

Abstract
Salinity is one of the important abiotic stress factors that limit crop production. Common bean, Phaseolus vulgaris L., a major protein source in developing countries, is highly affected by soil salinity and the information on genes that play a role in salt tolerance is scarce. We aimed to identify differentially expressed genes (DEGs) and related pathways by comprehensive analysis of transcriptomes of both root and leaf tissues of the tolerant genotype grown under saline and control conditions in hydroponic system. We have generated a total of 158 million high-quality reads which were assembled into 83,774 all-unigenes with a mean length of 813 bp and N50 of 1,449 bp. Among the all-unigenes, 58,171 were assigned with Nr annotations after homology analyses. It was revealed that 6,422 and 4,555 all-unigenes were differentially expressed upon salt stress in leaf and root tissues respectively. Validation of the RNA-seq quantifications (RPKM values) was performed by qRT-PCR (Quantitative Reverse Transcription PCR) analyses. Enrichment analyses of DEGs based on GO and KEGG databases have shown that both leaf and root tissues regulate energy metabolism, transmembrane transport activity, and secondary metabolites to cope with salinity. A total of 2,678 putative common bean transcription factors were identified and classified under 59 transcription factor families; among them 441 were salt responsive. The data generated in this study will help in understanding the fundamentals of salt tolerance in common bean and will provide resources for functional genomic studies.
Introduction
Soil salinity is one of the most severe abiotic stress factors limiting the productivity of agriculture. Although most plants are glycophytes that are highly sensitive to saline environment, halophytes are plants that naturally grow under saline conditions throughout their life cycle. Salinity effects nearly 20% of all irrigated lands worldwide [1] and expected to reach around 50% in the near future [2]. A soil is considered saline if the electrical conductivity of its saturation (EC) is above 4 dS/m [3] which is equivalent to approximately 40 mM NaCl.
As a member of grain legumes and a glycophyte crop, common bean (Phaseolus vulgaris L.) is a major source of human dietary protein, minerals, vitamins, and represents nearly half of the consumed grain legumes worldwide [4]. Common bean is also vital in agriculture as it forms root nodules via symbiotic associations with nitrogen fixing bacteria [4]. Nearly 60% (360 Mt) of the annual fresh bean production in Turkey (FAO:http://faostat.fao.org/faostat) is highly dominated in Black Sea region where the soil salinity levels can reach up to 2–4 dS/m [5]. However, it is known that even at 1 dS/m salinity level, the productivity of common bean can be reduced up to 20% [6].Thus, understanding the fundamentals of salt tolerance in common bean, eventual development of improved varieties and their introduction to saline environments are imperative in agriculture.
Although halophytes may use avoidance mechanisms, glycophytes tolerate salinity by minimizing ion disequilibrium and the consequent secondary effects [7]. In other words, tolerance mechanisms require concerted actions of mechano-receptors, ion transport channels, and secondary signal molecules to maintain ion homeostasis as well as cascades of gene activations for hormonal metabolism, signal transduction pathways, and stress responses [8]–[12].
Considering the multifactorial nature of tolerance responses, development of tolerant plants for the benefit of sustainable crop improvement still awaits accumulation of additional knowledge about the identity of components that are involved in this process.
Recent developments in high-throughput approaches to study gene expression profile have emerged as an important tool to understand how plants respond to biotic and abiotic stresses. In the last few years, there have been accumulating reports on RNA-sequencing data and expression profiling on both model plants and agriculturally important crops [13]–[21] to identify genes involved in stress responses. Although, recently such high throughput transcriptome assemblies have been started in legumes [22]–[30], there are still only a handful of studies regarding the transcriptome analysis under abiotic stress conditions in these species. These studies include the effects of drought, saline-alkaline conditions and salt stress in gene expression profiling of chickpea [31], soybean [32], Medicago truncatula [33], and alfa alfa [21] respectively. However, there is no such transcriptome analysis under abiotic stress available yet for common bean. In combination with continuously generated reference genome sequences for diverse legume species [34]–[39], next generation RNA sequence analyses will provide valuable information for both identification and cloning of stress tolerance genes which can be used to improve varieties with enhanced tolerance mechanisms.
In this study, we used the Illumina high-throughput RNA-sequencing platform for transcriptomic analysis of a salt tolerant common bean, Ispir genotype. We aimed to identify differentially expressed genes (DEGs) and related pathways by comprehensive analysis of data from both root and leaf tissues of the tolerant genotype grown under saline and control conditions in a hydroponic system. De novo assemblies of the sequencing data, functional annotations of unigenes, and their characterization with gene ontology and metabolic pathway analysis provided potential lists of candidate genes. Functional identification of these candidates using reverse genetics approaches in our ongoing studies will contribute to the understanding of salt tolerance mechanisms.
Methods
Plant growth and salt treatments
The seeds of salt tolerant “Ispir” variety were kindly supplied by Prof. H. Yildiz Dasgan (Cukurova University Department of Horticulture, Adana, Turkey). The seeds were surface sterilized in a solution containing 5% (v/v) hypochlorite for 5 min and rinsed three times with distilled water. The seeds were germinated in vermiculite containing plug trays at 24/20°C cycle under a 16-h light/8-h dark photoperiod with 300 μmol m−2 s−1 light intensity, and 50–60% relative humidity up to the fully expanded foliage stage by daily watering with hydroponic nutrient solution containing 3 mM Ca(NO3)2, 1 mM MgSO4, 0.9 mM K2SO4, 0.2 mM KH2PO4, 0.1 mM FeEDTA, 10 nM H3BO3, 1 nM MnSO4, 1 nM ZnSO4, 0.1 nM CuSO4, and 0.01 nM (NH)6Mo7O24 [40]. Eight seedlings were transferred to two pots (four seedlings each) containing hydroponic nutrient solution that was replenished daily. During salt treatment, the photoperiod was also kept the same as in the germination conditions. Once plants reached trifoliate stage (five days post transfer), one pot was left as control and the other was exposed to gradually increasing NaCl concentrations. In this study, the hydroponics system was preferred to keep the nutrients and NaCl levels under strict control to achieve homogenous growth of the plants. Furthermore to minimize the risk of plasmolysis due to the osmotic shock [41] during salt treatments “gradual step acclimation” method was used [42], thus salt application was started with 50 mM in the first day, followed by 100 mM in the second day, and reached to 125 mM between days three to five in hydroponic nutrient solution.
Sample collection and RNA isolation
The root and the leaf tissues from salt treated and control plants were sampled at the fifth day of the salt treatment. Samples were frozen in liquid nitrogen and stored at −80°C prior to RNA extractions. Total RNAs were extracted with RNeasy Plant kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. The concentrations of RNA samples were determined by NanoDrop 1000 spectrophotometer (Thermo Fisher Scientific, Wilmington, DE). RNA quality was assessed by 1% denaturing agarose gel electrophoresis.
Initially, RNA samples from root tissues of two control (RC: Root Control; RC1 and RC2) as well as two salt treated plants (RS: Root-Salt treated; RS1 and RS2) were isolated as two biological replicates, and they were pooled as described in Figure 1A and 1B. Forty μg of total RNA from the two biological replicates were sequenced by Illumina HiSeqTM 2000 system (BGI, Shenzen, China). Pearson correlation coefficients between the RPKM (reads per kilobase per million reads) values of the two biological replicates were calculated as 0.99 and 0.97 for control and treated samples respectively (Figure 1C and 1D). Due to the observation of high correlation within the biological replicates of root tissues, we pooled RNA samples directly from leaf tissues of the same four plants from both control (LC: Leaf Control) and treated samples as a cost effective strategy [43], [44].
Figure 1. Schematic representation of the sample pooling strategy and correlation analysis of root samples RPKM values.

