

Abstract
Background
Emerging bacterial zoonoses in bats and rodents remain relatively understudied. We conduct the first comparative host–pathogen coevolutionary analyses of bacterial pathogens in these hosts, using Bartonella spp. and Leptospira spp. as a model.
Methodology/Principal Findings
We used published genetic data for 51 Bartonella genotypes from 24 bat species, 129 Bartonella from 38 rodents, and 26 Leptospira from 20 bats. We generated maximum likelihood and Bayesian phylogenies for hosts and bacteria, and tested for coevoutionary congruence using programs ParaFit, PACO, and Jane. Bartonella spp. and their bat hosts had a significant coevolutionary fit (ParaFitGlobal = 1.9703, P≤0.001; m2 global value = 7.3320, P≤0.0001). Bartonella spp. and rodent hosts also indicated strong overall patterns of cospeciation (ParaFitGlobal = 102.4409, P≤0.001; m2 global value = 86.532, P≤0.0001). In contrast, we were unable to reject independence of speciation events in Leptospira and bats (ParaFitGlobal = 0.0042, P = 0.84; m2 global value = 4.6310, P = 0.5629). Separate analyses of New World and Old World data subsets yielded results congruent with analysis from entire datasets. We also conducted event-based cophylogeny analyses to reconstruct likely evolutionary histories for each group of pathogens and hosts. Leptospira and bats had the greatest number of host switches per parasite (0.731), while Bartonella and rodents had the fewest (0.264).
Conclusions/Significance
In both bat and rodent hosts, Bartonella exhibits significant coevolution with minimal host switching, while Leptospira in bats lacks evolutionary congruence with its host and has high number of host switches. Reasons underlying these variable coevolutionary patterns in host range are likely due to differences in disease-specific transmission and host ecology. Understanding the coevolutionary patterns and frequency of host-switching events between bacterial pathogens and their hosts will allow better prediction of spillover between mammal reservoirs, and ultimately to humans.
Author Summary
Bats and rodents are important hosts for emerging human diseases. While a large body of research has focused on viral pathogens in these hosts, the diversity, evolution, and transmission of their bacterial pathogens remains relatively unstudied. We conducted co-evolutionary analyses of two bacterial genera know to be pathogenic in humans, Bartonella and Leptospira, along with their bat and rodent hosts. We found that Bartonella had a significant pattern of coevolution with both bat and rodent hosts, while Leptospira in bats showed a lack of congruence with its bat hosts and a high number of host switching events. Our statistically driven approach to understand the frequency of host switching events in these mammal–bacterial systems can be easily applied to other host–pathogen systems, including viruses, to assess the likelihood of zoonotic spillover.
Introduction
Bats and rodents are the two most diverse and geographically widespread orders of mammals [1], [2], and are important reservoirs for a growing number of emerging infectious diseases (EIDs) with significant impacts on public health. Bats are reservoir hosts of several viral pathogens of high consequence, including Henipaviruses, Ebola and Marburg viruses, lyssaviruses, Severe Acute Respiratory Syndrome coronavirus, and likely Middle Eastern Respiratory Syndrome coronavirus [3]–[5]. Rodents are known reservoirs of hantaviruses, arenaviruses, Lassa fever virus, plague and other bacterial zoonoses [6]. Over the last two decades, the majority of research on bat and rodent zoonotic diseases has focused on viral infections (Figure S1). While the number of virus-related publications for bats has had a marked rise over the past decade, research on bacteria in bats has remained consistently low (Figure S1). The evolutionary relationships between these important mammalian hosts and their known bacterial pathogens has been little studied to date [7], [8].
Bats and rodents are evolutionarily ancient orders of mammals, with periods of diversification dating back 75 and 85 million years ago, respectively, thus allowing ample time for pathogens and hosts to coevolve [9]. Bats and rodents make up 60% of all extant mammal species while exhibiting a wide-range of life-history and ecological traits. Ecological, evolutionary, and life-history traits can influence pathogen richness and cross species transmission, or spillover, in these bat and rodent hosts [5], [8]–[11]. The peridomestic habits of these mammals also likely increase the frequency of human contact and facilitate disease spillover [12], [13]. Anthropogenic alterations that increase exposure to bats and rodents, including expanding agricultural operations, bushmeat hunting, and climate change, may increase the opportunity for diseases to emerge in human populations in the future [14]. How these ecological and life history factors may affect the coevolutionary patterns between reservoir host species and their associated pathogens is an open question, but will depend on characteristics related to pathogen transmission and host ecology.
The evolutionary patterns of hosts and their known pathogens can be used to quantify the frequency of spillover events within and between reservoir hosts, and is a crucial first step for developing predictive models for zoonotic disease emergence. Previous research has demonstrated how these coevolutionary studies can shed light on specific instances of host switching, cospeciation, and other events in coronaviruses and their bat hosts [15], as well as malaria parasites and their avian hosts [16]. However, to our knowledge, no comparative cophylogenetic analysis of bacterial pathogens has been applied yet to bat and rodent hosts. Here we examine host-pathogen evolution in bats and rodents using two bacterial genera, Bartonella spp. and Leptospira spp., known to cause neglected tropical diseases in humans.
The genus Bartonella consists of globally distributed and highly diverse alpha-proteobacteria that infects a wide-range of mammals. After infection, the bacteria eventually enters erythrocytes and endothelial cells and can persist asymptomatically in a wide range of mammalian reservoir hosts [17]. The disease is mainly transmitted through arthropod vectors including fleas, flies, lice, mites, and ticks [18]–[21]. Thus, the transmission and evolution of Bartonella species in mammals is the result of a complex relationship between multiple hosts, vectors, and pathogens. Bartonella has been reported with high prevalence and genetic diversity from numerous recent studies in bats [22]–[25] and rodents [26]–[30]. Bartonella is recognized as a neglected tropical disease, and there are indications of human infections derived from neighboring wildlife populations. In Thailand, genetic studies have indicated highly similar Bartonella strains between infected humans and nearby rodent populations [31]–[34]. Neighboring rodents have also been implicated as a possible source for bartonellosis in the United States and Nigeria [35], [36].
Leptospira is a genus of spirochete bacteria which also has a wide geographical distribution [37], and has been recognized as an important emerging pathogen due to its increasing incidence in both developing and developed countries [38]. Leptospires are maintained in nature by a large variety of wild and domestic animal hosts, and the bacteria colonize their kidneys and are excreted in their urine [39]. Rodents were the first recognized carriers, though the bacteria has been isolated from almost all screened mammals. Recently, bats have been found to carry Leptospira in Madagascar, Australia, Peru, and Brazil, and seroprevalence has been recorded to be as high as 35% [40]–[44]. Unlike Bartonella spp., Leptospira spp. are not vector-borne, and transmission to humans and other hosts is primarily through contact with water and environments contaminated with infected animal urine [45]. While most research has focused on rodents reservoirs of leptospirosis, recent genetic studies have also indicated bats as carriers of the bacteria [41], [44].
