

Abstract
Blood flow data from contracting muscle in humans indicates that adenosine (ADO) stimulates the production of nitric oxide (NO) and vasodilating prostaglandins (PG) to produce arteriolar vasodilatation in a redundant fashion such that when one is inhibited the other can compensate. We sought to determine whether these redundant mechanisms are employed at the microvascular level. First, we determined whether PGs were involved in active hyperaemia at the microvascular level. We stimulated four to five skeletal muscle fibres in the anaesthetized hamster cremaster preparation in situ and measured the change in diameter of 2A arterioles (maximum diameter 40 μm, third arteriolar level up from the capillaries) at a site of overlap with the stimulated muscle fibres before and after 2 min of contraction [stimulus frequencies: 4, 20 and 60 Hz at 15 contractions per minute (CPM) or contraction frequencies of 6, 15 or 60 CPM at 20 Hz; 250 ms train duration]. Muscle fibres were stimulated in the absence and presence of the phospholipase A2 inhibitor quinacrine. Further, we applied a range of concentrations of ADO (10−7–10−5 m) extraluminally, (to mimic muscle contraction) in the absence and presence of l-NAME (NO synthase inhibitor), indomethacin (INDO, cyclooxygenase inhibitor) and l-NAME + INDO and observed the response of 2A arterioles. We repeated the latter experiment on a different level of the cremaster microvasculature (1A arterioles) and on the microvasculature of a different skeletal muscle (gluteus maximus, 2A arterioles). We observed that quinacrine inhibited vasodilatation during muscle contraction at intermediate and high contraction frequencies (15 and 60 CPM). l-NAME, INDO and l-NAME + INDO were not effective at inhibiting vasodilatation induced by any concentration of ADO tested in 2A and 1A arterioles in the cremaster muscle or 2A arterioles in the gluteus maximus muscle. Our data show that PGs are involved in the vasodilatation of the microvasculature in response to muscle contraction but did not obtain evidence that extraluminal ADO causes vasodilatation through NO or PG or both. Thus, we propose that PG-induced microvascular vasodilation during exercise is independent of ADO.
Introduction
Local blood flow regulation in contracting skeletal muscle is the result of a complex release of vasodilators on to a vasculature system that varies in its responsive nature along its length. The ability to match blood flow to metabolic demand appears to be built on redundant, fail-safe systems to ensure the proper co-ordination of blood flow (Joyner & Wilkins, 2007). Vasodilatating prostaglandins (PGs) are part of this redundant system but their contribution to exercise hyperaemia during muscle contraction remains unresolved. Studies have shown that inhibition of PGs increase resistance (decrease conductance) and reduce the level of blood flow to contracting muscle (Kilbom & Wennmalm, 1976; Cowley et al. 1985; Wilson & Kapoor, 1993; Duffy et al. 1999; Farouque & Meredith, 2003; Schrage et al. 2004, 2010; Win & Marshall, 2005), while others have not (Beaty & Donald, 1979; Einstein & Goodman, 1980; Young & Sparks, 1980; Shoemaker et al. 1996; Saunders et al. 2005; Mortensen et al. 2007; Crecelius et al. 2011). The controversial role of PG in exercise hyperaemia extends to the microvascular level where some studies have shown that PG inhibition decreases contraction-induced vasodilatation (McKay et al. 1998; Nuttle et al. 1999) while others have not (Hammer & Boegehold, 2005).
The difficulties isolating the effects of PGs are at least two-fold. First, there may be specific contraction conditions [such as stimulus frequency, contraction frequency, contraction intensity (force), contraction duration, etc.] under which PGs are more readily released and most effective. We have previously shown that the effectiveness or presence of vasodilators involved in exercise hyperaemia [such as nitric oxide (NO), adenosine (ADO) and potassium] is dependent on the skeletal muscle contractile parameters (Dua et al. 2009). An intensity dependent release of PGs from contracting muscle has been shown (Symons et al. 1991; Wilson & Kapoor, 1993; Karamouzis et al. 2001a). Further, a difference in PG release during static and dynamic contractions has been demonstrated (Karamouzis et al. 2001b). Thus, there may be specific contractile parameters under which PGs are optimally produced and effective. Second, the difficulty in isolating the effects of PGs in exercise hyperaemia may be due to the interplay between ADO, NO and PGs. ADO has a long history of being implicated as an important vasodilator involved in blood flow regulation in contracting skeletal muscle by accumulating in the intracellular space and promoting arteriolar vasodilatation (for review see Marshall, 2007). It has recently been suggested that ADO causes vasodilatation through the production of both NO and PGs, and that PG and NO interact in a complex manner whereby the inhibition of NO (Radegran & Saltin, 1999; Frandsenn et al. 2001) or PGs (Shoemaker et al. 1996; Schrage et al. 2004; Mortensen et al. 2007) individually does not decrease the hyperaemic response but only the inhibition of both will result in a decrease in the hyperaemia during contraction (Boushel et al. 2002; Kalliokoski et al. 2006; Mortensen et al. 2007, 2009). In human studies, the measured changes in blood flow in response to intraluminal ADO infusion has been shown to decrease vascular resistance through mechanisms, including NO and PG formation (Mortensen et al. 2009; Nyberg et al. 2010) but whether extracellular accumulation of ADO causes vasodilatation through the same NO/PG-dependent system is unknown.
To further complicate the understanding of the actions of ADO, NO and PGs is that this complex trio of vasodilators acts on the vascular tree, which in itself has different reactivities and sensitivities along its length (Anderson & Faber, 1991; Ohyanagi et al. 1991; Hester et al. 1993; Tateishi & Faber, 1995; Hammer et al. 2001; Newcomer et al. 2008; Twynstra et al. 2012). This differentiation of function within the vasculature helps to co-ordinate the delivery of blood flow to muscle and distribute blood flow within a working muscle. Simplistically, larger arteries and arterioles control blood flow delivery to a muscle whereas smaller, more terminal arterioles control distribution of blood flow within a muscle (Klitzman et al. 1982; Lindbom & Arfors, 1984; Sarelius, 1986; Sweeney & Sarelius, 1989). Studies that implicate a redundant interaction between ADO, NO and PGs have used blood flow as the metric to measure responses of the vasculature. However, gross measurements in blood flow (or reflected by conductance or resistance calculations) cannot distinguish between the behaviour of large, proximal arterioles and the behaviour of small, distal arterioles given that the control of vascular resistance resides in both large (>100 um) and small (<100 um) arteries and arterioles (for review see Pohl et al. 2000). Further, we know that ADO and NO both play a significant role in microvascular vasodilatation that results from muscle contraction (Murrant & Sarelius, 2002; Dua et al. 2009) and that PGs have been shown to affect the microvasculature (McKay et al. 1998; Nuttle et al. 1999) but whether the effects of NO and PGs are secondary to the action of ADO at the microvascular level is unknown. In addition, whether the terminal microvasculature responds to ADO, NO and PGs in an integrated and redundant manner has not been tested.
Therefore, we designed a study to, first, determine if PGs are important in contraction-induced vasodilatation of the microvasculature, and secondly, to determine the contractile parameters under which PGs are effective at causing vasodilatation of the terminal microvasculature in response to muscle contraction. We studied 2A arterioles (maximum diameter 40 μm), which are, in part, responsible for the distribution of blood flow to capillaries within a muscle. Further, we sought to determine whether the PGs responsible for contraction-induced vasodilatation could be synthesized secondary to ADO release. We used extracellular application of ADO on the microvasculature in the absence and presence of NO synthase inhibition and cyclooxygenase inhibition to determine whether ADO causes vasodilatation that is dependent on NO or PGs, or both. We hypothesized that PGs are involved in contraction-induced vasodilatation and that ADO-induced vasodilatation is dependent on PGs.
Methods
All experimental procedures were approved by the Institutional Animal Care and Use Committee and conducted in accordance with the guidelines of the Canadian Council on Animal Care as set out in the Guide to the Care and Use of Experimental Animals. Following all experimental protocols, animals were killed with an overdose of sodium pentobarbital (0.26 mg ml−1 i.v. to effect).
In situ protocols
Adult male golden Syrian hamsters (100–130 g) were anaesthetized with sodium pentobarbital (70 mg kg−1 intraperitoneally) and tracheotomized. Polyethylene catheters (outer tip diameter ∼0.5 mm) were placed in the left femoral artery (to monitor mean arterial pressure) and left femoral vein for supplemental sodium pentobarbital infusion (10 mg ml−1 saline, 0.56 ml h−1) throughout the experimental protocol. Hamster oesophageal temperature was maintained at 37°C via convective heat from a coiled water-filled glass tube (42°C) secured under the hamster. The right cremaster was prepared for in situ microscopy as previously described (Baez, 1973) and modified (Murrant, 2005). Briefly, the cremaster was isolated, cut longitudinally, separated from the testis and epididymis and gently spread over a semicircular Lucite platform. The edges of the tissue were secured with insect pins to maintain tension but not stretch the muscle. During surgery and the experimental protocols, muscles were constantly superfused with a bicarbonate-buffered salt solution containing (in mm): NaCl, 131.9; KCl, 4.7; CaCl2, 2.0; MgSO4, 1.2; NaHCO3, 30 (all chemicals from Fisher Scientific, Waltham, MA, USA) and 0.3 mg l−1 (4 × 10−6 m) tubarine (curare) (Sigma-Aldrich, St Louis, MO, USA) equilibrated with gas containing 5% CO2–95% N2 (pH 7.35–7.45). Cremaster muscle temperature was maintained by heating the superfusion solution to 42°C and adjusting the drip rate to achieve 34°C. After surgery, preparations were allowed to equilibrate for 45–60 min before data collection.
The cremaster microvasculature was visualized by transillumination with a tungsten lamp and with an Olympus BX51WI microscope (Olympus Canada Inc., Richmond Hill, ON, Canada) using a ×20 long working distance water immersion objective (numerical aperture 0.50) and ×1.6 magnification changer. The microscope image was displayed via a video camera (Sony DXC-390; Sony Canada Ltd., Toronto, ON, Canada) on a monitor and recorded on a videotape recorder (Sony, SVO-9600MD) or collected digitally to an IBM computer using EZGrabber video compressor and software (Geniatech, Shenzhen, China). Final magnification of the site was approximately ×2000. Diameter measurements were reproducible to within ±0.3 μm (n = 10). Transverse arterioles (TA; 2A) of ∼40 μm maximum diameter were observed. These arterioles were identified by counting up three branch orders from capillary vessels as previously described (Sweeney & Sarelius, 1989; Murrant & Sarelius, 2000). The only selection criteria used was that muscle fibres associated with the TA run approximately perpendicular to the arteriole.
