23–25). Stabilized NIK phosphorylates and activates IKKα, which leads to p100 processing and non-canonical NF-κB activation. IKKα also phosphorylates active NIK to stimulate its degradation (27). The precise phosphorylation-dependent mechanism of NIK turnover is unclear, but this negative feedback loop is crucial to eliminate NIK and terminate non-canonical NF-κB signaling.
Noncanonical NF-κB plays a key role in immunoglobulin (Ig) class-switch recombination in activated B cells to generate the IgA isotype (28). Prolonged stability of NIK enhances non-canonical NF-κB signaling, and mice with increased NIK abundance display high serum concentrations of IgA (28). Notably, a subset of patients with inherited mutations in NEMO exhibits increased amounts of IgA, a condition known as hyper-IgA syndrome (29). These patients have a complex immunodeficiency termed NEMO-ID, which is a result of defects in classical NF-κB signaling; however, the mechanisms underlying the unusual clinical presentation of hyper-IgA have not been explained by the lack of classical NF-κB activity (29). Here, we showed that non-canonical NF-κB signaling was enhanced in resting peripheral blood mononuclear cells (PBMCs) from NEMO-ID patients and in cell lines lacking functional NEMO. Our findings define a crucial role for the intact classical IKK complex and classical NF-κB signaling in the regulation of basal NIK abundance to maintain the quiescent state of the non-canonical NF-κB pathway.
Results
NEMO inhibits the non-canonical NF-κB signaling pathway
To assess the status of the non-canonical NF-κB pathway in samples from NEMO-ID patients previously shown to have impaired classical NF-κB activation and increased concentrations of serum IgA (29, 30), we compared p100 processing in unstimulated PBMCs from two patients to that in PBMCs from a healthy donor. The abundance of p52 in comparison to the abundance of p100 was greater in the PBMCs of both of the NEMO-ID patients (Q403X and C417R) than in the PBMCs of the healthy donor or in human umbilical vein endothelial cells (HUVECs) stimulated with the TNF superfamily member LIGHT (also known as TNFSF14) (31) (Fig. 1A). We did not detect p100 protein in PBMCs from a patient with the Q403X mutation, suggesting that cellular p100 was completely processed to p52 in these cells. These data indicate that basal non-canonical NF-κB signaling was enhanced in the context of NEMO-ID.
Fig. 1. The extent of p100 processing is enhanced in NEMO-ID cells and cells lacking NEMO.

(A) Lysates of PBMCs from patients with distinct NEMO mutations (Q403X and C417R), PBMCs from a healthy donor (Control) and HUVECs that were either untreated (−) or were stimulated with LIGHT for 8 hours (+) were analyzed by Western blotting with antibodies against the indicated proteins. Values reported here (in numbers beneath the blots) for the quantification of the ratio of the intensity of the p52 bands to the intensity of the p100 bands are arbitary units relative to the intensity of the tubulin bands. (B) WT, IKKαKO, and NEMOKO MEFs were either untreated (−) or were stimulated with LIGHT for 12 hours (+). Cell lysates were then analyzed by Western blotting with antibodies against the indicated proteins. (C) WT, IKKαKO, and NEMOKO MEFs were either left untreated (−) or were incubated with anti-LTβR antibody (α-LTβR) for 12 hours (+). Cytoplasmic and nuclear extracts were then prepared and analyzed by Western blotting with antibodies against the indicated proteins. Western blots are representative of three independent experiments.
To understand how NEMO affected non-canonical NF-κB signaling, we examined basal and LIGHT-induced p100 processing in NEMO-deficient (NEMOKO) mouse embryonic fibroblasts (MEFs). Basal p100 processing was substantially increased in resting (unstimulated) NEMOKO MEFs compared to that in resting wild-type MEFs (Fig. 1B and fig. S1A). Consistent with earlier studies (17, 18, 21), p100 processing in NEMOKO MEFs was further enhanced in response to LIGHT or an agonistic anti-LTβR antibody (Fig. 1B and fig. S1, B and C); however, in some experiments, basal amounts of p52 protein were already maximal in NEMO-deficient cells and were not further increased in response to activating ligands (fig. S1, B and C).
To further examine the activation status of non-canonical NF-κB signaling in the absence of NEMO, we analyzed cytoplasmic and nuclear fractions from resting and anti-LTβR–stimulated MEFs by Western blotting. We found that p52 was absent from the nuclear samples of unstimulated wild-type and IKKαKO MEFs (Fig. 1C), and, as expected, that the anti-LTβR antibody stimulated the nuclear translocation of p52 in wild-type, but not IKKαKO, cells. Consistent with the enhanced processing of p100 in cells lacking NEMO (Fig. 1B and fig. S1), nuclear p52 was present in unstimulated NEMOKO MEFs and was increased in abundance in response to activation of LTβR (Fig. 1C and fig. S2A). Furthermore, more nuclear RelB was present in resting NEMOKO MEFs than in unstimulated wild-type and IKKαKO cells, and the extent of RelB nuclear translocation was markedly enhanced by anti-LTβR antibody (Fig. 1C and fig S2A). These data reveal that the basal amounts of p52 and RelB in the nuclei of resting NEMOKO MEFs were similar to those in wild-type cells stimulated through LTβR (fig. S2A). Consistent with the increased amounts of nuclear p52 and RelB, transcription of the non-canonical NF-κB gene target cxcl12 (18, 31, 32) was substantially increased in unstimulated NEMOKO MEFs compared to that in resting wild-type cells (fig. S2B). Together, these findings indicate that the non-canonical NF-κB signaling pathway is active in NEMOKO MEFs.
To confirm that these findings were not unique to MEFs, we assessed p100 processing in the Jurkat T cell line 8321, which contains a hypomorphic NEMO fragment that only retains partial function (33). As in the NEMO-ID PBMCs and NEMOKO MEFs (Fig. 1), p52 protein abundance was increased in abundance in unstimulated 8321 cells compared to that in the parental 3T8 line, which contains wild-type NEMO (fig. S3A). Reconstitution of 8321 cells with wild-type NEMO (8321WT) (33) reduced the extent of p100 processing to that seen in the parental cell line (fig. S3A). Similarly, reconstitution of NEMOKO MEFs with wild-type NEMO substantially reduced the ratio of p52 protein to p100 protein (fig. S3, B and C). Together, these findings suggest that intact NEMO maintains the inactive state of non-canonical NF-κB signaling in resting cells.
NIK is present in cells that lack NEMO
Noncanonical NF-κB activation requires ligand-induced stabilization of NIK (17, 18, 34). Because genetic loss of NEMO resulted in the increased processing of p100 (Fig. 1), we asked whether NIK protein amounts were also dysregulated in the absence of NEMO. As expected, NIK was undetected in resting wild-type MEFs, but was stabilized in response to LIGHT (Fig. 2A). Consistent with the recently reported role for IKKα in mediating NIK turnover (27), NIK was present in unstimulated IKKα-deficient cells, and its abundance was further increased in response to LIGHT (Fig. 2A). NIK was also present in resting NEMOKO MEFs (Fig. 2A), and its abundance was either unchanged or minimally enhanced in response to LIGHT. Despite the presence of a substantially increased amount of NIK protein in NEMOKO MEFs compared to that in wild-type MEFs (Fig. 2B), quantitative, reverse transcription polymerase chain reaction (RT-PCR) assays showed that the abundance of map3k14 mRNA was similar in wild-type, IKKαKO, and NEMOKO cells (Fig. 2C), indicating that the increased amount of NIK protein in NEMO-deficient MEFs was not a result of increased expression of map3k14, which encodes NIK.
Fig. 2. NEMO suppresses the accumulation of NIK protein.

