

Abstract
Depressed heart rate variability in severe inflammatory diseases can be partially explained by the lipopolysaccharide (LPS)-dependent modulation of cardiac pacemaker channels. Recently, we showed that LPS inhibits pacemaker current in sinoatrial node cells and in HEK293 cells expressing cloned pacemaker channels, respectively. The present study was designed to verify whether this inhibition involves LPS-dependent intracellular signalling and to identify structures of LPS responsible for pacemaker current modulation. We examined the effect of LPS on the activity of human hyperpolarization-activated cyclic nucleotide-gated channel 2 (hHCN2) stably expressed in HEK293 cells. In whole-cell recordings, bath application of LPS decreased pacemaker current (IhHCN2) amplitude. The same protocol had no effect on channel activity in cell-attached patch recordings, in which channels are protected from the LPS-containing bath solution. This demonstrates that LPS must interact directly with or close to the channel protein. After cleavage of LPS into lipid A and the polysaccharide chain, neither of them alone impaired IhHCN2, which suggests that modulation of channel activity critically depends on the integrity of the entire LPS molecule. We furthermore showed that β-cyclodextrin interfered with LPS-dependent channel modulation predominantly via scavenging of lipid A, thereby abrogating the capability of LPS to intercalate into target cell membranes. We conclude that LPS impairs IhHCN2 by a local mechanism that is restricted to the vicinity of the channels. Furthermore, intercalation of lipid A into target cell membranes is a prerequisite for the inhibition that is suggested to depend on the direct interaction of the LPS polysaccharide chain with cardiac pacemaker channels.
Introduction
In severe sepsis and multi-organ dysfunction syndrome, the regulation of cardiac function is impaired. In addition to a reduction in contractility, the cardiac cycle is more regular. This depression in heart rate variability (HRV) is a strong predictor of an unfavourable prognosis in patients with systemic inflammation (Werdan et al. 2009). Although this is of high clinical relevance, the mechanisms underlying the impairment in HRV are incompletely understood.
Experimental evidence suggests lipopolysaccharide (LPS) is involved. LPS (also called endotoxin) is liberated from the outer walls of Gram-negative bacteria in patients suffering from severe sepsis. Injection of LPS decreased HRV in healthy human volunteers (Godin et al. 1996). Furthermore, LPS was shown to modulate the sympathetic–vagal balance that strongly affects HRV (Huang et al. 2010) and reduces the responsiveness of sinoatrial node (SAN) cells to vagal stimuli (Gholami et al. 2012). These effects predominantly impair the autonomous regulation of cardiac pacemakers. Additionally, LPS may interfere with the mechanism of pacemaking itself because the application of LPS was found to reduce beating rate variability in cultured neonatal heart cells (Schmidt et al. 2007). Native pacemaker current If is generated by ion flux through hyperpolarization-activated cyclic nucleotide-gated (HCN) channels (Biel et al. 2009). In mammalian cardiac cells four HCN channels (HCN1–4) are expressed (Herrmann et al. 2011). HCN1, HCN2 and HCN4 are expressed in the cardiac conduction pathway, whereas HCN3 is expressed in ventricular myocytes that provide a background conductance that regulates the kinetics of repolarization (Fenske et al. 2011). Patch-clamp experiments with human atrial myocytes as well as HL-1 cells have demonstrated the activity of f-channels to be reduced by LPS (Zorn-Pauly et al. 2007; Wondergem et al. 2010).
Very recently, we conducted a study to determine the structural parts of LPS essential to modulate the activity of heterologously expressed human HCN2 channels (hHCN2) (Klöckner et al. 2011). Amphiphilic LPS is composed of three structural regions: the O-specific polysaccharide (O-chain), which acts as a serological determinant of various bacteria; the central core, and a lipid component named lipid A, which has been identified as the LPS component possessing the inflammatory capacity responsible for LPS-induced biological effects (Raetz, 1990; Rietschel et al. 1994).
We observed that only LPS types containing an intact O-chain inhibit hHCN2-mediated current (IhHCN2) amplitude. This inhibition of channel activity takes place within a few seconds and is caused by a reduction in maximal conductance and a shift in the channel open probability curve to more negative voltages. Furthermore, the deactivation kinetics of IhHCN2 activity are accelerated. By contrast, proinflammatory LPS-types lacking the O-chain did not reduce IhHCN2 or modulate its kinetic attributes. These results strongly suggest that the O-chain is essential for reduction of IhHCN2 by LPS and that pacemaker channels may act as a target for endotoxin. Similarly to the heterologous expression system, impairment of native If by LPS in isolated SAN cells critically depended on the presence of the O-chain (Klöckner et al. 2011).
The present study was conducted to address the question of whether LPS-induced modulation of hHCN2 channel activity depends on a relatively close and direct interaction or whether a distant and indirect interaction via intracellular signalling cascades is involved. Additionally, we aimed to further characterize the structural requirements for this channel modulation by LPS. Our results suggest that LPS affects IhHCN2 by a mechanism that is spatially restricted to the vicinity of the channels. Furthermore, we provide evidence that only the integral LPS molecule consisting of intact lipid A and the polysaccharide chain can modulate IhHCN2. β-cyclodextrin (βCD) blocked LPS-dependent hHCN2 channel modulation by scavenging the lipid A moiety of LPS. Therefore, we conclude that the intercalation of LPS into the target cell membrane via acyl chains of lipid A is a prerequisite for the subsequent interaction of the polysaccharide chain with extracellular structures of the pacemaker channel protein.
Methods
Stably transfected HEK293 cells
The stable cell line expressing hHCN2 was a gift from Dr Stieber (Stieber et al. 2005). Cells were cultured in Dulbecco's modified Eagle's medium (DMEM)/Ham's F12 medium (Biochrom AG, Berlin, Germany) supplemented with 200 μg ml−1 geneticin (Life Technologies, Inc., Carlsbad, CA, USA), 2 mm l-glutamine and 10% fetal bovine serum (FCS; Biochrom AG, Berlin, Germany), at 37°C and 5% CO2.
Electrophysiological recordings and data analysis
Currents were recorded from transfected cells with the whole-cell or cell-attached patch-clamp technique using an Axopatch 200 patch-clamp amplifier (Axon Instruments, Inc., Burlingame, CA, USA). Patch pipettes were pulled from thick wall borosilicate glass (Hilgenberg GmbH, Malsfeld, Germany). Electrical resistances of fire-polished electrodes were 1–2 MΩ for cell-attached patch-clamp recordings and 3–5 MΩ for whole-cell voltage-clamp recordings, respectively. The pipette solution for whole-cell voltage-clamp recordings consisted of 140 mm KCl, 5 mm Na2ATP, 6 mm MgCl2, 1 mm EGTA and 10 mm Hepes/KOH (pH 7.2). The bath solution for whole-cell recordings contained 110 mm NaCl, 30 mm KCl, 1.8 mm CaCl2, 0.5 mm MgCl2, 10 mm glucose and 10 mm Hepes/NaOH (pH 7.4). In order to zero the membrane potential during cell-attached patch-clamp recordings, a high potassium bath solution was used containing 140 mm KCl, 1 mm MgCl2, 1.8 mm CaCl2, 5 mm glucose and 10 mm Hepes/KOH (pH 7.4). The pipette solution for cell-attached patch-clamp recordings was comprised of the same composition as the bath solution used in the whole-cell voltage-clamp experiments. To allow fast LPS application in cell-attached recordings, a second patch pipette (∼50 μm in diameter) was placed near the investigated cell. Flow could be switched on and off using pinch valves.
For whole-cell voltage-clamp recordings, the input capacitance of HEK293 cells was estimated by integrating the capacitive current at the end of a 100 ms long voltage step from −60 mV to −50 mV (ISO-2; MFK, Niedernhausen, Germany). Series resistances between 6 MΩ and 10 MΩ were measured before maximal compensation (>95%). Currents were low-pass filtered at 1 kHz with an eight-pole Bessel filter built in the amplifier and sampled at 5 kHz. All measurements were conducted at room temperature (20–24°C) and not corrected for liquid junction potentials.
