

Abstract
We combined fluorogen activating protein (FAP) technology with high-throughput flow cytometry to detect real-time protein trafficking to and from the plasma membrane in living cells. The hybrid platform allows drug discovery for trafficking receptors, such as G-protein coupled receptors, receptor tyrosine kinases and ion channels, that were previously not suitable for high throughput screening by flow cytometry.. The system has been validated using the β2-adrenergic receptor (β2AR) system and extended to other GPCRs. When a chemical library containing ~1,200 off-patent drugs was screened against cells expressing FAP tagged β2AR, all known β2AR active ligands in the library were successfully identified, together with a few compounds that were later confirmed to regulate receptor internalization in a non-traditional manner. The unexpected discovery of new ligands by this approach indicates the potential of using this protocol for GPCR de-orphanization. In addition, screens of multiplexed targets promise improved efficiency with minor protocol modification.
Keywords: High throughput flow cytometer, Fluorogen activating protein, G protein coupled receptor, Receptor trafficking, Live cell assay
INTRODUCTION
Focus on the total number of druggable protein targets has increased since the completion of the human genome project.(1,2). Based on the most recent update, G-protein coupled receptors (GPCRs) are still the largest targeted family, representing 19% of all drug targets, and 34% of current drugs (3). Of the 350 or so druggable GPCRs, approximately 100 are orphans with no endogenous ligands. While the discovery of new ligands for both liganded and orphan GPCRs remains active, the apparent gap between high value drug targets in GPCR family and the number of drugs discovered so far calls for the development of new detection methods that can efficiently identify new GPCR ligands with the potential to be converted to drugs.
Common approaches to search for GPCR ligands involve fluorescent labeling of either the receptor, a known ligand or a secondary messenger such as β-arrestin. The signal change arising from the translocation of the labeled receptor or 2nd messenger is usually subtle and requires high resolution imaging or high-content screening, and is not suitable for flow cytometry measurements. Flow cytometry can be used to detect the binding of fluorescent ligand to the surface receptor, but is not compatible with deorphanization of an orphan GPCR or when the fluorescent ligand is not available. High levels of non-specific binding may also be a concern.
High throughput (HT) screening is routinely the starting point of drug discovery, and can be a rapid and efficient way to prioritize hit molecules (hits) found in the screens for drug leads. HT primary screening is often used to evaluate compound libraries consisting of hundreds of thousands to several million compounds. Hits from the primary screen may be examined with a series of single point counter screens and dose response screens to validate high priority hits via analysis of structural activity relationships (SAR). A typical work flow to identify high potential drug candidates in an academic environment (such as in our facility at University of New Mexico Center for Molecular Discovery) can be found in Figure 1. Assays suitable for HT screening generally require the assay: 1) to be robust with stable results: 2) to be miniaturized to several micro liters per sample; 3) to provide adequate signal to noise ratio; 4) and to have minimal processing steps for easy automation. Particle or suspension cell based assays can benefit more from HTFC, while adherent cell based assays are mostly performed with a plate reader because these require additional step(s) for cell detachment and resuspension.
Figure 1.

Typical High Throughput screening workflow in UNM Center for Molecular Discovery (UNMCMD).
We described a novel assay compatible with HTFC employing the newly available fluorogen activating protein (FAP) biosensor for the real-time measurement of protein trafficking to and from the cell plasma membrane. Fluorogens are a special group of small molecules that fluoresce on binding to their specific FAP binding partner. Depending on the fluorogen-FAP binding pair, the fluorescent signal can increase up to 105 fold (Figure 2A), with excitation/emission spectra similar to commercial available fluorophores such as FITC and APC (http://www.mbic.cmu.edu/materials.html). Fluorescent signals from FAP bound fluorogen can be easily detected by conventional flow cytometry or fluorescent microscopy, and the high binding specificity ensures that the background signal is low. The FAP tag can be fused to a non-reactive domain of the target protein, GPCR or other sensory proteins, and stably expressed in a cell line such as the U937 cells described here (4,5). The assay described is designed to monitor the real-time trafficking of proteins to and from the plasma membrane by measuring receptor expression on the cell surface by flow cytometry. The experimental design is critical for assays suitable for flow cytometric measurement, and assays intended to identify compounds that induce or prevent surface receptor endocytosis, or yield intracellular protein extocytosis. An example of the design to measure the amount of surface expressing receptors after agonist treatment by flow cytometry is illustrated in Figure 2B.