In order to better understand the evolutionary dynamics of Bartonella and Leptospira in bat and rodent hosts, we compiled available genetic information from hosts and bacterial pathogens to determine cophylogenetic patterns on a global scale. Evidence of cophylogeny can be used to test hypotheses of coevolution, and a lack of congruence between host and pathogen phylogenies can identify pathogen spillover, or interspecific transmission, events [46]. Long associations through evolutionary time can lead to reciprocal adaptations in both the hosts and their parasites, as well as concurrent divergence events in the two lineages. Evolutionary events including strict codivergence, parasite duplication, parasite extinction, and parasite host switching, will either strengthen or diminish the congruence between host and parasite [47]. Patterns of host-parasite or host-pathogen congruence may also vary geographically. For example, host specificity of Bartonella was observed in Old World bats in Kenya [24], while bats in Peru and Guatemala in the New World appeared to have no specific Bartonella-bat relationships [22], [48]. In contrast, consistency is observed for Bartonella in rodents, with host-specificity apparent in both Old and New World [49]–[51].
The primary goal of this paper is to examine the global co-evolutionary patterns of bats, rodents and their associated bacterial pathogens – using Bartonella and Leptospira as case studies. We specifically test for evolutionary congruence between bat host species and Bartonella and Leptospira, as well as rodent host species and Bartonella. Analysis of rodent Leptospira was unfortunately excluded due to a lack of comparable sequence datasets and host taxonomic diversity. Although there is a long history of research on leptospirosis in rodents, the publicly available sequence data that has been obtained thus far covers only a handful of rodent host species distributed across 3 genes: secY, flab,and lipL3 [52]–[56]. We also test whether evolutionary patterns and bacterial host specificity differ between the New World and Old World bat and rodent hosts, as was previously observed for Bartonella [22], [24], [48]. Finally, we conduct event-based cophylogeny analyses to reconstruct likely evolutionary histories for each group of pathogens and hosts.
Materials and Methods
Compiled sequence data
Sequence data used for analyses were obtained by searching for relevant papers from 1900–2013 through online sources PubMed, Web of Science, and Google Scholar using keywords “Bartonella*” and “Leptospir*” combined with “bat OR Chiroptera*” or “rodent*”. All Bartonella and Leptospira sequences from bat or rodent hosts identified to the species level were compiled into our datasets (Tables S1, S2, S3). Bat hosts include individuals in the Artibeus, Brachyphylla, Carollia, Coleura, Desmodus, Eidolon, Glossophaga. Hipposideros, Lonchophylla, Micronycteris, Mimon, Miniopterus, Monophyllus, Myotis, Nyctalus, Otomops, Phyllostomus, Promops, Pteronotus, Rousettus, Rhinophylla, Sturnira, Triaenops, Uroderma, and Vampyressa genera (Table S1, S3). Rodent hosts include individuals in the Acomys, Aethomys, Apodemus, Callosciurus, Clethrionomys, Dryomys, Gerbillus, Glaucomys, Jaculus, Mastomys, Microtus, Mus, Myodes, Niviventer, Pachyuromys, Peromyscus, Psammomys, Rattus, Rhabdomys, Sekeetamys, Spermophilus, Tamias, Tamiasciurus, Tatera, and Urocitellus genera (Table S2). Only unique genotypes were included in the dataset. The largest comparable genetic datasets consisted of the partial citrate synthase gene (gltA) for Bartonella and 16S rRNA gene for Leptospira, and these were selected for analysis. Cytochrome b gene sequences from all bat and rodent host species were obtained from GenBank (Tables S4, S5), as this mitochondrial gene has proven to be useful for within Order, species-level resolution of mammalian phylogenies [57]–[59]. For host species that did not have an available cytochrome b sequence, the most closely related species with available sequence was used as a substitute for host-parasite associations. For bats, we made four substitutions: Hipposideros armiger for Hipposideros commersoni, Phyllostomus hastatus for Phyllostomus discolor, Promops centralis for Promops nasutus, and Triaenops persicus for Triaenops menamena. Our results suggest that these genus-level host substitutions do not disrupt overall co-phylogenetic patterns. For all species, host taxonomy was synonymized according to Mammal Species of the World 3rd Edition [60].
In total, we compiled sequences from 51 Bartonella genotypes (38 New World, 13 Old World) from 24 bat species (15 New World, 9 Old World), and 129 (20 New World, 109 Old World) Bartonella genotypes from 38 rodent species (4 New World, 35 Old World). We also compiled sequences from 26 Leptospira genotypes (19 New World, 7 Old World) from 20 bat species (14 New World, 6 Old World). Insufficient genetic data of one gene for Leptospira in rodents precluded their use in the analysis; therefore only Leptospira in bat hosts was examined.
Phylogenetic analysis of sequence data
Bacterial and host species sequences were imported from GenBank into Geneious Pro 5.0.4. Sequences for each bacterial genus and their corresponding bat and rodent hosts were each aligned using default parameters in MUSCLE [61] as implemented in Geneious [62]. Outgroup taxa, obtained from GenBank, were included in each alignment, and were chosen based on previous species-level phylogenies. The outgroup for Bartonella was Brucella melitensis [25], for Leptospira was Leptonema illini [44], and for the bat and rodent hosts was the duck-billed platypus, Ornithorhynchus anatinus HQ379861 [63]. In order analyze the difference in host-specificity between Old and New World geographic regions, each alignment was further divided into Old and New World. Alignments were inspected visually and ends were trimmed and gaps found in only one non-outgroup sequence were deleted due to high likelihood of sequencing error. After these edits, this resulted in 1,133 base pairs (bp) for cytb bat sequences, 338 bp for gltA Bartonella sequences, and 1,246 bp for 16S Leptospira sequences.
Maximum likelihood (ML) phylogenetic trees were generated using RAxML 7.0.4 [64] implemented with the Cyberinfrastructure for Phylogenetic Research (CIPRES) Portal (www.phylo.org) using the substitution model GTRMIX, which determines an optimal tree by comparing likelihood scores under a GTR+G model. The number of bootstrap replicates were determined using the previously described stopping criteria. In order to corroborate the phylogenies as determined through ML, Bayesian inference (BI) host phylogenies were also generated using MrBayes 3.1.2 [65]. We utilized a GTR+I+G substitution model, with 10,000,000 generations, sampling every 5000th generation with 4 heated chains and a burn in length of 1,000,000.
Comparison of host and bacterial phylogenies
To visualize host-bacteria associations, tanglegrams were generated from the best ML trees in TreeMap 3.0 [66]. For cophylogenetic analyses, we utilized both global fit as well as event-based methods. We selected programs that are capable of accounting for evolutionary patterns given association of parasite species to multiple hosts, as well as the presence of multiple parasites in a single host.
Global-fit methods were used to quantify the degree of congruence between two given host and parasite topologies, and identify the individual associations contributing to the cophylogenetic structure [67]. First, global-fit analysis was tested using distance-based ParaFit [68], using matrices of patristic distances calculated from maximum likelihood host and parasite phylogenies in R 3.0.1 [69]. With an additional matrix of host-parasite links, ParaFit analyses [68] were also performed in R using package ape [70] with 999 permutations to implement a global test as well as individual links. Each individual host-bacteria interaction is determined to be significant if either its ParaFit 1 or Parafit 2 p-value≤0.05, and these significant interactions are shown in solid lines in the tanglegrams.