For stimulation, a silver wire microelectrode (tip diameter ∼250 μm) was placed in close proximity to, but not touching muscle fibres running approximately perpendicular to the arteriole. The microelectrode was positioned at least 1000 μm away from the site of the arteriole-stimulated muscle fibre intersection (observation site). The ground electrode was placed in the superfusate around the outer rim of the cremaster preparation and secured with pins on the pedestal. Curare was added to the superfusate, as previously described (Dua et al. 2009), to ensure direct electrode stimulation of the skeletal muscle cells and not nervous stimulation of the muscle fibres. Each stimulus consisted of a square wave pulse of 0.4 ms duration. Voltage (4–8 V) was chosen to maximally stimulate three to five muscle fibres and then kept constant throughout the duration of the experiment.
In situ skeletal muscle contraction experiments
To determine the contractile parameters under which PGs were effective at producing vasodilatation, muscle fibres were stimulated with either a range of stimulus frequencies (changing the number of impulses per contraction which will therefore change force of contraction) while contraction frequency was held constant or a range of contraction frequencies (changing the number of contractions per minute) while the stimulus frequency was held constant. Muscle fibre bundles were stimulated for 2 min at stimulus frequencies of 4, 20 or 60 Hz while contraction frequency was held constant at 15 contractions per minute (CPM) or a range of contraction frequencies (6, 15 or 60 CPM) with the stimulus frequency constant at 20 Hz. This stimulus and contraction frequency protocol was designed with one parameter the same in both protocols, 15 CPM, 20 Hz. This design helps us differentiate between whether stimulus frequency and contraction frequency is the more critical variable in the PG response (see discussion). For each experiment, the train duration was 250 ms, which allowed twitch and tetanic contractions to be compared within the stimulus frequency protocol, as 4 Hz for 250 ms train duration will result in a single twitch and 20 and 60 Hz for 250 ms train duration will result in submaximal and maximal tetanic contractions. The order of stimulus or contraction frequencies was randomized and a 5 min rest period was given between contraction bouts. The preparation was then superfused with the phospholipase A2 (PLA2) inhibitor quinacrine (Sigma-Aldrich). PLA2 is the enzyme that facilitates the breakdown of phosphatidylcholine to arachadonic acid, which is the precursor to the enzyme cyclooxygenase required for the production of PGs. Quinacrine (3 × 10−6 m) was superfused over the preparation for 1 h and the contraction protocol was repeated. The inhibitor was then washed out by superfusing the preparation for 20 min with normal salt solution and the contraction protocol was repeated to ensure that the preparation maintained its responsiveness. One hour of 3 × 10−6 m quinacrine exposure has been shown to be effective at blocking thrombin-induced vasodilatations in this preparation (Nuttle et al. 1999; personal observations, data not shown).
In situ extraluminal adenosine application experiments
To determine whether PG-induced vasodilatations were initiated by ADO, we exposed the cremaster preparation to a range of concentrations of ADO in the absence and presence of 10−6 m indomethacin (INDO), a cyclooxygenase inhibitor. Specifically, ADO was applied in a dose-dependent manner, added to the superfusate solution, exposing the entire preparation to 10−7, 10−6, 5 × 10−6 and 10−5 m ADO (RBI, Natick, MA, USA) starting from the lowest to the highest dose, for 2 min intervals. ADO was then washed out and the preparation was superfused with 10−6 m INDO (Sigma-Aldrich) (n = 10) for 30 min and then ADO was re-applied as described above in the presence of INDO. INDO was then washed out by superfusing the preparation for 30 min with normal salt solution and the ADO protocol was repeated to ensure the preparation maintained its responsiveness. The effectiveness of 10−6 m INDO was tested by showing that the vasodilatation in response to arachadonic acid (21.3 ± 8.3 μm) (n = 3) (Sigma-Aldrich) was completely abolished in the presence of INDO (−0.1 ± 0.9 μm).
NO pathways have been implicated in ADO-dependent vasodilatation. It is possible that if the cyclooxygenase pathway is blocked then a greater effect via NO would be produced, thus masking any effect that INDO inhibition may have on ADO-induced vasodilatation. To explore this possibility, we repeated the ADO dose response in the absence and presence of 10−6 m l-NAME, an NO synthase inhibitor. This concentration has been previously shown to inhibit vasodilatations induced by 10−4 m acetylcholine (Dua et al. 2009). Given that l-NAME can cause a decrease in resting diameter, presumably through the inhibition of endogenous NO production at rest, we added 10−7 m S-nitroso-N-acetylpenicillamine (SNAP; Sigma-Aldrich), an NO donor, during l-NAME application to maintain the NO contribution to resting diameter. Therefore, ADO dose responses were repeated in the presence of l-NAME (n = 8) and l-NAME + SNAP (n = 9).
Given the potential redundant nature of the NO and PG response to ADO, it was critical to repeat the ADO dose response in the presence of both NO and PG inhibition. Therefore, we repeated the ADO dose response in the absence and presence of l-NAME + SNAP and INDO (n = 7).
We performed the ADO dose response on the feed arteriole (1A), a larger branch order, to confirm that our results were not specific to the branch order under observation. Specifically, the feed arteriole, which is the parent blood vessel to the TA, is located one branch order up from our previous TA (2A) observation site and has been shown to be more sensitive to NO (Hester et al. 1993). We repeated the ADO dose response in the absence and the presence of l-NAME + SNAP and INDO while observing the feed arteriole (n = 8). To confirm further that our observations were not specific to the cremaster muscle microvasculature, we repeated the ADO dose response in the absence and presence of l-NAME + SNAP and INDO while observing the arteriolar response in the hamster gluteus maximus 2A arteriole (n = 6). The gluteus maximus preparation was prepared as described elsewhere in mice (Bearden et al. 2004) and rats (Al-Khazraji et al. 2012) and adapted to the hamster. Briefly, hamsters were anaesthetized, tracheotomized and cannulated as described above and then oriented ventral side down on the warming coil on the plexiglas platform. The left gluteus maximus muscle was exposed and dissected free of its origin along the spine and free from connective tissue along the caudal and rostral borders. The muscle was reflected on to the plexiglas viewing platform with the vascular supply remaining intact. The preparation was superfused as described above for the cremaster preparation.
Following each experiment, arteriolar diameters were recorded after 2 min superfusion with 10−4 m sodium nitroprusside (NO donor; Sigma-Aldrich), considered to produce maximal dilatation. Calcium-free superfusate solution (44.6 ± 5.6 μm) and 10−4 m sodium nitroprusside (44.4 ± 6.3 μm) produce maximal dilatations that do not significantly differ when compared on a subset of vessels (n = 6).
Fibre typing of cremaster and gluteus maximus
Fibre type distribution of hamster skeletal muscles was determined through electrophoretic separation of myosin heavy chains from isolated myofibrils via SDS-PAGE. Muscles were excised and immediately frozen in liquid nitrogen and stored at −80°C. Myofibrils were isolated from homogenates using a protocol adapted from Murphy & Solaro (1990). Briefly, muscles were homogenized with a glass pestle and mortar in ice-cold buffer: 60 mm KCl, 30 mm imidazole (pH 7.0) and 2 mm MgCl2. Isolated myofibrils were then separated on an 8% polyacrylamide gel for 31 h at 72 V in an ice bath (adapted from Agbulut et al. 2003). Gels were silver stained (Blum et al. 1987) followed by densitometric analysis to determine relative myosin heavy chain distribution.
In vitro skeletal muscle contraction protocols
During the in situ muscle contraction protocols, skeletal muscle cells were exposed to quinacrine, which could alter skeletal muscle function and, thus, result in an altered vasodilatory response to muscle contraction. To determine if there were any effects of quinacrine on the force of contraction of the skeletal muscle fibres, following in situ experiments the left cremaster muscle was exteriorized and cut into two strips, each were tied at the ends with 5.0 gauge suture silk and hung in a tissue chamber containing Krebs–Henseleit solution consisting of (in mm): NaCl, 118; KCl, 4.75; CaCl2, 2.54; KH2PO4, 1.18; MgSO4, 1.18; NaHCO3, 24.8; glucose, 10 (all chemicals from Fisher Scientific, Waltham, MA, USA). The solution contained 0.3 mg l−1 tubocurarine and 10 U insulin l−1 and was aerated continuously with 95% O2–5% CO2 at 27°C (pH 7.4). The cremaster strips were attached to a fixed point at one end and a force transducer (either model FT03, Grass Medical Instruments, Quincy, MA, USA or model 60–2995, Harvard Apparatus, Southmatic, MA, USA) on the other. Stimulating electrodes were fixed at each side of the muscle. Each muscle was adjusted to optimal length for force development. After an hour equilibration, force of contraction was measured as muscles were stimulated for 2 min at 4, 20 and 60 Hz (15 CPM, 250 ms train duration with a super maximal voltage; 100 V) or 6, 15 and 60 CPM (20 Hz, 250 ms train duration with a super maximal voltage; 100 V). For each experiment, the order of stimulus and contraction frequencies was randomized and there was a 20 min recovery period between contraction bouts. One of the pair of muscle strips (experimental) was then incubated with 3 × 10−6 m quinacrine (n = 7–8) for 60 min, the other of the pair was incubated in Krebs–Henseleit solution for 60 min to act as an animal and time-matched control. Following the incubation period, muscles were stimulated again over the above range of stimulus and contraction frequencies. Data were collected and analysed using the MP100WSW data acquisition system and Acqknowledge III software (Biopac Systems Inc., Goleta, CA, USA) on an IBM computer. Following each experiment, muscle length and weight were measured.
Data analysis and statistics
In situ protocols
Arteriolar diameter at the observation site was continuously recorded for 1 min before skeletal muscle contraction or drug stimulation, during stimulation and for 2 min after stimulation. Arteriolar diameter was measured every 10 s during the recording period.