The steady state voltage-dependence of IhHCN2 activation was determined from tail currents at −80 mV following 8 s long test pulses ranging from −50 mV to −150 mV in −10 mV steps (holding potential of −40 mV). Plots of tail current amplitude versus membrane potential were fitted using a least-squares method (Origin 8.0; MicroCal LLC, Northampton, MA, USA) to a Boltzmann distribution given by Itail(V) = A2 + (A1 − A2)/{1 + exp[(V − V0.5)/s]}, where A1 is the maximal amplitude, A2 is the current offset, V is the hyperpolarizing pulse potential, V0.5 is the half-maximal activation voltage and s is the slope factor. Boltzmann fits and tail current amplitudes were normalized with respect to the maximal tail current amplitude A1–A2.
The dose–response relationship for the shift in V0.5 (ΔV0.5) as a function of LPS was obtained by paired comparison experiments. Activation curves were constructed from averaged leakage-corrected current traces (n = 3 or n = 4) recorded before and 4 min after bath application of LPS. Data were fit in SigmaPlot using the hyperbolic function: ΔV0.5 = ΔV0.5max · [LPS]/(KD + [LPS]), where ΔV0.5max is the maximal shift in V0.5 and KD is the LPS concentration required to achieve 50% of the maximum shift in V0.5.
Deglycosylation
In order to verify a role of HCN channel glycosylation in LPS-dependent modulation of channel function, we treated hHCN2-expressing HEK293 cells with peptide N-glycosidase F (PNGase F; New England BioLabs, Inc., Ipswich, MA, USA). Cells were washed with bath solution and incubated with PNGase F (1000 U ml−1 in bath solution) at 37°C for 1 h (Noma et al. 2009).
Degradation of LPS
Lipopolysaccharide was degraded by mild acid hydrolysis. LPS (5 mg ml−1) was hydrolysed with 1% (v/v) acetic acid at 100°C for 90 min according to Leone et al. (2006). This standard procedure, originally established by Freeman & Anderson (1941), cleaves the acid labile ketosidic linkage between the polysaccharide moiety and lipid A, leaving the polysaccharide chain intact. The insoluble lipid A was separated from the polysaccharide component of LPS by centrifugation (15,000 g, 10 min). The pH level was adjusted to 7.4 (NaOH) and the aqueous solution containing the polysaccharide moiety used for the experiments. The fraction containing lipid A was prepared as stock solution in dimethyl sulphoxide (DMSO). Before use, lipid A stock solution was dissolved in physiological salt solution (PSS). The final concentration of the solvent had no effect on mean V0.5 (control: −92.3 ± 3.0 mV, n = 4; 0.5% DMSO: −91.3 ± 1.0 mV, n = 4; P = 0.8).
For control purposes, untreated and hydrolysed LPS samples were subjected to a 12% SDS-PAGE and visualized by silver staining as described elsewhere (Nesterenko et al. 1994). Electromobility of the LPS molecule critically depends on lipid A moiety, whereas silver staining exclusively detects the polysaccharide chains (Jann et al. 1975; Tsai & Frasch, 1982). Therefore, only intact LPS can be separated and visualized by this method.
Cholesterol depletion
For cholesterol depletion, cells were incubated with 10 mm βCD. As prolonged exposure of HEK293 cells to 5–10 mm βCD causes the cells to detach from the culture dish and subsequently results in cell death (Barbuti et al. 2004), we limited the preincubation time of cells to 60–120 min (37°C, serum-free DMEM or bath solution).
In order to verify a physical interaction of βCD with LPS, we used a βCD polymer (βCD monomers cross-linked by epichlorohydrin; sc-296103; Santa Cruz Biotechnology, Inc., Dallas, TX, USA). This polymer is insoluble and hence forms sediments in aqueous solutions.
Incubations were carried out in the bath solution used in current recordings. FITC-labelled LPS (50 μg ml−1) was incubated with βCD polymer (24 mm; calculated from the molecular weight fraction of the monomers) at room temperature for 3 h. Thereafter, the supernatant was collected and the sediment of the βCD polymer was washed three times with bath solution before FITC fluorescence was measured in a microplate reader. Using this approach, we were able to show a reduction of fluorescence in the supernatant by 36%. Concomitantly, FITC-LPS fluorescence appeared in the βCD polymer sediment, whereas the βCD polymer itself is non-fluorescent. As the βCD polymer has a crystalline structure, presentation of βCD molecules is restricted to the polymer surface and hence is rather limited. This might have prevented a more complete removal of FITC-LPS from the supernatant. Nevertheless, our data indicate that βCD and LPS physically interact with one another.
Reagents
Unless otherwise noted, all reagents and LPS preparations used in this study were purchased from Sigma-Aldrich Chemie GmbH (Deisenhofen, Germany). LPS from Escherichia coli (O111:B4) was either purified by gel-filtration chromatography (L3012) or isolated by phenol extraction (L2630). A synthetic lipid A was purchased from PeptaNova GmbH (Sandhausen, Germany) and dissolved in 0.1% triethylamine to make a 2 mg ml−1 stock solution.
Statistical analysis
The data are presented as the mean ± s.e.m. Statistical comparisons were carried out using Student's t test. Differences were considered significant at P < 0.05. To minimize dispersion of data, mostly day-matched cells were used.
Results
In our previous study, we showed that the application of LPS reduces the activity of hHCN2 channels heterologously expressed in HEK293 cells. This modulation involves an attenuation of the maximal conductance, a shift in the activation curve to more negative potentials and an acceleration of the deactivation time course of IhHCN2.
Assuming that all aspects of LPS-dependent hHCN2 channel modulation are mediated by the same mechanism of interaction and to facilitate comparison of our data with those in the literature, in the present study we used the shift of the half-maximal activation potential (V0.5) as a readout for the effect of LPS on IhHCN2. This parameter has proved to be most robust in experiments concerning the effect of LPS on pacemaker channel activity (Zorn-Pauly et al. 2007; Klöckner et al. 2011). Initially using paired experiments, we established a dose–response curve for the LPS-induced shift in V0.5. Averaged ΔV0.5 values (n = 3 or n = 4) were fitted with a hyperbolic function using a maximal ΔV0.5 of −26.5 mV and a KD value of 6.3 μg ml−1.
In order to elicit maximal responses, we used 50 μg ml−1 LPS throughout this study.
No evidence for the involvement of LPS-induced cytosolic second messengers in the effect of LPS on IhHCN2
Figure 1A shows the inhibitory effect of LPS on IhHCN2, recorded in the whole-cell configuration of the patch-clamp technique. To differentiate between direct and indirect long-distance interactions between LPS and hHCN2 channels, similar experiments were performed in the cell-attached configuration (Fig. 1B). Here, the investigated hHCN2 channels are located within the patch pipette and are therefore protected from a direct interaction with LPS that was added to bath solution. However, in this configuration, modulation of hHCN2 channel activity via LPS-induced intracellular signalling cascades would still be possible. As it has been demonstrated previously that LPS reduced the amplitude of IhHCN2 with a time constant of ∼8 s (Klöckner et al. 2011), cells were exposed for at least 20 s to LPS before the amplitude of IhHCN2 was estimated. In the absence of LPS, the amplitude of IhHCN2 at the end of the 8 s long hyperpolarizing voltage step amounted to –87 pA, whereas after application of 50 μg ml−1 LPS to the same cell an inward current with an amplitude of –92 pA was recorded. We observed IhHCN2 with mean amplitudes of −41.0 ± 10.5 pA (n = 8) and −42.5 ± 10.6 pA (n = 8) under the control condition and after application of LPS, respectively (P = 0.22).
Figure 1.