As ParaFit tends to be liberal with its values, we also implemented newly developed program Procrustean Approach to Cophylogeny (PACo) [71] in R using packages ape and vegan [72] in order to obtain, and potentially corroborate, comparable global goodness-of-fit statistics with Parafit global values. PACo differs from ParaFit by utilizing Procrustean superimposition, in which the parasite matrix is rotated and scaled to fit the host matrix. Thus, PACo explicitly tests the dependence of the parasite phylogeny upon the host phylogeny.
We then used event-based program Jane 4 [73] to determine the most probable coevolutionary history of the associated host and parasites, again using the ML host and bacteria trees as input. We assigned different relative costs to 5 possible evolutionary events, in a method similar to previous research efforts [74]. We performed analyses with 100 generations, population sizes of 100, and a default cost setting matrix of 0 for cospeciation, 1 for duplication of parasites, 2 for duplication and host switch, 1 for loss of parasite, and 1 for failure to diverge. In further runs, we changed one of the possible events to a cost of 10 each time, rendering that event prohibitively expensive. By further exploring the parameter space this way, we determined how these changes affected the overall costs of the optimal evolutionary history.
Results
Phylogenetic analysis
The topology of the BI tree was identical to that of the ML tree, except for a few branches with low support values. Thus, only the ML trees are presented here and used for further cophylogenetic analyses. Phylogenies tend to be well supported for more recent divergence events, but not deeper nodes. Nodes with bootstrap values ≥50 are labeled on all tanglegrams (Figures 1–6).
Figure 1. Tanglegram of cophylogenetic relationships between New World bat hosts and Bartonella.

Maximum likelihood phylogenies for Bartonella bacteria (yellow) and their New World bat hosts (blue), with bootstrap support values ≥50 labeled, rooted with outgroups. All host-pathogen associations are shown in the tanglegram as gray and black connecting lines. Black lines indicate significant individual cospeciation links between Bartonella and their hosts as indicated by ParaFit (P≤0.05), while gray lines represent non-significant links. Bat species that did not have an available cytochrome b sequence on GenBank, is substituted with a closely related species. Phyllostomus hastatus substituted for Phyllostomus discolor.
Figure 6. Tanglegram of cophylogenetic relationships between Old World bat hosts and Leptospira.
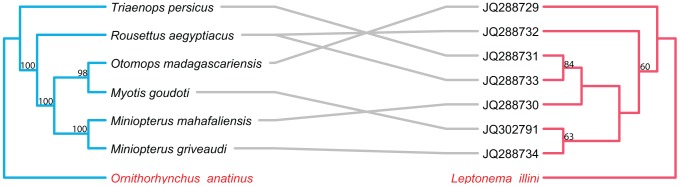
Maximum likelihood phylogenies for Leptospira bacteria (pink) and their Old World bat hosts (blue), with bootstrap support values ≥50 labeled, rooted with outgroups. All host-pathogen associations are shown in the tanglegram as gray connecting lines, and are significant as indicated by ParaFit (P≤0.05). Bat species that did not have an available cytochrome b sequence on GenBank, is substituted with a closely related species. Triaenops persicus substituted for Triaenops menamena.
Global-fit cophylogeny
Bats–Bartonella
Overall, both ParaFit and PACo analyses of Bartonella and bats provided evidence for significant co-evolution between Bartonella and bat hosts (ParaFitGlobal = 1.9703, P≤0.001; m2 global value = 7.3320, P≤0.0001). Twenty-six of the 51 individual host-parasite links are significant based on either a ParaFit1 or Parafit2 value of P≤0.05. While there was evidence of overall cospeciation, a substantial proportion of specific host-parasite links were non-significant.
The separate analyses of New World and Old World bat associated Bartonella both indicated evidence for overall coevolution with host species. In the New World dataset, global values from both ParaFit and PACo were significant (ParaFitGlobal = 0.4762, P≤0.001; m2 global value = 2.3313, P≤0.0001) and 29/38 individual were significant (Figure 1). For the Old World dataset, (ParaFitGlobal = 0.0871, P = 0.029; m2 global value = 0.7385, P = 0.0004) and 4/13 individual links were significant (Figure 2).
Figure 2. Tanglegram of cophylogenetic relationships between Old World bat hosts and Bartonella.

Maximum likelihood phylogenies for Bartonella bacteria (yellow) and their Old World bat hosts (blue), with bootstrap support values ≥50 labeled, rooted with outgroups. All host-pathogen associations are shown in the tanglegram as gray and black connecting lines. Black lines indicate significant individual cospeciation links between Bartonella and their hosts as indicated by ParaFit (P≤0.05), while gray lines are non-significant links. Bat species that did not have an available cytochrome b sequence on GenBank, is substituted with a closely related species. Triaenops persicus substituted for Triaenops menamena, and Hipposideros armiger for Hipposideros commersoni.
Rodents–Bartonella
Both ParaFit and PACo analyses of Bartonella and rodent phylogenies also indicated strong overall patterns of cospeciation (ParaFitGlobal = 102.4409, P≤0.001; m2 global value = 86.532, P≤0.0001). For the entire rodent-Bartonella dataset, 94/140 individual host-parasite links were significant based on ParaFit1 values and P≤0.05.
The separate analyses for the New World and Old World rodent hosts with associated Bartonella both also indicated evidence for significant overall coevolution. In the New World dataset, global values from both ParaFit and PACo were significant (ParaFitGlobal = 0.156, P≤0.001; m2 global value = 0.3548, P≤0.0001) and all 20 individual links were significant (Figure 3). For the Old World dataset, (ParaFitGlobal = 42.8037, P≤0.001; m2 global value = 72.31235, P≤0.0001) and 79/120 individual links were significant (Figure 4).
Figure 3. Tanglegram of cophylogenetic relationships between New World rodent hosts and Bartonella.

Maximum likelihood phylogenies for Bartonella bacteria (yellow) and their New World rodent hosts (green), with bootstrap support values ≥50 labeled, rooted with outgroups. All host-pathogen associations are shown in the tanglegram as black connecting lines and are significant as indicated by ParaFit (P≤0.05).
Figure 4. Tanglegram of cophylogenetic relationships between Old World rodent hosts and Bartonella.

Maximum likelihood phylogenies for Bartonella bacteria (yellow) and their Old World rodent hosts (green), with bootstrap support values ≥50 labeled, rooted with outgroups. All host-pathogen associations are shown in the tanglegram as gray and black connecting lines. Black lines indicate significant individual cospeciation links between Bartonella and their hosts as indicated by ParaFit (P≤0.05), while gray lines are non-significant links.
Bats–Leptospira
In contrast to Bartonella in mammalian hosts, ParaFit and PACo analyses of Leptospira and bats were unable to reject the hypothesis of independence of speciation events (ParaFitGlobal = 0.0042, P = 0.84; m2 global value = 4.6310, P = 0.5629). Based on ParaFit1 values, only 1 of 26 individual host-parasite links is significant based on P≤0.05. The separate analyses for the New World and Old World bat hosts with associated Bartonella both also indicated no evidence for overall coevolution. In the New World dataset, global values from both ParaFit and PACo were non-significant (ParaFitGlobal = 0.0012, P = 0.858; m2 global value = 4.631, P = 0.563) and none of the 19 individual links were significant (Figure 5). For the Old World dataset, (ParaFitGlobal = 0.0002, P = 0.269; m2 global value = 2.0802, P = 0.7587) and none of the 7 individual links were significant (Figure 6).