Baseline diameter was defined as the diameter just before muscle contraction or ADO stimulation. Only one arteriole was observed per cremaster preparation to collect data and n indicates the number of arterioles observed. All in situ experiments were videotaped and analysed off line. Images were digitized and arteriolar diameters measured via ImageJ software (NIH freeware, http://rsbweb.nih.gov/ij/). Arteriolar diameter was measured immediately following stimulation and at various time points depending upon the experimental protocol.
For data collected over time, group means were compared with the repeated-measures ANOVA. Baseline and maximal diameter were compared with an ANOVA. When the ANOVA identified significant differences, a protected least squares difference test was used post hoc to determine if diameter changes were significantly different. Differences were considered significant when P < 0.05.
In vitro protocols
For data collected over time, group means were compared with the repeated-measures ANOVA. When the ANOVA identified significant differences, a protected least squares difference test was used post hoc to determine if diameter changes were significantly different. Differences were considered significant when P < 0.05.
Results
Baseline and maximal diameters for arterioles observed in in situ muscle contraction experiments in the absence and presence of quinacrine are shown in Table 1. We observed that quinacrine did not affect the vasodilatation at stimulus frequencies of 4 Hz but did attenuate the vasodilatation produced at 20 Hz (Fig. 1). Vasodilatation at 60 Hz was attenuated but not significantly [P = 0.11 at greatest difference (90 s)], whereby six of the 10 experiments showed a decrease in vasodilation in the presence of quinacrine, three showed no effect and one increased vasodilatation. This is the cause of the higher variability at 60 Hz not observed at the other stimulus frequencies. This higher variability at 60 Hz indicates a potentially smaller, less consistent contribution of PGs to 60 Hz and resulted in the lack of significance of the effect of quinacrine. Further, we observed that quinacrine significantly inhibited the contraction-induced vasodilatation at contraction frequencies of 15 and 60 CPM but not 6 CPM (Fig. 2) and did not affect the rate of recovery (data not shown). In vitro studies showed that quinacrine did not affect the absolute force or fatigue rates of cremaster strips at any stimulus or contraction frequency (Table 2). Therefore, we assume there were no changes in force of contraction during the in situ experiments in the presence of quinacrine.
Table 1.
Resting baseline and maximal diameters of arterioles used in in situ contraction experiments investigating the role of prostaglandins in the vasodilatation in response to skeletal muscle contraction. Baseline diameters are reported before (control) and following 60 min incubation with the phospholipase A2 inhibitor quinacrine (experimental)
| Contraction parameters | Control baseline diameter (μm) | Experimental baseline diameter (μm) | Maximum diameter (μm) | n |
|---|---|---|---|---|
| 4 Hz 15 CPM | 19.8 ± 1.8 | 15.5 ± 1.8 | 45.2 ± 5.7 | 10 |
| 20 Hz 15 CPM | 19.4 ± 1.8 | 14.2 ± 1.4* | 45.2 ± 5.7 | 10 |
| 60 Hz 15 CPM | 16.6 ± 1.7 | 15.0 ± 1.5 | 45.2 ± 5.7 | 10 |
| 6 CPM 20 Hz | 22.2 ± 2.2 | 17.7 ± 1.7 | 42.4 ± 2.9 | 10 |
| 15 CPM 20 Hz | 22.1 ± 2.0 | 20.0 ± 2.9 | 42.4 ± 2.9 | 10 |
| 60 CPM 20 Hz | 23.6 ± 2.2 | 19.2 ± 2.2 | 42.4 ± 2.9 | 10 |
Values are means ± SE, n = number of arterioles.
Experimental baseline diameters were significantly different from controls.
Figure 1.
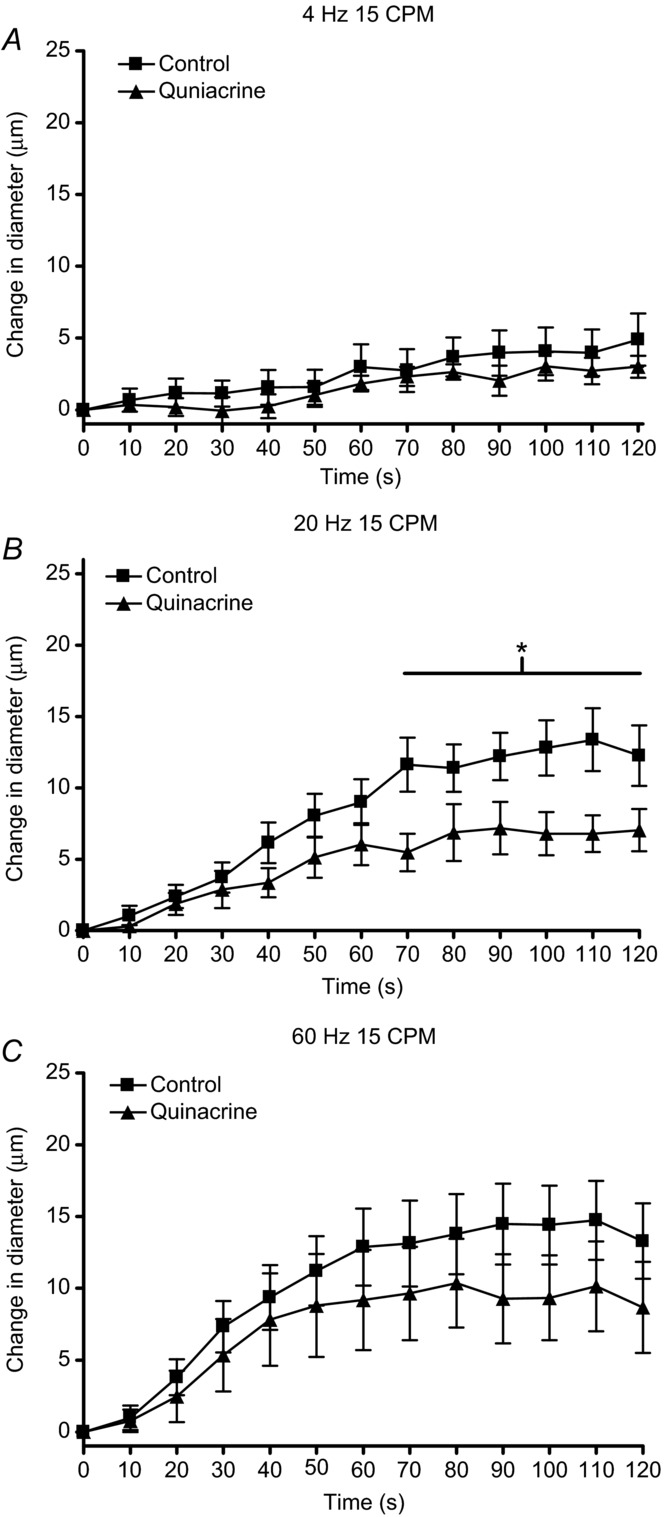
The change in diameter over 2 min of contractions at stimulus frequencies of (A) 4 Hz, (B) 20 Hz and (C) 60 Hz at a constant contraction frequency of 15 CPM in the absence (▪) and presence (▴) of the phospholipase A2 inhibitor quinacrine. The line with the * indicates the range over which the dilatation in the presence of quinacrine was significantly different from control dilatation.
Figure 2.
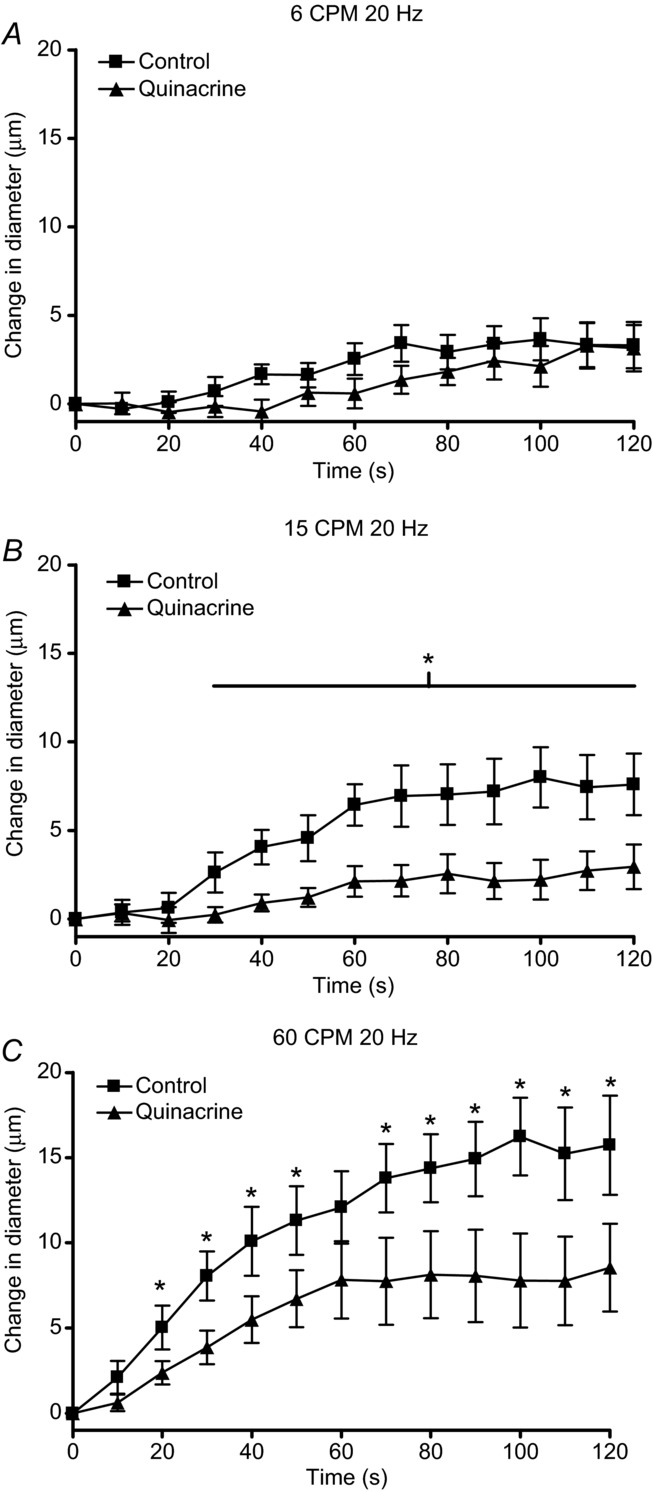
The change in diameter over 2 min of contractions at contraction frequencies of (A) 6 CPM, (B) 15 CPM and (C) 60 CPM at a constant stimulus frequency of 20 Hz in the absence (▪) and presence (▴) of the phospholipase A2 inhibitor quinacrine. The line with the * indicates the range over which the dilatation in the presence of quinacrine was significantly different from control dilatation.