Black lines indicate the pooling of leaf and root samples from control plants (A) and salt treated plants (B). Graphs show the correlation analysis within root control (C) and salt treated (D) samples. Nomenclature: LC: Leaf Control; LS: Leaf Salt treated; RC1 and RC2: Root Control 1 and 2; RS1 and RS2: Root Salt treated 1 and 2.
cDNA library construction and Illumina sequencing
RNA quality and quantity were verified using Nanodrop 1000 spectrophotometer and Agilent 2100 bioanalyzer before cDNA library generation at BGI (Shenzen, China). Total RNAs were treated with DNase I before poly (A+) mRNA enrichment with oligo dT magnetic beads. Poly (A+) RNAs were digested into 200–700 nt fragments by RNA Fragmentation Reagent, and random hexamer primed poly (A+) RNA fragments were transcribed into first-strand cDNAs. Subsequently in the presence of dNTPs, RNase H, and DNA polymerase I, the second strands were synthesized, purified using QiaQuick PCR purification kit (Qiagen, Hilden, Germany), and used for end repair single adenine nucleotide addition. The sequencing adaptors were ligated to the fragments. The paired end library constructions were finalized by size selection with agarose gel electrophoresis and selective enrichment of the cDNA fragments with PCR amplification. The libraries were sequenced on a flow cell using Illumina HiSeq2000 sequencing instrument after quality control with Agilent 2100 bioanalyzer and qPCR to detect fragment size and concentration.
De novo assembly and data analyses
Subsequent to adapter trimming, the raw data were filtered for reads with more than 5% ambigous bases and/or low quality reads with bases 20% of which has a Phred score less than 10. The clean reads from each library were de novo assembled into contigs with Trinity software [45] (http://sourceforge.net/projects/trinityrnaseq/files) setting k-mer length to 25. This has generated a total of six subtranscriptomes. Contigs of the subtranscriptomes were pooled, clustered, and assembled using the Trinity software to obtain sequences that can no longer be extended on either end, and they were referred as all-unigenes. All-unigene sequences were aligned using BLASTx against NCBI non-redundant (Nr) protein, Swiss-Prot protein, the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway, and Cluster of Orthologous Groups (COG) databases to determine sequence orientations and protein coding region predictions. Proteins with the highest ranks in BLASTx alignment results were used to predict coding region sequences. All-unigenes that could not be aligned to sequences in any of the databases mentioned above were scanned by ESTScan software [46] to predict sequence orientations and coding regions. For annotations, all-unigenes were searched against the Nr database using BLASTx with 10−5 as E-value cut-off point and sequences with the highest similarities were retrieved. To obtain Gene Ontology (GO) terms regarding biological process, molecular function and cellular component [47] descriptions, the resulting BLASTx hits were analyzed by Blast2GO software [48]. The GO annotations were functionally classified by WEGO [49] software for gene function distributions of common bean species at macro level. BLASTx analysis against the KEGG pathway database was also performed to assign putative metabolic pathways to all-unigenes.
To estimate gene expression levels, six (four from the root libraries, two from the leaf libraries) RPKM values were calculated for every all-unigene by mapping six subtranscriptomes with SOAP2 software [50], applying three mismatches as threshold. The clean reads mapped to more than one all-unigene were not used to calculate RPKM. Corrections for false positive and false negative errors were performed by calculating the FDR (false discovery rate) values [51]. The DEGs were selected by using a FDR-value ≤0.001 and the absolute ratio of log2 (RPKM-tr/RPKM-cont) ≥1 as threshold values. The GO terms and the KEGG pathways that were enriched within the DEGs were identified by publicly available agriGO [52] and FatiGO [53] software respectively. To analyze the functional significance of enriched terms hypergeometric tests were employed by using the common bean transcriptome as background, setting the FDR and the Adjusted P-values lower than 0.05 for the agriGO and FatiGO software respectively.
qRT-PCR analyses
Quantitative Reverse Transcription PCR (qRT-PCR) analyses were performed for 43 unigenes (Table S1) using the insulin degrading enzyme (IDE, Unigene29213) [GenBank: FE702602.1] and the actin-11 (Act-11, CL442.Contig3) [GenBank: CV529679.1] genes of common bean as stably expressed internal references under salt stress [54]. Four individual plants from both control and treated groups were used in qRT-PCR analyses as biological replicates. For each biological replicates, three qRT-PCR reactions were performed as technical replicates. Single stranded cDNAs were synthesized from one μg of total RNA using RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, Wilmington, DE). Gene-specific primers were designed using the Primer Design module of CLC Main Workbench (version 6.0) software. qRT-PCR analyses were performed by PikoReal 96 Real-time PCR system (Thermo Fisher Scientific, Wilmington, DE) using 10 ng first strand cDNAs and AccuPower 2X Greenstar master mix (Bioneer, Daejon, Korea) with 0.25 pmol forward and reverse primers in a 10 μl solution. Upon initial denaturation at 95°C for five min., 40 cycles of denaturation at 95°C for 10 s and 60 s of annealing, and extension steps were performed at specific temperatures (Table S2) optimized for each primer pair. The relative expression levels were calculated using the 2–ΔΔCt method for each gene and were normalized to the geometric average of Ct (threshold cycle) values of the internal reference genes. The Student's t-test was applied to determine the significant differences in expression levels between treated and control samples collected at the fifth day of salt treatment.
Identification of putative transcription factors
Sequences for Hidden Markov Model (HMM) motifs of the transcription factors (TFs) belonging to Glycine max, Medicago truncatula and Lotus japonicus were acquired from the Legume Transcription Factor Database (Legume TFDB) [55], [56] and combined to create a single legume motif database consisting of 61 TF families. An all-unigene was accepted as a putative TF if it has shown 90% or more sequence homology (E-value ≤10−10) with any of the HMM motifs of at least one of the three legume species. The putative TFs within the all-unigenes and the DEGs were categorized based on the TF families and their presence in root or leaf tissues.
Results
De novo assembly of the sequencing data
Previous reports have emphasized that during NaCl applications, Na+ concentrations reach to toxic levels and plants start to react to salt stress rather than osmotic shock only after 24–72 hours gradual step acclimation [41], [42]. In our studies with NaCl treatment on Ispir variety have also shown that detectable physiological effects of salt stress become significant at the fifth day (72 hours after gradual step acclimation) of treatment in hydroponics cultures (data not shown). Therefore, the sequencing was performed on the leaf and the root RNA samples collected at this time point. The Illumina sequencing was performed separately for each RNA sample (LC, LS, RC1, RC2, RS1, and RS2) and six subtranscriptomes (Figure 1A and 1B) were generated with 90-bp raw reads. More than 158 million clean reads remained from the six subtranscriptomes (Table 1) with ∼97% Q20 bases (i.e. percentage of sequences with sequencing error rate lower than 1%) that constituted over 14 GBase (7.05×109 nt in control, and 7.23×109 nt in salt treated samples) of data after quality assessment and data filtering. All clean reads were deposited in the NCBI Short Read Archive (SRA) database and can be accessed with SRP029243 accession number.
Table 1. Statistics of raw data for P.vulgaris L. transcriptome.
| Samples | Total Reads | Total Nucleotides (nt) | Q20 percentage | GC percentage |
| 1LC | 25 907 332 | 2 331 659 880 | 97.02% | 46.02% |
| 2RC | 26 481 724 | 2 383 355 160 | 96.91% | 46.70% |
| 3RC | 25 998 386 | 2 339 854 740 | 96.94% | 46.97% |
| 1LS | 27 191 626 | 2 447 246 340 | 97.12% | 46.19% |
| 2RS | 27 280 462 | 2 455 241 580 | 96.91% | 47.08% |
| 3RS | 25 821 046 | 2 323 894 140 | 96.83% | 47.30% |
The clean reads from LC, LS, RC1, RC2, RS1, and RS2 subtranscriptomes were de novo assembled using the Trinity software, which generated 52,858, 51,564, 60,590, 64,986, 59,510, and 58,174 unigenes, respectively (Table 2). Further assembly of the subtranscriptomes generated the common bean transcriptome consisting of 83,774 all-unigenes. The total length of all-unigenes was 68,147,816 bp with a mean length of 813 bp and N50 of 1,449 bp (i.e 50% of the assembled bases were incorporated into all-unigenes of 1,449 bp or longer) (Table 2). The lengths of all-unigenes ranged between 150 and 17,022 bp. In total, 55,471 (66.72%) all-unigenes were unique and the remaining 28,303 (33.8%) fell into 5,403 clusters in which the number of unigenes ranged between 2 and 42. Among the 83,774 all-unigenes, 63,011 (71% Nr-annotated) were expressed in the leaf samples whereas 73,523 (69% Nr-annotated) were expressed in the root samples. Randomly distributed clean reads from the six subtranscriptomes evenly covered the common bean transcriptome with an average of ∼20 fold coverage depth (Figure S1).