(A) WT, IKKαKO, and NEMOKO MEFs were either left untreated (−) or incubated with LIGHT for 12 hours (+). Cell lysates were then analyzed by Western blotting with antibodies against the indicated proteins. (B) Quantification of the abundance of NIK protein relative to that of tubulin in the experiments represented by the Western blot in (A). Data are means ± SEM from three independent experiments. *P < 0.05 by one-way ANOVA and Dunnett’s post-test. (C) Analysis of the abundance of map3k14 mRNA (which encodes NIK) in WT, IKKαKO, and NEMOKO MEFs. Data are means ± SEM from four independent experiments. (D and E) Lysates from (D) WT and NEMO-deficient Jurkat cells and (E) Rat1R fibroblasts were analyzed by Western blotting with antibodies against the indicated proteins. (F) WT, NEMOKO, and NEMOKO MEFs retrovirally transduced with either control MigR1 vector or with MigR1 encoding WT NEMO or a truncated mutant NEMO (86–419) were either untreated (−) or incubated with LIGHT for 12 hours (+) before cell lysates were analyzed by Western blotting with antibodies against the indicated proteins. Western blots are representative of four independent experiments.
We next analyzed NIK in NEMO-mutant 8321 cells, which exhibited enhanced processing of p100 (fig. S3). The amount of NIK in unstimulated NEMO-mutant 8321 cells was markedly increased compared to that in unstimulated parental 3T8 cells (Fig. 2D). Similarly, NIK abundance was increased in resting NEMO-deficient Rat5R fibroblasts compared to that in parental Rat1 cells (Fig. 2E). These data from separate cell types and different species suggest that the presence of NEMO suppresses the accumulation of NIK protein. To confirm this function of NEMO, we reconstituted NEMOKO MEFs with wild-type NEMO (NEMOWT) and found that NIK was not aberrantly stabilized (Fig. 2F). Moreover, stimulation of NEMOWT MEFs with LIGHT led to NIK stabilization, analogous to the activation of wild-type MEFs (Fig. 2F). Hence, exogenous NEMO rescued the basal regulation of NIK in NEMOKO MEFs.
We next questioned whether the role of NEMO in regulating NIK abundance was related to its ability to interact with the catalytic IKKs. To address this, we reconstituted NEMOKO MEFs with a NEMO truncation mutant that is unable to bind to either IKKα or IKKβ (NEMO86–419) and is thereby unable to rescue classical NF-κB activation (fig. S4). Notably, NIK remained detectable in unstimulated NEMO86–419 MEFs, similar to NEMOKO cells (Fig. 2F). These data indicate that NEMO must associate with either IKKα or IKKβ to properly constrain basal NIK abundance.
The association of NEMO with IKKα is dispensable to regulate non-canonical NF-κB signaling
To determine whether activation of non-canonical NF-κB signaling required the association of NEMO with IKKα, we transduced IKKαKO MEFs with retrovirus containing MigR1 plasmid alone or MigR1 plasmid encoding either wild-type IKKα (IKKαWT) or a mutant IKKα that lacks the NEMO-binding domain (IKKαΔNBD) to generate stable cell lines (fig. S5) (35). Stimulation of wild-type MEFs with LIGHT led to transient stabilization of NIK and processing of p100 (Fig. 3A). Consistent with previous reports (27), NIK was detected in IKKαKO cells that were transduced with retrovirus containing the control MigR1 plasmid and was further stabilized in response to LIGHT (Fig. 3A). Reconstitution of IKKαKO cells with either IKKαWT or IKKαΔNBD resulted in NIK becoming undetectable under resting conditions. In addition, stimulation of either IKKαWT or IKKαΔNBD MEFs with LIGHT induced the stabilization of NIK and processing of p100 to generate p52 (Fig. 3A). Thus, we conclude that the association of NEMO with IKKα is dispensable for regulating basal NIK abundance, and that binding to NEMO is not required for IKKα to regulate p100 processing in response to LIGHT.
Fig. 3. An association between NEMO and IKKα is dispensable to regulate non-canonical NF-κB.
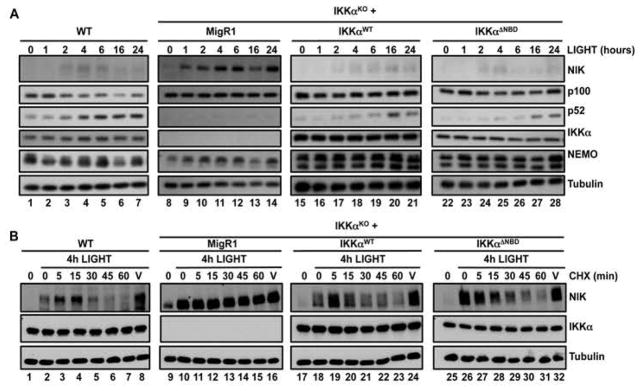
(A) WT and IKKα-deficient (IKKαKO) MEFs retrovirally transduced with either MigR1 alone or MigR1 expressing IKKαWT or IKKαΔNBD were incubated with LIGHT for the indicated times. Cell lysates were then analyzed by Western blotting with antibodies against the indicated proteins. (B) WT, IKKαKO, and IKKαKO MEFs transduced with the indicated constructs were incubated for 4 hours with LIGHT and then were treated either with cycloheximide (CHX) for the indicated times or with ethanol for 60 min as a vehicle control (V). Lysates were then analyzed by Western blotting with antibodies against the indicated proteins. Western blots are representative of four independent experiments.
We next questioned whether the association of IKKα with NEMO was required for the IKKα-dependent turnover of active NIK. To address this, we incubated MEFs with LIGHT for 4 hours to stabilize NIK and then incubated the cells with cycloheximide for up to 1 hour to prevent new protein synthesis before visualizing NIK turnover by Western blotting. Supporting the model of IKKα-dependent feedback regulation (27), LIGHT-induced NIK protein was rapidly lost in wild-type MEFs, whereas NIK in IKKα-deficient cells remained detectable 60 min after incubation with cycloheximide (Fig. 3B). Furthermore, both IKKαWT and IKKαΔNBD restored NIK turnover in IKKαKO cells, which demonstrated that the ability of active IKKα to destabilize LIGHT-induced NIK protein (27) is independent of its association with NEMO (Fig. 3B). The aberrant stabilization of NIK in unstimulated NEMO-deficient cells was therefore not a result of the loss of the NEMO-IKKα interaction.
The IKK complex is required for the regulation of basal NIK abundance
Because dissociation of NEMO from IKKα alone did not affect the basal extent of p100 processing or NIK stability (Fig. 3), our earlier findings (Fig. 2F) led us to ask whether IKKβ might be involved in the NEMO-dependent control of NIK abundance. To address this, we examined non-canonical NF-κB signaling in resting IKKβ-deficient (IKKβKO) MEFs and found that, similar to the situation in NEMOKO cells, basal p52 abundance was increased (Fig. 4A). Consistent with previous reports (18), p100 processing in IKKβKO MEFs was further enhanced by LIGHT, confirming that ligand-induced, non-canonical NF-κB signaling was intact in the absence of IKKβ (Fig. 4A). Similar to that in NEMOKO cells, NIK abundance was substantially increased in resting IKKβKO MEFs compared to that in wild-type cells, and treatment with LIGHT or anti-LTβR led to additional stabilization of NIK (Fig. 4B and fig. S6A). Examination of a second independently generated IKKβKO MEF line confirmed these findings (fig. S6B). Furthermore, constitutive IKKα phosphorylation was detected in IKKβKO cells before they were stimulated (Fig. 4B), indicating that the non-canonical NF-κB pathway was basally active in these cells.
Fig. 4. The catalytically active IKK complex is required to suppress accumulation of NIK.

(A) WT, IKKαKO, NEMOKO, and IKKβKO MEFs were either untreated (−) or incubated with LIGHT for 12 hours (+) before cell lysates were analyzed by Western blotting with antibodies against the indicated proteins. (B) The indicated MEF cell lines were either untreated (−) or were stimulated with LIGHT (L) or anti-LTβR antibody (Ab) for 12 hours before cell lysates were analyzed by Western blotting with antibodies against the indicated proteins. Active IKK was detected with an antibody specific for phosphorylated IKKα/β (p-IKKα/β). (C) WT, IKKβKO, and IKKβKO MEFs retrovirally transduced with LZRS or LZRS expressing either IKKβWT or a catalytically inactive mutant IKKβ (IKKβK44M) were either untreated (−) or incubated with LIGHT for 12 hours (+) before cell lysates were analyzed by Western blotting with antibodies against the indicated proteins. Western blots are representative of four independent experiments.