A, whole-cell configuration. Application of 50 μg ml−1 LPS decreased mean whole-cell current IhHCN2 at a test potential of −100 mV to about 65% (n = 4 each). B, cell-attached recordings (macro-patch) with LPS applied via the bath solution. The patch electrode protects the channels under investigation from direct interaction with LPS. However, LPS remained able to induce receptor-mediated intracellular signalling, thereby modulating channel activity. Representative IhHCN2 traces were recorded in the absence of LPS (control) and after application of 50 μg ml−1 LPS. Mean current amplitude of IhHCN2 did not differ between control and LPS treatments (n = 8 each). C, cell-attached recordings with LPS applied via the pipette solution. Action of LPS is spatially restricted to the vicinity of the channels under investigation. This experimental configuration does not allow for paired experiments. Therefore, activation curves were recorded with or without 50 μg ml−1 LPS included in the pipette solution (n = 3 each). Potential of half-maximal activation (V0.5) is shifted by LPS to more negative potentials. *P < 0.05 versus control.
Figure 1C illustrates further cell-attached experiments. However, by contrast with the experiments described earlier, here LPS is applied via the electrode solution inside the patch pipette. Similarly to the whole-cell experiments, the channels under investigation are accessible to the applied LPS. In this patch-clamp configuration paired recordings of IhHCN2 before and after application of LPS are impossible and unpaired measurements would suffer from large variations as a result of the differences in expression levels of hHCN2 channels between individual cells. To circumvent these difficulties, we recorded activation curves. We found that 50 μg ml−1 LPS shifted the activation curve to more negative potentials as indicated by the significantly more negative V0.5 (control: −83.4 ± 1.9 mV, n = 3; LPS: −103.0 ± 1.4 mV, n = 3; P < 0.05). Thus, LPS must act in close proximity to the channel protein in order to modulate IhHCN2.
Role of channel glycosylation in LPS-mediated modulation of IhHCN2
The state of glycosylation has been reported to modulate channel function (Watanabe et al. 2007; Veldhuis et al. 2012; Weiss et al. 2013). Therefore, we proved the necessity of channel glycosylation for LPS-dependent modulation of IhHCN2. The results of these experiments are depicted in Fig. 2A. Treatment of cells with PNGase F did not modify V0.5 of IhHCN2 (control: −84.7 ± 0.9 mV, n = 7; PNGase F: −84.9 ± 0.9 mV, n = 7). Additionally, deglycosylation did not prevent an LPS-dependent shift in V0.5 (PNGase F + LPS: −116.9 ± 1.4 mV, n = 7; P < 0.05 versus PNGase F alone). Moreover, this shift is larger than that induced by LPS application without preceding PNGase F treatment (LPS: −105.9 ± 1.9 mV, n = 7; P < 0.05 versus PNGase F + LPS). This indicates that channel glycosylation is not a prerequisite for LPS-dependent modulation. Channel deglycosylation was examined using immunoblotting (Fig. 2B).
Figure 2.
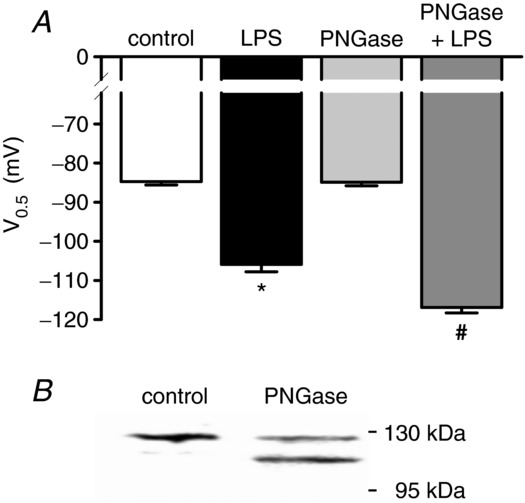
A, effect of channel deglycosylation on LPS-dependent modulation of IhHCN2. Cells were left untreated (control) or were incubated with peptide N-glycosidase F (PNGase F, 1000 U ml−1, 37°C, 1 h). Mean V0.5 recorded under control conditions (open bar, n = 7), in the presence of 50 μg ml−1 LPS (black bar, n = 7), after incubation of cells with PNGase F (light grey bar, n = 7) and in the presence of 50 μg ml−1 LPS after incubation with PNGase F (dark grey bar, n = 7). LPS-dependent modulation of IhHCN2 is not attenuated by channel deglycosylation. Rather, it sensitizes channels to LPS-dependent modulation because the shift in V0.5 is augmented after PNGase F treatment of cells. B, a representative Western blot of total lysates from control and PNGase F-treated hHCN2-expressing HEK293 cells. The intensity of the upper band is reduced, whereas a lower band appears by PNGase F treatment, indicating channel deglycosylation. *P < 0.05 versus control; #P < 0.05 versus PNGase F; §P < 0.05 versus LPS.
Impact of isolated polysaccharide chains or lipid A on IhHCN2
In previous experiments, we showed that mutant LPS types lacking an O-chain did not affect the amplitude of IhHCN2 (Klöckner et al. 2011). This suggests a crucial role for the O-antigen of LPS in the modulation of IhHCN2. To test this hypothesis more directly, we degraded LPS into lipid A and the polysaccharide moiety by mild acidic hydrolysis (Fig. 3A). The efficacy of degradation was controlled by SDS-PAGE and silver staining (Fig. 3B). Compared to an equal amount of untreated LPS, hydrolysis reduced the intensity of silver staining to only 10%, indicating that 90% of LPS was degraded.
Figure 3.
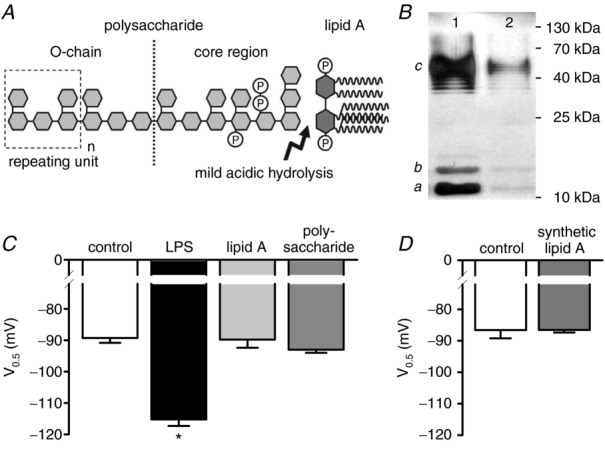
A, schematic chemical structure of lipopolysaccharide (LPS) (according to Rietschel et al. 1994). The arrow indicates the chemical bond, which is cleaved by mild acetic acid hydrolysis, resulting in a separation of the polysaccharide moiety containing the O-chain from the hydrophobic lipid A. B, 7.5 μg of untreated (lane 1) or hydrolysed (lane 2) LPS were subjected to 12% SDS-PAGE and silver-stained. The typical staining pattern of S-type LPS is characterized by intermediates of the full-length LPS representing lipid A with the core oligosaccharide only (a), lipid A and core with one repeating unit (b), and lipid A and core with different numbers of the repeating unit (c) (according to Palva & Makela, 1980). Because only intact LPS can be separated and visualized by this method, the reduction in staining intensity indicates 90% hydrolysis of LPS. C, mean V0.5 recorded under control conditions (open bar, n = 11), in the presence of 50 μg ml−1 LPS (black bar, n = 11) and in the presence of degradation products derived from 50 μg ml−1 LPS exposed to mild acidic hydrolysis (lipid A: light grey bar, n = 10; polysaccharide: dark grey bar, n = 10). Only the intact LPS molecule affects V0.5 of IhHCN2. D, compared to control (open bar, n = 6), V0.5 of IhHCN2 is not modulated by 10 μg ml−1 synthetic lipid A (grey bar, n = 10). *P < 0.05 versus control.
Figure 3C depicts the effect of intact LPS and its isolated degradation products on V0.5 of IhHCN2. Compared to control (−89.4 ± 1.6 mV, n = 9), uncleaved LPS significantly shifted mean V0.5 (−115.2 ± 2.1 mV, n = 7; P < 10−7). By contrast, neither the isolated polysaccharide chains (−93.0 ± 1.0 mV, n = 10; P = 0.07) nor the isolated lipid A (−89.8 ± 2.6 mV, n = 6; P = 0.87) affected mean V0.5. Similarly, 10 μg ml−1 of synthetic proinflammatory lipid A had no modulatory effect on V0.5 of hHCN2 channels (control: −86.6 ± 2.6 mV, n = 6; synthetic lipid A: −86.6 ± 0.8 mV, n = 10) (Fig. 3D). These data demonstrate that neither isolated polysaccharide chains nor isolated lipid A modulate IhHCN2.