Figure 5. Tanglegram of cophylogenetic relationships between New World bat hosts and Leptospira.

Maximum likelihood phylogenies for Leptospira bacteria (pink) and their New World bat hosts (blue), with bootstrap support values ≥50 labeled, rooted with outgroups. All host-pathogen associations are shown in the tanglegram as gray connecting lines, and are insignificant as indicated by ParaFit (P≤0.05). Bat species that did not have an available cytochrome b sequence on GenBank, is substituted with a closely related species. Phyllostomus hastatus substituted for Phyllostomus discolor, Promops centralis for Promops nasutus.
Event-based cophylogeny
Based on a default cost setting, we calculated the optimal number of each type of coevolutionary event, to minimize total cost, for each host-pathogen association (Table 1). In order to account for the different sample sizes of each of the phylogenies, we divided the number of cospeciation and host switch events by the number of parasites in each association (Table 1). The resulting ratios can then be compared across the different associations in order to see the overall impact of each event given the number of parasites. Based on these calculations, Leptospira and bat host associations have the greatest number of host switches per parasite (0.731), while Bartonella and rodent host associations have the fewest (0.264). Leptospira and bat host associations also have the greatest number of cospeciations per parasite (0.231), while Bartonella and rodent host associations have the fewest (0.132).
Table 1. Results from event-based cophylogeny, using default cost settings of 0, 1, 2, 1, 1 in Jane.
| Cospeciations | Duplications | Duplications and host switches | Losses | Failures to diverge | Total Cost | # of hosts | # of parasites | Host switches/parasites | Cospeciations/parasites | |
| Bats-Bartonella | 10 | 21 | 24 | 4 | 0 | 73 | 23 | 56 | 0.429 | 0.179 |
| New World | 5 | 14 | 18 | 3 | 0 | 53 | 14 | 38 | 0.474 | 0.132 |
| Old World | 6 | 6 | 5 | 1 | 0 | 17 | 9 | 18 | 0.278 | 0.333 |
| Bats-Leptospira | 6 | 0 | 19 | 3 | 0 | 41 | 20 | 26 | 0.731 | 0.231 |
| New World | 5 | 1 | 12 | 2 | 0 | 27 | 14 | 19 | 0.632 | 0.263 |
| Old World | 2 | 0 | 4 | 1 | 0 | 9 | 6 | 7 | 0.571 | 0.286 |
| Rodents-Bartonella | 16 | 77 | 35 | 127 | 11 | 285 | 39 | 129 | 0.264 | 0.132 |
| New World | 1 | 15 | 3 | 0 | 0 | 21 | 4 | 20 | 0.150 | 0.050 |
| Old World | 17 | 54 | 37 | 75 | 11 | 214 | 35 | 109 | 0.339 | 0.156 |
We also compared cophylogenetic fit between bacteria and Old World vs. New World host species. Bartonella had nearly twice as many host switches per parasite in New World (0.474) as compared to Old World bats (0.278). Bartonella in rodents had the opposite trend, with more than twice as many host switches per parasite in Old World (0.339) as compared to New World rodents (0.150). There were also approximately three times as many cospeciation events per parasite for Bartonella in the Old World for both groups of hosts (bats: 0.333, rodents: 0.156) compared to the New World (bats: 0.132, rodents: 0.050). For Leptospira, the differences between Old and New World cophylogenetic patterns for both host switches and cospeciations were minimal.
We explored a wide-range of cost parameters in order to determine the effect of removing different evolutionary event options from each analysis (Table S6). Increasing the cost of host switching events had the greatest overall impact on total cost. In fact, the role of the host switching events in the overall coevolutionary pattern was so strong that even with a potentially prohibitive cost of 10, the solution still proposed between 2–4 host switch events for Bartonella in New World bats and Old World rodents.
Discussion
We found significant coevolutionary congruence between Bartonella and both their rodent and bat hosts at a global level, while the relationship between Leptospira and their bat hosts was non-significant. Event-cost results support the global-fit findings, with the rodent-Bartonella and bat-Bartonella associations having the least number of host switches per parasite, which indicates greater evolutionary congruence over time. Co-evolution of bartonellae and their mammalian hosts also remains significant when New and Old World datasets are analyzed separately. The evolutionary pattern in bat hosts is driven mostly by a few strong host-parasite interactions, with 51% of individual associations significant. In comparison, a greater proportion, 67%, of the individual rodent-bacteria associations are significant, indicating stronger coevolutionary interactions throughout these lineages. In fact, in the New World association of rodents and Bartonella, a full 100% of the host-parasite links were significant. In contrast, for Leptospira and their bat hosts, there is only 1 significant individual in analysis of the entire data set, and no significant host-parasite links when the data are analyzed separately as Old vs. New World. We note that the sample sizes for Bartonella in New World rodents and Leptospira in Old World bats are both small, and that the observed patterns could change with the addition of more data. Similarly, the relatively short sequence available of only one gene for both Bartonella and Leptospira may limit the resolution and nodal support for the pathogen phylogenies we obtained. These issues can only be addressed with additional sampling and genetic sequencing to complement these sparse datasets. For example, while our analysis of Bartonella-host relationships was limited by the availability of gltA fragments, the use of multi-gene phylogenies would be a more robust approach given the confounding effect of recombination [75]. Despite these potentially confounding factors, our preliminary analysis suggests a strong signal was present for some host-pathogen relationships and at a host order and pathogen genus level these trends were generalizable.
Event-cost methods corroborate the non-significant coevolutionary history of Leptospira and bats. Interestingly, the number of cospeciations per parasite is also the highest for Leptospira and bats, although they also have the highest number of host switches per parasite. Since their overall coevolutionary relationship is nonsignificant, this suggests that for the bat-Leptospira system, coevolutionary relationships are driven mostly strongly by the host switching events rather than cospeciation. Exploring the parameter space of cost structures further supports our findings. For all associations, maximizing the cost of host switching results in the largest overall change in the total cost (Table S6). This indicates that host switching is an “expensive” evolutionary event, and our finding of frequent and well-supported host switching in the bat-Leptospira system suggest that there are intrinsic ecological and transmission factors driving this.
One explanation for the different coevolutionary patterns between Bartonella and Leptopira may be differences in the modes of transmission and infection dynamics for each pathogen. As a vector transmitted parasite, Bartonella has an additional evolutionary step in adapting to an arthropod organism as well as a mammalian host. Combined, this can exert greater evolutionary selection and act as a selective force driving speciation. Further, Bartonella forms persistent, often asymptomatic, infections in its hosts [17], and some evidence even suggests that Bartonella may be acting as a symbiont more than a pathogen [18], [76], [77]. Many Bartonella species are also likely transmitted by only one arthropod species [78], and this specificity can then be translated to a greater coevolutionary pattern between the disease and eventual mammalian host. In bats, the arthropod vectors include blood-feeding bat flies, from which Bartonella has been sequenced and cultured [77], [79]. Host specificity of these arthropods may help to maintain the high diversity of Bartonella and long-term coevolutionary patterns between bat flies and their Bartonella parasites [77]. However no in-depth cophylogenetic analyses have been conducted for these bacteria and their known arthropod vectors, and this is an area for future exploration. Additional studies on arthropod ecology, e.g. bat fly, population structure, dispersal, ecology, and host specificity will also help to clarify the role of bat hosts vs. arthropod vectors in the evolution of Bartonella [77], [80]. Additionally, Bartonella is an intracellular bacteria which can survive only within erythrocytes and endothelial cells [17]. This requires a finer adaptation to the host's cells in order for bacterial penetration. In summary, Bartonella infection dynamics favor vector transmission, and the specific host-vector relationships, potential vertical transmission in vectors, and intracellular nature of the bacteria allow for co-evolutionary relationships to develop over time.