Figure 2.

Principle of FAP biosensor (A) and experimental design of flow cytometry assays to measure ISO induced internalization of surface AM2.2-β2AR. Both the FAP and fluorogen remain dark until binding occurs. Cell membrane permeable and non-permeable versions of fluorogens are available, and by choosing the cell membrane impermeable fluorogen TO1-2p, we are capable of designing an experiment to measure only the cell surface expressing AM2.2-GPCR as indicated in Figure 2B.
We describe two basic protocols aimed at the discovery of compounds that induce receptor internalization, and prevent ligand induced receptor internalization, respectively. The protocols are specifically miniaturized for high-throughput flow cytometry. The design of primary/single point counter screens is listed in basic protocols, and the design for dose response screens is listed in support protocols.
BASIC PROTOCOL 1: High-throughput flow cytometry measurement of agonist induced GPCR internalization
We describe an assay combining the recently available FAP based biosensor with high-throughput flow cytometry specifically designed for this purpose. The hybrid platform takes advantages of the high sensitivity and low background features of the FAP biosensor integrated with quantitative flow cytometry that is suitable for multiplexing. Together they provide throughput up to 20,000 compounds per HT flow cytometer (HTFC) per 8 hr work day in 384 well plate format. In this specific protocol, we chose β2AR as our target protein. As one of the most studied GPCRs, β2AR is the target of 47 approved small molecule drugs and continues to be actively studied. Traditionally, β2AR ligands have been widely used in the treatment of respiratory disease and cardiovascular diseases. β2AR was recently found to be directly associated with cancer, cancer metastasis, and neurological diseases such as Parkinson’s and Alzheimer’s. These newly discovered clinical needs call for the design of novel therapeutic drugs including non-canonical ligands of β2AR with biased signaling pathways.
For these studies, a FAP tag, AM2.2, is fused to the extracellular, non-functional N-terminus of the β2AR. Upon binding to fluorogen Thiazole Orange derivatives (TO1-2p), the presence of AM2.2 can be detected using the FL1 (FITC) channel of a flow cytometer. The AM2.2-β2AR fusion protein is stably expressed in U937 cell lines, and the function of the receptor has been validated (6). Surface expression of β2AR and most GPCRs can be modulated by two known groups of molecules: receptor agonists that induce the internalization of the surface receptor, and antagonists that prevent agonist induced receptor internalization. A well-known β2AR agonist that induces β2AR internalization is isopreterenol (ISO), and has been chosen to be the positive control in this protocol. The protocol aims to identify molecules that induce receptor internalization regardless of their signaling pathways. The HTFC readout from a single 384 well plate screen is presented in Figure 3 where fluorescent signal from FL1 channel is plotted against time. The inset expands results from the first row of the screen. As shown in the figure, the fluorescent signals from sample wells are separated by air bubbles, and the signal from positive control wells (the 1st and 23rd bin in the insert) is lower than that from negative control wells (2nd and 24th bin in the insert) and most sample wells.
Figure 3.
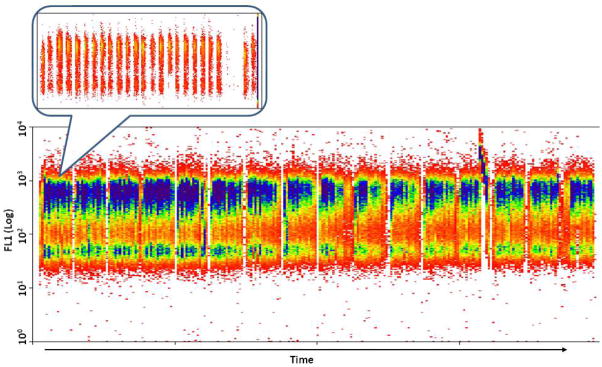
Screen shot of the read out form a single plate prepared following Basic Protocol 1. The fluorescent signal from the FL1 channel is plotted vs. time. The inset shows the time resolved fluorescent signal from the first 24 wells containing samples (cells). The fluorescent signals from individual wells are recorded with unique time stamps and are separated by air bubbles. The data is de-convoluted based on the time stamps and the order of samples read (in the order of consecutive rows, consecutive columns or other format), and mapping it to the data table containing compound information from each well. The first two bins in the magnified image represent signal from cells from PCntrl and NCntrl wells A1 and A2. The following 20 bins represent signal from cells exposed to 20 different compounds from wells A3 to A22 of the compound plate. As indicated in Basic Protocol 1, the bigger gap following bin 22 indicates “signal” from sample-free wells A23 and A24.