βCD directly blocks the capability of LPS to impair IhHCN2
As the localization of pacemaker channels in lipid rafts has been reported to regulate their biophysical properties (Barbuti et al. 2004), we sought to evaluate the importance of these specialized membrane domains for LPS-dependent modulation of hHCN2 channels. Therefore, we disrupted lipid rafts by cholesterol depletion using βCD. Figure 4 shows current traces recorded in the presence of either 10 mm βCD (Fig. 4A) or 10 mm βCD and 50 μg ml−1 LPS (Fig. 4B). As depicted in Fig. 4C, without βCD, incubation with LPS (60–120 min) shifted the mean V0.5 of IhHCN2 from −86.0 ± 0.3 mV (control, n = 15) to −104.1 ± 1.1 mV (n = 8). Incubation with βCD alone (60–120 min) induced a shift of V0.5 to less negative potentials (−73.2 ± 0.4 mV, n = 10) as expected from the literature (Barbuti et al. 2004). However, incubation of cells with LPS in the presence of βCD did not further modify mean V0.5 (−74.0 ± 0.6 mV, n = 13). This seems to suggest that the presence of lipid rafts is a prerequisite for LPS-dependent modulation of hHCN2 channels.
Figure 4.
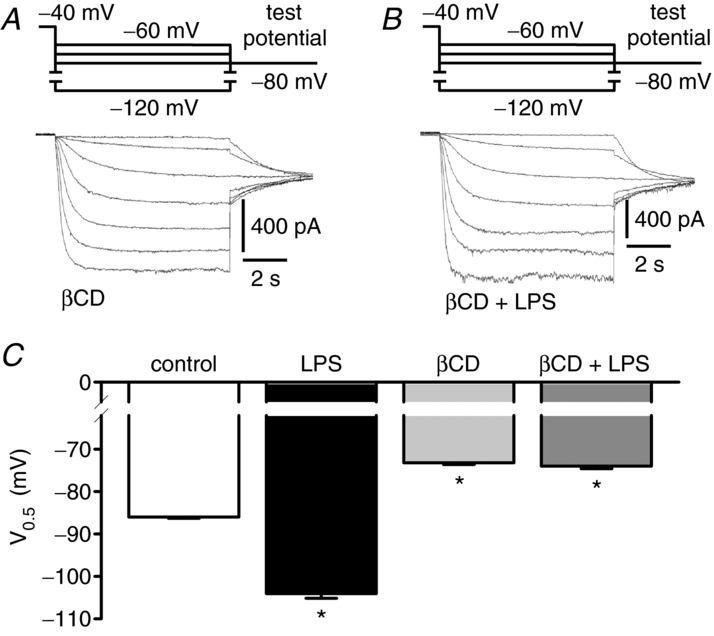
A, current traces recorded after incubation of cells with 10 mm βCD (90 min). B, current traces obtained after co-incubation of cells with 10 mm βCD and 50 μg ml−1 LPS (90 min). C, mean V0.5 recorded under control conditions (open bar, n = 15), in the presence of 50 μg ml−1 LPS (black bar, n = 8), after incubation of cells with 10 mm βCD (60–120 min, light grey bar, n = 10) and after co-incubation of cells with 10 mm βCD and 50 μg ml−1 LPS (60–120 min, dark grey bar, n = 13). βCD shifts V0.5 of IhHCN2 to less negative potentials. However, in the presence of βCD, LPS does not modulate IhHCN2. *P < 0.05 versus control.
This conclusion was challenged by a recent report showing that βCD may act as a scavenger for endotoxin (Sakata et al. 2011). To examine the interplay between βCD, LPS and cells in more detail, we conducted two different sets of experiments.
Firstly, we re-evaluated the effect of LPS on IhHCN2 in cholesterol-depleted cells using a slightly different protocol. Again, cholesterol depletion by βCD (60–120 min) caused a shift in mean V0.5 from −87.6 ± 1.1 mV (n = 6) to −75.9 ± 1.4 mV (n = 9) (Fig. 5A, C). By contrast with the experiments reported above, βCD-containing bath solution was then exchanged for normal bath solution. About 1 h later, mean V0.5 remained at less negative potentials (−77.2 ± 1.0 mV, n = 7), indicating that after washout of βCD, the replenishment of membranes with cholesterol takes considerable time. This time delay enabled us to examine the effects of LPS on IhHCN2 in cholesterol-depleted cells but in the absence of βCD. Under these conditions LPS significantly shifted mean V0.5 to −91.1 ± 0.9 mV (n = 9) (Fig. 5B, C).
Figure 5.
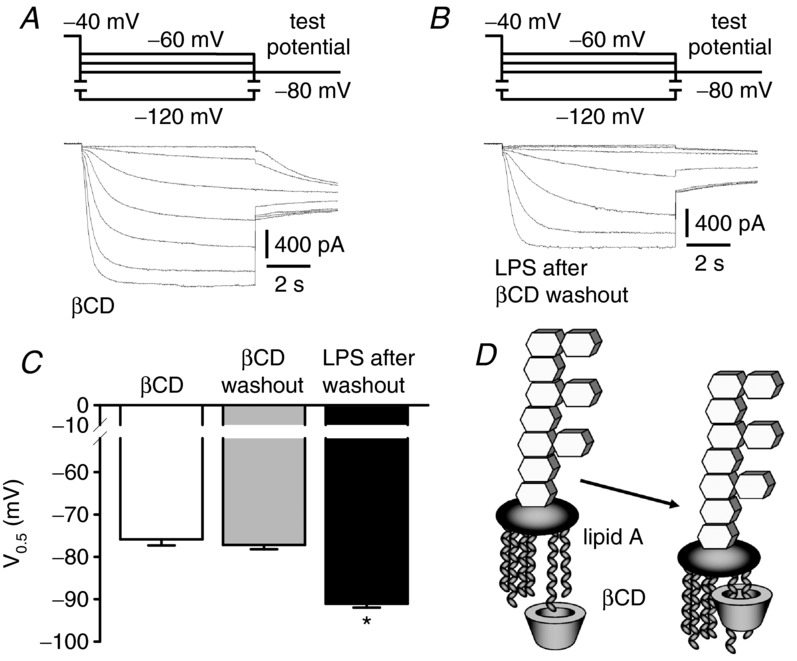
A, current traces recorded in the presence of 10 mm βCD. B, current traces recorded in the presence of 50 μg ml−1 LPS, 60 min after washout of βCD. C, mean V0.5 obtained in the presence of 10 mm βCD (white bar, n = 9), 60 min after washout of βCD (grey bar, n = 7) and in the presence of 50 μg ml−1 LPS, 60 min after washout of βCD (black bar, n = 9). The effect of βCD on V0.5 of IhHCN2 is still present 60 min after βCD washout, indicating that cell membranes are still cholesterol-depleted. The LPS-dependent shift of V0.5 to more negative potentials is reduced but still detectable after βCD is washed out. D, schematic diagram of the interaction of the acyl chains of lipid A with the hydrophobic cavity of βCD (according to Sakata et al. 2011). *P < 0.05 versus βCD washout.
Secondly, we tried to verify the scavenging effect of βCD on LPS. We co-incubated 50 μg ml−1 LPS and 10 mm βCD in bath solution (90 min). Subsequently we exposed cells to this βCD-pretreated LPS. During the first 30 min of incubation, mean V0.5 (−87.8 ± 2.6 mV, n = 4) did not differ from the value obtained with control cells (−87.6 ± 1.1 mV, n = 6). After prolonged exposure (60–120 min), mean V0.5 was shifted to −77.8 ± 1.4 mV (n = 9), a value indistinguishable from that obtained with βCD alone (−75.9 ± 1.4 mV, n = 9; P = 0.37). However, the typical LPS-induced shift of V0.5 to more negative potentials could not be detected with βCD-pretreated LPS.