Table 2. Statistics analysis for unigene assembly of P. vulgaris L.
| Length of Unigenes (bp) | Number of unigenes | ||||||
| LC | RC1 | RC2 | LS | RS1 | RS2 | All | |
| 150–500 | 31659 | 37290 | 41636 | 29684 | 38096 | 37683 | 44558 |
| 500–1000 | 11135 | 12780 | 12836 | 10819 | 12194 | 11878 | 15702 |
| 1000–1500 | 5104 | 5499 | 5514 | 5280 | 4939 | 4612 | 9754 |
| 1500–2000 | 2622 | 2675 | 2678 | 2972 | 2352 | 2202 | 6115 |
| ≥ 2000 | 2338 | 2346 | 2322 | 2809 | 1929 | 1799 | 7645 |
| Total | 52858 | 60590 | 64986 | 51564 | 59510 | 58174 | 83774 |
| Mean length | 644 | 613 | 585 | 685 | 579 | 568 | 813 |
| N50 (bp) | 1016 | 946 | 906 | 1121 | 866 | 843 | 1449 |
| Total Length (bp) | 34018974 | 37138056 | 38042659 | 35328894 | 34441515 | 33036044 | 68147816 |
Annotation and classification of common bean transcriptome
The BLASTx searches (E-value ≤ 10−5) revealed that out of 83,774 all-unigenes, 58,171 (69.4%), 44,174 (52.7%), and 28,564 (34.1%) showed significant similarity to the sequences in Nr, Swiss-Prot and KEGG databases respectively. Among the all-unigenes, 43,519 (51.9%) were commonly annotated in Nr and Swiss-Prot; 41,087 (49%) in Nr and KEGG; 27,116 (32.4%) in KEGG and Swiss-Prot, and 27,078 (32.3%) were annotated in all three databases. For the 91% of the Nr -annotated sequences, the E-values were less than or equal to 10−10. In addition, 24,909 (29.7%) all-unigenes did not match significantly to any of the sequences in these databases.
More than 46% of all-unigenes smaller than 500 bp had BLASTx hits in the Nr database whereas for those longer than 500 bp the ratio was over 90.4% (Figure 2A). This has indicated that the longer the all-unigenes were the more they were likely to have Nr annotations and the lower E-values than the shorter all-unigenes (Figure 2B). The great majority (over 96%) of Nr annotated all-unigenes showed the highest homology to the plant proteins (Figure 2C). Furthermore, 64.4% were specifically annotated with sequences from legume species (Figure 2C), which indicated the reliability of our transcriptome analysis. In our data set, we also found few all-unigenes, which were annotated by phytoplankton and plant pathogens proteins, most probably due to the hydroponic growth conditions used in this study. Similar to the observation in an earlier report [57], 96 all-unigenes expressed exclusively in roots were Nr annotated from Phytophthora sojae, which is a soybean pathogen that causes rotting of stem and roots [58]. This observation indicates that our transcriptome analysis detected transcripts even in trace amounts.
Figure 2. Reliability analysis of all-unigene annotations.
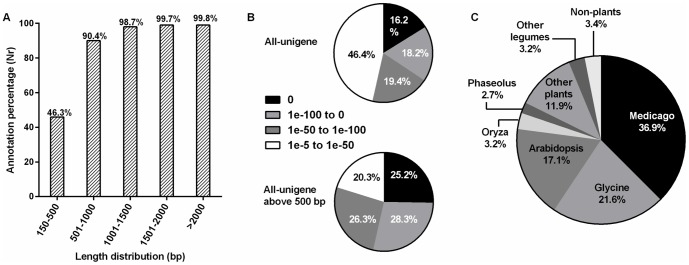
Histogram of all-unigenes according to the Nr database annotations with 500 bp intervals (A). E-value distributions for Nr annotations based on BLASTx analysis; panel B1 represent the percentage distributions of all-unigenes and panel B2 represent all-unigenes longer than 500 bp. Genera distribution of all-unigenes according to the Nr database annotations (C).
Using WEGO software the 27,959 all-unigenes were assigned to a total of 98,948 GO annotations which fall into three main categories; 19,154 all-unigenes were within 28 terms of the “Biological Process”category, 18,175 all-unigenes were within 15 terms of the “Cellular Component” category, and 22,190 all-unigenes were within 12 terms of the “Molecular Function” category (Figure S2).
All-unigenes were assigned to KEGG pathway and COG databases, a total of 28,564 all-unigenes were annotated in 125 individual pathways (Table S3) and a total of 23,141 all-unigenes were assigned to 25 functional classes respectively (Figure S3).
qRT-PCR verification of RPKM based gene expression
We performed qRT-PCR analysis for 43 selected all-unigenes (Table S1) from root and leaf samples with specific primers (Table S2) to assess the reliability of our sequencing results (Figure 3). Among these selected all-unigenes, 15 were upregulated (log2 (RPKM tr/cont) ranged between 1.57 and 12.28), 17 were non-differentially expressed (log2 (RPKM tr/cont) ranged between −0.77 and 0.89), and 11 were downregulated (log2 (RPKM tr/cont) ranged between −1.18 and −13.13). The comparison of qRT-PCR and sequencing results revealed a high correlation for the selected unigenes (Pearson r = 0.91, Figure 3). In addition to high correlation between qRT-PCR and RNA-seq results, the production of expected fragment sizes using designed primers have also confirmed the reliability of de novo assembly.
Figure 3. Relative expression level comparisons of qRT-PCR and RNA-seq results for selected all-unigenes.

The qRTPCR results of 43 all-unigenes were compared to the RPKM values (log2 (RPKM-tr/RPKM-cont) ≥1).
Identification and functional classification of DEGs
Upon comparison against control groups, all-unigenes with more than or equal to two-fold change (│log2 (RPKM tr/cont) │≥1) in their gene expression level with a FDR value below 10-3 were defined as DEGs. Based on these criteria, the number of DEGs in leaves and roots were 6,422 and 4,555, respectively (Figure 4; Table S4 and S5). Out of 6,422 DEGs, 3,024 (88% Nr-annotated) were upregulated, and 3,398 (88% Nr-annotated) were downregulated in the leaf samples, whereas among the 4,555 DEGs of the roots, 1,237 (89% Nr-annotated) were upregulated, and 3,318 (76% Nr-annotated) were downregulated upon salt treatment (Figure 4).
Figure 4. Venn diagram showing the number of DEGs.
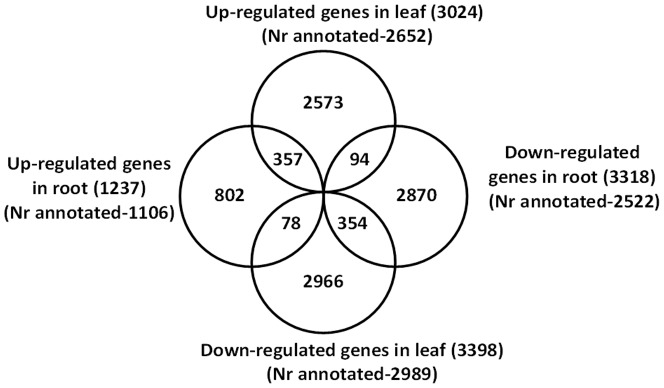
The sums of the numbers in each circle were indicated within the parantheses of the corresponding title.
In both leaf and root tisssues, the enriched terms in the GO database (Figure 5, Table S6 and S7) were in agreement with the KEGG metabolic pathways (Figure 6, Table S8 and S9). Within the leaf tissues upregulated genes, the terms related with the secondary metabolite metabolism and the membrane transport activity were mostly enriched, however the macromolecular energy metabolism related terms were highly enriched within the downregulated genes (Figure 5A and 6A). Although, the DEGs in the root tissues have also shown a similar pattern with the leaf tissues for the terms related with the secondary metabolite metabolism and the membrane transport activity, the terms related with the macromolecular energy metabolism were only slightly enriched within the upregulated genes. Additionally, in root tissues, the transcription/translation related terms were notably enriched within the downregulated genes (Figure 5B and 6B).