Table 2.
Control and experimental initial forces and fatigue rates of cremaster muscle strips used in in vitro contraction experiments investigating the role of prostaglandins in the vasodilatation in response to skeletal muscle contraction. Forces and fatigue rates reported were following 60 min incubation with either Krebs solution (control) or the phospholipase A2 inhibitor quinacrine (experimental)
| Contraction parameters | Control force (g) | Experimental force (g) | Control fatigue rate (g min−1) | Experimental fatigue rate (g min−1) | n |
|---|---|---|---|---|---|
| 4 Hz 15 CPM | 1.0 ± 0.2 | 1.2 ± 0.2 | 0.01 ± 0.01 | 0.04 ± 0.02 | 7 |
| 20 Hz 15 CPM | 2.5 ± 0.5 | 2.6 ± 0.6 | −0.006 ± 0.02 | −0.006 ± 0.07 | 7 |
| 60 Hz 15 CPM | 3.4 ± 0.7 | 3.6 ± 0.9 | 0.02 ± 0.04 | −0.02 ± 0.03 | 7 |
| 6 CPM 20 Hz | 2.5 ± 0.5 | 3.6 ± 0.6 | −0.03 ± 0.05 | 0.07 ± 0.05 | 7 |
| 15 CPM 20 Hz | 2.8 ± 0.6 | 3.4 ± 0.5 | −0.04 ± 0.04 | 0.05 ± 0.04 | 8 |
| 60 CPM 20 Hz | 2.2 ± 0.5 | 3.1 ± 0.6 | −0.57 ± 0.2 | −0.69 ± 0.3 | 8 |
Values are means ± SE, n = number of muscle strips.
Baseline and maximal diameters for arterioles observed in in situ ADO application experiments in the absence and presence of specific inhibitors are shown in Table 3. To determine whether the PG release was the result of ADO stimulation, we applied a range of concentrations of ADO in the absence and presence of 10−6 m INDO and found no significant inhibition of the ADO vasodilatation at any concentration (Fig. 3A). To determine whether the vasodilatation via ADO through NO was masking the effects of PGs, we applied a range of concentrations of ADO in the absence and presence of l-NAME or l-NAME + SNAP. We did not observe a significant effect of either condition on the ADO dilatation at any concentration (Fig. 3B). To ensure that the PG or NO pathways were not compensating for each other with the single inhibitor present, we repeated the ADO dose response in the presence of INDO and L-NAME + SNAP and, again, saw no effect of the double block on the vasodilatation produced by any concentration of extraluminal ADO tested (Fig. 3C).
Table 3.
Resting baseline and maximal diameters of arterioles used in experiments investigating the role of prostaglandins and NO in vasodilatations produced by a range of concentrations of ADO on different arterioles of different muscles. Baseline diameters are reported before (control) and following 30 min of incubation (experimental) with the cyclooxygenase inhibitor INDO or NO synthase inhibitor l-NAME without and with the addition of NO by adding SNAP
| Preparation | Experimental condition | Control baseline diameter (μm) | Experimental baseline diameter (μm) | Maximum diameter (μm) | n |
|---|---|---|---|---|---|
| Cremaster TA | ADO + INDO | 17.8 ± 2.6 | 13.4 ± 2.0 | 41.4 ± 3.6 | 10 |
| Cremaster TA | ADO + l-NAME | 13.8 ± 2.1 | 12.2 ± 2.4 | 44.2 ± 4.7 | 8 |
| Cremaster TA | ADO + l-NAME + SNAP | 14.2 ± 1.7 | 12.9 ± 1.7 | 36.6 ± 2.7 | 9 |
| Cremaster TA | ADO + INDO + l-NAME + SNAP | 13.6 ± 2.7 | 10.7 ± 2.1 | 37.6 ± 3.9 | 7 |
| Cremaster feed arteriole | ADO + INDO + l-NAME + SNAP | 79.1 ± 4.5 | 74.0 ± 4.1 | 95.0 ± 4.9 | 8 |
| Gluteus maximus arteriole | ADO + INDO + l-NAME + SNAP | 38.9 ± 4.3 | 39.6 ± 4.6 | 54.1 ± 4.0 | 6 |
Values are mean ± SE, n = number of arterioles. Abbreviations: ADO, adenosine; INDO, indomethacin; l-NAME, nitro-l-arginine methyl ester; NO, nitric oxide; TA, transverse arteriole; SNAP, S-nitroso-N-acetylpenicillamine.
Figure 3.
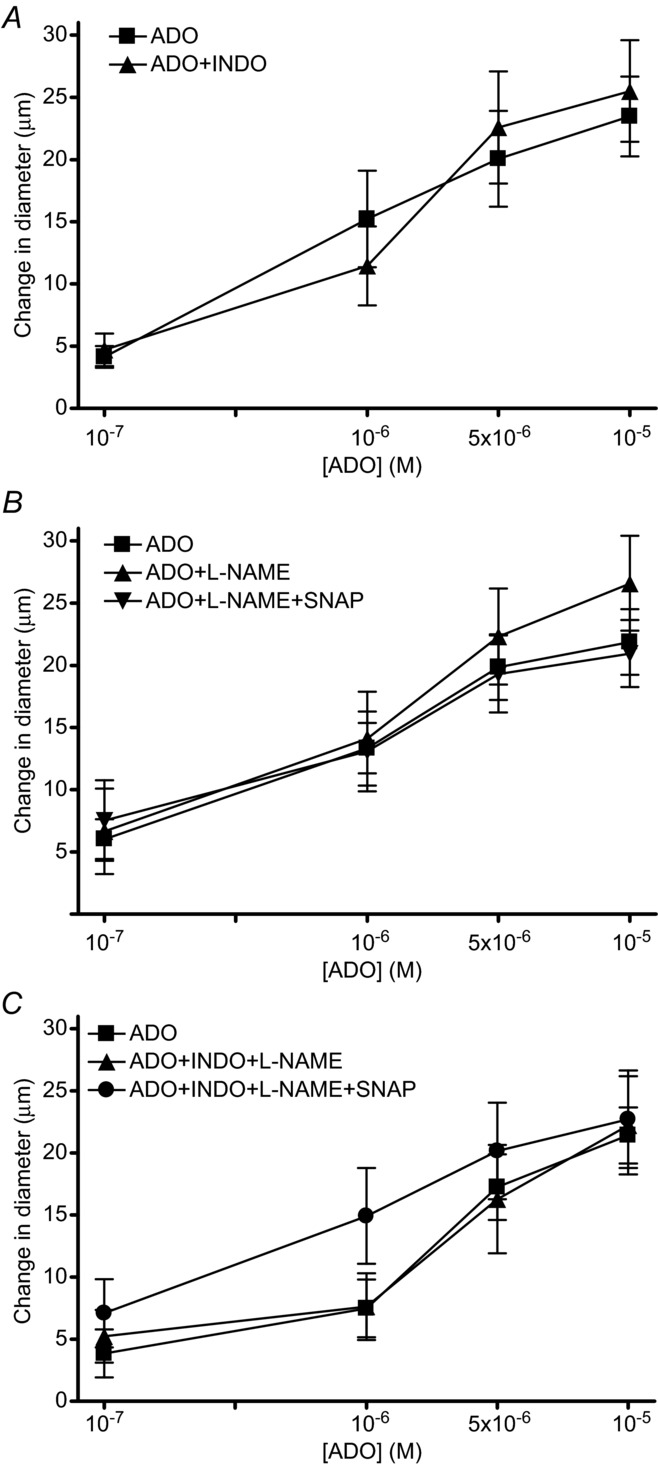
The change in diameter in response to incremental doses of ADO (A) in absence (▪) and presence (▴) of INDO, (B) absence (▪) and presence of 10−5 m l-NAME alone (▴) or 10−5 m l-NAME in presence of 10−7 m SNAP (▾) and (C) absence (▪) and presence of INDO + 10−5 m l-NAME (▴) or INDO + 10−5 m l-NAME + 10−7 m SNAP (•). ADO, adenosine; INDO, indomethacin; l-NAME, nitro-l-arginine methyl ester; SNAP, S-nitroso-N-acetylpenicillamine.
To determine whether this lack of involvement of either NO or PGs in ADO-induced vasodilatations was specific to the TA (2A) level of the vasculature, we repeated the ADO dose response in the absence and presence of the double block (INDO and l-NAME + SNAP) one branch order up from the TA, on the feed arteriole (1A arterioles) and found that the inhibition of PGs and NO did not interfere with the magnitude of the vasodilatation induced at all ADO concentrations tested (Fig. 4A).
Figure 4.
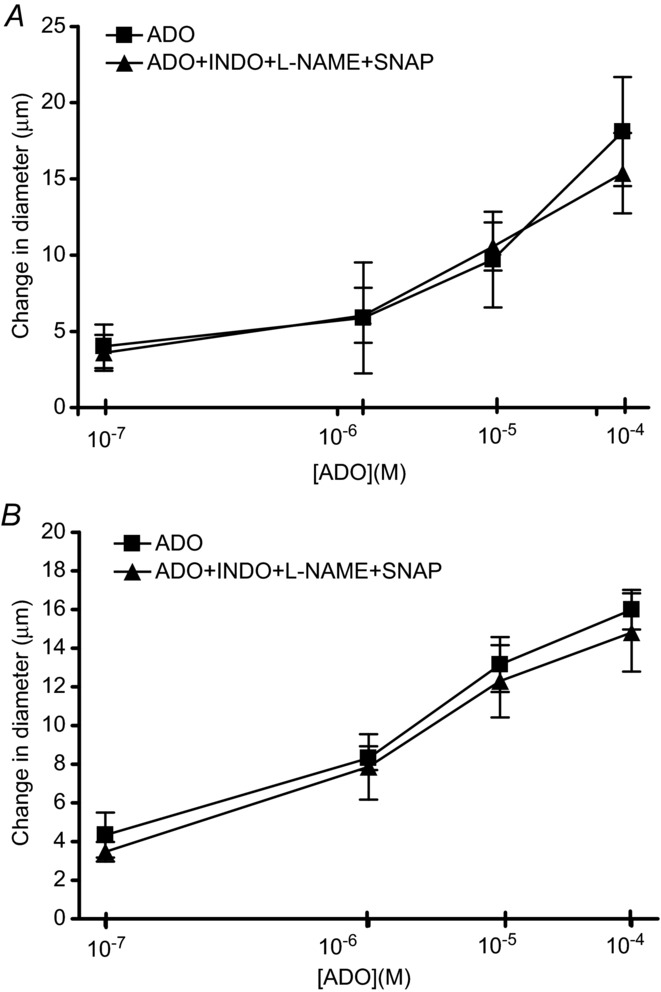
A, change in diameter in response to incremental doses of ADO in the absence (▪) and presence of INDO + 10−5 m l-NAME + 10−7 m SNAP (▴). B, change in diameter in response to incremental doses of ADO in absence (▪) and presence of INDO + 10−5 m l-NAME + 10−7 m SNAP (▴). ADO, adenosine; INDO, indomethacin; l-NAME, nitro-l-arginine methyl ester; SNAP, S-nitroso-N-acetylpenicillamine.