These data suggest that LPS-dependent modulation of hHCN2 channels does not depend on the integrity of lipid rafts or the localization of hHCN2 channels in these cholesterol-rich membrane domains. Rather, βCD interferes with LPS-dependent channel modulation by its capability to scavenge LPS putatively via interaction with the acyl chains of the lipid A moiety (Fig. 5D).
Discussion
Accessibility of hHCN2 membrane microdomains is required for LPS-induced inhibition of IhHCN2
In addition to regulation by cAMP, HCN channels are known to be regulated by additional factors including phosphorylation, interacting proteins and low molecular weight factors (Biel et al. 2009). Given the large number of putatively involved signalling cascades, we first wanted to establish whether signalling cascades were involved at all. Therefore, we chose the cell-attached mode of patch-clamp technique. It allows for the analysis of ion channels that are physically protected by the patch electrode from bath-applied agents, thus preventing direct interaction. However, the modulation of f-channel activity via intracellular signalling cascades and diffusible second messengers has been demonstrated in a cell-attached configuration (DiFrancesco, 1986). Therefore, modulation of hHCN2 channel activity via LPS-induced intracellular signalling cascades will not be compromised in cell-attached recordings. In these experiments, LPS did not modulate IhHCN2 when it was applied via the bath solution, but did affect channel activity when it was applied via the electrode solution. Therefore, we conclude that LPS must directly interact with the channel protein in order to impair activity. The short time constant for LPS-dependent channel blocking of ∼8 s may reflect such a direct mechanism (Klöckner et al. 2011).
Channel glycosylation is not a prerequisite for LPS-induced inhibition of IhHCN2
The state of channel glycosylation is reported to modulate the activity of a variety of ion channels, including potassium channels, transient receptor potential (TRP) channels and T-type calcium channels (Watanabe et al. 2007; Veldhuis et al. 2012; Weiss et al. 2013). In addition, HCN channels are affected by alterations in glycosylation. Mutation of N-glycosylation sites was shown to almost completely prevent surface expression of HCN2 channels, whereas surface expression of HCN1 channels remained almost unchanged (Hegle et al. 2010).
We wondered whether modulation of IhHCN2 by LPS also critically depended on channel glycosylation. We used PNGase F to deglycosylate hHCN2 channels. As a result, a new protein band approximately 20 kDa smaller appeared in the Western blot. A similar difference in molecular weight between glycosylated and unglycosylated HCN2 channels was reported by Much et al. (2003) and by Hegle et al. (2010). However, channel deglycosylation did not change V0.5 of IhHCN2. This is in line with the report by Hegle et al. (2010) on the effect of glycosylation on HCN1 channel function. Moreover, deglycosylation did not prevent the shift of V0.5 induced by subsequent application of LPS. Rather, it sensitized HCN channels to LPS because the shift in V0.5 was more pronounced after PNGase treatment of the cells. Putatively, the removal of negative charges from the channel surface by deglycosylation promotes the interaction of negatively charged LPS with the channel protein. We conclude that the N-linked glycosylation of hHCN2 channels is not a prerequisite for LPS-dependent modulation.
Only intact LPS affects IhHCN2
In our previous study, we identified the O-chain of LPS as a critical component responsible for HCN2 channel modulation (Klöckner et al. 2011). Here, we tried to further substantiate this crucial role of polysaccharide moiety. Therefore, we degraded LPS by mild acid hydrolysis, which cleaves the 2-keto 3-deoxy-octulosonate bond linking lipid A to the core polysaccharide (Morrison & Ulevitch, 1978).
Surprisingly, the isolated polysaccharide had no impact on the activity of IhHCN2, which suggests that polysaccharide moiety is necessary but not sufficient to modulate pacemaker activity. This conclusion is further supported by our experiments using βCD-pretreated LPS. It is proposed that βCD may shield the lipid A moiety of LPS by interaction with its fatty acids (Sakata et al. 2011). Regardless of the resultant block of lipid A function, LPS-dependent inhibition of IhHCN2 should still be possible if the polysaccharide chains were sufficient to mediate this effect of LPS. However, no hHCN2 channel modulation was detected using βCD-pretreated LPS.
Additionally, neither isolated lipid A after hydrolysis nor synthetic lipid A modulated hHCN2 channel activity. Therefore, we conclude that the polysaccharide chains and lipid A moiety can inhibit IhHCN2 only as integral parts of the whole LPS molecule. Similarly, LPS-dependent inhibition of low-density lipoprotein (LDL) uptake into hepatoblastoma (HepG2) cells has been shown to critically depend on the integrity of the LPS molecule (Liao & Floren, 1992).
LPS-dependent modulation of IhHCN2 and lipid rafts
Lipid rafts are specialized dynamic membrane microdomains enriched in cholesterol and sphingomyelin. These lipid rafts may form flask-like membrane invaginations, the caveolae, with the help of additional structural proteins (i.e. caveolins) (Allen et al. 2007).
Caveolin-3 has been shown to regulate cardiac pacemaker channels and heterologously expressed HCN4 channels (Ye et al. 2008). In addition, Barbuti et al. (2012) showed that loss of interaction between caveolin-1 and heterologously expressed HCN4 channels reduces surface expression of the channels and, more importantly, shifts the curve of channel activation to less negative potentials. As LPS shifts V0.5 in the opposite direction, it is rather unlikely that LPS modulates HCN channel function via the attenuation of caveolin–HCN channel interaction.
However, even localization of ion channels within lipid rafts may modulate their function (Strain et al. 1983; Allen et al. 2007). Indeed, this has been shown for native pacemaker channels in SAN cells, as well as HCN4 channels expressed in HEK293 cells (Barbuti et al. 2004).
We used βCD to assess the roles of these membrane microdomains in the LPS-dependent modulation of IhHCN2. Cyclodextrins are water-soluble oligosaccharides which form a hydrophobic cavity that can accommodate hydrophobic molecules (Stella & He, 2008). βCD, which has the highest affinity to form inclusion complexes with cholesterol (Zidovetzki & Levitan, 2007), can interfere with lipid raft integrity and function by removing cholesterol from the plasma membrane (Ohtani et al. 1989). In the present study, βCD shifted the activation curve of hHCN2 channels to less negative potentials. A similar modulation of channel gating was demonstrated for methyl-βCD-treated cells expressing native pacemaker channels or recombinant HCN4 channels (Barbuti et al. 2004). These alterations in biophysical properties would translate into an augmented activation of pacemaker channels at physiological membrane potentials. This is in agreement with the general observation that an increase in the cholesterol concentration of plasma membranes leads to a rigidification of the membrane and a decrease in the activity of ion channels and vice versa (Levitan et al. 2010).
Regardless of this basic activating effect of βCD on hHCN2 channel activity, we sought to verify the role of lipid rafts in LPS-dependent channel inhibition. Therefore, we co-incubated cells with βCD and LPS. In the presence of βCD LPS was no longer able to shift the activation curve to more negative potentials. Assuming that βCD is a specific tool to manipulate lipid rafts, this result may suggest that the integrity of lipid rafts is a prerequisite for the inhibitory effect of LPS on hHCN2 channel activity. However, recently it has been reported that cyclodextrins remove endotoxin from DNA solutions (Sakata et al. 2011). It was proposed that the fatty acids of lipid A may be partially adsorbed into the hydrophobic cavity of βCD. Thus, LPS scavenging by βCD would provide an alternative explanation for the lack of LPS-dependent channel modulation in our co-incubation experiments.
To elucidate the attenuation of LPS-dependent channel modulation by βCD, we modified our experimental set-up. Now, after cholesterol depletion of membranes, βCD was washed off the cells before LPS incubation was started. Again, βCD alone shifted the activation curve to less negative potentials. This effect was stable for at least 1 h after washout of βCD, indicating that the cell membranes were still depleted of cholesterol. Within this period, the LPS-dependent inhibition of hHCN2 channel activity could be readily detected, although to a lesser extent. This demonstrates that the integrity of lipid rafts is not an absolute prerequisite for the modulation of hHCN2 channel activity by LPS.