Figure 5. Gene Ontology functional enrichment analysis of DEGs.

Panel A represents the DEGs in leaf tissues and panel B represents the DEGs in root tissues. (FDR-value <0.05).
Figure 6. Pathway enrichment analysis of DEGs.

Panel A represents the DEGs in leaf tissues and panel B represents the DEGs in root tissues. (Adjusted-P-value <0.05).
Identification of putative transcription factors (TFs)
In this study, 2,678 putative TFs were identified based on the homology analysis of the all-unigenes against a combined legume database which was constructed from Glycine max, Medicago truncatula and Lotus japonicus HMM motifs. The putative TFs were classified under 59 out of the 61 TF families present in this database. The most abundant 10 TF families were AP2_EREBP, bHLH, PHD, HB, (R1)R2R3_Myb, WRKY_Zn, NAC, bZIP, C3H-TypeI, and Myb_related (Table 3, Table S10). A total of 441(16%) TFs, 331 in the leaf, and 161 in the root tissues (Table 3 and Table S10) identified as salt responsive. The composition of most abundant TF families were preserved within the leaf DEGs except the C3H-Type I family (replaced by C2C2-Zn-CO like) however in the root DEGs, the bZIP, C3H-TypeI, and Myb_related HSF were replaced by the C2C2_Zn-Dof, and C2H2_Zn TF families (Table S10).
Table 3. Number of total and differentially expressed bean TFs on family basis.
| All-unigene | DEGs | ||||
| TFs | All | Leaf | Root | Leaf | Root |
| AP2_EREBP | 222 | 163 | 173 | 42 | 25 |
| bHLH | 212 | 160 | 171 | 29 | 18 |
| PHD | 172 | 157 | 162 | 14 | 8 |
| HB | 157 | 115 | 112 | 18 | 7 |
| (R1)R2R3_Myb | 154 | 114 | 122 | 28 | 22 |
| WRKY_Zn | 144 | 117 | 122 | 20 | 14 |
| NAC | 122 | 95 | 104 | 20 | 13 |
| bZIP | 111 | 75 | 85 | 14 | 3 |
| C3H-TypeI | 97 | 90 | 91 | 7 | 3 |
| Myb_related | 94 | 79 | 78 | 16 | 3 |
| TOTAL | 2678 | 2141 | 2196 | 331 | 161 |
Discussion
Common bean is an important grain legume that provides 30% of the protein intake in developing countries (FAO: http://faostat.fao.org/faostat), and the production suffers drastically even in slightly saline soils [6]. Presence of genetic diversity within the species toward salt tolerance [59] offers a valuable opportunity to identify the key elements that might play a role in salt tolerance. The common bean genotype “Ispir” used in this study is known to be a very old local variety named after Ispir county of Erzurum province where it has been produced by more than 150 years. The variety has been registered in 2008 and patented as “Geographic Trademark” by Turkish patent office (www.tpe.gov.tr). The ability of the variety to tolerate high salt concentrations even at germination stage without compromising from germination time was well documented [59].
There exists a substantial amount of reports regarding the physiological responses to salt stress in common bean [59], [60]. Also there is a considerable collection of ESTs from cDNA libraries obtained from different organs and root nodules of legumes [61], [62] including sequences related to abiotic stresses such as phosphorus starvation, rust disease resistance, and drought stress. Within the last few years, there has been increasing number of studies on transcriptome analysis of legume species using high-throughput RNA sequencing approach, among those regarding the effects of salinity were recently reported on Medicago [21], [33], Glycine [32], Cicer [31], [63], and Millettia [57]. However, there are only three transcriptomic studies in the literature on common bean conducted by high-throughput RNA-sequencing approach, one of which is a general transcriptome [24]assembly, the other is a data-mining of host resistance gene like sequences [64] within the transcriptome and the third one is on the sulfur metabolism in developing seeds [65]. Therefore, we aimed to perform a large-scale comparative transcriptome analysis in two different tissues of a salt tolerant common bean variety under salt stress to identify key elements and their related functional pathways, which may play a critical role in stress tolerance responses.
In our study, the assembly of the transcriptome relied on the de novo method. The reliability and the sensitivity of our transcriptome were demonstrated by the high percentage of annotations (i.e. Nr-annotations, 69.4%), the ability to detect the trace amounts of transcripts from plant pathogens as well as phytoplankton contaminations (due to hydroponic system), and the correlation between the qRT-PCR results and the RPKM values. Functional annotation of common bean transcriptome also revealed that the highly represented terms in the GO (Figure S2), KEGG (Table S3), and COG (Figure S3) databases were also commonly represented in previous legume transcriptome studies [57], [66], [67]. Additionally the two previous common bean transcriptome studies [24], [65] generated comparable number of all-unigenes (94,623 and 77,448 all-unigenes) with our transcriptome assembly (83,774 all-unigenes), although they used pyrosequencing platform.
Comparative transcriptional level analysis of salt treated and control samples using RPKM values revealed that 10094 (12%) all-unigenes were salt responsive (Figure 4, Table S4 and S5). Among them 8457 (84%) were Nr-annotated. Although we used the same (or higher) stringent threshold values (│log2 (RPKM tr/cont) │≥1, FDR ≤0.001) with the previous studies in legumes [19], [21], [33], a higher number of salt responsive genes were identified in this study except the recently reported study in Milletia pinnata [57]. The reasons which may contribute to these disparities could be the differences in sensitivity and resolution between the hybridization [19] and sequencing based platforms as well as the differences between the assembly software. Moreover, the differences between salt tolerance characteristics of Milletia pinnata, a transitional species between halophytes and glycophytes [57], and common bean, a glycophyte [6] might be another reason. Additionally, other contributing factors might include the plant growth conditions, direct exposure of plants to high salt concentrations, and the differences in sample collection times. The previous transcriptome studies focused on earlier time points and sudden applications of elevated salt concentrations, whereas our transcriptome analysis was performed using the “gradual step acclimated” plant material and thus reflected the salt responses rather than osmotic shock responses [41].
During salt stress within plant, all the major processes such as photosynthesis, protein synthesis, energy and lipid metabolisms are affected [68].Therefore, in order to tolerate this stress, plants require to initiate protective and survival actions by balancing cellular ion concentrations and minimizing ion toxicity through Na+/H+, K+ and Cl- transporter/antiporter activities to eliminate water and nutrient deficits, regulate osmotic potential changes via synthesis of osmoprotectant and osmoregulants such as sugars, amino acids, amines, and also minimize tissue damage through activation of scavenging pathways to eliminate antioxidant productions [1], [69], [70].
Based on the enrichment analysis of the DEGs, both root and leaf tissue genes were closely related with stress tolerance mechanisms. Major categories of the stress related functions were transmembrane transport activities (such as GO:0031224, GO:0015698, GO:0015103, GO:0015893, ko02010), carbohydrate metabolism (such as GO:0005976, GO:0015925, G0:15926, ko00040, ko00500, ko00052), lipid metabolism (such as GO:0010876, GO:006629, ko00591, ko00565, ko00600), secondary metabolite metabolism and oxidoreductase activity (such as GO:0006720, GO:0016884, GO:0006528 GO:16491 GO:0016679, ko00900, ko00940, ko00944), and transcriptional/translational activities (such as GO:0034728, GO:0022613, GO:0006412, GO:0010467,ko03020, ko03010, ko03013) (Figure 5 and 6, Table S6, S7, S8, and S9).