Materials
U937 cells stably transfected with AM2.2-β2AR are used as a model system. The cells are cultured in RPMI full media (see Reagents and Solutions section for recipe of the media) before harvesting.
RPMI 1640 media (RPMI, Life Technologies); isoproterenol (ISO, Sigma-Aldrich, MO); Prestwick Chemical Library (PCL) in 384 well-format; shallow well 384 well plates (Greiner Bio-One, Germany); Breathe-Easy sealing membrane (Research Products International Corp. IL); BD Falcon 70 μm cell strainers (Becton-Dickinson, NJ); FITC MESF beads (Becton-Dickinson, NJ)
Forma Series II water jacketed CO2 incubator (Thermo Fisher, MA); Eppendorf MixMate plate shaker; Biotek Multiflow liquid dispenser (Multiflow, BioTek Instruments, VT); Biomek FXP or NXP laboratory automation work station equipped with a pin tool for compound transfer transfer (FX or NX, Beckman Coulter, CA); CyanADP flow cytometer (Beckman Coulter, CA) or Accuri C6 flow cytometer (Becton-Dickinson, NJ); HyperCyt® autosampler (IntelliCyt, NM); centrifuge; hemocytometer
Protocol steps
Prepare 650 nM fluorogen TO1-2p from 5 mM stock in RPMI 1640 media and store the solution in 4°C refrigerator until time of use. The diluted TO1-2p solution can be refrigerated for at least a month with no apparent loss of affinity and fluorescent intensity.
Carefully count the cells using a hemocytometer or similar cell counting device.
Calculate the amount of cells needed for harvest, plus 10% extra.
In a typical screening day, assay plates are prepared in batches of 40. For each batch of plates, spin down 3×108 AM2.2-β2AR cells, discard supernatant, re-suspend in 60 mL of fresh RPMI 1640 medium, and store in 37°C incubator until time of experiment. The cells are filtered through a 70 μm cell strainer before dispensing them into assay plates to remove any cell clusters and debris. The cell density will be 5×106 cells/mL, and the left over cells from each batch can be stored in a 37°C incubator and used by the end of the day.
Prepare 5 mL of 32 μM isopreterenol (ISO) from 20 mM ISO stock (dissolved in DMSO) in RPMI media immediately before making each batch of assay plates. Do not reuse the thawed ISO stock or the 32 μM ISO.
Add 5 μL RPMI to columns 2 to 24 of each assay plate by Biotek Multiflow or a similar liquid dispenser of choice.
Add 5 μL of freshly prepared 32 μM ISO to Column 1 of the plates as, in-plate positive controls (PCntrls) by Multiflow.
Transfer 100 nL of library compounds (typical compound concentration for single point experiment is 1 mM) from library compound plates to assay plates by FX or NX. Library compounds are located at columns 3–22 of the compound plates, cells in column 2 are not exposed to compounds or ISO, and are considered in-plate negative controls (NCntrls).
Add 3 μL of cells as described in step 3 to Columns 1 – 22 of the assay plates by Multiflow, columns 23 and 24 are left without cells intentionally for smoother data analysis.
Cover the plates with breathable plate covers, shake the plates, and keep them in 37°C incubator for 90 minutes before transferring them to 4°C refrigerator.
The plates are refrigerated for 30 minutes, and 3 μL 650 nM TO1-2p are added to all wells of the assay plates by Multiflow.
Compound treated cells are exposed to TO1-2p for 30 minutes at 4°C, and the plates are read by high-throughput flow cytometry (CyanADP or Accuri C6 flow cytometer connected with the HyperCyt platform). Cells are excited with a 488 nm laser; front scatter, side scatter, time stamp and the fluorescent signal collected using a 530±20 nm bandpass filter are recorded.
Data is analyzed using the in-house HyperView, (developed by Dr. Bruce Edwards from the University of New Mexico), and is available from IntelliCyt (Albuquerque, NM). This software automatically resolves data clusters, analyzes each bin to determine mean or median channel forward scatter, side scatter, and fluorescence intensity (MCF), and number of gated events from each well.