To determine whether this phenomenon was cremaster muscle specific, we repeated the above experiment on 2A arterioles in the hamster gluteus maximus muscle. Again, we observed that the double block (INDO and L-NAME + SNAP) did not influence the magnitude of the ADO-induced vasodilatation at any concentration tested (Fig. 4B). Gluteus maximus fibre type composition was similar to the composition of the cremaster muscle. Figure 5 shows the percentage myosin heavy chain composition of gluteus maximus (∼6% type 1, 76% type IIA and 18% type IIB) is similar to cremaster muscle (∼17% type 1, 77% type IIA and 6% type IIB) being primarily type IIA. To confirm the accuracy of our assay, we also ran Western blot analyses on muscles of known fibre type composition and found that soleus (∼75% type 1, 25% type IIA) is primarily type 1 and extensor digitorum longus (∼7% type 1, 78% type IIA and 15% type IIB) and diaphragm (∼12% type 1, 80% type IIA and 8% type IIB) are primarily type IIA, and all conformed to known compositions (Mattson et al. 2002).
Figure 5.
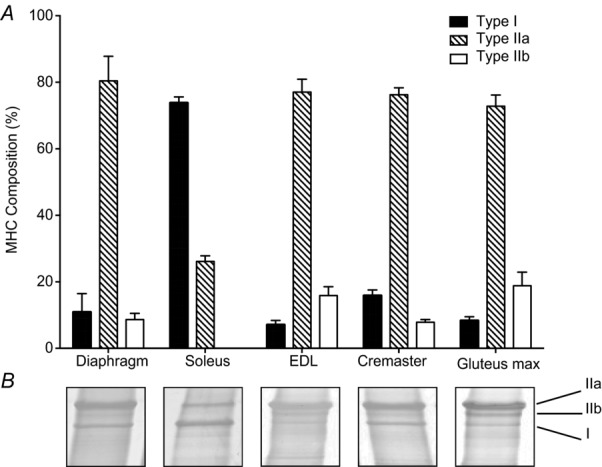
The relative fibre-type distribution of hamster skeletal muscles as determined by electrophoretic separation of MHC isoforms. EDL, extensor digitorum longus; MHC, myosin heavy chain.
Discussion
The important new findings in this study are that PGs are involved in contraction-induced vasodilatation of the terminal vasculature under specific contractile conditions and extraluminal ADO induces arteriolar vasodilatation independently of PGs. Extraluminal ADO did not induce vasodilatation at any concentration that was dependent on PG production, NO production or a combination of the two. This phenomenon was not unique to the 2A arterioles in the cremaster muscle as ADO acted independently of PGs and NO in different arterioles (1A arterioles of the cremaster muscle) and different muscles (2A arterioles in the gluteus maximus). Therefore, our data show that the terminal microvasculature does not use the ADO/PG/NO pathway to co-ordinate vasodilatation and implies that the PG-induced vasodilatation in response to muscle contraction is independent of extraluminal ADO production.
We demonstrated that the importance of PGs in arteriolar vasodilatation during muscle contraction was dependent on how the skeletal muscle cells were stimulated to contract. We used different stimulus frequencies and contraction frequencies to determine which stimulation component was more important in producing a PG-dependent vasodilatation. Stimulus frequency, or the number of impulses used per contraction, is an important determinant of force generated by the muscle. Physiologically, two different strategies are used to increase the force generated by a muscle, either increase the stimulus frequency of the recruited motor units or recruit more motor units. Within this study, we increased the stimulus frequency, which increased force of the recruited skeletal muscle cells and did not increase the number of muscle cells recruited. We also changed contraction frequency, or the number of contractions per minute, which affects the rate at which the processes for contraction and relaxation are repeated. In the present study, we observed that PG synthesis inhibition attenuated arteriolar dilatation evoked by muscle contraction at intermediate and high contraction frequencies (15 and 60 CPM at 20 Hz) and at intermediate stimulus frequencies (20 Hz at 15 CPM), but not dilatation induced at low or high stimulus frequencies (4 or 60 Hz at 15 CPM). We therefore reasoned that the effect of PG synthesis inhibition on dilatation evoked by intermediate stimulus frequencies was probably an effect of the contraction frequency rather than the stimulus frequency. Therefore, we conclude that contraction frequency is a more important determinant of the contribution of PGs than stimulus frequency.
There are two potential mechanisms that may link contraction frequency to the production of PGs; one is the direct release of PGs from skeletal muscle that is influenced by how the muscle is contracted and the other is through flow-dependent responses. An influence of contraction frequency, rather than stimulus frequency, indicates that the number of times the muscle is stimulated, rather than the amount of force generated per contraction is an important determinant of PG production or effectiveness. Thus, the number of impulses of calcium/minute was important in the production of PG rather than the calcium/contraction. PLA2 isoforms are calcium dependent (for review see Burke & Dennis, 2009). In addition, there is evidence that skeletal muscle cells contain PLA2 (McLennan & Macdonald, 1991) and cyclooxygenase (Testa et al. 2007) and that PGs accumulate in muscle interstitium during exercise (Karamouzis et al. 2001a,b2001b) but there is little evidence to suggest that contracting skeletal muscle cells release PGs (Vandenburgh et al. 1990, 1995). However, endothelial cells can release PGs (for review see Davidge, 2001) and release from both adluminal and abluminal surfaces of vascular endothelium could explain the increase in interstitial PGs and in venous efflux of PGs. Although endothelial cells have been shown to release PGs in response to ADO in culture (Nyberg et al. 2010) endothelial cells also release PGs in response to increases in flow and shear stress (Koller & Kaley, 1990; Ward, 1999; Osanai et al. 2000; Giles et al. 2012). During the 2 min of our contraction protocol, we assume that blood flow increases in the microcirculation and it is probable that an increase in shear stress occurred in association with each new period of contraction. Thus, it is possible that the PGs that contributed to the arteriolar dilatation were released from the vascular endothelium by increased flow and shear stress and that this release was proportional to contraction frequency. We recognize that PGs released from venular endothelium have been shown to cause vasodilatation of the arteriole that runs alongside it during muscle contraction (MacKay et al. 1998) but this is not a key mechanism for the arteriolar dilatation recorded in the present study as the arterioles we observed were not paired with venules.
Prostaglandins involved in contraction-induced vasodilatations
As indicated in the Introduction, the contribution of PGs to exercise hyperaemia may not be clear, as there are reports that show a role for PGs during exercise (Kilbom & Wennmalm, 1976; Cowley et al. 1985; Wilson & Kapoor, 1993; Duffy et al. 1999; Farouque & Meredith, 2003; Schrage et al. 2004, 2010; Win & Marshall, 2005) while others do not (Beaty & Donald, 1979; Einstein & Goodman, 1980; Young & Sparks, 1980; Shoemaker et al. 1996; Saunders et al. 2005; Mortensen et al. 2007; Crecelius et al. 2011). A similar controversy exists at the microvascular level where a role for PGs in contraction-induced vasodilatation has been observed in cremaster arterioles paired with veins (McKay et al. 1998; Nuttle et al. 1999) but not in unpaired arterioles of the spinotrapezius muscle (Hammer & Boegehold, 2005). However, our data now suggest that whether PGs contribute to dilatation at the microvascular level depends on how the muscle is stimulated to contract. Thus, Hammer and Boeghold (2005) did not find a role for PGs in arteriolar dilatation when they evoked twitch contractions at 0.5, 1 or 2 Hz for 2 min, while Nuttle et al. (1999) only observed a role for PGs when using 1 Hz twitch contractions for 2 min and observing arterioles that were paired with a venule. Similarly, McKay et al. (1998) observed PG-dependent dilatations in arterioles that were paired with a venule in response to twitch contractions at 1 Hz. Our observation that PG-dependent dilatation did not occur in arterioles that were not paired with a venule when we used twitch contractions at 4 Hz, 15 CPM is therefore consistent with those findings. On the other hand, our finding that PG-dependent dilatation occurred in arterioles that were not paired with venules at higher frequency tetanic contractions (20 Hz, 15 CPM) allow us to suggest that low-frequency twitch contractions are only effective in inducing PG-dependent dilatation in arterioles that are paired with venules; this may be due to the proximity of the venular endothelium as a source of PGs. As intermediate frequency stimuli (20 Hz) at 60 CPM did cause PG-dependent dilatation in arterioles that were not paired with venules, we can assume that this type of contraction did release sufficient PGs from the microvascular endothelium, as discussed above. Previous studies have shown that the magnitude of the PG release into venous efflux and interstitium during muscle contraction is dependent on the contraction intensity (Young & Sparks, 1980; Symons et al. 1991; Wilson & Kapoor, 1993; Karamouzis et al. 2001a) and that there are differences in their relative concentrations of different PGs in static and dynamic contractions (Karamouzis et al. 2001b). In light of the present findings, it seems likely that discrepancies in the literature as to whether PGs do or do not contribute to exercise hyperaemia may be dependent on characteristics of the muscle contraction. This should be tested in future studies.