Ionic imbalance causes ion toxicity due to replacement of K+ by Na+ ions via competitive transport of Na+ through potassium channels [71]. Although cytosolic K+ is essential as cofactor for several enzyme activities, Na+ has toxic effects and thus has to be excluded or sequestrated into vacuoles in the cells [71]. Therefore to achieve a high K+/Na+ ratio for improved salt tolerance, increased expression of Na+/H+ antiporters with driving force of vacuolar H+-ATPases and H+ pyrophosphatases are important. Importance of these antiporter activities was also implicated once more in our study as the leaf tissues have shown distinctive upregulation in Na+/H+ antiporter (CL4908.Contig3_All) as well as vacuolar H+ATPase genes (CL3536.Contig4_All). Additionally, upregulation in the nonselective cyclic nucleotide gated cation channels, CNGC2 (CL5050.Contig1_All) in root tissues were striking in our salt tolerant variety, considering the previously implicated role of the CNGC channels in the main pathway of Na+ entry to roots [72]. However, studies in Arabidopsis has shown that when the K+ level was limited due to elevated Na+ during stress, activation of nonselective channel AtCNGC family members were still crucial to increase K+ influx to the root cells even at the expense of Na+ influx [73], [74]. More strikingly, the CNGC2 (CL5050.Contig1_All) was shown to be the only member that preferentially conducts K+ without the transport of Na+ [75].
Similar to Na+, Cl− is also toxic to the cells, thus Cl− homeostasis by endosomal compartmentalization is critical for plant adaptations to salt stress [76]. Although Cl− transport genes have been poorly studied, the attention mainly focused on voltage-dependent Cl− channels (Cl−/H+ antiporters) localized in endosomal membranes which may function in vacuolar sequestration of Cl− to minimize the tissue necrosis and long distance transport in shoots as well as Cl− exclusion from roots [77], [78]. Our enrichment analyses have also revealed upregulation of Cl− channels and Cl−/NO3 − transporter genes in the leaf (CL5256.Contig5_All) and the root tissues (CL5256.Contig6_All) respectively. The actions of Cl− channels were correlated with NO3 −, another macronutrient univalent anion in plants, and Cl−/NO3 − interactions show analogy to Na+/K+ interactions and selectivity to Na+ exclusion [78] during salt tolerance.
Osmotic potential change in plants is a typical outcome of ionic imbalance during salt stress, which creates domino effect in the activation of multiple metabolic processes. One of these metabolic processes includes increase in the accumulation of highly hydrophilic organic compounds (osmolytes) and hydrophylic proteins [68]. Upregulation of both osmolyte biosynthesis enzymes (CL7986.Contig1_All, CL2135.Contig2_All, CL3755.Contig1_All, Unigene14850_All, CL6414.Contig1_All, Unigene27033_All) and the hydrophilic LEA proteins (CL3431.Contig2_All, Unigene12424_All) in both leaf and root transcriptomes were a good indication of their protective role of cytosolic components of cells during osmotic imbalance. Activation of metabolic processes also enhances demands on energy resources, which are supplied by cellular respirations [79]. Increases in the expression of the genes related with respiration pathway enzymes in both tissues were indicative of such catabolic activities (CL8031.Contig1_All, Unigene25787_All, Unigene20031_All, and CL237.Contig7_All). Catabolic activities bring high risk of oxidative stress in plants which is associated with antioxidant production enzymes [80] to eliminate free oxygen radical accumulation in cells. Several different antioxidant biosynthesis related genes were upregulated within our transcriptome (CL1487.Contig1_All, Unigene1099_All, Unigene20329_All, and Unigene5947_All). During the battle against the osmotic as well as ionic stress in salinity, plant tolerance requires considerable efforts in maintenance of cell structure integrity by reinforcements or reorganizations in membranes, and cell wall components [81 and references therein]. It was not surprising to observe considerable amount of upregulation of genes regarding such activities (CL8348.Contig1_All, Unigene29739_All, Unigene20233_All, and CL7042.Contig1_All) in our transcriptome.
When we evaluated the common bean transcriptome under salt stress the results suggested that tolerance responses requires highly obvious efforts regarding ionic and osmotic homeostasis through transmembrane transport activities, mobilization and utilization of energy reserves for protection and preservation of structural integrity via metabolite biosynthesis while avoiding energy consumption for growth related transcriptional/translational activities (Figure 5 and 6).
Several transcription factors play important roles in translating stress signals into changes in gene expression. Based on the BLASTx analysis against the combined Legume TFDB, we have identified a total of 2,678 TF genes, in our common bean transcriptome (Table 3, and S10). In an earlier report by Kalavacharla et al. (2011), a similar number of TFs (2,516) were also identified.
Almost all of the most abundant TF families identified in our study within the DEGs (Table 3 and Table S10) have been well studied and implicated to play a role in both biotic and abiotic stress responses in plants [82 and references therein].
DREB genes are well studied members of AP2-ERF family, suggested to function in drought, salt, heat, and cold stresses by both ABA dependent or ABA-independent signaling pathways [83 and references therein]. Overexpression of DREB genes in diverse species of plants under various promoters have provided improved tolerance in abiotic stresses.
Although the biological function of most bHLH family members has not yet been studied in plants [84], increase in the bHLH transcription in tolerant wheat variety during salt stress, and regulation of drought tolerance response in Arabidopsis were elucidated by the bHLH regulated ABA signaling [84]. Increased focus on bHLH genes might shed light on its involvement in salinity stress as observed from overexpressions of bHLH92 in Arabidopsis [85], and OrbHLH001 in wild rice phloem tissues [86] conferring improved salt tolerance.
After the first sequencing of NAC family gene RD26 [87] NAC domain has been characterized based on the consensus sequences from NAM, CUC2, and ATAF1/2 proteins [88]. Since then differential expressions and involvement of several NAC genes in abiotic stress responses have been demonstrated [89].
As one of the largest TF families, MYB is involved in several physiological and biochemical processes during abiotic stress responses [90]. Among the members, TaMYB33 TF, which shows high similarity to R2R3-MYB proteins in rice and maize, [91] has been indicated in salt stress tolerance via ROS detoxification and osmotic balance reconstruction in wheat [92].
The regulatory role of WRKY family was also demonstrated in high salinity responses, Arabidopsis WRKY8 was predominantly expressed in roots and functions of it was consistent with the changes in Na+/K+ concentrations [86].
Several members of PHD finger protein family were observed to respond to abiotic stresses differentially. Especially soybean PHD2 was shown to be uniquely expressed in tolerant variety and suspected to provide tolerance by diminishing the oxidative stress through regulation of downstream genes [93]. Functional relevance of PHD family as chromatin-mediated transcriptional mediator was suggested through their involvement in activation or repression of ING1 [94], Pf1 [95], TIF1 [96], and KAP1 [97] genes.
The Homeobox-zip (HB family) TF members were suggested as excellent candidates to generate stress responses in transgenic plants and cotton root development regulation under salt stress involved GmHB1 gene expression [98], [99].
Certain members of C3H-type family TFs (AtSZF1 and AtSZF2) were shown to be involved in salt responses transiently within the first few minutes of salt exposure [100], however in our study the C3H-type TFs were not abundant in salt responsive genes (Table 3), most probably due to the differences in sample collection time.
C2C2-Dof TFs are involved in the photosynthetic gene expression of plants and Dof2-domains play a role as tissue-specific repressors in PEP carboxylase promoters, which suggest their regulatory activities in photosynthesis [101]. The ERF-domain of C2H2-Zn TFs were also shown to be important transcriptional repressors in responses to abiotic stresses, a key role of Zat7 protein in defense response during salinity was indicated in Arabidopsis [102].
A vast number of HSF family members of TFs were compiled from nine species; major aspects of functions implied their role in gene expression of chaperons for stability, localization of cellular components as well as in regulation of abiotic stress responses [103].
Conclusion
Our comprehensive transcriptome analysis of a salinity tolerant common bean variety by Illumina sequencing is the first transcriptomic study on the response of common bean under salt stress. The identified all-unigenes have been observed to be involved in similar pathways as in previous reports of legumes according to the GO, KEGG and COG analyses. Substantial upregulation of transmembrane transport activities indicates the efforts of common bean to maintain ionic and osmotic homeostasis. To do so, energy consumption seems to be shifted from growth related transcriptional/translational activities towards maintaining current structural integrity through metabolite biosynthesis. Analysis of TFs among DEGs has implicated well studied TF families with known roles in abiotic and biotic stress as well as those that were not strongly associated with salinity stress previously, such as bHLH family. Although many of the DEGs identified has been annotated in publicly available databases, there were DEGs with dramatic expression differences that has not been annotated or implicated in abiotic stress responses, which are awaiting functional characterization. Overall, transcription profiling and identification of DEGs have provided valuable information for salinity tolerance mechanisms that is indispensable to maximize yield and utilization of arid lands.