Intercalation of LPS into membranes is critical for modulation of IhHCN2
Lipopolysaccharide stimulates target cells via the activation of toll-like receptor 4 (TLR4) (Mitchell et al. 2007). However, HEK293 cells do not express TLR4 endogenously (Medvedev & Vogel, 2003). Furthermore, TLR4 can be activated by lipid A or LPS variants lacking the O-chain, whereas these agents did not modulate IhHCN2 in HEK293 or SAN cells in our previous study (Klöckner et al. 2011). In conjunction with the data from our cell-attached IhHCN2 recordings, which argue against an intracellular signal transduction, these findings indicate that it is unlikely that LPS mediates its effect on IhHCN2 via TLR4.
Alternatively, intercalation of LPS molecules into the plasma membrane has been shown to involve a different mechanism of cell activation by LPS (Schromm et al. 1996; Gutsmann et al. 2007; Mitchell et al. 2007; Brandenburg et al. 2010). Intercalation of LPS into membranes via hydrophobic interactions can take place even in the absence of known binding partners of LPS (i.e. CD14 or LPS-binding protein) (Alam & Yamazaki, 2011). Therefore, we suggest that LPS must intercalate into the cell membrane via lipid A in order to modulate IhHCN2. Co-incubation of cells with LPS and βCD completely attenuates this critical step because βCD neutralizes LPS via βCD-dependent scavenging of the acyl chains of lipid A (Sakata et al. 2011). Interestingly, intercalation of LPS is not prevented but reduced after cholesterol depletion of membranes using cyclodextrin (Ciesielski et al. 2012). In agreement with this finding, LPS sequentially applied to βCD actually modulated IhHCN2, although to a lesser extent.
Linking f-channel inhibition with HRV
In severe sepsis and multi-organ dysfunction syndrome, HRV is reduced (Werdan et al. 2009). Experimental evidence suggests LPS is involved (Godin et al. 1996). We hypothesized that this LPS-dependent reduction in HRV could be at least partially mediated by an inhibition of f-channels.
Is it reasonable to relate a reduced activity of f-channels to a reduced HRV (i.e. in sepsis)? To answer this question, one might consider data from HCN knockout models. Ludwig et al. (2003) reported an increased HRV in HCN2 knockout mice. Concerning HCN4, Herrmann et al. (2007) showed that in mice harbouring a global knockout, sinus pauses occur that increase HRV, whereas in mice with a cardiac-specific knockout, HRV is unchanged (Baruscotti et al. 2011). These results of HCN knockout models might argue in favour of a negative correlation between HCN channel activity and HRV. However, HCN knockout models may not adequately reflect the situation in sepsis for two reasons. Firstly, by contrast with the acute effect of LPS on HCN channels, knockout models alter the activity of HCN channels on a much larger time scale, providing the opportunity for counter-regulatory or compensatory processes either on the level of sinus node cells or on the level of the autonomic nervous system. Secondly, knockout models affect only a single HCN isoform. As native f-channels represent hetero-tetramers of different isoforms, a single isoform knockout changes the composition and concurrently the properties of the remaining channels. This may have effects that differ strikingly from the reduction in overall HCN channel activity (i.e. by LPS during sepsis) or selective HCN channel blockers. Therefore, pharmacological inhibition of all HCN channels using selective blockers seems to be a better reference concerning putative modulation of HRV by LPS-mediated HCN inhibition. Using such an approach, Khaykin et al. (1998) and Joannides et al. (2006) demonstrated a reduction in HRV in healthy volunteers after infusion of the selective HCN channel inhibitors zatebradine and ivabradine, respectively. Additionally, after blockade of the autonomic nervous system, infusion of ivabradine reduced HRV in conscious rats (Mangin et al. 1998). These data at least open the possibility that HRV can be reduced as a result of LPS-mediated HCN channel inhibition.
In summary, we demonstrate that LPS inhibits cardiac pacemaker channel activity by a mechanism different from that responsible for its proinflammatory properties. In our recombinant expression system, intracellular signalling cascades seem not to contribute to the impairment of IhHCN2 by LPS. We propose that intercalation of the acyl chains of lipid A into target cell membranes is the first critical step. This lipid A-dependent anchorage near the channel protein now allows the direct interaction of the polysaccharide chains with extracellular hHCN2 channel structures, which, in turn, modulate channel activity. Conclusively, the integrity of the LPS molecule is a prerequisite for LPS-dependent channel modulation.
Lipopolysaccharide is known to affect the function of cardiac pacemakers via modulation of the autonomic nervous system, which provides an explanation for the depression of HRV in severe sepsis (Godin et al. 1996). However, the direct effect of LPS on pacemaker channel activity described in the present study represents a new mechanism that putatively contributes to this hallmark of severe inflammatory disease.
Key points
The regulation of cardiac function is seriously impaired in severe inflammatory diseases. One characteristic of this dysfunction is a strong reduction in heart rate variability (HRV) so that the cardiac cycle is more regular. This phenomenon is strongly correlated with an unfavourable prognosis in patients with systemic inflammation.
Although the depression in HRV can be partially explained by the interplay between cardiac pacemaker channels and lipopolysaccharide (LPS) liberated from the outer walls of Gram-negative bacteria, the underlying mechanism is still elusive.
Using HEK293 cells stably expressing a cardiac pacemaker channel, we demonstrate that only intact LPS molecules can intercalate into target cell membranes and then directly interact with extracellular parts of pacemaker channels. Intracellular signalling cascades do not contribute to LPS-dependent channel modulation.
The present results help to elucidate how LPS interacts with pacemaker channels to attenuate the regularity of the cardiac cycle.
Acknowledgments
The authors thank Dr J. Stieber for kindly supplying stable HEK293 cell lines expressing hHCN2.
Glossary
- βCD
β-cyclodextrin
- CD14
cluster of differentiation 14
- ΔV0.5
shift of V0.5
- ΔV0.5max
maximal shift of V0.5
- DMEM
Dulbecco's modified Eagle's medium
- HCN
hyperpolarization-activated cyclic nucleotide-gated channel
- hHCN2
human hyperpolarization-activated cyclic nucleotide-gated channel 2
- HRV
heart rate variability
- IhHCN2
hHCN2-mediated current
- LPS
lipopolysaccharide
- PNGase F
peptide N-glycosidase F
- SAN
sinoatrial node
- TLR4
toll-like receptor 4
- V0.5
half-maximal activation voltage
Additional information
Competing interests
None declared.
Author contributions
All experiments were performed at the Julius-Bernstein Institut für Physiologie, Martin-Luther Universität Halle-Wittenberg, Halle, Germany. Authors contributed to the study as follows: study concept and design of experiments: U.K., U.R., M.G., C.G. and U.M.-W.; data collection and analysis: U.K., S.M. and K.S.; interpretation of data: U.K., S.M., U.R., M.G. and C.G.; drafting of the manuscript: U.K., U.R. and M.G.; critical revision of the manuscript: C.G., H.E., U.M.-W., H.L. and K.W. All authors approved the final version of the manuscript.
Funding
This work was supported by grants provided by the Deutsche Forschungsgemeinschaft (DFG) (grants GE 905/18-1 and MU 1010/4-1) and the Roux-Program of the Medical Faculty of the University of Halle-Wittenberg.