The clarity for the role of PGs in contraction-induced vasodilatation is further confounded by the potential for interactions of PGs with other vasodilators such as NO and ADO. There are data that support, at the whole muscle level measuring bulk blood flow, that ADO is an important mediator of exercise hyperaemia (for review see Marshall, 2007) and may promote an increase in blood flow through the production of both NO and PGs (Mortensen et al. 2009; Nyberg et al. 2010). We could not find evidence to support that extraluminal ADO produced vasodilatation through PGs, NO or both in the microvascular network, specifically 2A arterioles of the cremaster muscle, larger vessels in the cremaster muscle, the 1A arteriole, or in different muscles, 2A arterioles in the gluteus maximus muscle. The cremaster and gluteus maximus muscle are of similar fibre type but represent muscles that are functionally different, being non-locomotory and locomotory muscles, respectively. Our data indicate that the redundant interaction between extraluminal ADO and NO and PG is not a mechanism that either muscle employs at the microvascular level for the action of extraluminal ADO.
The method of application of ADO is an important consideration when experimentally testing the dependence of ADO on NO and PGs. Studies that involve infusion of ADO have shown an increase in the synthesis of NO (Ray & Marshall, 2005) and NO-and PG-dependent vasodilations (Ray et al. 2002; Mortensen et al. 2009) whereas ADO generated extracellularly during muscle contraction (Hellsten et al. 1998) may not increase muscle blood flow via NO (Ray et al. 2002; Ray & Marshall, 2009). Indeed, it has been shown that the ADO generated during muscle contraction does not increase muscle blood flow by acting via NO but ADO infused intraluminally does (Ray & Marshall, 2009) and causes arteriolar dilatation by acting via NO (Baker & Sutton, 1993). In our protocol, we applied ADO extraluminally, as would occur during skeletal muscle contraction, and find that extraluminal ADO does not vasodilatate via NO or PGs. Thus, it is possible that infused ADO activates vasodilatatory pathways primarily through endothelial cells and involves NO and PG production, whereas ADO produced extraluminally by skeletal muscle contraction, or applied extraluminally, primarily affects smooth muscle of the microvasculature and acts in an NO/PG independent manner. We acknowledge that ADO infused by microdialysis into muscle interstitial space caused an increase in the production of NO and PG (Nyberg et al. 2010) but this could have been via capillary or venular endothelium rather than arteriolar endothelium.
In further support of our conclusions that there is little interaction between extraluminal ADO, and NO and PGs at the arteriolar level, we have previously shown, using a protocol similar to that used in the present study, that ADO made a contribution to arteriolar dilatation at low and intermediate stimulus frequencies and low, intermediate and high contraction frequencies. (Dua et al. 2009). On the other hand, NO contributed to arteriolar dilatation at low, intermediate and high stimulus frequencies, but at only low and intermediate contraction frequencies. Both of these sets of parameters differ from those at which we found PGs to be contributing in the present study: intermediate stimulus frequencies and intermediate and high contraction frequencies. Taken together these findings indicate there are conditions in which ADO acts independently of PGs and/or NO to produce arteriolar vasodilatation and conditions in which PGs act independently of ADO and/or NO. Even at intermediate stimulus frequencies and intermediate contraction frequencies where theoretically all three could be acting interdependently, the present findings demonstrate this is not the case.
Experimental limitations
We used two different inhibitors of PG production, indomethacin, a specific inhibitor of cyclooxygenase, and quinacrine, a PLA2 inhibitor, which inhibits synthesis of the precursor of PGs. The muscle contraction and ADO application experiments were completed at different times and inhibitors were used when we had gained confidence and experience with each. The use of two different inhibitors does not detract from our overall observations and conclusions. We showed that PLA2 inhibition attenuated the arteriolar vasodilatation evoked by muscle contraction using specific contractile parameters. We used a PLA2 inhibitor to block the first step in PG synthesis by inhibition of the production of arachadonic acid, which acts as the substrate for the production of vasodilatory PGs through cyclooxygenase activity. Arachadonic acid is also the precursor for the production of epoxyeicosatrienoic and hypdoxyeicosastetraenioc acids through the activity of cytochrome P450 epoxygenase. Cytochrome P450 products have been shown to be vasoactive in skeletal muscle (Bolz et al. 2000; Frisbee et al. 2000; Huang et al. 2000, 2001; Wu et al. 2001) and have been identified as a potential endothelial-derived hyperpolarizing factor (EDHF) (Busse et al. 2002; Campbell & Falck, 2007). Inhibition of PLA2 will block the production of both PG and EDHF. The role of EDHF in the regulation of blood flow during muscle contraction is uncertain (Hillig et al. 2003; Mortensen et al. 2007). EDHF appears to mediate flow-induced dilatation in skeletal muscle arterioles in the absence of NO (Huang et al. 2001). Indeed, NO appears to inhibit the effectiveness of EDHF in that the effects of EDHF are difficult to observe unless NO synthesis is inhibited (Bauersachs et al. 1996; Nishikawa et al. 2000; Huang et al. 2001; Hillig et al. 2003). During our muscle contraction experiments, the NO synthesis pathways were intact, so we assume there was a minimal EDHF contribution. Further, the arteriolar vasodilatation evoked by arachadonic acid, the precursor for both cyclooxygenase and cytochrome P450-dependent vasoactive products, was completely abolished by INDO, the cyclooxygenase inhibitor in this tissue in the presence of NO synthase activity. Thus, we believe it is reasonable to assume that the inhibition of contraction-induced vasodilatation we observed in the presence of PLA2 was primarily due to the blockage of PG synthesis, although a small role for cytochrome P450 products cannot be completely ruled out.
Conclusions
We have shown that PGs are an important component of vasodilatation associated with muscle contraction at the microvascular level and the extent of their importance was dependent on the contractile characteristics of skeletal muscle. Importantly, we could not find evidence to support that PG and NO vasodilatations were secondary to extraluminal ADO, implying that contraction-induced vasodilatation through PG is not secondary to skeletal muscle production of ADO.
Key points
The role of prostaglandins in the changes in blood flow and microvascular vasodilation associated with exercise and muscle contraction is controversial.
Whether prostaglandins are produced independently during muscle contraction or whether their production is dependent on the production of adenosine is not well understood.
We show that prostaglandins are an important component of the microvascular vasodilation associated with muscle contraction but only under specific contractile conditions. Further, we show that microvascular vasodilation in response to adenosine is not dependent on prostaglandins.
Therefore, we conclude that there are specific contractile conditions under which prostaglandins are an important component of the vasodilation induced by muscle contraction and we propose that prostaglandin-induced microvascular vasodilation during exercise is independent of adenosine.
Acknowledgments
None.
Glossary
- ADO
adenosine
- INDO
indomethacin
- l-NAME
nitro-l-arginine methyl ester
- NO
nitric oxide
- PG
prostaglandin
- PLA2
phospholipase A2
- SNAP
S-nitroso-N-acetylpenicillamine
- TA
transverse arteriole
Additional information
Competing interests
None.
Author contributions
C.M. was responsible for the conception and experimental design, data analysis and interpretation and drafting the article. J.D., K.I., F.M., D.R. and J.S. were responsible for the conception and design of the experiments, collection, analysis and interpretation of the data. A.F. and J.S. were responsible for the collection, analysis and interpretation of fibre typing data. All authors have approved the final version of the manuscript and all persons designated as authors qualify for authorship.
Funding
This work was funded by NSERC, Canada.
References
- Agbulut O, Noirez P, Beaumont F, Butler-Browne G. Myosin heavy chain isoforms in postnatal muscle development of mice. Biol Cell. 2003;95:399–406. doi: 10.1016/s0248-4900(03)00087-x. [DOI] [PubMed] [Google Scholar]
- Al-Khazraji BK, Novielli NM, Goldman D, Medeiros PJ, Jackson DN. A simple ‘streak length method’ for quantifying and characterizing red blood cell velocity profiles and blood flow in rat skeletal muscle arterioles. Microcirculation. 2012;19:327–335. doi: 10.1111/j.1549-8719.2012.00165.x. [DOI] [PubMed] [Google Scholar]
- Anderson KM, Faber JE. Differential sensitivity of arteriolar alpha 1-and alpha 2-adrenoceptor constriction to metabolic inhibition during rat skeletal muscle contraction. Circ Res. 1991;69:174–184. doi: 10.1161/01.res.69.1.174. [DOI] [PubMed] [Google Scholar]
- Baez S. An open cremaster muscle preparation for the study of blood vessels by in vivo microscopy. Microvasc Res. 1973;5:384–394. doi: 10.1016/0026-2862(73)90054-x. [DOI] [PubMed] [Google Scholar]