Step annotations
The assay described uses β2AR as a model system. It can be generalized to the discovery of compounds that induce the internalization of any surface GPCRs (6). The system has already been adapted, targeting chemokine receptors CCR5 (7), CXCR4, adrenergic receptors α1BAR and α2BAR, and orphan GPCR GPR32. It is common for a small population of the FAP-GPCR transfected cells to have no expression of the receptors on the surface. However, these cells are still carrying the gene for the receptor, and the expression will recover over time, but the gene has been silenced for reasons unknown at the time of measurement. Therefore, no action (such as sorting) needs to be taken unless > 50% of the cells appeared to lose their receptor expression or the signal to noise ratio does not meet the experimental requirements. Please see Figure 4 for the fluorescent distribution from a normal batch of cells and a batch of cells that requires sorting.
Figure 4.
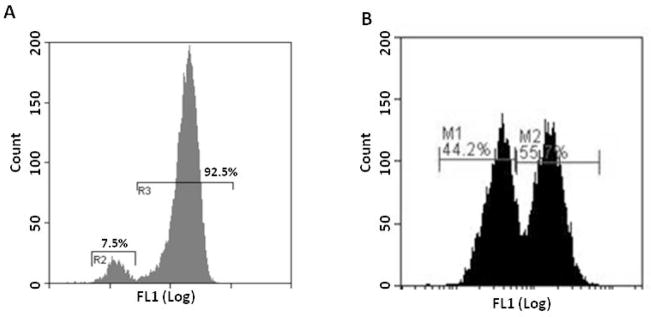
Histogram of a batch of normal β2AR cells (A) and β2AR cells requiring sorting (B). Notice the small proportion of the cells that appear to have no surface expression of FAP-β2AR cells in R2 of Figure 4A. This phenomenon is commonly observed in all FAP-GPCR cells we have worked with and will not affect the cell function/experimental result. However, the cells will require sorting if a significant proportion of the cells appear to lose surface expression as indicated in Figure 4B.
Cells stably transfected with FAP tagged receptors are sensitive to the tissue culture environment, and one needs to optimize tissue culture conditions for each cell line before assay development. AM2.2-β2AR cells required to be maintained at a cell density of less than 500,000/mL at all times to minimize the number of cells with the silenced FAP-β2AR genes.
FCS files from HTFC are converted to ASCII data, the time stamps and air gaps assist the separation of samples on the plate. Fluorescent intensity and standard deviation obtained from PCntrl and NCntrl wells are used to determine the quality of the assay (Z′), and set the baseline to determine hits (positives) from the library. Where Z′ is determined by the mean fluorescent intensities and standard deviatioσ) of the signal from PCntrl and NCntrls as described below in Equation 1(8):
| (1) |
It is widely accepted in the HTS community that any HT assay with a Z′ of 0.5 or higher is of exceptional quality that provides reliable data output although a lower Z′ score may also be acceptable according to the nature of the assay. As indicated in the equation, a higher Z′ score can be achieved by increasing the separation between PCntrl and NCntrl signals or minimizing the standard deviation of the controls, especially the later. The assay described in the protocol is robust with small variations, and a signal to background ratio (or MeanNCntrl/MeanPCntl in this case) of ~ 2:1 or higher is usually sufficient to achieve the Z′ requirement for high quality HTS assays.
SUPPORT PROTOCOL 1 (optional): High throughput dose response screen for β2AR agonists
High-throughput screening has been widely used in the drug discovery field, and often involves a single point, primary screen followed by several high-throughput counter screens and 2nd screens. One of the standard screens to validate the “hit” molecules from the primary screen is the dose response screen. The purpose of this screen is to validate the acitvity of these compounds with the target protein (affinity for β2AR, in our case), and determine high priority hits from the results. The assay employs the same materials and instruments as used in the primary screen, but the compound plates are set up in a different format.
Materials
ICI 118, 551 (ICI, Sigma-Aldrich, MO); Please refer to the Materials described in Basic Protocol 1 for other required materials and instrumentations.
Protocol steps
Steps 1 – 5 are the same as described in Basic Protocol 1.
-
6
Add 5 μL RPMI to Columns 1 to 10, 12 to 22 and 24 of each assay plate by Biotek Multiflow or a similar liquid dispenser of choice.
-
7
Add 5 μL of freshly prepared 32 μM ISO to Column 11 and 23 of the plates as, in-plate positive controls by Multiflow.