Supporting Information
Random distribution of clean reads. The x-axis describes the number of reads and the y-axis indicates the number of clean reads mapped to relative positions in unigenes for the six subtranscriptomes. The orientations of the genes are in 5′ to 3′direction and the gene lengths are normalized.
(TIF)
GO annotations of all-unigenes. The annotations were performed with Blast2GO software. The length of each bar indicated the percentage of all unigenes falls under each GO terms. The x-axis is in logarithmic scale.
(TIF)
COG function prediction of all-unigenes. The possible functions of all-unigenes were predicted by alignment to COG database. Each letter in the x-axis represented the COG categories listed on the right of the graph.
(TIFF)
List of selected all-unigenes for comparison of qRT-PCR and RNA-seq analysis.
(XLSX)
List of primers to amplify the selected all-unigenes for qRT-PCR analysis.
(XLSX)
KEGG pathway annotations for all-unigenes.
(XLSX)
Differentially expressed unigenes in leaf tissues.
(XLSX)
Differentially expressed unigenes in root tissues.
(XLSX)
Gene Ontology funtional enrichment analysis of leaf DEGs. (FDR-value <0.05).
(XLSX)
Gene Ontology functional enrichment analysis of root DEGs. (FDR-value <0.05).
(XLSX)
Pathway enrichment analysis of leaf DEGs. (Adjusted-P-value <0.05).
(XLSX)
Pathway enrichment analysis of root DEGs. (Adjusted-P-value <0.05).
(XLSX)
The number of genes in TF families.
(XLSX)
Acknowledgments
We thank Prof. H. Yildiz Dasgan from Cukurova University Department of Horticulture, Adana, Turkey for Ispir seed supply. We are also grateful to Prof. Kuyas Bugra for assistance with manuscript editing and Dr. Ersen Kavak for his guidance in the statistical analysis.
Funding Statement
This work was financially supported by The Scientific and Technological Research Council of Turkey (TUBITAK 110T922) and Bogazici University Research Funds (BAP 6674). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Munns R, Tester M (2008) Mechanisms of salinity tolerance. Annu Rev Plant Biol 59: 651–681. [DOI] [PubMed] [Google Scholar]
- 2. Mahajan S, Tuteja N (2005) Cold, salinity and drought stresses: an overview. Arch Biochem Biophys 444: 139–158. [DOI] [PubMed] [Google Scholar]
- 3.Hide JC (1954) Diagnosis and improvement of saline and alkali soils. Washington, D.C.: U.S. Dept. of Agriculture. 160 p. [Google Scholar]
- 4. Broughton WJ, Hernandez G, Blair M, Beebe S, Gepts P, et al. (2003) Beans (Phaseolus spp.) - model food legumes. Plant and Soil 252: 55–128. [Google Scholar]
- 5. Madakbas SY, Ellialtioglu S (2005) Taze fasulyede dayaniklilik islahinin kavrami, mekanizmasi ve kalitimi. Hasad Bitkisel Uretim 21: 80–86. [Google Scholar]
- 6. Chinnusamy V, Jagendorf A, Zhu JK (2005) Understanding and improving salt tolerance in plants. Crop Sci 45: 437–448. [Google Scholar]
- 7. Touchette B, Rhodes K, Smith G, Poole M (2009) Salt spray induces osmotic adjustment and tissue rigidity in smooth cordgrass, Spartina alterniflora (Loisel.). Estuar Coast 32: 917–925. [Google Scholar]
- 8. Huang L, Zhao A, Lew JL, Zhang T, Hrywna Y, et al. (2003) Farnesoid X receptor activates transcription of the phospholipid pump MDR3 . J Biol Chem 278: 51085–51090. [DOI] [PubMed] [Google Scholar]
- 9. Magnan F, Ranty B, Charpenteau M, Sotta B, Galaud JP, et al. (2008) Mutations in AtCML9, a calmodulin-like protein from Arabidopsis thaliana, alter plant responses to abiotic stress and abscisic acid. Plant J 56: 575–589. [DOI] [PubMed] [Google Scholar]
- 10. Wang H, Liang X, Wan Q, Wang X, Bi Y (2009) Ethylene and nitric oxide are involved in maintaining ion homeostasis in Arabidopsis callus under salt stress. Planta 230: 293–307. [DOI] [PubMed] [Google Scholar]
- 11. Xu P, Xiang Y, Zhu H, Xu H, Zhang Z, et al. (2009) Wheat cryptochromes: subcellular localization and involvement in photomorphogenesis and osmotic stress responses. Plant Physiol 149: 760–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Silva P, Geros H (2009) Regulation by salt of vacuolar H+-ATPase and H+-pyrophosphatase activities and Na+/H+ exchange. Plant Signal Behav 4: 718–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Seki M, Narusaka M, Ishida J, Nanjo T, Fujita M, et al. (2002) Monitoring the expression profiles of 7000 Arabidopsis genes under drought, cold and high-salinity stresses using a full-length cDNA microarray. Plant J 31: 279–292. [DOI] [PubMed] [Google Scholar]
- 14. Wang H, Miyazaki S, Kawai K, Deyholos M, Galbraith DW, et al. (2003) Temporal progression of gene expression responses to salt shock in maize roots. Plant Mol Biol 52: 873–891. [DOI] [PubMed] [Google Scholar]
- 15. Rabbani MA, Maruyama K, Abe H, Khan MA, Katsura K, et al. (2003) Monitoring expression profiles of rice genes under cold, drought, and high-salinity stresses and abscisic acid application using cDNA microarray and RNA gel-blot analyses. Plant Physiol 133: 1755–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jiang Y, Deyholos MK (2006) Comprehensive transcriptional profiling of NaCl-stressed Arabidopsis roots reveals novel classes of responsive genes. BMC Plant Biol 6: 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Walia H, Wilson C, Zeng L, Ismail AM, Condamine P, et al. (2007) Genome-wide transcriptional analysis of salinity stressed japonica and indica rice genotypes during panicle initiation stage. Plant Mol Biol 63: 609–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Qing DJ, Lu HF, Li N, Dong HT, Dong DF, et al. (2009) Comparative profiles of gene expression in leaves and roots of maize seedlings under conditions of salt stress and the removal of salt stress. Plant Cell Physiol 50: 889–903. [DOI] [PubMed] [Google Scholar]
- 19. Ge Y, Li Y, Zhu YM, Bai X, Lv DK, et al. (2010) Global transcriptome profiling of wild soybean (Glycine soja) roots under NaHCO3 treatment. BMC Plant Biol 10: 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xu P, Liu Z, Fan X, Gao J, Zhang X, et al. (2013) De novo transcriptome sequencing and comparative analysis of differentially expressed genes in Gossypium aridum under salt stress. Gene 525: 26–34. [DOI] [PubMed] [Google Scholar]
- 21. Postnikova OA, Shao J, Nemchinov LG (2013) Analysis of the alfalfa root transcriptome in response to salinity stress. Plant Cell Physiol 54: 1041–1055. [DOI] [PubMed] [Google Scholar]
- 22. Libault M, Farmer A, Joshi T, Takahashi K, Langley R, et al. (2010) An integrated transcriptome atlas of the crop model Glycine max, and its use in comparative analyses in plants. Plant J 63: 86–99. [DOI] [PubMed] [Google Scholar]
- 23. Garg R, Patel RK, Tyagi AK, Jain M (2011) De novo assembly of chickpea transcriptome using short reads for gene discovery and marker identification. DNA Res 18: 53–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kalavacharla V, Liu Z, Meyers BC, Thimmapuram J, Melmaiee K (2011) Identification and analysis of common bean (Phaseolus vulgaris L.) transcriptomes by massively parallel pyrosequencing. BMC Plant Biol 11: 135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kaur S, Cogan N, Pembleton L, Shinozuka M, Savin K, et al. (2011) Transcriptome sequencing of lentil based on second-generation technology permits large-scale unigene assembly and SSR marker discovery. BMC Genomics 12: 265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Moe K, Chung J-W, Cho Y-I, Moon J-K, Ku J-H, et al. (2011) Sequence information on simple sequence repeats and single nucleotide polymorphisms through transcriptome analysis of mungbean. J Integr Plant Biol 53: 63–73. [DOI] [PubMed] [Google Scholar]
- 27. Franssen S, Shrestha R, Bräutigam A, Bornberg-Bauer E, Weber A (2011) Comprehensive transcriptome analysis of the highly complex Pisum sativum genome using next generation sequencing. BMC Genomics 12: 227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Parra-González L, Aravena-Abarzúa G, Navarro-Navarro C, Udall J, Maughan J, et al. (2012) Yellow lupin (Lupinus luteus L.) transcriptome sequencing: molecular marker development and comparative studies. BMC genomics 13: 425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang J, Liang S, Duan J, Wang J, Chen S, et al. (2012) De novo assembly and characterisation of the transcriptome during seed development, and generation of genic-SSR markers in peanut (Arachis hypogaea L.). BMC Genomics 13: 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wu N, Matand K, Wu H, Li B, Li Y, et al. (2013) De novo next-generation sequencing, assembling and annotation of Arachis hypogaea L. Spanish botanical type whole plant transcriptome. Theor Appl Genet 126: 1145–1149. [DOI] [PubMed] [Google Scholar]
- 31. Hiremath P, Farmer A, Cannon S, Woodward J, Kudapa H, et al. (2011) Large-scale transcriptome analysis in chickpea (Cicer arietinum L.), an orphan legume crop of the semi-arid tropics of Asia and Africa. Plant Biotechnol J 9: 922–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fan X-D, Wang J-Q, Yang N, Dong Y-Y, Liu L, et al. (2013) Gene expression profiling of soybean leaves and roots under salt, saline-alkali and drought stress by high-throughput Illumina sequencing. Gene 512: 392–402. [DOI] [PubMed] [Google Scholar]
- 33. Zahaf O, Blanchet S, de Zelicourt A, Alunni B, Plet J, et al. (2012) Comparative transcriptomic analysis of salt adaptation in roots of contrasting Medicago truncatula genotypes. Mol Plant 5: 1068–1081. [DOI] [PubMed] [Google Scholar]
- 34.Phytozome v9.1 website. Available: http://www.phytozome.org/commonbean.php. Accessed 3 Feb 2014.