We hypothesized that reconstitution of IKKβKO MEFs with wild-type IKKβ might restore the normal regulation of NIK abundance. As expected, stable transduction of IKKβKO MEFs with retrovirus containing plasmid encoding wild-type IKKβ (IKKβWT), but not a catalytically inactive mutant kinase (IKKβK44M), rescued classical NF-κB signaling (fig. S7, A and B). Furthermore, the presence of IKKβWT, but not IKKβK44M, in IKKβKO MEFs led to the reduced abundance of NIK (Fig. 4C), demonstrating that the catalytic activity of IKKβ was required to suppress the accumulation of NIK in resting cells. Furthermore, reconstitution of IKKβ-deficient cells with wild-type IKKβ reduced the extent of phosphorylation of IKKα (fig. S7C), suggesting that increased NIK abundance was required for the robust activation of IKKα seen in these cells. Collectively, these findings suggest that basal regulation of the non-canonical NF-κB signaling pathway requires the intact and catalytically active IKK complex.
NIK is phosphorylated in cells lacking NEMO or IKKβ
The recently described negative feedback loop that leads to the degradation of NIK requires the phosphorylation of NIK by IKKα (27). This mechanism prevents prolonged non-canonical NF-κB signaling, and can be monitored by the detection of a single band corresponding to unphosphorylated NIK in IKKαKO MEFs (Fig. 4B). Because the band corresponding to NIK that we detected in cells lacking NEMO or IKKβ resembled that detected in wild-type MEFs stimulated by LIGHT or anti-LTβR antibody (Fig. 4B), we reasoned that this slower migrating band corresponded to phosphorylated NIK. Treatment of LIGHT-stimulated MEFs with λ-phosphatase caused NIK in wild-type, NEMOKO, and IKKβKO cells to migrate with the same electrophoretic mobility as that of unphosphorylated NIK in IKKαKO MEFs (Fig. 5A). Moreover, λ-phosphatase reduced the apparent molecular mass of stabilized NIK in TRAF3KO MEFs, in which basal degradation of NIK is defective (Fig. 5B) (36). These data suggest that active NIK is phosphorylated by IKKα in MEFs lacking NEMO, IKKβ, or TRAF3, similar to that in wild-type cells.
Fig. 5. The IKK complex does not mediate the IKKα-dependent turnover of active NIK protein.

(A) Left: The indicated cells were incubated with LIGHT for 4 hours, and then cell lysates were prepared and either left untreated (−) or incubated with λ-phosphatase (ptase; +) before being analyzed by Western blotting with antibodies against the indicated proteins. The asterisk indicates the band corresponding to phosphorylated NIK. Right: Loading controls to show the abundances of NIK, IKKα, IKKβ, and NEMO in lysates of the indicated LIGHT-stimulated cells before treatment with phosphatase. (B) TRAF3KO MEFs were processed and analyzed as described in (A). (C) WT, NEMOKO, IKKβKO, DKO, and TRAF3KO MEFs were incubated for 4 hours with LIGHT and then were either treated with cycloheximide (CHX) for the indicated times or were treated for 60 min with ethanol as a vehicle control (V). Cell lysates were analyzed by Western blotting with antibodies against the indicated proteins. (D) Unstimulated IKKαKO, NEMOKO, IKKβKO, and DKO MEFs were incubated with CHX for the indicated times or with ethanol (V) for 120 min before cell lysates were prepared and analyzed by Western blotting with antibodies against the indicated proteins. Western blots are representative of four independent experiments.
Because our results suggest that both IKKα and IKKβ are required for the overall regulation of NIK abundance, we assessed NIK protein stability in IKKα and IKKβ double-knockout (DKO) MEFs. Consistent with our earlier findings, NIK was detectable in unstimulated DKO MEFs (fig. S8A). Similar to what we observed with IKKαKO MEFs, there was no electrophoretic shift indicating IKKα-mediated phosphorylation of NIK in DKO MEFs when compared to NIK in stimulated wild-type cells (fig. S8A). Treatment with λ-phosphatase enhanced the electrophoretic mobility of NIK in unstimulated NEMOKO and IKKβKO MEFs, but not in DKO MEFs (fig. S8B). These data demonstrate that NIK is phosphorylated in an IKKα-dependent manner in cells lacking either NEMO or IKKβ.
IKKα-mediated turnover of NIK is independent of the IKK complex
Our accumulated findings establish that NIK is present and phosphorylated by IKKα under resting conditions in NEMO- or IKKβ-deficient MEFs. Because NIK in these resting cells resembled NIK in ligand-stimulated wild-type MEFs (Fig. 5A), we asked whether the intact IKK complex was required for the IKKα-dependent degradation of active NIK (27). Consistent with the inducible activation of the non-canonical pathway in NEMOKO and IKKβKO cells, NIK abundance was enhanced in these cells in response to LIGHT, and this active NIK was degraded with similar kinetics to that of NIK in wild-type MEFs (Fig. 5C). As expected, NIK remained stable in DKO MEFs in the presence of cycloheximide, which confirmed that negative feedback in the absence of the intact IKK complex required IKKα. Furthermore, activation-induced turnover of NIK was independent of the basal NIK regulatory complex, because active NIK in TRAF3KO cells was degraded similarly to NIK in wild-type, NEMOKO, and IKKβKO MEFs (Fig. 5C). These data indicate the intact IKK complex is not required for the IKKα-mediated turnover of activated NIK.
We next questioned whether the IKKα-phosphorylated NIK present in resting NEMOKO and IKKβKO MEFs resulted from defective protein turnover. We found that treatment of unstimulated NEMOKO and IKKβKO MEFs with cycloheximide led to loss of basal NIK (Fig. 5D). In contrast unphosphorylated NIK in both resting IKKαKO and DKO cells was stable and remained detectable two hours after treatment with cycloheximide (Fig. 5D). Thus, these data suggest that the IKK complex does not regulate IKKα-mediated NIK turnover, but instead plays an obligate role in limiting the basal pool of newly synthesized NIK.
Classical NF-κB activity is required for the regulation of basal NIK abundance
To determine whether the IKK complex regulated NIK abundance through classical, NF-κB-driven transcription, we assessed non-canonical NF-κB activity in MEFs lacking the prototypic classical NF-κB subunit p65 (p65KO). Similar to unstimulated NEMOKO and IKKβKO MEFs, NIK was increased in abundance in p65KO MEFs before stimulation with LIGHT or anti-LTβR antibody (Fig. 6A). Quantitative RT-PCR analysis of p65KO MEFs showed that classical NF-κB signaling did not control map3k14 expression directly (Fig. 6B). Consistent with experiments with NEMOKO and IKKβKO MEFs (Fig. 5A), treatment with λ-phosphatase revealed that active NIK was phosphorylated in p65KO MEFs (Fig. 6C). Furthermore, the normal reduced abundance of NIK in resting cells was partially restored by stable reconstitution of p65KO MEFs with wild-type p65 (Fig. 6D).
Fig. 6. Classical NF-κB–dependent gene expression is required to regulate basal NIK abundance.

(A) WT or p65KO MEFs were left unstimulated or were treated with either LIGHT (L) or the anti-LTβR antibody (Ab) for 4 hours. Cell lysates were then analyzed by Western blotting with antibodies against the indicated proteins. (B) RNA from WT or p65KO MEFs was isolated and analyzed by quantitative RT-PCR to determine the abundance of map3k14 mRNA in comparison to that of actb mRNA (which encodes β-actin). Data are means ± SEM from three independent experiments. (C) WT and p65KO MEFs were stimulated with LIGHT for 4 hours. Whole-cell extracts were then left untreated or were treated with λ phosphatase and analyzed by Western blotting with antibodies against the indicated proteins. The asterisk indicates the band corresponding to phosphorylated NIK. (D) p65KO MEFs were transduced with empty pBABE vector or the pBABE vector encoding WT p65. Cell lysates were then analyzed by Western blotting with antibodies against the indicated proteins. (E) Whole-cell extracts from WT or p65KO MEFs were analyzed by Western blotting with antibodies against the indicated proteins. (F) NEMOKO MEFs stably transduced with either MigR1 or MigR1 encoding WT IKKβ were transfected with the pBIIx-κB firefly luciferase (FFL) and the TK renilla luciferase (RL) vectors. Twenty-four hours later, classical NF-κB transcriptional activity was determined as the fluorescence ratio of FFL:RL. Data are means ± SEM of six replicates from two independent experiments. **P < 0.01 by student’s two-tailed t-test. (G) WT or NEMOKO MEFs stably transduced with either MigR1 or MigR1 encoding WT IKKβ were analyzed by Western blotting with antibodies against the indicated proteins. Western blots are representative of four independent experiments.