- Baker CH, Sutton ET. Antagonism of acetylcholine and adenosine rat cremaster arteriolar vasodilation by combination of NO antagonists. Int J Microcirc Clin Exp. 1993;12:275–286. [PubMed] [Google Scholar]
- Bauersachs J, Popp R, Hecker M, Sauer E, Fleming I, Busse R. Nitric oxide attenuates the release of endothelium-derived hyperpolarizing factor. Circulation. 1996;94:3341–3347. doi: 10.1161/01.cir.94.12.3341. [DOI] [PubMed] [Google Scholar]
- Bearden SE, Payne GW, Chisty A, Segal SS. Arteriolar network architecture and vasomotor function with ageing in mouse gluteus maximus muscle. J Physiol. 2004;561:535–545. doi: 10.1113/jphysiol.2004.068262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaty O, 3rd, Donald DE. Contribution of prostaglandins to muscle blood flow in anaesthetized dogs at rest, during exercise, and following inflow occlusion. Circ Res. 1979;44:67–75. doi: 10.1161/01.res.44.1.67. [DOI] [PubMed] [Google Scholar]
- Blum H, Beier H, Gross HJ. Improved silver staining of plant proteins, RNA and DNA in polyacrylamide gels. Electrophoresis. 1987;8:93–99. [Google Scholar]
- Bolz SS, Fisslthaler B, Pieperhoff S, De Wit C, Fleming I, Busse R, Pohl U. Antisense oligonucleotides against cytochrome P450 2C8 attenuate EDHF-mediated Ca2+ changes and dilation in isolated resistance arteries. FASEB J. 2000;14:255–260. doi: 10.1096/fasebj.14.2.255. [DOI] [PubMed] [Google Scholar]
- Boushel R, Langberg H, Gemmer C, Olesen J, Crameri R, Scheede C, Sander M, Kjaer M. Combined inhibition of nitric oxide and prostaglandins reduces human skeletal muscle blood flow during exercise. J Physiol. 2002;543:691–698. doi: 10.1113/jphysiol.2002.021477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke JE, Dennis EA. Phospholipase A2 structure/function, mechanism, and signalling. J Lipid Res. 2009;50(Suppl):S237–S242. doi: 10.1194/jlr.R800033-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busse R, Edwards G, Feletou M, Fleming I, Vanhoutte PM, Weston AH. EDHF: bringing the concepts together. Trends Pharmacol Sci. 2002;23:374–380. doi: 10.1016/s0165-6147(02)02050-3. [DOI] [PubMed] [Google Scholar]
- Campbell WB, Falck JR. Arachidonic acid metabolites as endothelium-derived hyperpolarizing factors. Hypertension. 2007;49:590–596. doi: 10.1161/01.HYP.0000255173.50317.fc. [DOI] [PubMed] [Google Scholar]
- Cowley AJ, Stainer K, Rowley JM, Wilcox RG. Effect of aspirin and indomethacin on exercise-induced changes in blood pressure and limb blood flow in normal volunteers. Cardiovasc Res. 1985;19:177–180. doi: 10.1093/cvr/19.3.177. [DOI] [PubMed] [Google Scholar]
- Crecelius AR, Kirby BS, Voyles WF, Dinenno FA. Augmented skeletal muscle hyperaemia during hypoxic exercise in humans is blunted by combined inhibition of nitric oxide and vasodilating prostaglandins. J Physiol. 2011;589:3671–3683. doi: 10.1113/jphysiol.2011.209486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidge ST. Prostaglandin H synthase and vascular function. Circ Res. 2001;89:650–660. doi: 10.1161/hh2001.098351. [DOI] [PubMed] [Google Scholar]
- Dua AK, Dua N, Murrant CL. Skeletal muscle contraction-induced vasodilator complement production is dependent on stimulus and contraction frequency. Am J Physiol Heart Circ Physiol. 2009;297:H433–H442. doi: 10.1152/ajpheart.00216.2009. [DOI] [PubMed] [Google Scholar]
- Duffy SJ, New G, Tran BT, Harper RW, Meredith IT. Relative contribution of vasodilator prostanoids and NO to metabolic vasodilation in the human forearm. Am J Physiol Heart Circ Physiol. 1999;276:H663–H670. doi: 10.1152/ajpheart.1999.276.2.H663. [DOI] [PubMed] [Google Scholar]
- Einstein R, Goodman AH. Prostaglandins in the control of blood flow in canine skeletal muscle. Clin Exp Pharmacol Physiol. 1980;7:129–138. doi: 10.1111/j.1440-1681.1980.tb00054.x. [DOI] [PubMed] [Google Scholar]
- Farouque HM, Meredith IT. Relative contribution of vasodilator prostanoids, NO, and KATP channels to human forearm metabolic vasodilation. Am J Physiol Heart Circ Physiol. 2003;284:H2405–H2411. doi: 10.1152/ajpheart.00879.2002. [DOI] [PubMed] [Google Scholar]
- Frandsenn U, Bangsbo J, Sander M, Hoffner L, Betak A, Saltin B, Hellsten Y. Exercise-induced hyperaemia and leg oxygen uptake are not altered during effective inhibition of nitric oxide synthase with NG-nitro-l-arginine methyl ester in humans. J Physiol. 2001;531:257–264. doi: 10.1111/j.1469-7793.2001.0257j.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisbee JC, Roman RJ, Falck JR, Linderman JR, Lombard JH. Impairment of flow-induced dilation of skeletal muscle arterioles with elevated oxygen in normotensive and hypertensive rats. Microvasc Res. 2000;60:37–48. doi: 10.1006/mvre.2000.2245. [DOI] [PubMed] [Google Scholar]
- Giles TD, Sander GE, Nossaman BD, Kadowitz PJ. Impaired vasodilation in the pathogenesis of hypertension: focus on nitric oxide, endothelial-derived hyperpolarizing factors, and prostaglandins. J Clin Hypertens (Greenwich) 2012;14:198–205. doi: 10.1111/j.1751-7176.2012.00606.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer LW, Boegehold MA. Functional hyperemia is reduced in skeletal muscle of aged rats. Microcirculation. 2005;12:517–526. doi: 10.1080/10739680591003396. [DOI] [PubMed] [Google Scholar]
- Hammer LW, Ligon AL, Hester RL. Differential inhibition of functional dilation of small arterioles by indomethacin and glibenclamide. Hypertension. 2001;37:599–603. doi: 10.1161/01.hyp.37.2.599. [DOI] [PubMed] [Google Scholar]
- Hellsten Y, Maclean D, Radegran G, Saltin B, Bangsbo J. Adenosine concentrations in the interstitium of resting and contracting human skeletal muscle. Circulation. 1998;98:6–8. doi: 10.1161/01.cir.98.1.6. [DOI] [PubMed] [Google Scholar]
- Hester RL, Eraslan A, Saito Y. Differences in EDNO contribution to arteriolar diameters at rest and during functional dilation in striated muscle. Am J Physiol Heart Circ Physiol. 1993;265:H146–H151. doi: 10.1152/ajpheart.1993.265.1.H146. [DOI] [PubMed] [Google Scholar]
- Hillig T, Krustrup P, Fleming I, Osada T, Saltin B, Hellsten Y. Cytochrome P450 2C9 plays an important role in the regulation of exercise-induced skeletal muscle blood flow and oxygen uptake in humans. J Physiol. 2003;546:307–314. doi: 10.1113/jphysiol.2002.030833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang A, Sun D, Smith CJ, Connetta JA, Shesely EG, Koller A, Kaley G. In eNOS knockout mice skeletal muscle arteriolar dilation to acetylcholine is mediated by EDHF. Am J Physiol Heart Circ Physiol. 2000;278:H762–H768. doi: 10.1152/ajpheart.2000.278.3.H762. [DOI] [PubMed] [Google Scholar]
- Huang A, Sun D, Carroll MA, Jiang H, Smith CJ, Connetta JA, Falck JR, Shesely EG, Koller A, Kaley G. EDHF mediates flow-induced dilation in skeletal muscle arterioles of female eNOS-KO mice. Am J Physiol Heart Circ Physiol. 2001;280:H2462–H2469. doi: 10.1152/ajpheart.2001.280.6.H2462. [DOI] [PubMed] [Google Scholar]
- Joyner MJ, Wilkins BW. Exercise hyperaemia: is anything obligatory but the hyperaemia. J Physiol. 2007;583:855–860. doi: 10.1113/jphysiol.2007.135889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalliokoski KK, Langberg H, Ryberg AK, Scheede-Bergdahl C, Doessing S, Kjaer A, Kjaer M, Boushel R. Nitric oxide and prostaglandins influence local skeletal muscle blood flow during exercise in humans: coupling between local substrate uptake and blood flow. Am J Physiol Regul Integr Comp Physiol. 2006;291:R803–R809. doi: 10.1152/ajpregu.00808.2005. [DOI] [PubMed] [Google Scholar]
- Karamouzis M, Karamouzis I, Vamvakoudis E, Ampatzidis G, Christoulas K, Angelopoulou N, Mandroukas K. The response of muscle interstitial prostaglandin E2 (PGE2), prostacyclin I2 (PGI2) and thromboxane A2 (TXA2) levels during incremental dynamic exercise in humans determined by in vivo microdialysis. Prostaglandins Leukot Essent Fatty Acids. 2001a;64:259–263. doi: 10.1054/plef.2001.0269. [DOI] [PubMed] [Google Scholar]
- Karamouzis M, Langberg H, Skovgaard D, Bulow J, Kjaer M, Saltin B. In situ microdialysis of intramuscular prostaglandin and thromboxane in contracting skeletal muscle in humans. Acta Physiol Scand. 2001b;171:71–76. doi: 10.1046/j.1365-201X.2001.00775.x. [DOI] [PubMed] [Google Scholar]
- Kilbom A, Wennmalm A. Endogenous prostaglandins as local regulators of blood flow in man: effect of indomethacin on reactive and functional hyperaemia. J Physiol. 1976;257:109–121. doi: 10.1113/jphysiol.1976.sp011358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klitzman B, Damon DN, Gorczynski RJ, Duling BR. Augmented tissue oxygen supply during striated muscle contraction in the hamster. Relative contributions of capillary recruitment, functional dilation, and reduced tissue PO2. Circ Res. 1982;51:711–721. doi: 10.1161/01.res.51.6.711. [DOI] [PubMed] [Google Scholar]
- Koller A, Kaley G. Prostaglandins mediate arteriolar dilation to increased blood flow velocity in skeletal muscle microcirculation. Circ Res. 1990;67:529–534. doi: 10.1161/01.res.67.2.529. [DOI] [PubMed] [Google Scholar]
- Lindbom L, Arfors KE. Non-homogeneous blood flow distribution in the rabbit tenuissimus muscle. Differential control of total blood flow and capillary perfusion. Acta Physiol Scand. 1984;122:225–233. doi: 10.1111/j.1748-1716.1984.tb07505.x. [DOI] [PubMed] [Google Scholar]
- Marshall JM. The roles of adenosine and related substances in exercise hyperaemia. J Physiol. 2007;583:835–845. doi: 10.1113/jphysiol.2007.136416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson JP, Miller TA, Poole DC, Delp MD. Fiber composition and oxidative capacity of hamster skeletal muscle. J Histochem Cytochem. 2002;50:1685–1692. doi: 10.1177/002215540205001214. [DOI] [PubMed] [Google Scholar]