- 35. Sato S, Nakamura Y, Kaneko T, Asamizu E, Kato T, et al. (2008) Genome structure of the legume, Lotus japonicus . DNA Res 15: 227–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schmutz J, Cannon S, Schlueter J, Ma J, Mitros T, et al. (2010) Genome sequence of the palaeopolyploid soybean. Nature 463: 178–183. [DOI] [PubMed] [Google Scholar]
- 37. Young N, Debellé F, Oldroyd G, Geurts R, Cannon S, et al. (2011) The Medicago genome provides insight into the evolution of rhizobial symbioses. Nature 480: 520–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Varshney R, Chen W, Li Y, Bharti A, Saxena R, et al. (2012) Draft genome sequence of pigeonpea (Cajanus cajan), an orphan legume crop of resource-poor farmers. Nature biotechnology 30: 83–89. [DOI] [PubMed] [Google Scholar]
- 39. Varshney R, Song C, Saxena R, Azam S, Yu S, et al. (2013) Draft genome sequence of chickpea (Cicer arietinum) provides a resource for trait improvement. Nat Biotechnol 31: 240–246. [DOI] [PubMed] [Google Scholar]
- 40. Dasgan HY, Koc S (2009) Evaluation of salt tolerance in common bean genotypes by ion regulation and searching for screening parameters. J Food Agric Environ 7: 363–372. [Google Scholar]
- 41. Shavrukov Y (2013) Salt stress or salt shock: which genes are we studying? J Exp Bot 64: 119–127. [DOI] [PubMed] [Google Scholar]
- 42. Sanchez D, Lippold F, Redestig H, Hannah M, Erban A, et al. (2008) Integrative functional genomics of salt acclimatization in the model legume Lotus japonicus . Plant J 53: 973–987. [DOI] [PubMed] [Google Scholar]
- 43. Liu S, Lin L, Jiang P, Wang D, Xing Y (2011) A comparison of RNA-Seq and high-density exon array for detecting differential gene expression between closely related species. Nucleic Acids Res 39: 578–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Everett MV, Grau ED, Seeb JE (2011) Short reads and nonmodel species: exploring the complexities of next-generation sequence assembly and SNP discovery in the absence of a reference genome. Mol Ecol Resour 11 Suppl 193–108. [DOI] [PubMed] [Google Scholar]
- 45. Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, et al. (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29: 644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Iseli C, Jongeneel CV, Bucher P (1999) ESTScan: a program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. Proc Int Conf Intell Syst Mol Biol: 138–148. [PubMed]
- 47. Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, et al. (2000) Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25: 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talon M, et al. (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21: 3674–3676. [DOI] [PubMed] [Google Scholar]
- 49. Ye J, Fang L, Zheng H, Zhang Y, Chen J, et al. (2006) WEGO: a web tool for plotting GO annotations. Nucleic Acids Res 34: W293–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Li R, Yu C, Li Y, Lam TW, Yiu SM, et al. (2009) SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 25: 1966–1967. [DOI] [PubMed] [Google Scholar]
- 51. Benjamini Y, Yekutieli D (2001) The control of the false discovery rate in multiple testing under dependency. Annals of Statistics 29: 1165–1188. [Google Scholar]
- 52. Du Z, Zhou X, Ling Y, Zhang Z, Su Z (2010) agriGO: a GO analysis toolkit for the agricultural community. Nucleic Acids Res 38: W64–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Al-Shahrour F, Díaz-Uriarte R, Dopazo J (2004) FatiGO: a web tool for finding significant associations of Gene Ontology terms with groups of genes. Bioinformatics 20: 578–580. [DOI] [PubMed] [Google Scholar]
- 54. Borges A, Tsai SM, Caldas DG (2012) Validation of reference genes for RT-qPCR normalization in common bean during biotic and abiotic stresses. Plant Cell Rep 31: 827–838. [DOI] [PubMed] [Google Scholar]
- 55. Mochida K, Yoshida T, Sakurai T, Yamaguchi-Shinozaki K, Shinozaki K, et al. (2009) In silico analysis of transcription factor repertoire and prediction of stress responsive transcription factors in soybean. DNA Res 16: 353–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mochida K, Yoshida T, Sakurai T, Yamaguchi-Shinozaki K, Shinozaki K, et al. (2010) LegumeTFDB: an integrative database of Glycine max, Lotus japonicus and Medicago truncatula transcription factors. Bioinformatics 26: 290–291. [DOI] [PubMed] [Google Scholar]
- 57. Huang J, Lu X, Yan H, Chen S, Zhang W, et al. (2012) Transcriptome characterization and sequencing-based identification of salt-responsive genes in Millettia pinnata, a semi-mangrove plant. DNA Res 19: 195–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tyler BM (2007) Phytophthora sojae: root rot pathogen of soybean and model oomycete. Mol Plant Pathol 8: 1–8. [DOI] [PubMed] [Google Scholar]
- 59. Gulduren S, Elkoca E (2012) Salinity tolerance at germination stage of some bean (Phaseolus vulgaris L.) genotypes collected from north East Anatolia Region and Coruh Valley. J of Agricultural Faculty of Ataturk Univ 43: 29–41. [Google Scholar]
- 60. Gama PBS, Inanaga S, Tanaka K, Nakazawa R (2007) Physiological response of common bean (Phaseolus vulgaris L.) seedlings to salinity stress. Afr J Biotechnol 6: 79–88. [Google Scholar]
- 61. Ramirez M, Graham MA, Blanco-Lopez L, Silvente S, Medrano-Soto A, et al. (2005) Sequencing and analysis of common bean ESTs. Building a foundation for functional genomics. Plant Physiol 137: 1211–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Melotto M, Monteiro-Vitorello CB, Bruschi AG, Camargo LE (2005) Comparative bioinformatic analysis of genes expressed in common bean (Phaseolus vulgaris L.) seedlings. Genome 48: 562–570. [DOI] [PubMed] [Google Scholar]
- 63.Molina C, Zaman-Allah M, Khan F, Fatnassi N, Horres R, et al. (2011) The salt-responsive transcriptome of chickpea roots and nodules via deepSuperSAGE. BMC Plant Biology 11.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Liu Z, Crampton M, Todd A, Kalavacharla V (2012) Identification of expressed resistance gene-like sequences by data mining in 454-derived transcriptomic sequences of common bean (Phaseolus vulgaris L.). BMC Plant Biol 12: 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Liao D, Cram D, Sharpe A, Marsolais F (2013) Transcriptome profiling identifies candidate genes associated with the accumulation of distinct sulfur γ-glutamyl dipeptides in Phaseolus vulgaris and Vigna mungo seeds. Front Plant Sci 4: 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wang C, Chen H, Hao Q, Sha A, Shan Z, et al. (2012) Transcript profile of the response of two soybean genotypes to potassium deficiency. PloS ONE 7: e39856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Pang T, Ye C-Y, Xia X, Yin W (2013) De novo sequencing and transcriptome analysis of the desert shrub, Ammopiptanthus mongolicus, during cold acclimation using Illumina/Solexa. BMC Genomics 14: 488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Parida A, Das A (2005) Salt tolerance and salinity effects on plants: a review. Ecotoxicol Environ Saf 60: 324–349. [DOI] [PubMed] [Google Scholar]
- 69.Carillo P, Annunziata MG, Pontecorvo G, Fuggi A, Woodrow P (2011) Salinity stress and salt tolerance. In: Arun S, editor. Abiotic Stress in Plants - Mechanisms and Adaptations. Croatia: InTech. pp. 22–38.