-
8
Transfer 100 nL of compounds (typical compound concentrations range from 1.5 μM to 10 mM) from compound dose response plates to assay plates by FX or NX. Hit compounds are located at columns 2 to 10, and 14 to 22 of the compound plates, with the highest concentration located at columns 10 and 22, and the lowest at columns 2 and 14. The compounds are subsequently diluted 3 fold from the highest concentration (10 mM in our case). Cells in columns 1 and 13 are not exposed to compounds or ISO, and are considered in-plate negative controls.
-
9
Add 3μL of cells as described in step 3 to columns 1 to 11, and 13 to 23 of the assay plates by Multiflow. Columns 12 and 24 are left without cells intentionally for smoother data analysis.
Steps 10 to 13 are the same as described in Basic Protocol 1.
Step annotations
BASIC PROTOCOL 2 (optional): High-throughput flow cytometry measurement of antagonist stabilized surface GPCR
Another important feature of the recently developed FAP based HTFC assay is to identify antagonists of the surface receptor, or, compounds that prevent agonist induced internalization of the surface GPCR. Both agonists and antagonists of GPCRs may serve a profound role in drug discovery. Beta adrenergic receptor agonists are widely used in the treatment of asthma, allergy, chronic obstructive pulmonary disease and hypotension. Beta-blockers or antagonists have anti-hypertension and anti-arrhythmic effects. Some well known drugs that are GPCR antagonists include: Maraviroc (9)(CCR5), Cozaar (10) (Angiotensin receptor) and Zantac (11) (Histamine receptor). Recently, beta-blockers were also found to be associated with the prevention of cancer metastasis (12). Using AM2.2-β2AR as our model system, we developed a HT assay that specifically targets the antagonists of the receptor.
Materials
Please refer to the Materials described in Basic Protocol 2 for the required materials and instrumentations.
Protocol steps
Steps 1 – 4 are the same as described in Basic Protocol 1.
-
5
Prepare 30 μM of ICI 118, 551 (ICI) in RPMI media from stock ICI. The diluted form of ICI can be stored at 4°C and used until the end of the day. Prepare 60 mL of 3 μM ISO from 20 mM ISO stock (dissolved in DMSO) in RPMI media immediately before making each batch of assay plates. Do not reuse the thawed ISO stock or the 3 μM ISO.
-
6
Add 3 μL RPMI to Columns 2–24 of each assay plate by Biotek Multiflow or a similar liquid dispenser of choice.
-
7
Add 3 μL of freshly prepared 30 μM ICI to column 1 of the plates as in-plate positive controls by Multiflow.
-
8
Transfer 100 nL of library compounds (typical compound concentration for single point experiment is 1 mM) from library compound plates to assay plates by FX or NX. Library compounds are located at columns 3–22 of the compound plates.
-
9
Add 3 μL of cells as described in step 3 to columns 1 – 22 of the assay plates by Multiflow. Columns 23 and 24 are left without cells intentionally for smoother data analysis.
-
10
Add 3 μL of freshly prepared 3 μM ISO as described in step 5 to all wells. Cells in column 2 are exposed only to 1 μM ISO, and are considered in-plate negative controls. Cells in other columns of the plates are exposed to both the library compounds (ICI in column 1 as PCntrls) and 1 μM ISO.
Steps 11 to 14 are the same as described in steps 10 to 13 in Basic Protocol 1.
Step annotations
SUPPORT PROTOCOL 2 (optional): High throughput dose response screen for β2AR antagonists
High-throughput screening has been widely used in the drug discovery field, and often involves a single point, primary screen followed by several high-throughput counter screens and 2nd screens. One of the standard screens to validate the “hit” molecules from primary screen is to perform a dose response screen. The purpose of this screen is to confirm the activity of these compounds with the target protein (affinity for β2AR, in our case), and determine high priority hits from the results. The assay employs the same materials and instruments as used in the primary screen, but the compound plates are set up in a different format.
Materials
Please refer to the Materials described in Basic Protocol 2 for the required materials and instrumentations.
Protocol steps
Steps 1 – 5 are the same as described in Basic Protocol 2.
-
6
Add 3 μL RPMI to columns 1 to 10, 12 to 22 and 24 of each assay plate by Biotek Multiflow or a similar liquid dispenser of choice.
-
7
Add 3 μL of freshly prepared 30 μM ICI to column 11 and 23 of the plates as, in-plate positive controls by Multiflow.