Because classical NF-κB activity did not directly affect the expression of map3k14 (Figs. 2C and 6B), we sought to determine whether any of the known modulators of NIK abundance were dysregulated in p65KO MEFs. The molecular components that reduce the basal abundance of NIK form the TRAF2:TRAF3:cIAP1:cIAP2 E3 ubiquitin ligase complex (20). We found that the amounts of TRAF2, TRAF3, cIAP1, and cIAP2 were similar or increased in p65KO MEFs compared to those in wild-type cells (Fig. 6E), consistent with a potential role for non-canonical NF-κB signaling in regulating the abundance of TRAF3 (37). Because birc3 (which encodes cIAP2) is a classical NF-κB target gene (38), we assessed birc3 transcripts in p65KO MEFs and found that they were present in similar amounts in p65KO and wild-type MEFs (fig. S9A). In addition, birc3 expression was intact in NEMOKO MEFs, suggesting that disruption of the IKK complex did not affect basal birc3 expression. We therefore conclude that cIAP2 is not the molecular target of classical NF-κB signaling that controls basal NIK abundance. The amounts of cIAP1, TRAF2, and TRAF3 proteins were similar, if not increased, among the cell lines that we studied (fig. S9B), and TRAF3 stability was unaffected by loss of classical NF-κB activity (fig. S9C). In addition, exogenous NIK physically associated with endogenous TRAF3, TRAF2, and cIAP1 even in the absence of NEMO or p65 (fig. S10). Together, these results suggest that aberrant NIK detected in the absence of NEMO, IKKβ, or p65 does not arise because of changes in the currently known NIK regulatory machinery.
Our results indicated that the transcriptional activity of classical NF-κB was required to actively suppress basal non-canonical NF-κB signaling, because loss of p65 enabled the stabilization of NIK and the processing of p100 in resting cells, similar to the case when the upstream signaling components NEMO or IKKβ are lost (Figs. 1, 2, and 4). We therefore hypothesized that enforced classical NF-κB activity could rescue aberrant non-canonical NF-κB signaling in NEMOKO MEFs. To test this, we transduced NEMOKO MEFs with retrovirus containing control MigR1 plasmid alone or MigR1 encoding wild-type IKKβ to generate stable cell lines over-expressing IKKβ. Over-expression of wild-type IKKβ alone was sufficient to drive classical NF-κB transcriptional activity, even in the absence of NEMO (Fig. 6F). Furthermore, we observed reduced NIK amounts and decreased processing of p100 in NEMOKO MEFs that over-expressed wild-type IKKβ compared to control cells (Fig. 6G). Collectively, these results confirm that classical NF-κB transcriptional activity is required to actively prevent the accumulation of NIK and thus to prevent non-canonical NF-κB signaling in resting cells.
NEMO-ID mutants do not restore regulation of NIK abundance
Our studies in MEFs revealed a previously uncharacterized level of cross talk between classical NF-κB signaling and the regulation of NIK abundance. Because of the scarcity of patient-derived material, we could not definitively determine the status of basal NIK abundance in NEMO-ID PBMCs. Thus, we reconstituted NEMOKO MEFs with the hyper-IgA–associated Q403X and C417R mutant NEMO proteins to further evaluate the effects of NEMO-ID mutations on non-canonical NF-κB signaling. Unlike the NEMO86–419 truncation mutant that cannot bind to IKKα or IKKβ (fig. S4), NEMOQ403X and NEMOC417R each associated with the IKKs to form a heterotrimeric IKK complex in reconstituted cells (Fig. 7A). Despite being able to form IKK complexes, neither NEMOQ403X nor NEMOC417R rescued TNF-α–induced NF-κB activation in NEMOKO MEFs, confirming that these mutants ablate classical IKK activity (Fig. 7B).
Fig. 7. Noncanonical NF-κB signaling is enhanced in cells containing hypomorphic ikbkg mutations.

(A) WT, NEMOKO, and NEMOKO MEFs retrovirally transduced with MigR1 or MigR1 encoding WT NEMO or the indicated NEMO mutants were lysed and then subjected to immunoprecipitation (IP) with anti-IKKα and anti-NEMO antibodies. Cell lysates and the immunoprecipitated samples were analyzed by Western blotting with antibodies against the indicated proteins. (B) WT and NEMOKO cells were transfected with the pBIIx-κB firefly luciferase (FFL) and the TK Renilla luciferase (RL) vectors. Twenty-four hours later, cells were either left untreated or were treated with TNF-α for 5 hours. NF-κB transcriptional activity was determined by measuring the fluorescence ratio of FFL:RL, and the extent of κB luciferase induction was defined as the fold-difference between the TNF-α–stimulated samples and the controls. Data are means ± SD of four replicates from one of three independent experiments. (C) WT and NEMOKO MEFs retrovirally transduced with MigR1 or MigR1 encoding WT NEMO or the indicated NEMO mutants were either untreated (−) or incubated with anti-LTβR antibody for 8 hours (+) before being lysed and analyzed by Western blotting with antibodies against the indicated proteins. Western blots are representative of four independent experiments.
Whereas normal regulation of NIK abundance was restored in NEMOWT MEFs, increased amounts of NIK were maintained in NEMOKO MEFs reconstituted with either NEMOQ403X or NEMOC417R (Fig. 7C). We further generated an additional NEMO-ID mutant MEF cell line containing a NEMO variant (NEMOL153R) with a mutation in a distinct functional domain of the protein. Previous reports showed that this mutation does not enable classical NF-κB signaling (30, 39), and we found that NEMOL153R failed to restore control of basal NIK abundance in transfected NEMOKO cells (Fig. 7C). These findings suggest that the ability of NEMO to constitute part of the IKK complex is insufficient to properly regulate the non-canonical NF-κB pathway. Rather, a signal-competent IKK complex and intact classical NF-κB activity are essential to maintain the quiescent state of the non-canonical NF-κB pathway.
Discussion
Our findings reveal an essential role for the NEMO-dependent classical NF-κB pathway in inhibiting the basal activity of the non-canonical NF-κB pathway. We showed that naturally occurring NEMO-ID mutations prevented inhibition of the non-canonical NF-κB pathway by the classical IKK complex. Patients with hypomorphic mutations in ikbkg exhibit a complex immunologic phenotype that results in widespread immunodeficiency (40). The clinical presence of enhanced amounts of serum IgA in patients with NEMO-ID is intriguing, because non-canonical NF-κB signaling directly controls class switching to the IgA isotype (28). B cell–specific deletion of the gene encoding TRAF-associated NF-κB activator (TANK)-binding kinase 1 (TBK1) in mice leads to NIK stabilization and nephritis because of prolonged non-canonical signaling and increased amounts of IgA (28). We found that unstimulated PBMCs from patients with NEMO mutations associated with hyper-IgA syndrome exhibited enhanced processing of p100, supporting the existence of a defect in the basal regulation of non-canonical NF-κB. Thus, we conclude that in a subset of NEMO-ID patients, defective activation of classical NF-κB signaling leads to the increased basal activity of the non-canonical NF-κB pathway. This finding provides a potential mechanism for the aberrant class switching that occurs in NEMO-ID, and represents a demonstration of the dysregulated non-canonical NF-κB signaling pathway in humans.