In contrast, Leptospira spp. are not vector-transmitted and instead are transmitted via environmental contamination. Leptospires are able to survive outside of their hosts, and can persist in water bodies when shed in animal urine [81]. Although the vast majority of Leptospira infections are mild, a small proportion involve multiple organ systems and develop various complications resulting in a case fatality in human patients of about 40% [45]. As contact with urine and contaminated water is the main form of disease transmission, physical proximity to environmental sources can play a large role in influencing host-pathogen interactions [82]. Thus it is possible that geographic overlap of the host species will better predict similarity in the bacteria they carry rather than the phylogenetic relatedness of the hosts. Overlapping geographic distribution of host species has been found to be an important determinant of pathogen sharing in primates [83]. The role of environmental transmission is most likely why we observed frequent host switching events and a lack of coevolutionary patterns in the Leptospira lineages we studied. Further investigations of Leptospirosis disease dynamics, including shedding, transmission, and immunity, in bat populations is warranted, as well as their zoonotic potential given the propensity towards cross-species transmission.
We originally hypothesized a difference in the strength of coevolutionary relationships between Old and New World host species, since previous research in bats had indicated host specificity in the Old World but not New World for Bartonella [24]. While the mechanism for this observation was not clear, it may be hypothesized that a greater degree of congruence between host-bacteria phylogenies in the Old World may be due to longer evolutionary time for the establishment of mutualistic relationships with mammalian hosts [24]. Yet, in our larger datasets, we did not see this pattern emerge, and our results indicated that coevolutionary patterns are generalizable globally. For Bartonella, significant coevolutionary congruence with hosts was evident globally and across host ranges, while for Leptospira, the lack of a coevolutionary relationship in bat hosts was evident in both the Old and New World. However, it is interesting that we observed a stronger relationship between rodents and Bartonella than between bats and Bartonella. There are two possible explanations for this. First, in mammalian evolutionary history, rodents existed for a longer period with 4.1 million years earlier time of origin and a 10 million year difference in time of basal diversification between the two [9]. Thus it is possible that there has been a longer time for parasite-host relations to coevolve in rodents and create stronger patterns. However it is not clear that these hosts have been infected with the two pathogens in question over their entire evolutionary history, and further detecting such deep evolutionary divergences is confounded by genetic saturation and nucleotide homoplasy. A second explanation is that the ecological differences between bats and rodents may explain the observed differences in host-specificity. Unlike rodents, a number of bat species form large multi-species gatherings and are more likely to have direct ecological overlap between host species (e.g. many thousands of individuals from >8 bat species roosting together in a single cave site in Mexico [84]). Similarly, at sites across the tropics, an extraordinary numbers of bat species can exist in sympatry, e.g. >70 species sharing tropical forest habitat in Krau Wildlife Reserve, Malaysia [85]. The gregarious aggregations of highly mobile individuals, often between multiple species, may help to explain differences in the global coevolutionary patterns observed between bats and rodents. While there has been growing scientific interest in these ecological and life-history host traits to explain viral sharing in bats and rodents [5], [8], [10], the role that these traits may play in bacterial pathogen diversification and spillover has been little investigated to date.
Overall, it is likely that the interplay of multiple factors, including geographic overlap, pathogen transmission pathways, infection dynamics, and host ecological and evolutionary history, that contribute to the contrasting coevolutionary patterns evident in mammal-bacteria interactions we observed. Further research is warranted to better understand and tease apart these contributing factors, and we recognize some of the limitations of this preliminary study. First, our analysis was limited by the availability of comparable data sets for a given gene and host taxonomic group. This precluded us from examining Leptospira in rodents; and resulted in low support values from some nodes in our phylogenies. In the future, using multiple genes or full genome data, for a greater number of bat and rodent taxonomic groups and bacterial microbes once they are available, will allow for more robust taxonomic analyses. Also, in addition to host phylogeny that we examine here, future data collection and analyses should focus on arthropod vector host specificity and phylogenetic relationships to better predict specificity within Bartonella. Future investigations should also consider the role of host geographic range and niche overlap to explain pathogen sharing between hosts. The application of spatial analyses of wildlife hosts for both Bartonella and Leptospira will provide valuable information on transmission potential based on the role of contact vs. cophylogeny. We predict that species with overlapping ranges will share more similar communities of Leptospira than non-overlapping bat species, regardless of their phylogenetic relatedness. For Bartonella there is also likely to be a geographic effect, as interaction among bats of different species within multi-species roosts, or shared habitats, could be an important factor for bacterial pathogen sharing.
Lastly, this work is particularly important because it involves two emerging, neglected tropical diseases with known, sylvatic wildlife reservoirs. Bartonella has been of concern as an emerging zoonoses due to its ability to induce life-threatening illnesses such as endocarditis, myocarditis, meningoencephalitis, and contributing to chronic debilitating disease, all while being difficult to diagnose in humans as well as animals [86]. Leptospirosis is a constant concern to public health authorities, and annual global incidence of severe leptospirosis has been estimated as 500,000 [87]. Elucidating the diversity and coevolutionary patterns of these bacteria in their natural hosts and understanding the frequency and causes of host-switching events, will help us better predict spillover from the mammal reservoirs into humans. Disruption of strict coevolutionary patterns, as we observed for both bacterial genera, to varying degrees, provides a framework to forecast pathogen spillover potential to any mammalian host, including humans [88]. The methods that we employed here to study bacterial disease in bats and rodent hosts are broadly applicable to a wide range of other disease types, including viruses in their mammalian hosts. By expanding these tools to better understand the evolutionary past of pathogens within and among wildlife hosts, we gain information to better predict the outbreaks of the future.
Supporting Information
Disease-related publications for bats and rodents over the past 20 years. The number of publications pertaining to viral (red) and bacterial (blue) disease research in bats (dotted) and rodents (solid) in each year from 1993 to 2012. Data from Web of Science keyword search for all publications from 1993 onwards using combinations of keywords bat OR Chiroptera*, rodent*, bacteria*, virus* OR vira*. Bacterial studies in bats are the most understudied category to date.
(DOCX)
gltA GenBank accession numbers for studied Bartonella sequences in bat hosts.
(DOCX)
gltA GenBank accession numbers for studied Bartonella sequences in rodent hosts.
(DOCX)
16S GenBank accession numbers for studied Leptospira sequences in bat hosts.
(DOCX)
Cytochrome b GenBank accession numbers of bat species host to studied bacteria.
(DOCX)
Cytochrome b GenBank accession numbers of rodent species host to studied Bartonella.
(DOCX)
Results from event-based cophylogeny Jane, using different cost-settings.
(DOCX)
Acknowledgments
BL thanks the scientists and staff at EcoHealth Alliance, including Peter Daszak and Jonathan Epstein, for invaluable discussions and support in developing this manuscript. We thank Katharina Dittmar for critical input on the role of bat flies and other potential vectors in bats.