-
8
Transfer 100 nL of compounds (typical compound concentrations range from 1.5 μM to 10 mM) from compound dose response plates to assay plates by FX or NX. Hit compounds are located at columns 2 to 10, and 14 to 22 of the compound plates, with the highest concentration located at columns 10 and 22, and the lowest at columns 2 and 14. The compounds are subsequently diluted 3 fold from the highest concentration (10 mM in our case). Cells in columns 1 and 13 do not expose to compounds or ISO, and are considered in-plate negative controls.
-
9
Add 3 μL of cells to columns 1 to 11, and 13 to 23 of the assay plates by Multiflow. Columns 12 and 24 are left without cells intentionally for smoother data analysis.
-
10
Add 3 μL of freshly prepared 3 μM ISO to wells 1 to 11 and 13 to 23. Cells in column 1 and 22 are exposed only to 1 μM ISO, and are considered in-plate negative controls. Cells in other columns of the plates are exposed to both the library compounds (except for ICI in columns 11 and 23 as PCntrls) and 1 μM ISO.
Steps 11 to 14 are the same as described in steps 10 to 13 in Basic Protocol 1.
REAGENTS AND SOLUTIONS
RPMI full media for tissue culture (stored in 4°C refrigerator): RPMI 1640, 10% heat inactivated fetal bovine serum, 100 units/mL penicillin, 100 μg/mL streptomycin, 10 mM HEPES, pH 7.4, 20 μg/mL ciprofloxacin, 2 mM L-glutamine.
COMMENTARY
Background Information
The FAP based technology was first reported by Szent-Gyorgyi et. al. from Carnegie Mellon University in 2008 (13), followed by the FAP tagged receptor trafficking monitored by confocol microscopic imaging (4,14). We then published the combined platform of the FAP biosensors and high throughput flow cytometry (6,7), and this protocol expands on those published experimental procedure sections.
Critical Parameters
FAP tagged protein transfected cells are extremely sensitive to environmental conditions and require evaluation of optimal growth conditions of each new cell line expressing the tagged receptors. If cells appear to lose expression of the FAP tagged receptors, spin down the cells and resuspend them in fresh RPMI 1640 full media (RPMI 1640 with 10% fetal bovine serum, 100 mM HPSM buffer and 25 mM L-glutamine) at a cell density of 100,000/mL to 200,000/mL and check for the recovery of cell surface receptor expression before sorting.
Tolerance levels of DMSO vary from cell line to cell line. The cell line used in the present protocol, U937 cells, function normally in the presence of ~ 1% DMSO or less. Perform a DMSO tolerance test to ensure the cells function in the presence of DMSO or other solvents that the library compounds are dissolved in.
The threshold determination to filter out hits from the screen is based on a general guideline that hits fall outside of 3 times the standard deviation from the baseline. The threshold should be tailored for each assay to ensure there are not too many or too few compounds are chosen to enter the next step of the screen. A practical approach is to include ~0.5% – 1% of compounds when screening a large, non-focused library. The percentage should be increased accordingly if screening a focused library or the size of the library is relatively small (e.g. 103 of compounds in compare with 105 compounds or more). The benefit to emphasis on minimizing false negatives at the expense of increasing the amount of false positives is because false positives can be easily removed in the following screens but false negatives will be lost forever after the primary screen.
Troubleshooting
Anticipated Results
The assay is robust with high sensitivity. When the surface expression of AM2.2-β2AR is within acceptable ranges, one should expect to see consistent results from PCntrl and NCntrl wells with minimal variation and have the capability to identify compounds that induce or prevent surface β2AR internalization.
Time Considerations
The preparation time for tissue culture, cell counting, and reagent solution will be ~ 30 mins. Cell harvesting/re-suspension, and ISO preparation will be 15 min (repeated every 4 hrs). The estimated rate of assay plate preparation and stamping will be 20 plates per hour. The throughput of a single HTFC is 1 plate per 10–12 mins. An assay plate should be ready for HTFC analysis ~ 3 hrs after media dispensing (Step 6 in Basic Protocol 1), and can be stored in a 4°C refrigerator for up to 4 hrs. Because the assay measures signaling in live cells, we do not recommend keeping the plates in the refrigerator for longer than 4 hrs.
Acknowledgments
This research was supported by US National Institute of Health 1U54MH084690-02 (LAS), 5U54MH084690-03 CDP2 (YW and LAS), and 5U54RR022241 (ASW).