To investigate the mechanism underlying defective regulation of non-canonical NF-κB signaling, we used a panel of IKK-deficient MEF cell lines. In light of the prevailing model in which the IKK complex plays no role in the non-canonical NF-κB pathway (19), we were surprised to detect enhanced p100 processing in cells lacking NEMO or IKKβ. In both cases, p100 processing was further enhanced by stimulation of the cells with LIGHT or anti-LTβR antibody; however, in some instances, p52 amounts in NEMO- or IKKβ-deficient cells appeared to be maximal. Moreover, the ratio of p52 to p100 in untreated NEMOKO MEFs was similar to that in maximally stimulated wild-type MEFs, suggesting that non-canonical signaling was fully active in the absence of NEMO. These findings are consistent with earlier studies (17, 18, 41); however, the catalytically active IKKα and enhanced basal p100 processing previously observed in NEMOKO cells has never been highlighted or addressed.
In seeking to determine the mechanism for enhanced p100 processing, we found that NIK abundance was substantially increased in resting cells lacking either NEMO or IKKβ. Consistent with active p100 processing, the NIK protein detected in the absence of NEMO or IKKβ resembled that induced by stimulation of wild-type MEFs with LIGHT or anti-LTβR antibody. This result further corroborates the finding that non-canonical NF-κB signaling is basally active in these cells. We also found that NEMO and IKKβ must physically interact, and that the catalytic activity of IKKβ is required for regulation of basal NIK abundance. Hence, we conclude that, in resting cells, the intact and catalytically competent IKK complex maintains basal amounts of NIK and prevents the constitutive processing of p100 to generate p52.
Because the limited availability of clinical samples precluded a definitive determination of the status of NIK in PBMCs from patients with NEMO-ID, we established stable MEF cell lines that expressed pathological ikbkg mutations. In each case, the NEMO-ID mutants assembled with endogenous IKKα and IKKβ to form a heterotrimeric IKK complex; however, consistent with previous studies, these complexes were unable to activate classical NF-κB signaling (42, 43). Furthermore, we detected increased amounts of NIK protein in the NEMO-mutant cells. Hence, our data establish that functional mutations of NEMO that do not prevent IKK complex assembly alter the basal regulation of the non-canonical NF-κB pathway. We have only been able to assess this defect retrospectively in clinical samples, but our studies support future assessment of non-canonical NF-κB signaling in NEMO-ID patients. Moreover, these accumulated findings prompt a re-evaluation of the current view that the IKK complex plays no role in regulating non-canonical NF-κB signaling.
Unlike the stable NIK protein present in IKKαKO MEFs, the NIK protein that we detected in IKKβ-or NEMO-deficient cells was phosphorylated. Feedback inhibition of NIK requires its IKKα-mediated phosphorylation (27), which we showed to occur in the absence of NEMO binding to IKKα. Signal-induced p100 processing was also intact in IKKα-deficient cells reconstituted with an IKKα mutant that cannot bind to NEMO. Our findings therefore provide definitive biochemical support for the tenet that IKKα alone, independent of its association with the IKK complex, is sufficient to both activate the processing of p100 and inhibit non-canonical NF-κB signaling.
We found that NIK was basally phosphorylated in cells lacking NEMO or IKKβ to the same extent as it was in stimulated wild-type cells. This led us to question whether the heterotrimeric IKK complex “licenses” IKKα-mediated turnover of NIK protein, enabling phosphorylated NIK to accumulate in NEMO- or IKKβ-deficient cells. Our experiments with cycloheximide revealed that although NIK turnover in both resting and stimulated IKKαKO MEFs was defective, turnover occurred normally in cells lacking IKKβ or NEMO. We thus conclude that the IKK complex does not regulate IKKα-mediated NIK turnover, but instead plays an obligate role in limiting the basal pool of newly synthesized NIK. In support of this interpretation, we found that the stable NIK in TRAF3KO MEFs (23) was phosphorylated and turned over normally. Hence, basal NIK in cells lacking the intact IKK complex was similar to newly synthesized NIK that had escaped the TRAF2:TRAF3:cIAP1:cIAP2 regulatory machinery. These findings also confirm that TRAF3 plays no role in the IKKα-driven degradation of active NIK and imply that a previously uncharacterized regulatory mechanism is responsible for this phase of deactivation of non-canonical NF-κB signaling.
IKKα (27) and TBK1 (28) phosphorylate NIK, leading to its controlled turnover. Although our results do not support a role for classical NF-κB signaling in inhibition of the non-canonical NF-κB pathway, NIK is clearly subject to multiple layers of regulation by IKK or IKK-like kinases. We have not directly ruled out a role for a previously uncharacterized catalytic function by the heterotrimeric IKK complex; however, our studies in p65KO MEFs revealed that classical NF-κB activity was required to maintain undetectable amounts of NIK in resting cells. To accomplish this, classical NF-κB signaling may regulate a specific gene or genes whose products regulate the basal amount of NIK. Likely candidate genes include those encoding TRAF2, TRAF3, the cIAPs or OTUD7B, a de-ubiquitinase that promotes TRAF3 stability (44). However, we found that, compared to those in wild-type MEFs, the amounts of TRAF2, TRAF3, cIAP1, and cIAP2 were similar or somewhat increased among all of the MEF cell lines that we tested. Furthermore, over-expressed NIK associated with endogenous TRAF3, TRAF2, and cIAP1 in wild-type MEFs and in cells lacking NEMO or p65, suggesting that defective classical NF-κB signaling does not prevent the basal NIK regulatory complex from forming in these cells.
Mounting evidence has shown that cIAP1 and cIAP2 are functionally redundant in their roles as E3 ubiquitin ligases that lead to NIK degradation; individual knockout cells must be complemented with siRNA or Smac mimetics to degrade the other cIAP and stabilize NIK (24, 25, 45). We observed similar amounts of birc3 mRNA among p65KO and NEMOKO MEFs compared to wild-type MEFs, confirming the presence of this E3 ubiquitin ligase that is required for regulation of NIK abundance. Our data further suggest that OTUD7B (44) is not involved in basal NIK regulation by the classical IKK complex, because TRAF3 stability was unaffected by the loss of NEMO or IKKβ. We therefore surmise that classical NF-κB signaling drives the expression of a unique gene or set of genes whose products are involved in regulating non-canonical NF-κB signaling. Although the exact gene targets remain to be determined, our results establish an obligate role for classical NF-κB activity to facilitate the quiescent state of non-canonical NF-κB in unstimulated cells.
In conclusion, we demonstrated that classical NF-κB signaling limits the size of the basal pool of NIK, identifying a previously uncharacterized function for the IKK complex in inhibiting the non-canonical NF-κB pathway. Disruption of the heterotrimeric IKK complex through genetic loss of NEMO or IKKβ, catalytic inactivity of IKKβ, failure to form a heterotrimeric IKK complex, or naturally occurring mutations in NEMO that prevent classical NF-κB activity resulted in the increased abundance of NIK and aberrant non-canonical NF-κB signaling. Collectively, these findings establish a model of NF-κB pathway crosstalk that regulates basal non-canonical signaling and provide a potential mechanism to explain the perplexing hyper-IgA phenotype observed in patients with NEMO-ID.
Materials and Methods
Cell lines and culture
NIH 3T3 MEFs were purchased from ATCC. The 3T8 Jurkat T cell line has been described elsewhere (33) and was previously mutagenized to generate the NEMOKO cell line 8321 (33). These 8321 cells were transfected with plasmid encoding full-length NEMO to generate 8321WT cells (30). All fibroblasts and Plat-E cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Invitrogen) supplemented with 10% fetal bovine serum (FBS, Invitrogen), 2 mM L-glutamine, penicillin (50 U/ml), and streptomycin (50 U/ml). Jurkat T cells were cultured in RPMI medium (Invitrogen) supplemented with 10% FBS, 2 mM L-glutamine, penicillin (50 U/ml), and streptomycin (50 U/ml). Cells were passaged with 0.25% trypsin (Invitrogen). Unless otherwise noted, cells were stimulated with TNF-α (10 ng/ml), LIGHT (100 ng/ml), or anti-LTβR antibody (300 ng/ml) when they reached 80% confluence.
Patient samples
Patient PBMCs were obtained with parental consent and with approval by the Children’s Hospital Committee on Clinical Investigation, Children’s Hospital Boston, as described previously (29). Clinical presentations and immunological findings from these patients were previously reported (29, 30).