- McKay MK, Gardner AL, Boyd D, Hester RL. Influence of venular prostaglandin release on arteriolar diameter during functional hyperemia. Hypertension. 1998;31:213–217. doi: 10.1161/01.hyp.31.1.213. [DOI] [PubMed] [Google Scholar]
- McLennan IS, Macdonald RE. Prostaglandin synthetase and prostacyclin synthetase in mature rat skeletal muscles: immunohistochemical localisation to arterioles, tendons and connective tissues. J Anat. 1991;178:243–253. [PMC free article] [PubMed] [Google Scholar]
- Mortensen SP, Gonzalez-Alonso J, Damsgaard R, Saltin B, Hellsten Y. Inhibition of nitric oxide and prostaglandins, but not endothelial-derived hyperpolarizing factors, reduces blood flow and aerobic energy turnover in the exercising human leg. J Physiol. 2007;581:853–861. doi: 10.1113/jphysiol.2006.127423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortensen SP, Nyberg M, Thaning P, Saltin B, Hellsten Y. Adenosine contributes to blood flow regulation in the exercising human leg by increasing prostaglandin and nitric oxide formation. Hypertension. 2009;53:993–999. doi: 10.1161/HYPERTENSIONAHA.109.130880. [DOI] [PubMed] [Google Scholar]
- Murphy AM, Solaro RJ. Developmental difference in the stimulation of cardiac myofibrillar Mg2+-ATPase activity by calmidazolium. Pediatr Res. 1990;28:46–49. doi: 10.1203/00006450-199007000-00011. [DOI] [PubMed] [Google Scholar]
- Murrant CL. Stimulation characteristics that determine arteriolar dilation in skeletal muscle. Am J Physiol Regul Integr Comp Physiol. 2005;289:R505–R513. doi: 10.1152/ajpregu.00571.2004. [DOI] [PubMed] [Google Scholar]
- Murrant CL, Sarelius IH. Local and remote arteriolar dilations initiated by skeletal muscle contraction. Am J Physiol Heart Circ Physiol. 2000;279:H2285–H2294. doi: 10.1152/ajpheart.2000.279.5.H2285. [DOI] [PubMed] [Google Scholar]
- Murrant CL, Sarelius IH. Multiple dilator pathways in skeletal muscle contraction-induced arteriolar dilations. Am J Physiol Regul Integr Comp Physiol. 2002;282:R969–R978. doi: 10.1152/ajpregu.00405.2001. [DOI] [PubMed] [Google Scholar]
- Newcomer SC, Taylor JC, McAllister RM, Laughlin MH. Effects of chronic nitric oxide synthase inhibition on endothelium-dependent and-independent relaxation in arteries that perfuse skeletal muscle of swine. Endothelium. 2008;15:17–31. doi: 10.1080/10623320802092211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa Y, Stepp DW, Chilian WM. Nitric oxide exerts feedback inhibition on EDHF-induced coronary arteriolar dilation in vivo. Am J Physiol Heart Circ Physiol. 2000;279:H459–H465. doi: 10.1152/ajpheart.2000.279.2.H459. [DOI] [PubMed] [Google Scholar]
- Nuttle LC, Ligon AL, Farrell KR, Hester RL. Inhibition of phospholipase A2 attenuates functional hyperemia in the hamster cremaster muscle. Am J Physiol Heart Circ Physiol. 1999;276:H1289–H1294. doi: 10.1152/ajpheart.1999.276.4.H1289. [DOI] [PubMed] [Google Scholar]
- Nyberg M, Mortensen SP, Thaning P, Saltin B, Hellsten Y. Interstitial and plasma adenosine stimulate nitric oxide and prostacyclin formation in human skeletal muscle. Hypertension. 2010;56:1102–1108. doi: 10.1161/HYPERTENSIONAHA.110.161521. [DOI] [PubMed] [Google Scholar]
- Ohyanagi M, Faber JE, Nishigaki K. Differential activation of alpha 1-and alpha 2-adrenoceptors on microvascular smooth muscle during sympathetic nerve stimulation. Circ Res. 1991;68:232–244. doi: 10.1161/01.res.68.1.232. [DOI] [PubMed] [Google Scholar]
- Osanai T, Fujita N, Fujiwara N, Nakano T, Takahashi K, Guan W, Okumura K. Cross talk of shear-induced production of prostacyclin and nitric oxide in endothelial cells. Am J Physiol Heart Circ Physiol. 2000;278:H233–H238. doi: 10.1152/ajpheart.2000.278.1.H233. [DOI] [PubMed] [Google Scholar]
- Pohl U, De Wit C, Gloe T. Large arterioles in the control of blood flow: role of endothelium-dependent dilation. Acta Physiol Scand. 2000;168:505–510. doi: 10.1046/j.1365-201x.2000.00702.x. [DOI] [PubMed] [Google Scholar]
- Radegran G, Saltin B. Nitric oxide in the regulation of vasomotor tone in human skeletal muscle. Am J Physiol Heart Circ Physiol. 1999;276:H1951–H1960. doi: 10.1152/ajpheart.1999.276.6.H1951. [DOI] [PubMed] [Google Scholar]
- Ray CJ, Marshall JM. Measurement of nitric oxide release evoked by systemic hypoxia and adenosine from rat skeletal muscle in vivo. J Physiol. 2005;568:967–978. doi: 10.1113/jphysiol.2005.094854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray CJ, Marshall JM. Nitric oxide (NO) does not contribute to the generation or action of adenosine during exercise hyperaemia in rat hindlimb. J Physiol. 2009;587:1579–1591. doi: 10.1113/jphysiol.2008.163691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray CJ, Abbas MR, Coney AM, Marshall JM. Interactions of adenosine, prostaglandins and nitric oxide in hypoxia-induced vasodilatation: in vivo and in vitro studies. J Physiol. 2002;544:195–209. doi: 10.1113/jphysiol.2002.023440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarelius IH( Cell flow path influences transit time through striated muscle capillaries. Am J Physiol Heart Circ Physiol. 1986;250:H899–H907. doi: 10.1152/ajpheart.1986.250.6.H899. [DOI] [PubMed] [Google Scholar]
- Saunders NR, Dinenno FA, Pyke KE, Rogers AM, Tschakovsky ME. Impact of combined NO and PG blockade on rapid vasodilation in a forearm mild-to-moderate exercise transition in humans. Am J Physiol Heart Circ Physiol. 2005;288:H214–H220. doi: 10.1152/ajpheart.00762.2004. [DOI] [PubMed] [Google Scholar]
- Schrage WG, Joyner MJ, Dinenno FA. Local inhibition of nitric oxide and prostaglandins independently reduces forearm exercise hyperaemia in humans. J Physiol. 2004;557:599–611. doi: 10.1113/jphysiol.2004.061283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrage WG, Wilkins BW, Johnson CP, Eisenach JH, Limberg JK, Dietz NM, Curry TB, Joyner MJ. Roles of nitric oxide synthase and cyclooxygenase in leg vasodilation and oxygen consumption during prolonged low-intensity exercise in untrained humans. J Appl Physiol. 2010;109:768–777. doi: 10.1152/japplphysiol.00326.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker JK, Naylor HL, Pozeg ZI, Hughson RL. Failure of prostaglandins to modulate the time course of blood flow during dynamic forearm exercise in humans. J Appl Physiol. 1996;81:1516–1521. doi: 10.1152/jappl.1996.81.4.1516. [DOI] [PubMed] [Google Scholar]
- Sweeney TE, Sarelius IH. Arteriolar control of capillary cell flow in striated muscle. Circ Res. 1989;64:112–120. doi: 10.1161/01.res.64.1.112. [DOI] [PubMed] [Google Scholar]
- Symons JD, Theodossy SJ, Longhurst JC, Stebbins CL. Intramuscular accumulation of prostaglandins during static contraction of the cat triceps surae. J Appl Physiol. 1991;71:1837–1842. doi: 10.1152/jappl.1991.71.5.1837. [DOI] [PubMed] [Google Scholar]
- Tateishi J, Faber JE. Inhibition of arteriole alpha 2-but not alpha 1-adrenoceptor constriction by acidosis and hypoxia in vitro. Am J Physiol Heart Circ Physiol. 1995;268:H2068–H2076. doi: 10.1152/ajpheart.1995.268.5.H2068. [DOI] [PubMed] [Google Scholar]
- Testa M, Rocca B, Spath L, Ranelletti FO, Petrucci G, Ciabattoni G, Naro F, Schiaffino S, Volpe M, Reggiani C. Expression and activity of cyclooxygenase isoforms in skeletal muscles and myocardium of humans and rodents. J Appl Physiol. 2007;103:1412–1418. doi: 10.1152/japplphysiol.00288.2007. [DOI] [PubMed] [Google Scholar]
- Twynstra J, Ruiz DA, Murrant CL. Functional coordination of the spread of vasodilations through skeletal muscle microvasculature: implications for blood flow control. Acta Physiol (Oxf) 2012;206:229–241. doi: 10.1111/j.1748-1716.2012.02465.x. [DOI] [PubMed] [Google Scholar]
- Vandenburgh HH, Hatfaludy S, Sohar I, Shansky J. Stretch-induced prostaglandins and protein turnover in cultured skeletal muscle. Am J Physiol Cell Physiol. 1990;259:C232–C240. doi: 10.1152/ajpcell.1990.259.2.C232. [DOI] [PubMed] [Google Scholar]
- Vandenburgh HH, Shansky J, Solerssi R, Chromiak J. Mechanical stimulation of skeletal muscle increases prostaglandin F2 alpha production, cyclooxygenase activity, and cell growth by a pertussis toxin sensitive mechanism. J Cell Physiol. 1995;163:285–294. doi: 10.1002/jcp.1041630209. [DOI] [PubMed] [Google Scholar]
- Ward ME. Dilation of rat diaphragmatic arterioles by flow and hypoxia: roles of nitric oxide and prostaglandins. J Appl Physiol. 1999;86:1644–1650. doi: 10.1152/jappl.1999.86.5.1644. [DOI] [PubMed] [Google Scholar]
- Wilson JR, Kapoor SC. Contribution of prostaglandins to exercise-induced vasodilation in humans. Am J Physiol Heart Circ Physiol. 1993;265:H171–H175. doi: 10.1152/ajpheart.1993.265.1.H171. [DOI] [PubMed] [Google Scholar]
- Win TS, Marshall JM. Contribution of prostaglandins to the dilation that follows isometric forearm contraction in human subjects: effects of aspirin and hyperoxia. J Appl Physiol. 2005;99:45–52. doi: 10.1152/japplphysiol.01289.2004. [DOI] [PubMed] [Google Scholar]
- Wu Y, Huang A, Sun D, Falck JR, Koller A, Kaley G. Gender-specific compensation for the lack of NO in the mediation of flow-induced arteriolar dilation. Am J Physiol Heart Circ Physiol. 2001;280:H2456–H2461. doi: 10.1152/ajpheart.2001.280.6.H2456. [DOI] [PubMed] [Google Scholar]
- Young EW, Sparks HV. Prostaglandins and exercise hyperemia of dog skeletal muscle. Am J Physiol Heart Circ Physiol. 1980;238:H191–H195. doi: 10.1152/ajpheart.1980.238.2.H191. [DOI] [PubMed] [Google Scholar]