Funding Statement
This study benefitted from intellectual developments from the PREDICT project of the United States Agency for International Development (USAID) Emerging Pandemic Threats Program, and was supported in part by a NIAID Non-Biodefense Emerging Infectious Disease Research Opportunities award R01 AI079231 (KJO). Partial funding also provided by a Herchel Smith Harvard Research Fellowship to BRL. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Simmons NB, Wilson D, Reeder D (2005) Order chiroptera. Mammal species of the world: a taxonomic and geographic reference 1: 312–529. [Google Scholar]
- 2. Pagel MD, May RM, Collie AR (1991) Ecological aspects of the geographical distribution and diversity of mammalian species. American Naturalist 137: 791–815. [Google Scholar]
- 3. Wong S, Lau S, Woo P, Yuen KY (2007) Bats as a continuing source of emerging infections in humans. Rev Med Virol 17: 67–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Memish ZA, Mishra N, Olival KJ, Fagbo SF, Kapoor V, et al. (2013) Middle East Respiratory Syndrome Coronavirus in Bats, Saudi Arabia. Emerging Infectious Diseases 19: 1819–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Olival KJ, Epstein JH, Wang LF, Field HE, Daszak P (2012) Are bats unique viral reservoirs? In: Aguirre AA, Ostfeld RS, Daszak P, editors. New Directions in Conservation Medicine: Applied Cases of Ecological Health. 2nd ed. Oxford: Oxford University Press. pp. 195–212. [Google Scholar]
- 6. Meerburg BG, Singleton GR, Kijlstra A (2009) Rodent-borne diseases and their risks for public health. Crit Rev Microbiol 35: 221–270. [DOI] [PubMed] [Google Scholar]
- 7. Muhldorfer K (2013) Bats and bacterial pathogens: a review. Zoonoses Public Health 60: 93–103. [DOI] [PubMed] [Google Scholar]
- 8. Turmelle AS, Olival KJ (2009) Correlates of viral richness in bats (Order Chiroptera). EcoHealth 6: 522–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bininda-Emonds OR, Cardillo M, Jones KE, MacPhee RD, Beck RM, et al. (2007) The delayed rise of present-day mammals. Nature 446: 507–512. [DOI] [PubMed] [Google Scholar]
- 10. Luis AD, Hayman DT, O'Shea TJ, Cryan PM, Gilbert AT, et al. (2013) A comparison of bats and rodents as reservoirs of zoonotic viruses: are bats special? Proceedings of the Royal Society B: Biological Sciences 280: 20122753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Calisher CH, Childs JE, Field HE, Holmes KV, Schountz T (2006) Bats: important reservoir hosts of emerging viruses. Clin Microbiol Rev 19: 531–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Plowright RK, Foley P, Field HE, Dobson AP, Foley JE, et al. (2011) Urban habituation, ecological connectivity and epidemic dampening: the emergence of Hendra virus from flying foxes (Pteropus spp.). Proceedings of the Royal Society B: Biological Sciences 278: 3703–3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. O'Shea TJ, Neubaum DJ, Neubaum MA, Cryan PM, Ellison LE, et al. (2011) Bat ecology and public health surveillance for rabies in an urbanizing region of Colorado. Urban Ecosystems 14: 665–697. [Google Scholar]
- 14. Murray KA, Daszak P (2013) Human ecology in pathogenic landscapes: two hypotheses on how land use change drives viral emergence. Current Opinion in Virology 3: 79–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cui J, Han N, Streicker D, Li G, Tang X, et al. (2007) Evolutionary relationships between bat coronaviruses and their hosts. Emerging infectious diseases 13: 1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ricklefs RE, Fallon SM, Bermingham E (2004) Evolutionary relationships, cospeciation, and host switching in avian malaria parasites. Systematic Biology 53: 111–119. [DOI] [PubMed] [Google Scholar]
- 17. Harms A, Dehio C (2012) Intruders below the Radar: Molecular Pathogenesis of Bartonella spp. Clinical Microbiology Reviews 25: 42–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Billeter SA, Levy MG, Chomel BB, Breitschwerdt EB (2008) Vector transmission of Bartonella species with emphasis on the potential for tick transmission. Medical and Veterinary Entomology 22: 1–15. [DOI] [PubMed] [Google Scholar]
- 19. Brown KJ, Bennett M, Begon M (2004) Flea-borne Bartonella grahamii and Bartonella taylorii in bank voles. Emerging Infectious Diseases 10: 684–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kernif T, Socolovschi C, Wells K, Lakim MB, Inthalad S, et al. (2012) Bartonella and Rickettsia in arthropods from the Lao PDR and from Borneo, Malaysia. Comparative Immunology Microbiology and Infectious Diseases 35: 51–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tsai YL, Chang CC, Chuang ST, Chomel BB (2011) Bartonella species and their ectoparasites: Selective host adaptation or strain selection between the vector and the mammalian host? Comparative Immunology Microbiology and Infectious Diseases 34: 299–314. [DOI] [PubMed] [Google Scholar]
- 22. Bai Y, Recuenco S, Gilbert AT, Osikowicz LM, Gomez J, et al. (2012) Prevalence and Diversity of Bartonella spp. in Bats in Peru. American Journal of Tropical Medicine and Hygiene 87: 518–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Concannon R, Wynn-Owen K, Simpson V, Birtles R (2005) Molecular characterization of haemoparasites infecting bats (Microchiroptera) in Cornwall, UK. PARASITOLOGY-CAMBRIDGE- 131: 489. [DOI] [PubMed] [Google Scholar]
- 24. Kosoy M, Bai Y, Lynch T, Kuzmin IV, Niezgoda M, et al. (2010) Bartonella spp. in Bats, Kenya. Emerging Infectious Diseases 16: 1875–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lin JW, Hsu YM, Chomel BB, Lin LK, Pei JC, et al. (2012) Identification of novel Bartonella spp. in bats and evidence of Asian gray shrew as a new potential reservoir of Bartonella. Veterinary Microbiology 156: 119–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Angelakis E, Khamphoukeo K, Grice D, Newton PN, Roux V, et al. (2009) Molecular detection of Bartonella species in rodents from the Lao PDR. Clin Microbiol Infect 15 Suppl 2: 95–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bai Y, Kosoy MY, Cully JF, Bala T, Ray C, et al. (2007) Acquisition of nonspecific Bartonella strains by the northern grasshopper mouse (Onychomys leucogaster). Fems Microbiology Ecology 61: 438–448. [DOI] [PubMed] [Google Scholar]
- 28. Brettschneider H, Bennett NC, Chimimba CT, Bastos ADS (2012) Bartonellae of the Namaqua rock mouse, Micaelamys namaquensis (Rodentia: Muridae) from South Africa. Veterinary Microbiology 157: 132–136. [DOI] [PubMed] [Google Scholar]
- 29. Inoue K, Kabeya H, Kosoy MY, Bai Y, Smirnov G, et al. (2009) Evolutional and Geographical Relationships of Bartonella grahamii Isolates from Wild Rodents by Multi-locus Sequencing Analysis (vol 57, pg 534, 2009). Erratum. Microbial Ecology 57: 534–541. [DOI] [PubMed] [Google Scholar]
- 30. Liu Q, Sun J, Lu L, Fu G, Ding G, et al. (2010) Detection of bartonella species in small mammals from Zhejiang Province, China. J Wildl Dis 46: 179–185. [DOI] [PubMed] [Google Scholar]
- 31. Kabeya H, Colborn JM, Bai Y, Lerdthusnee K, Richardson JH, et al. (2010) Detection of Bartonella tamiae DNA in Ectoparasites from Rodents in Thailand and Their Sequence Similarity with Bacterial Cultures from Thai Patients. Vector-Borne and Zoonotic Diseases 10: 429–434. [DOI] [PubMed] [Google Scholar]
- 32. Kosoy M, Bai Y, Morway C, Sheff K, Peruski L, et al. (2007) Identification of animal sources of human bartonellosis in Thailand: Comparison of Bartonella sequences from human patients and rodent hosts. American Journal of Tropical Medicine and Hygiene 77: 330.17690407 [Google Scholar]
- 33. Kosoy M, Bai Y, Sheff K, Morway C, Baggett H, et al. (2010) Identification of Bartonella Infections in Febrile Human Patients from Thailand and Their Potential Animal Reservoirs. American Journal of Tropical Medicine and Hygiene 82: 1140–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kosoy MY, Bai Y, Boonmar S, Sawatwong P, Jorakate P, et al. Bartonella Species in Bats from Thailand; 2012 March 11–14, 2012 Atlanta, GA.