Footnotes
- http://nmmlsc.health.unm.edu/ This is the homepage of UNMCMD, with detailed description of the specialties and facilities of the center. One can also find other HTFC assays performed in the center in addition to the one reported herein.
- http://www.mbic.cmu.edu/materials.html This website provides detail information regarding fluorogens Thiazole Orange (TO) and Malachite Green (MG) derivatives, as well as the FAP available in the center.
LITERATURE CITED
- 1.Overington JP, Al-Lazikani B, Hopkins AL. Opinion - How many drug targets are there? Nature Reviews Drug Discovery. 2006;5:993–996. doi: 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- 2.Hopkins AL, Groom CR. The druggable genome. Nature Reviews Drug Discovery. 2002;1:727–730. doi: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- 3.Rask-Andersen M, Almen MS, Schioth HB. Trends in the exploitation of novel drug targets. Nature Reviews Drug Discovery. 2011;10:579–590. doi: 10.1038/nrd3478. [DOI] [PubMed] [Google Scholar]
- 4.Fisher GW, Adler SA, Fuhrman MH, Waggoner AS, Bruchez MP, Jarvik JW. Detection and Quantification of beta 2AR Internalization in Living Cells Using FAP-Based Biosensor Technology. Journal of Biomolecular Screening. 2010;15:703–709. doi: 10.1177/1087057110370892. [DOI] [PubMed] [Google Scholar]
- 5.Jarvik JW, Fisher GW, Shi C, Hennen L, Hauser C, Adler S, Berget PB. In vivo functional proteomics: Mammalian genome annotation using CD-tagging. Biotechniques. 2002;33:852–866. doi: 10.2144/02334rr02. [DOI] [PubMed] [Google Scholar]
- 6.Wu Y, Tapia PH, Fisher GW, Simons PC, Strouse JJ, Foutz T, Waggoner AS, Jarvik JW, Sklar LA. Discovery of Regulators of Receptor Internalization by High Throughput Flow Cytometry. Molecular Pharmacology. 2012;82:645–657. doi: 10.1124/mol.112.079897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu Y, Tapia PH, Fisher GW, Waggoner AS, Jarvik J, Sklar LA. High-throughput flow cytometry compatible biosensor based on fluorogen activating protein technology. Cytometry Part A. 2013;83A:220–226. doi: 10.1002/cyto.a.22242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang JH, Chung TDY, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. Journal of Biomolecular Screening. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 9.Dickinson L, Khoo S, Back D. Pharmacokinetics and drug-drug interactions of antiretrovirals: An update. Antiviral Research. 2010;85:176–189. doi: 10.1016/j.antiviral.2009.07.017. [DOI] [PubMed] [Google Scholar]
- 10.Habashi JP, Judge DP, Holm TM, Cohn RD, Loeys BL, Cooper TK, Myers L, Klein EC, Liu G, Calvi C. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science. 2006;312:117–121. doi: 10.1126/science.1124287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cook D, Guyatt G, Marshall J, Leasa D, Fuller H, Hall R, Peters S, Rutledge F, Griffith L, McLellan A. A comparison of sucralfate and ranitidine for the prevention of upper gastrointestinal bleeding in patients requiring mechanical ventilation. New England Journal of Medicine. 1998;338:791–797. doi: 10.1056/NEJM199803193381203. [DOI] [PubMed] [Google Scholar]
- 12.Cole SW, Sood AK. Molecular Pathways: Beta-Adrenergic Signaling in Cancer. Clinical Cancer Research. 2012;18:1201–1206. doi: 10.1158/1078-0432.CCR-11-0641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Szent-Gyorgyi C, Schmidt BA, Creeger Y, Fisher GW, Zakel KL, Adler S, Fitzpatrick JAJ, Woolford CA, Yan Q, Vasilev KV, et al. Fluorogen-activating single-chain antibodies for imaging cell surface proteins. Nature Biotechnology. 2008;26:235–240. doi: 10.1038/nbt1368. [DOI] [PubMed] [Google Scholar]
- 14.Holleran J, Brown D, Fuhrman MH, Adler SA, Fisher GW, Jarvik JW. Fluorogen-Activating Proteins as Biosensors of Cell-Surface Proteins in Living Cells. Cytometry Part A. 2010;77A:776–782. doi: 10.1002/cyto.a.20925. [DOI] [PMC free article] [PubMed] [Google Scholar]