Reagents
Recombinant human TNF-α and LIGHT were purchased from R&D Systems, and the agonistic antibody against LTβR (clone 5G11) was from Abcam. Antibodies against NIK, histone H3, and murine p100/p52 were purchased from Cell Signaling Technology. Anti-NEMO antibody was purchased from MBL International. Anti-human p100/p52 was from Millipore. Antibodies against NEMO, IκBα, IKKα, TRAF3, TRAF2, cIAP-2, and TBK1 were purchased from Santa Cruz Biotechnology. Anti-IKKα and IKKβ antibodies were purchased from Imgenex. Monoclonal anti-cIAP1 antibody was purchased from Enzo Life Sciences. Anti–α-tubulin antibody (T5168), protein G Sepharose beads, anti-FLAG (M2) beads, and MG132 were from Sigma-Aldrich. Fugene 6 transfection reagent was from Promega. Horseradish peroxidase (HRP)–conjugated secondary antibodies and normal rabbit serum were from Jackson Immuno Research. Fluorescently labeled secondary antibodies were purchased from LI-COR Biosciences. Cycloheximide was purchased from Calbiochem and λ-phosphatase was from New England Biosciences.
Generation of stable cell lines
All cloning procedures were performed with the Pfu Turbo PCR procedure, as previously described (35). Briefly, complementary DNAs (cDNAs) encoding full-length wild-type and ΔNBD IKKα (encoding amino acid residues 1 to 734 lacking the NEMO-binding domain) were subcloned into the MigR1 vector, which contains an IRES-green fluorescent protein (GFP) (Addgene Plasmid #27490) (46). Platinum-E retroviral packaging cells were transfected with these vectors with Fugene 6, and retrovirus-containing supernatants were collected. IKKαKO MEFs were retrovirally transduced as previously described (35). IKKαWT and IKKαΔNBD cells were subjected to fluorescence-activated cell sorting (FACS) on the basis of GFP abundance, and sorted cells were analyzed by Western blotting to detect IKKα protein. Complementary DNAs encoding full-length NEMO, the NEMO mutant NEMO86–419, and full-length IKKβ were subcloned into the MigR1 vector, and NEMOKO MEFs were retrovirally transduced to generate stable NEMOWT, NEMO86–419, and NEMOKO-IKKβ cell lines, respectively, as described earlier. To reconstitute IKKβKO MEFs, Phoenix-ecotropic retroviral packaging cells were transfected with LZRS-EGFP, LZRS-IKKβWT, or LZRS-IKKβK44M as described previously (32). Twenty-four hours after transfection, cells were selected with puromycin (1μg/ml). Retrovirus-containing supernatants were collected and used to transduce IKKβKO MEFs. To reconstitute p65KO MEFs, a cDNA encoding full-length p65 was cloned into the pBABE retroviral vector. Phoenixecotropic retroviral packaging cells were transfected with pBABE-p65WT and selected with puromycin (1μg/ml). Retroviral supernatants were used as described earlier to transduce p65KO MEFs.
NIK overexpression
WT, NEMOKO, and p65KO MEFs were transiently transfected with the pFLAG-CMV2 NIK plasmid with a 3:2 ratio of Fugene 6 to DNA. Briefly, cells were transfected in 100-mm dishes. Twenty-four hours later, cells were harvested for immunoprecipitation with anti-FLAG (M2) beads and Western blotting analysis.
Western blotting analysis and immunoprecipitations
Cells were harvested on ice in lysis buffer consisting of 150 mM NaCl, 50 mM Tris-HCl (pH 7.5), 1% Triton X-100, and complete protease inhibitors (Roche). Proteins were resolved by SDS-PAGE and were transferred onto polyvinylidene difluoride (PVDF) membranes (Millipore). Unless otherwise noted, the Western blots presented in the figures are from one experiment and are representative of four independent experiments. PVDF membranes were developed with enhanced chemiluminescence. Immunoprecipitations were performed with 500 μg of total protein lysate, as previously described (35).
Luciferase reporter assays
Cells were transfected with 500 ng of total DNA [PBIIx kB Firefly Luciferase (FFL) and pRL TK Renilla Luciferase (RL) in a 10:1 ratio] as previously described (31). Twenty-four hours after transfection, cells were treated with TNF-α (10 ng/ml), lysed, and analyzed for dual luciferase activity as described previously (12). Mean relative luciferase units (FFL:RL) were determined from at least three independent experiments, unless otherwise noted, and P values were calculated with a student’s two-tailed t-test or one-way ANOVA in Prism software.
Preparation of nuclear and cytosolic extracts
Cells were scraped from dishes into phosphate-buffered saline (PBS) at 4°C and pelleted by centrifugation at 425g for 10 min. Cell pellets were processed for nuclear extraction as described previously (32). Lysates were analyzed by Western blotting as described earlier.
Isolation of mRNA and quantitative RT-PCR analysis
RNA was isolated with the RNase Easy Spin Kit (Qiagen) according to the manufacturer’s instructions. Samples were subjected to on-column DNase digestion. Semi-quantitative amplification of target genes was performed with Power SYBR Green (Applied Biosystems) according to the manufacturer’s instructions. PCR products were generated in quadruplicate and their abundance was normalized to that of the actb product (which encodes β-actin) with the ABI 7500 Real-Time PCR system. Relative quantitation (RQ) was derived from the difference in cycle threshold (Ct) between the gene of interest and actb with the equation RQ = 2−ΔΔCt and was analyzed with SDS v1.3 software. PCR product specificity was confirmed by performing a dissociation curve with each experiment. The map3k14 QuantiTect Primer Assay was purchased from Qiagen; the actb primer pair (5′-ACC CAC ACT GTG CCC ATC TA-3′ and 3′-ATC GGA ACC GCT CGT TGC-5′) and the birc3 primer pair (5′-CTG GCC AAA GCA GGC TTC TAC TAC-3′ and 3′-CAC GCT ACC CTT TGA CTC GTT GAC-5′) were purchased from Integrated DNA Technologies.
Protein quantification and statistical analyses
Western blots were quantified with background subtraction with the LI-COR Odyssey imaging system or ImageJ software (National Institutes of Health). For each method, the intensity of the band of interest was normalized to its corresponding control band (α-tubulin, β-actin, or histone-H3). Each value was normalized relative to the unstimulated (WT) control, and relative arbitrary units are shown. Statistical analyses of at least three independent experiments were performed with Prism software. A student’s two-tailed t-test or one-way ANOVA with Dunnett’s post-test to the WT control were performed for all data sets.
Supplementary Material
Fig. S1. Basal and anti-LTβR–induced p100 processing in IKKα- and NEMO-deficient cells.
Fig. S2. Quantification of nuclear p52 and RelB amounts and analysis of gene expression in IKK-deficient MEFs.
Fig. S3. Wild-type NEMO rescues basal p100 processing in NEMOKO cells.
Fig. S4. Classical NF-κB signaling in NEMO-reconstituted MEFs.
Fig. S5. Generation of IKKαWT and IKKαΔNBD MEF cell lines.
Fig. S6. Increased NIK abundance in IKKβ-deficient MEF cell lines.
Fig. S7. Classical NF-κB activity in reconstituted IKKβKO MEFs.
Fig. S8. Increased NIK abundance in IKKα/β DKO MEFs.
Fig. S9. Expression of birc3 and components of the basal NIK regulatory complex in various MEF cell lines.
Fig. S10. FLAG-NIK associates with the endogenous basal NIK regulatory complex in NEMOKO and p65KO MEFs.
Acknowledgments
We thank I. Verma for IKKαKO, IKKβKO, and IKKα/β DKO MEFs; M. Karin for NEMOKO and IKKβKO MEFs; C. Ware for TRAF3KO MEFs; A. Baldwin for p65KO MEFs; T. Kitamuri for Plat-E cells; S. Yamaoka for Rat1 and Rat5R fibroblasts; and W. Pear for the MigR1 retroviral vector.