- 35. Kamani J, Morick D, Mumcuoglu KY, Harrus S (2013) Prevalence and diversity of bartonella species in commensal rodents and ectoparasites from Nigeria, west Africa. PLoS Negl Trop Dis 7: e2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kosoy M, Murray M, Gilmore RD, Bai Y, Gage KL (2003) Bartonella strains from ground squirrels are identical to Bartonella washoensis isolated from a human patient. Journal of Clinical Microbiology 41: 645–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Waitkins S (1986) Leptospirosis as an occupational disease. British Journal of Industrial Medicine 43: 721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Meites E, Jay MT, Deresinski S, Shieh WJ, Zaki SR, et al. (2004) Reemerging leptospirosis, California. Emerg Infect Dis 10: 406–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Turner LH (1967) Leptospirosis. I. Trans R Soc Trop Med Hyg 61: 842–855. [DOI] [PubMed] [Google Scholar]
- 40. Bunnell JE, Hice CL, Watts DM, Montrueil V, Tesh RB, et al. (2000) Detection of pathogenic Leptospira spp. infections among mammals captured in the Peruvian Amazon basin region. Am J Trop Med Hyg 63: 255–258. [PubMed] [Google Scholar]
- 41. Lagadec E, Gomard Y, Guernier V, Pascalis H, Temmam S, et al. High infection prevalence and diversity of pathogenic Leptospira in bats from the Comoros and Madagascar. Soumis à. Emerg Infect Dis [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tulsiani SM, Cobbold RN, Graham GC, Dohnt MF, Burns MA, et al. (2011) The role of fruit bats in the transmission of pathogenic leptospires in Australia. Ann Trop Med Parasitol 105: 71–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bessa TA, Spichler A, Chapola EG, Husch AC, de Almeida MF, et al. (2010) The contribution of bats to leptospirosis transmission in Sao Paulo City, Brazil. Am J Trop Med Hyg 82: 315–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Matthias MA, Díaz MM, Campos KJ, Calderon M, Willig MR, et al. (2005) Diversity of bat-associated Leptospira in the Peruvian Amazon inferred by Bayesian phylogenetic analysis of 16S ribosomal DNA sequences. The American journal of tropical medicine and hygiene 73: 964. [PMC free article] [PubMed] [Google Scholar]
- 45. Vijayachari P, Sugunan AP, Shriram AN (2008) Leptospirosis: an emerging global public health problem. J Biosci 33: 557–569. [DOI] [PubMed] [Google Scholar]
- 46.Hafner MS, Demastes JW, Spradling TA, Reed DL (2003) Cophylogeny between pocket gophers and chewing lice. Tangled trees: phylogeny, cospeciation, and coevolution University of Chicago Press, Chicago: 195–220. [Google Scholar]
- 47. Page RD (1994) Parallel phylogenies: reconstructing the history of host-parasite assemblages. Cladistics 10: 155–173. [Google Scholar]
- 48. Bai Y, Kosoy M, Recuenco S, Alvarez D, Moran D, et al. (2011) Bartonella spp. in bats, Guatemala. Emerging infectious diseases 17: 1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kosoy MY, Regnery RL, Tzianabos T, Marston EL, Jones DC, et al. (1997) Distribution, diversity, and host specificity of Bartonella in rodents from the southeastern United States. American Journal of Tropical Medicine and Hygiene 57: 578–588. [DOI] [PubMed] [Google Scholar]
- 50. Ying B, Kosoy MY, Maupin GO, Tsuchiya KR, Gage KL (2002) Genetic and ecologic characteristics of Bartonella communities in rodents in southern China. American Journal of Tropical Medicine and Hygiene 66: 622–627. [DOI] [PubMed] [Google Scholar]
- 51. Castle KT, Kosoy M, Lerdthusnee K, Phelan L, Bai Y, et al. (2004) Prevalence and diversity of Bartonella in rodents of northern Thailand: A comparison with Bartonella in rodents from southern China. American Journal of Tropical Medicine and Hygiene 70: 429–433. [PubMed] [Google Scholar]
- 52. Gonçalves AT, Paiva C, Melo-Mota F, Vieira ML, Carreira T, et al. (2010) First isolation of human Leptospira strains, Azores, Portugal. International Journal of Infectious Diseases 14: e148–e153. [DOI] [PubMed] [Google Scholar]
- 53. Foronda P, Martin-Alonso A, del Castillo-Figueruelo B, Feliu C, Gil H, et al. (2011) Pathogenic Leptospira spp. in Wild Rodents, Canary Islands, Spain. Emerging Infectious Diseases 17: 1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Koizumi N, Muto M, Tanikawa T, Mizutani H, Sohmura Y, et al. (2009) Human leptospirosis cases and the prevalence of rats harbouring Leptospira interrogans in urban areas of Tokyo, Japan. Journal of medical microbiology 58: 1227–1230. [DOI] [PubMed] [Google Scholar]
- 55. Paiva-Cardoso MdN, Arent Z, Gilmore C, Hartskeerl R, Ellis WA (2012) Altodouro, a new Leptospira serovar of the Pomona serogroup isolated from rodents in northern Portugal. Infection, Genetics and Evolution 13: 211–217. [DOI] [PubMed] [Google Scholar]
- 56. Rahelinirina S, Léon A, Harstskeerl RA, Sertour N, Ahmed A, et al. (2010) First isolation and direct evidence for the existence of large small-mammal reservoirs of Leptospira sp. in Madagascar. PloS one 5: e14111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kocher TD, Thomas WK, Meyer A, Edwards SV, Pääbo S, et al. (1989) Dynamics of mitochondrial DNA evolution in animals: amplification and sequencing with conserved primers. Proceedings of the National Academy of Sciences 86: 6196–6200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bradley RD, Baker RJ (2001) A test of the genetic species concept: cytochrome-b sequences and mammals. Journal of Mammalogy 82: 960–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Agnarsson I, Zambrana-Torrelio CM, Flores-Saldana NP, May-Collado LJ (2011) A time-calibrated species-level phylogeny of bats (Chiroptera, Mammalia). PLoS Curr 3: RRN1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wilson D, Reeder D (2005) Mammal species of the world: a taxonomic and geographic reference. Washington D.C.: Smithsonian Institution Press. [Google Scholar]
- 61. Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32: 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, et al. (2012) Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28: 1647–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Pumo DE, Finamore PS, Franek WR, Phillips CJ, Tarzami S, et al. (1998) Complete mitochondrial genome of a neotropical fruit bat, Artibeus jamaicensis, and a new hypothesis of the relationships of bats to other eutherian mammals. Journal of Molecular Evolution 47: 709–717. [DOI] [PubMed] [Google Scholar]