References
- Alam JM, Yamazaki M. Spontaneous insertion of lipopolysaccharide into lipid membranes from aqueous solution. Chem Phys Lipids. 2011;164:166–174. doi: 10.1016/j.chemphyslip.2010.12.008. [DOI] [PubMed] [Google Scholar]
- Allen JA, Halverson-Tamboli RA, Rasenick MM. Lipid raft microdomains and neurotransmitter signalling. Nat Rev Neurosci. 2007;8:128–140. doi: 10.1038/nrn2059. [DOI] [PubMed] [Google Scholar]
- Barbuti A, Gravante B, Riolfo M, Milanesi R, Terragni B, DiFrancesco D. Localization of pacemaker channels in lipid rafts regulates channel kinetics. Circ Res. 2004;94:1325–1331. doi: 10.1161/01.RES.0000127621.54132.AE. [DOI] [PubMed] [Google Scholar]
- Barbuti A, Scavone A, Mazzocchi N, Terragni B, Baruscotti M, DiFrancesco D. A caveolin-binding domain in the HCN4 channels mediates functional interaction with caveolin proteins. J Mol Cell Cardiol. 2012;53:187–195. doi: 10.1016/j.yjmcc.2012.05.013. [DOI] [PubMed] [Google Scholar]
- Baruscotti M, Bucchi A, Viscomi C, Mandelli G, Consalez G, Gnecchi-Rusconi T, Montano N, Casali KR, Micheloni S, Barbuti A, DiFrancesco D. Deep bradycardia and heart block caused by inducible cardiac-specific knock-out of the pacemaker channel gene Hcn4. Proc Natl Acad Sci U S A. 2011;108:1705–1710. doi: 10.1073/pnas.1010122108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biel M, Wahl-Schott C, Michalakis S, Zong XG. Hyperpolarization-activated cation channels: from genes to function. Phys Rev. 2009;89:847–885. doi: 10.1152/physrev.00029.2008. [DOI] [PubMed] [Google Scholar]
- Brandenburg K, Schromm AB, Gutsmann T. Endotoxins: relationship between structure, function, and activity. Subcell Biochem. 2010;53:53–67. doi: 10.1007/978-90-481-9078-2_3. [DOI] [PubMed] [Google Scholar]
- Ciesielski F, Davis B, Rittig M, Bonev BB, O'Shea P. Receptor-independent interaction of bacterial lipopolysaccharide with lipid and lymphocyte membranes: the role of cholesterol. PLoS One. 2012;7:e38677. doi: 10.1371/journal.pone.0038677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiFrancesco D. Characterization of single pacemaker channels in cardiac sino-atrial node cells. Nature. 1986;324:470–473. doi: 10.1038/324470a0. [DOI] [PubMed] [Google Scholar]
- Fenske S, Mader R, Scharr A, Paparizos C, Cao-Ehlker X, Michalakis S, Shaltiel L, Weidinger M, Stieber J, Feil S, Feil R, Hofmann F, Wahl-Schott C, Biel M. HCN3 contributes to the ventricular action potential waveform in the murine heart. Circ Res. 2011;109:1015–1023. doi: 10.1161/CIRCRESAHA.111.246173. [DOI] [PubMed] [Google Scholar]
- Freeman GG, Anderson TH. The hydrolytic degradation of the antigenic complex of Bact. typhosum Ty 2. Biochem J. 1941;35:564–577. doi: 10.1042/bj0350564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gholami M, Mazaheri P, Mohamadi A, Dehpour T, Safari F, Hajizadeh S, Moore KP, Mani AR. Endotoxemia is associated with partial uncoupling of cardiac pacemaker from cholinergic neural control in rats. Shock. 2012;37:219–227. doi: 10.1097/SHK.0b013e318240b4be. [DOI] [PubMed] [Google Scholar]
- Godin PJ, Fleisher LA, Eidsath A, Vandivier RW, Preas HL, Banks SM, Buchman TG, Suffredini AF. Experimental human endotoxemia increases cardiac regularity: results from a prospective, randomized, crossover trial. Crit Care Med. 1996;24:1117–1124. doi: 10.1097/00003246-199607000-00009. [DOI] [PubMed] [Google Scholar]
- Gutsmann T, Schromm AB, Brandenburg K. The physicochemistry of endotoxins in relation to bioactivity. Int J Med Microbiol. 2007;297:341–352. doi: 10.1016/j.ijmm.2007.03.004. [DOI] [PubMed] [Google Scholar]
- Hegle AP, Nazzari H, Roth A, Angoli D, Accili EA. Evolutionary emergence of N-glycosylation as a variable promoter of HCN channel surface expression. Am J Physiol Cell Physiol. 2010;298:C1066–C1076. doi: 10.1152/ajpcell.00389.2009. [DOI] [PubMed] [Google Scholar]
- Herrmann S, Stieber J, Stockl G, Hofmann F, Ludwig A. HCN4 provides a ‘depolarization reserve’ and is not required for heart rate acceleration in mice. EMBO J. 2007;26:4423–4432. doi: 10.1038/sj.emboj.7601868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann S, Fabritz L, Layh B, Kirchhof P, Ludwig A. Insights into sick sinus syndrome from an inducible mouse model. Cardiovasc Res. 2011;90:38–48. doi: 10.1093/cvr/cvq390. [DOI] [PubMed] [Google Scholar]
- Huang JA, Wang YL, Jiang DB, Zhou JA, Huang XK. The sympathetic–vagal balance against endotoxemia. J Neural Transm. 2010;117:729–735. doi: 10.1007/s00702-010-0407-6. [DOI] [PubMed] [Google Scholar]
- Jann B, Reske K, Jann K. Heterogeneity of lipopolysaccharides – analysis of polysaccharide chain lengths by sodium dodecylsulfate polyacrylamide gel-electrophoresis. Eur J Biochem. 1975;60:239–246. doi: 10.1111/j.1432-1033.1975.tb20996.x. [DOI] [PubMed] [Google Scholar]
- Joannides R, Moore N, Iacob M, Compagnon P, Lerebours G, Menard JF, Thuillez C. Comparative effects of ivabradine, a selective heart rate-lowering agent, and propranolol on systemic and cardiac haemodynamics at rest and during exercise. Br J Clin Pharmacol. 2006;61:127–137. doi: 10.1111/j.1365-2125.2005.02544.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaykin Y, Dorian P, Tang A, Green M, Mitchell J, Wulffhart Z, Newman D. The effect of sinus node depression on heart rate variability in humans using zatebradine, a selective bradycardic agent. Can J Physiol Pharmacol. 1998;76:806–810. doi: 10.1139/cjpp-76-7-8-806. [DOI] [PubMed] [Google Scholar]
- Klöckner U, Rueckschloss U, Grossmann C, Ebelt H, Müller-Werdan U, Loppnow H, Werdan K, Gekle M. Differential reduction of HCN channel activity by various types of lipopolysaccharide. J Mol Cell Cardiol. 2011;51:226–235. doi: 10.1016/j.yjmcc.2011.05.004. [DOI] [PubMed] [Google Scholar]
- Leone S, Molinaro A, Gerber IB, Dubery IA, Lanzetta R, Parrilli M. The O-chain structure from the LPS of the endophytic bacterium Burkholderia cepacia strain ASP B 2D. Carbohydr Res. 2006;341:2954–2958. doi: 10.1016/j.carres.2006.10.010. [DOI] [PubMed] [Google Scholar]
- Levitan I, Fang Y, Rosenhouse-Dantsker A, Romanenko V. Cholesterol and ion channels. Subcell Biochem. 2010;51:509–549. doi: 10.1007/978-90-481-8622-8_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao W, Floren CH. Both the polysaccharide and lipid A parts of endotoxins are needed for the inhibitory effects of endotoxins on cellular LDL uptake. Scand J Clin Lab Invest. 1992;52:183–188. doi: 10.3109/00365519209088783. [DOI] [PubMed] [Google Scholar]