Funding: M.J.M. is supported by NIH/NHLBI 1R01 HL096642. C.M.G. is supported by NIH/NIGMS T32 GM-07229 and The American Heart Association Great Rivers Predoctoral Fellowship 11PRE7540013. C.R. and E.D. are supported by a grant from the Fédération Contre le Cancer, Belgium.
Footnotes
Author contributions: C.M.G. and M.J.M. designed the experiments, analyzed the data, and wrote the manuscript; C.M.G. and C.R. performed experiments; K.A.M. and L.A.S. created cell lines; and E.D.J. and J.S.O. provided cell lines and patient samples, respectively.
Competing interests: The authors declare that they have no competing interests.
References and Notes
- 1.Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132:344–362. doi: 10.1016/j.cell.2008.01.020. published online EpubFeb 8. [DOI] [PubMed] [Google Scholar]
- 2.Hayden MS, Ghosh S. NF-kappaB in immunobiology. Cell Res. 2011;21:223–244. doi: 10.1038/cr.2011.13. published online EpubFeb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kawai T, Nishikomori R, Heike T. Diagnosis and treatment in anhidrotic ectodermal dysplasia with immunodeficiency. Allergology international: official journal of the Japanese Society of Allergology. 2012;61:207–217. doi: 10.2332/allergolint.12-RAI-0446. [DOI] [PubMed] [Google Scholar]
- 4.Solt LA, May MJ. The IkappaB kinase complex: master regulator of NF-kappaB signaling. Immunol Res. 2008;42:3–18. doi: 10.1007/s12026-008-8025-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rudolph D, Yeh WC, Wakeham A, Rudolph B, Nallainathan D, Potter J, Elia AJ, Mak TW. Severe liver degeneration and lack of NF-kappaB activation in NEMO/IKKgamma-deficient mice. Genes Dev. 2000;14:854–862. [PMC free article] [PubMed] [Google Scholar]
- 6.Zandi E, Rothwarf DM, Delhase M, Hayakawa M, Karin M. The IkappaB kinase complex (IKK) contains two kinase subunits, IKKalpha and IKKbeta, necessary for IkappaB phosphorylation and NF-kappaB activation. Cell. 1997;91:243–252. doi: 10.1016/s0092-8674(00)80406-7. [DOI] [PubMed] [Google Scholar]
- 7.Rothwarf DM, Zandi E, Natoli G, Karin M. IKK-gamma is an essential regulatory subunit of the IkappaB kinase complex. Nature. 1998;395:297–300. doi: 10.1038/26261. [DOI] [PubMed] [Google Scholar]
- 8.DiDonato JA, Hayakawa M, Rothwarf DM, Zandi E, Karin M. A cytokine-responsive IkappaB kinase that activates the transcription factor NF-kappaB. Nature. 1997;388:548–554. doi: 10.1038/41493. [DOI] [PubMed] [Google Scholar]
- 9.Li Q, Van Antwerp D, Mercurio F, Lee KF, Verma IM. Severe liver degeneration in mice lacking the IkappaB kinase 2 gene. Science. 1999;284:321–325. doi: 10.1126/science.284.5412.321. [DOI] [PubMed] [Google Scholar]
- 10.Li ZW, Chu W, Hu Y, Delhase M, Deerinck T, Ellisman M, Johnson R, Karin M. The IKKbeta subunit of IkappaB kinase (IKK) is essential for nuclear factor kappaB activation and prevention of apoptosis. J Exp Med. 1999;189:1839–1845. doi: 10.1084/jem.189.11.1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mercurio F, Murray BW, Shevchenko A, Bennett BL, Young DB, Li JW, Pascual G, Motiwala A, Zhu H, Mann M, Manning AM. IkappaB kinase (IKK)-associated protein 1, a common component of the heterogeneous IKK complex. Mol Cell Biol. 1999;19:1526–1538. doi: 10.1128/mcb.19.2.1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Solt LA, Madge LA, Orange JS, May MJ. Interleukin-1-induced NF-kappaB activation is NEMO-dependent but does not require IKKbeta. J Biol Chem. 2007;282:8724–8733. doi: 10.1074/jbc.M609613200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yamamoto Y, Verma UN, Prajapati S, Kwak YT, Gaynor RB. Histone H3 phosphorylation by IKK-alpha is critical for cytokine-induced gene expression. Nature. 2003;423:655–659. doi: 10.1038/nature01576. [DOI] [PubMed] [Google Scholar]
- 14.Anest V, Hanson JL, Cogswell PC, Steinbrecher KA, Strahl BD, Baldwin AS. A nucleosomal function for IkappaB kinase-alpha in NF-kappaB-dependent gene expression. Nature. 2003;423:659–663. doi: 10.1038/nature01648. [DOI] [PubMed] [Google Scholar]
- 15.Adli M, Merkhofer E, Cogswell P, Baldwin AS. IKKalpha and IKKbeta each function to regulate NF-kappaB activation in the TNF-induced/canonical pathway. PLoS One. 2010;5:e9428. doi: 10.1371/journal.pone.0009428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lawrence T, Bebien M, Liu GY, Nizet V, Karin M. IKKalpha limits macrophage NF-kappaB activation and contributes to the resolution of inflammation. Nature. 2005;434:1138–1143. doi: 10.1038/nature03491. [DOI] [PubMed] [Google Scholar]
- 17.Senftleben U, Cao Y, Xiao G, Greten FR, Krahn G, Bonizzi G, Chen Y, Hu Y, Fong A, Sun SC, Karin M. Activation by IKKalpha of a second, evolutionary conserved, NF-kappa B signaling pathway. Science. 2001;293:1495–1499. doi: 10.1126/science.1062677. [DOI] [PubMed] [Google Scholar]
- 18.Dejardin E, Droin NM, Delhase M, Haas E, Cao Y, Makris C, Li ZW, Karin M, Ware CF, Green DR. The lymphotoxin-beta receptor induces different patterns of gene expression via two NF-kappaB pathways. Immunity. 2002;17:525–535. doi: 10.1016/s1074-7613(02)00423-5. [DOI] [PubMed] [Google Scholar]
- 19.Sun SC. Non-canonical NF-kappaB signaling pathway. Cell Res. 2011;21:71–85. doi: 10.1038/cr.2010.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun SC. The noncanonical NF-kappaB pathway. Immunol Rev. 2012;246:125–140. doi: 10.1111/j.1600-065X.2011.01088.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Derudder E, Dejardin E, Pritchard LL, Green DR, Korner M, Baud V. RelB/p50 dimers are differentially regulated by tumor necrosis factor-alpha and lymphotoxin-beta receptor activation: critical roles for p100. J Biol Chem. 2003;278:23278–23284. doi: 10.1074/jbc.M300106200. [DOI] [PubMed] [Google Scholar]
- 22.Sun SC. Controlling the fate of NIK: a central stage in noncanonical NF-kappaB signaling. Sci Signal. 2010;3:pe18. doi: 10.1126/scisignal.3123pe18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liao G, Zhang M, Harhaj EW, Sun SC. Regulation of the NF-kappaB-inducing kinase by tumor necrosis factor receptor-associated factor 3-induced degradation. J Biol Chem. 2004;279:26243–26250. doi: 10.1074/jbc.M403286200. [DOI] [PubMed] [Google Scholar]
- 24.Vallabhapurapu S, Matsuzawa A, Zhang W, Tseng PH, Keats JJ, Wang H, Vignali DA, Bergsagel PL, Karin M. Nonredundant and complementary functions of TRAF2 and TRAF3 in a ubiquitination cascade that activates NIK-dependent alternative NF-kappaB signaling. Nat Immunol. 2008;9:1364–1370. doi: 10.1038/ni.1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zarnegar BJ, Wang Y, Mahoney DJ, Dempsey PW, Cheung HH, He J, Shiba T, Yang X, Yeh WC, Mak TW, Korneluk RG, Cheng G. Noncanonical NF-kappaB activation requires coordinated assembly of a regulatory complex of the adaptors cIAP1, cIAP2, TRAF2 and TRAF3 and the kinase NIK. Nat Immunol. 2008;9:1371–1378. doi: 10.1038/ni.1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Varfolomeev E, Blankenship JW, Wayson SM, Fedorova AV, Kayagaki N, Garg P, Zobel K, Dynek JN, Elliott LO, Wallweber HJ, Flygare JA, Fairbrother WJ, Deshayes K, Dixit VM, Vucic D. IAP antagonists induce autoubiquitination of c-IAPs, NF-kappaB activation, and TNFalpha-dependent apoptosis. Cell. 2007;131:669–681. doi: 10.1016/j.cell.2007.10.030. [DOI] [PubMed] [Google Scholar]