- 64. Stamatakis A (2006) RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22: 2688–2690. [DOI] [PubMed] [Google Scholar]
- 65. Ronquist F, Huelsenbeck JP (2003) MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19: 1572–1574. [DOI] [PubMed] [Google Scholar]
- 66. Charleston M, Robertson D (2002) Preferential host switching by primate lentiviruses can account for phylogenetic similarity with the primate phylogeny. Systematic biology 51: 528–535. [DOI] [PubMed] [Google Scholar]
- 67. Desdevises Y (2007) Cophylogeny: insights from fish-parasite systems. Parassitologia 49: 125. [PubMed] [Google Scholar]
- 68. Legendre P, Desdevises Y, Bazin E (2002) A statistical test for host-parasite coevolution. Syst Biol 51: 217–234. [DOI] [PubMed] [Google Scholar]
- 69.R Core Team (2013) R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing. [Google Scholar]
- 70. Paradis E, Claude J, Strimmer K (2004) APE: Analyses of Phylogenetics and Evolution in R language. Bioinformatics 20: 289–290. [DOI] [PubMed] [Google Scholar]
- 71. Balbuena JA, Míguez-Lozano R, Blasco-Costa I (2013) PACo: a Novel procrustes application to cophylogenetic analysis. PloS one 8: e61048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Oksanen J, Kindt R, Legendre P, O'Hara B, Stevens MHH, et al. (2007) vegan: Comunity Ecology Package. R package. 1.15-4 ed.
- 73. Conow C, Fielder D, Ovadia Y, Libeskind-Hadas R (2010) Jane: a new tool for the cophylogeny reconstruction problem. Algorithms for Molecular Biology 5: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Althoff DM, Segraves KA, Smith CI, Leebens-Mack J, Pellmyr O (2012) Geographic isolation trumps coevolution as a driver of yucca and yucca moth diversification. Molecular Phylogenetics and Evolution 62: 898–906. [DOI] [PubMed] [Google Scholar]
- 75. Paziewska A, Harris P, Zwolińska L, Bajer A, Siński E (2011) Recombination Within and Between Species of the Alpha Proteobacterium Bartonella Infecting Rodents. Microbial Ecology 61: 134–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Halos L, Jamal T, Maillard R, Girard B, Guillot J, et al. (2004) Role of Hippoboscidae flies as potential vectors of Bartonella spp. infecting wild and domestic ruminants. Applied and Environmental Microbiology 70: 6302–6305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Morse SF, Olival KJ, Kosoy M, Billeter SA, Patterson BD, et al. (2012) Global distribution and genetic diversity of Bartonella in bat flies (Hippoboscoidea, Streblidae, Nycteribiidae). Infection, Genetics and Evolution 12: 1717–1723. [DOI] [PubMed] [Google Scholar]
- 78.Vayssier-Taussat M, Le Rhun D, Bonnet S, Cotte V (2009) Insights in Bartonella Host Specificity. In: Hechemy KE, Brouqui P, Samuel JE, Raoult DA, editors. Rickettsiology and Rickettsial Diseases. pp. 127–132. [DOI] [PubMed] [Google Scholar]
- 79. Billeter SA, Hayman DT, Peel A, Baker K, Wood JL, et al. (2012) Detection and/or isolation of Bartonella species from African bat flies Cyclopodia greefi (Diptera: Nycteribiidae) including description of Candidatus Bartonella breitschwerdtii . Parasitology 139: 324–329. [DOI] [PubMed] [Google Scholar]
- 80. Olival KJ, Dick CW, Simmons NB, Morales JC, Melnick DJ, et al. (2013) Lack of population genetic structure and host specificity in the bat fly, Cyclopodia horsfieldi, across species of Pteropus bats in Souteast Asia. Parasites & Vectors 6: 231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Adler B, de la Pena Moctezuma A (2010) Leptospira and leptospirosis. Vet Microbiol 140: 287–296. [DOI] [PubMed] [Google Scholar]
- 82. Woolhouse ME, Webster JP, Domingo E, Charlesworth B, Levin BR (2002) Biological and biomedical implications of the co-evolution of pathogens and their hosts. Nature genetics 32: 569–577. [DOI] [PubMed] [Google Scholar]
- 83. Davies TJ, Pedersen AB (2008) Phylogeny and geography predict pathogen community similarity in wild primates and humans. Proceedings of the Royal Society B: Biological Sciences 275: 1695–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Vargas-Contreras J, Escalona-Segura G, Arroyo-Cabrales J, Rendón J, Navarro L (2012) Conservación de Murciélagos en Campeche. THERYA 3: 53–66. [Google Scholar]
- 85.Kingston T, Liat LB, Zubaid A (2006) Bats of Krau Wildlife Reserve. Bangi, Malaysia: Kuala Lumpur Penerbit Universiti Kebangsaan Malaysia. [Google Scholar]
- 86. Breitschwerdt EB, Maggi RG, Chomel BB, Lappin MR (2010) Bartonellosis: an emerging infectious disease of zoonotic importance to animals and human beings. Journal of Veterinary Emergency and Critical Care 20: 8–30. [DOI] [PubMed] [Google Scholar]
- 87. Hartskeerl RA, Collares-Pereira M, Ellis WA (2011) Emergence, control and re-emerging leptospirosis: dynamics of infection in the changing world. Clin Microbiol Infect 17: 494–501. [DOI] [PubMed] [Google Scholar]
- 88. Woolhouse ME, Haydon DT, Antia R (2005) Emerging pathogens: the epidemiology and evolution of species jumps. Trends in ecology & evolution 20: 238–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Disease-related publications for bats and rodents over the past 20 years. The number of publications pertaining to viral (red) and bacterial (blue) disease research in bats (dotted) and rodents (solid) in each year from 1993 to 2012. Data from Web of Science keyword search for all publications from 1993 onwards using combinations of keywords bat OR Chiroptera*, rodent*, bacteria*, virus* OR vira*. Bacterial studies in bats are the most understudied category to date.
(DOCX)
gltA GenBank accession numbers for studied Bartonella sequences in bat hosts.
(DOCX)
gltA GenBank accession numbers for studied Bartonella sequences in rodent hosts.
(DOCX)
16S GenBank accession numbers for studied Leptospira sequences in bat hosts.
(DOCX)
Cytochrome b GenBank accession numbers of bat species host to studied bacteria.
(DOCX)
Cytochrome b GenBank accession numbers of rodent species host to studied Bartonella.
(DOCX)
Results from event-based cophylogeny Jane, using different cost-settings.
(DOCX)