- Ludwig A, Budde T, Stieber J, Moosmang S, Wahl C, Holthoff K, Langebartels A, Wotjak C, Munsch T, Zong X, Feil S, Feil R, Lancel M, Chien KR, Konnerth A, Pape HC, Biel M, Hofmann F. Absence epilepsy and sinusdysrhythmia in mice lacking the pacemaker channel HCN2. EMBO J. 2003;22:216–224. doi: 10.1093/emboj/cdg032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangin L, Swynghedauw B, Benis A, Thibault N, Lerebours G, Carré F. Relationships between heart rate and heart rate variability: study in conscious rats. J Cardiovasc Pharmacol. 1998;32:601–607. doi: 10.1097/00005344-199810000-00012. [DOI] [PubMed] [Google Scholar]
- Medvedev AE, Vogel SN. Overexpression of CD14, TLR4, and MD-2 in HEK 293T cells does not prevent induction of in vitro endotoxin tolerance. J Endotoxin Res. 2003;9:60–64. doi: 10.1179/096805103125001360. [DOI] [PubMed] [Google Scholar]
- Mitchell JA, Paul-Clark MJ, Clarke GW, McMaster SK, Cartwright N. Critical role of toll-like receptors and nucleotide oligomerisation domain in the regulation of health and disease. J Endocrinol. 2007;193:323–330. doi: 10.1677/JOE-07-0067. [DOI] [PubMed] [Google Scholar]
- Morrison DC, Ulevitch RJ. Effects of bacterial endotoxins on host mediation systems. Am J Pathol. 1978;93:526–617. [PMC free article] [PubMed] [Google Scholar]
- Much B, Wahl-Schott C, Zong X, Schneider A, Baumann L, Moosmang S, Ludwig A, Biel M. Role of subunit heteromerization and N-linked glycosylation in the formation of functional hyperpolarization-activated cyclic nucleotide-gated channels. J Biol Chem. 2003;278:43781–43786. doi: 10.1074/jbc.M306958200. [DOI] [PubMed] [Google Scholar]
- Nesterenko MV, Tilley M, Upton SJ. A simple modification of Blum's silver stain method allows for 30 minute detection of proteins in polyacrylamide gels. J Biochem Biophys Methods. 1994;28:239–242. doi: 10.1016/0165-022x(94)90020-5. [DOI] [PubMed] [Google Scholar]
- Noma K, Kimura K, Minatohara K, Nakashima H, Nagao Y, Mizoguchi A, Fujiyoshi Y. Triple N-glycosylation in the long S5–P loop regulates the activation and trafficking of the Kv12.2 potassium channel. J Biol Chem. 2009;284:33139–33150. doi: 10.1074/jbc.M109.021519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtani Y, Irie T, Uekama K, Fukunaga K, Pitha J. Differential effects of α-, β-and γ-cyclodextrins on human erythrocytes. Eur J Biochem. 1989;186:17–22. doi: 10.1111/j.1432-1033.1989.tb15171.x. [DOI] [PubMed] [Google Scholar]
- Palva ET, Makela PH. Lipopolysaccharide heterogeneity in Salmonella Typhimurium analyzed by sodium dodecyl sulfate-polyacrylamide gel-electrophoresis. Eur J Biochem. 1980;107:137–143. doi: 10.1111/j.1432-1033.1980.tb04634.x. [DOI] [PubMed] [Google Scholar]
- Raetz CRH. Biochemistry of endotoxins. Annu Rev Biochem. 1990;59:129–170. doi: 10.1146/annurev.bi.59.070190.001021. [DOI] [PubMed] [Google Scholar]
- Rietschel ET, Kirikae T, Schade FU, Mamat U, Schmidt G, Loppnow H, Ulmer AJ, Zähringer U, Seydel U, Di Padova F, Schreier M, Brade H. Bacterial endotoxin: molecular relationships of structure to activity and function. FASEB J. 1994;8:217–225. doi: 10.1096/fasebj.8.2.8119492. [DOI] [PubMed] [Google Scholar]
- Sakata M, Yoshimura K, Sakamoto I, Todokoro M, Kunitske M. Selective removal of endotoxin from a DNA solution by cross-linked cyclodextrin beads. Anal Sci. 2011;27:213–216. doi: 10.2116/analsci.27.213. [DOI] [PubMed] [Google Scholar]
- Schmidt H, Saworski J, Werdan K, Müller-Werdan U. Decreased beating rate variability of spontaneously contracting cardiomyocytes after co-incubation with endotoxin. J Endotoxin Res. 2007;13:339–342. doi: 10.1177/0968051907086233. [DOI] [PubMed] [Google Scholar]
- Schromm AB, Brandenburg K, Rietschel ET, Flad HD, Carroll SF, Seydel U. Lipopolysaccharide-binding protein mediates CD14-independent intercalation of lipopolysaccharide into phospholipid membranes. FEBS Letters. 1996;399:267–271. doi: 10.1016/s0014-5793(96)01338-5. [DOI] [PubMed] [Google Scholar]
- Stella VJ, He Q. Cyclodextrins. Toxicol Pathol. 2008;36:30–42. doi: 10.1177/0192623307310945. [DOI] [PubMed] [Google Scholar]
- Stieber J, Stockl G, Herrmann S, Hassfurth B, Hofmann F. Functional expression of the human HCN3 channel. J Biol Chem. 2005;280:34635–34643. doi: 10.1074/jbc.M502508200. [DOI] [PubMed] [Google Scholar]
- Strain SM, Fesik SW, Armitage IM. Structure and metal-binding properties of lipopolysaccharides from heptoseless mutants of Escherichia coli studied by 13C and 31P nuclear magnetic resonance. J Biol Chem. 1983;258:13466–13477. [PubMed] [Google Scholar]
- Tsai CM, Frasch CE. A sensitive silver stain for detecting lipopolysaccharides in polyacrylamide gels. Anal Biochem. 1982;119:115–119. doi: 10.1016/0003-2697(82)90673-x. [DOI] [PubMed] [Google Scholar]
- Veldhuis NA, Lew MJ, Abogadie FC, Poole DP, Jennings EA, Ivanusic JJ, Eilers H, Bunnett NW, McIntyre P. N-glycosylation determines ionic permeability and desensitization of the TRPV1 capsaicin receptor. J Biol Chem. 2012;287:21765–21772. doi: 10.1074/jbc.M112.342022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe I, Zhu J, Sutachan JJ, Gottschalk A, Recio-Pinto E, Thornhill WB. The glycosylation state of Kv1.2 potassium channels affects trafficking, gating, and simulated action potentials. Brain Res. 2007;1144:1–18. doi: 10.1016/j.brainres.2007.01.092. [DOI] [PubMed] [Google Scholar]
- Weiss N, Black SA, Bladen C, Chen L, Zamponi GW. Surface expression and function of Cav3.2 T-type calcium channels are controlled by asparagine-linked glycosylation. Pflugers Arch. 2013;465:1159–1170. doi: 10.1007/s00424-013-1259-3. [DOI] [PubMed] [Google Scholar]
- Werdan K, Schmidt H, Ebelt H, Zorn-Pauly K, Koidl B, Hoke RS, Heinroth K, Müller-Werdan U. Impaired regulation of cardiac function in sepsis, SIRS, and MODS. Can J Physiol Pharmacol. 2009;87:266–274. doi: 10.1139/Y09-012. [DOI] [PubMed] [Google Scholar]
- Wondergem R, Graves BM, Ozment-Skelton TR, Li C, Williams DL. Lipopolysaccharides directly decrease Ca2+ oscillations and the hyperpolarization-activated nonselective cation current If in immortalized HL-1 cardiomyocytes. Am J Physiol Cell Physiol. 2010;299:C665–C671. doi: 10.1152/ajpcell.00129.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye B, Balijepalli RC, Foell JD, Kroboth S, Ye Q, Luo YH, Shi NQ. Caveolin-3 associates with and affects the function of hyperpolarization-activated cyclic nucleotide-gated channel 4. Biochemistry. 2008;47:12312–12318. [PMC free article] [PubMed] [Google Scholar]
- Zidovetzki R, Levitan I. Use of cyclodextrins to manipulate plasma membrane cholesterol content: evidence, misconceptions and control strategies. Biochim Biophys Acta. 2007;1768:1311–1324. doi: 10.1016/j.bbamem.2007.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorn-Pauly K, Pelzmann B, Lang P, Machler H, Schmidt H, Ebelt H, Werdan K, Koidl B, Müller-Werdan U. Endotoxin impairs the human pacemaker current If. Shock. 2007;28:655–661. [PubMed] [Google Scholar]