- 27.Razani B, Zarnegar B, Ytterberg AJ, Shiba T, Dempsey PW, Ware CF, Loo JA, Cheng G. Negative feedback in noncanonical NF-kappaB signaling modulates NIK stability through IKKalpha-mediated phosphorylation. Sci Signal. 2010;3:ra41. doi: 10.1126/scisignal.2000778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jin J, Xiao Y, Chang JH, Yu J, Hu H, Starr R, Brittain GC, Chang M, Cheng X, Sun SC. The kinase TBK1 controls IgA class switching by negatively regulating noncanonical NF-kappaB signaling. Nat Immunol. 2012;13:1101–1109. doi: 10.1038/ni.2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Orange JS, Jain A, Ballas ZK, Schneider LC, Geha RS, Bonilla FA. The presentation and natural history of immunodeficiency caused by nuclear factor kappaB essential modulator mutation. J Allergy Clin Immunol. 2004;113:725–733. doi: 10.1016/j.jaci.2004.01.762. [DOI] [PubMed] [Google Scholar]
- 30.Hanson EP, Monaco-Shawver L, Solt LA, Madge LA, Banerjee PP, May MJ, Orange JS. Hypomorphic nuclear factor-kappaB essential modulator mutation database and reconstitution system identifies phenotypic and immunologic diversity. J Allergy Clin Immunol. 2008;122:1169–1177. e1116. doi: 10.1016/j.jaci.2008.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Madge LA, Kluger MS, Orange JS, May MJ. Lymphotoxin-alpha 1 beta 2 and LIGHT induce classical and noncanonical NF-kappa B-dependent proinflammatory gene expression in vascular endothelial cells. J Immunol. 2008;180:3467–3477. doi: 10.4049/jimmunol.180.5.3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Madge LA, May MJ. Classical NF-kappaB activation negatively regulates noncanonical NF-kappaB-dependent CXCL12 expression. J Biol Chem. 2010;285:38069–38077. doi: 10.1074/jbc.M110.147207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.He KL, Ting AT. A20 inhibits tumor necrosis factor (TNF) alpha-induced apoptosis by disrupting recruitment of TRADD and RIP to the TNF receptor 1 complex in Jurkat T cells. Mol Cell Biol. 2002;22:6034–6045. doi: 10.1128/MCB.22.17.6034-6045.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xiao G, Harhaj EW, Sun SC. NF-kappaB-inducing kinase regulates the processing of NF-kappaB2 p100. Mol Cell. 2001;7:401–409. doi: 10.1016/s1097-2765(01)00187-3. [DOI] [PubMed] [Google Scholar]
- 35.Solt LA, Madge LA, May MJ. NEMO-binding domains of both IKKalpha and IKKbeta regulate IkappaB kinase complex assembly and classical NF-kappaB activation. J Biol Chem. 2009;284:27596–27608. doi: 10.1074/jbc.M109.047563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.He JQ, Zarnegar B, Oganesyan G, Saha SK, Yamazaki S, Doyle SE, Dempsey PW, Cheng G. Rescue of TRAF3-null mice by p100 NF-kappa B deficiency. J Exp Med. 2006;203:2413–2418. doi: 10.1084/jem.20061166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sasaki Y, Calado DP, Derudder E, Zhang B, Shimizu Y, Mackay F, Nishikawa S, Rajewsky K, Schmidt-Supprian M. NIK overexpression amplifies, whereas ablation of its TRAF3-binding domain replaces BAFF:BAFF-R-mediated survival signals in B cells. Proc Natl Acad Sci U S A. 2008;105:10883–10888. doi: 10.1073/pnas.0805186105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chu ZL, McKinsey TA, Liu L, Gentry JJ, Malim MH, Ballard DW. Suppression of tumor necrosis factor-induced cell death by inhibitor of apoptosis c-IAP2 is under NF-kappaB control. Proc Natl Acad Sci U S A. 1997;94:10057–10062. doi: 10.1073/pnas.94.19.10057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Orange JS, Brodeur SR, Jain A, Bonilla FA, Schneider LC, Kretschmer R, Nurko S, Rasmussen WL, Kohler JR, Gellis SE, Ferguson BM, Strominger JL, Zonana J, Ramesh N, Ballas ZK, Geha RS. Deficient natural killer cell cytotoxicity in patients with IKK-gamma/NEMO mutations. The Journal of clinical investigation. 2002;109:1501–1509. doi: 10.1172/JCI14858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Orange JS, Geha RS. Finding NEMO: genetic disorders of NF-[kappa]B activation. The Journal of clinical investigation. 2003;112:983–985. doi: 10.1172/JCI19960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Claudio E, Brown K, Park S, Wang H, Siebenlist U. BAFF-induced NEMO-independent processing of NF-kappa B2 in maturing B cells. Nat Immunol. 2002;3:958–965. doi: 10.1038/ni842. [DOI] [PubMed] [Google Scholar]
- 42.Makris C, Roberts JL, Karin M. The carboxyl-terminal region of IkappaB kinase gamma (IKKgamma) is required for full IKK activation. Mol Cell Biol. 2002;22:6573–6581. doi: 10.1128/MCB.22.18.6573-6581.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cordier F, Vinolo E, Veron M, Delepierre M, Agou F. Solution structure of NEMO zinc finger and impact of an anhidrotic ectodermal dysplasia with immunodeficiency-related point mutation. Journal of molecular biology. 2008;377:1419–1432. doi: 10.1016/j.jmb.2008.01.048. [DOI] [PubMed] [Google Scholar]
- 44.Hu H, Brittain GC, Chang JH, Puebla-Osorio N, Jin J, Zal A, Xiao Y, Cheng X, Chang M, Fu YX, Zal T, Zhu C, Sun SC. OTUD7B controls non-canonical NF-kappaB activation through deubiquitination of TRAF3. Nature. 2013;494:371–374. doi: 10.1038/nature11831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gardam S, Turner VM, Anderton H, Limaye S, Basten A, Koentgen F, Vaux DL, Silke J, Brink R. Deletion of cIAP1 and cIAP2 in murine B lymphocytes constitutively activates cell survival pathways and inactivates the germinal center response. Blood. 2011;117:4041–4051. doi: 10.1182/blood-2010-10-312793. [DOI] [PubMed] [Google Scholar]
- 46.Pear WS, Miller JP, Xu L, Pui JC, Soffer B, Quackenbush RC, Pendergast AM, Bronson R, Aster JC, Scott ML, Baltimore D. Efficient and rapid induction of a chronic myelogenous leukemia-like myeloproliferative disease in mice receiving P210 bcr/abl-transduced bone marrow. Blood. 1998;92:3780–3792. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Basal and anti-LTβR–induced p100 processing in IKKα- and NEMO-deficient cells.
Fig. S2. Quantification of nuclear p52 and RelB amounts and analysis of gene expression in IKK-deficient MEFs.
Fig. S3. Wild-type NEMO rescues basal p100 processing in NEMOKO cells.
Fig. S4. Classical NF-κB signaling in NEMO-reconstituted MEFs.
Fig. S5. Generation of IKKαWT and IKKαΔNBD MEF cell lines.
Fig. S6. Increased NIK abundance in IKKβ-deficient MEF cell lines.
Fig. S7. Classical NF-κB activity in reconstituted IKKβKO MEFs.
Fig. S8. Increased NIK abundance in IKKα/β DKO MEFs.
Fig. S9. Expression of birc3 and components of the basal NIK regulatory complex in various MEF cell lines.
Fig. S10. FLAG-NIK associates with the endogenous basal NIK regulatory complex in NEMOKO and p65KO MEFs.