- 70. Tomoaki H, Ichirou K, Maki K (2012) Salinity tolerance mechanisms in glycophytes: An overview with the central focus on rice plants. Rice 5: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Faïçal B, Khaled M (2012) Ion transporters and abiotic stress tolerance in plants. ISRN Mol Biol 2012: ID927436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Tester M, Davenport R (2003) Na+ tolerance and Na+ transport in higher plants. Ann Bot 91: 503–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Guo K-M, Babourina O, Christopher D, Borsics T, Rengel Z (2008) The cyclic nucleotide-gated channel, AtCNGC10, influences salt tolerance in Arabidopsis. Physiol Plant 134: 499–507. [DOI] [PubMed] [Google Scholar]
- 74.Yuen CYL, Christopher DA (2010) The role of cyclic nucleotide-gated channels in cation nutrition and abiotic stress. In: Demidchik V, Maathuis F, editors. Ion Channels and Plant Stress Responses: Springer Berlin Heidelberg. pp. 137–157.
- 75. Leng Q, Mercier R, Hua B-G, Fromm H, Berkowitz G (2002) Electrophysiological analysis of cloned cyclic nucleotide-gated ion channels. Plant Physiol 128: 400–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Teakle N, Flowers T, Real D, Colmer T (2007) Lotus tenuis tolerates the interactive effects of salinity and waterlogging by ‘excluding’ Na+ and Cl− from the xylem. J Exp Bot 58: 2169–2180. [DOI] [PubMed] [Google Scholar]
- 77. Samir A, Sameh S, Chedly A (2008) Growth, nitrogen fixation and ion distribution in Medicago truncatula subjected to salt stress. Plant Soil 312: 59–67. [Google Scholar]
- 78. Teakle N, Tyerman S (2010) Mechanisms of Cl- transport contributing to salt tolerance. Plant Cell Environ 33: 566–589. [DOI] [PubMed] [Google Scholar]
- 79. Jacoby RP, Taylor NL, Millar AH (2011) The role of mitochondrial respiration in salinity tolerance. Trends Plant Sci 16: 614–623. [DOI] [PubMed] [Google Scholar]
- 80. Mariana Lins de Oliveira C, Bety Shiue de H, João Antônio de Almeida G, Rafaela Moura C, Jarcilene Silva de A-C, et al. (2012) Photosynthesis and antioxidant activity in Jatropha curcas L. under salt stress. Braz J Plant Physiol 24: 55–67. [Google Scholar]
- 81. Ghosh D, Xu J (2014) Abiotic stress responses in plant roots: a proteomics perspective. Front Plant Sci 5: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Lindemose S, O'Shea C, Jensen MK, Skriver K (2013) Structure, function and networks of transcription factors involved in abiotic stress responses. Int J Mol Sci 14: 5842–5878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Lata C, Prasad M (2011) Role of DREBs in regulation of abiotic stress responses in plants. J Exp Bot 62: 4731–4748. [DOI] [PubMed] [Google Scholar]
- 84. Li H, Sun J, Xu Y, Jiang H, Wu X, et al. (2007) The bHLH-type transcription factor AtAIB positively regulates ABA response in Arabidopsis. Plant Mol Biol 65: 655–665. [DOI] [PubMed] [Google Scholar]
- 85. Jiang Y, Yang B, Deyholos MK (2009) Functional characterization of the Arabidopsis bHLH92 transcription factor in abiotic stress. Mol Genet Genomics 282: 503–516. [DOI] [PubMed] [Google Scholar]
- 86. Chen Y, Li F, Ma Y, Chong K, Xu Y (2013) Overexpression of OrbHLH001, a putative helix-loop-helix transcription factor, causes increased expression of AKT1 and maintains ionic balance under salt stress in rice. J Plant Physiol 170: 93–100. [DOI] [PubMed] [Google Scholar]
- 87. Yamaguchi-Shinozaki K, Koizumi M, Urao S, Shinozaki K (1992) Molecular cloning and characterization of 9 cDNAs for genes that are responsive to desiccation in Arabidopsis thaliana: Sequence analysis of one cDNA clone that encodes a putative transmembrane channel protein. Plant Cell Physiol 33: 217–224. [Google Scholar]
- 88. Aida M, Ishida T, Fukaki H, Fujisawa H, Tasaka M (1997) Genes involved in organ separation in Arabidopsis: an analysis of the cup-shaped cotyledon mutant. Plant Cell 9: 841–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Nakashima K, Takasaki H, Mizoi J, Shinozaki K, Yamaguchi-Shinozaki K (2012) NAC transcription factors in plant abiotic stress responses. Biochim Biophys Acta 1819: 97–103. [DOI] [PubMed] [Google Scholar]
- 90. Dai X, Xu Y, Ma Q, Xu W, Wang T, et al. (2007) Overexpression of an R1R2R3 MYB gene, OsMYB3R-2, increases tolerance to freezing, drought, and salt stress in transgenic Arabidopsis. Plant Physiol 143: 1739–1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Li-Chao Z, Guang-Yao Z, Ji-Zeng JIA, Xiu-Ying K (2009) Cloning and Analysis of Salt Stress-Related Gene TaMYB32 in Wheat. Acta Agron Sin 35: 1181–1187. [Google Scholar]
- 92. Qin Y, Wang M, Tian Y, He W, Han L, et al. (2012) Over-expression of TaMYB33 encoding a novel wheat MYB transcription factor increases salt and drought tolerance in Arabidopsis. Mol Biol Rep 39: 7183–7192. [DOI] [PubMed] [Google Scholar]
- 93. Wei W, Huang J, Hao YJ, Zou HF, Wang HW, et al. (2009) Soybean GmPHD-type transcription regulators improve stress tolerance in transgenic Arabidopsis plants. PLoS One 4: e7209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Skowyra D, Zeremski M, Neznanov N, Li M, Choi Y, et al. (2001) Differential association of products of alternative transcripts of the candidate tumor suppressor ING1 with the mSin3/HDAC1 transcriptional corepressor complex. J Biol Chem 276: 8734–8739. [DOI] [PubMed] [Google Scholar]
- 95. Yochum G, Ayer D (2001) Pf1, a novel PHD zinc finger protein that links the TLE corepressor to the mSin3A-histone deacetylase complex. Mol Cell Biol 21: 4110–4118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Venturini L, You J, Stadler M, Galien R, Lallemand V, et al. (1999) TIF1gamma, a novel member of the transcriptional intermediary factor 1 family. Oncogene 18: 1209–1217. [DOI] [PubMed] [Google Scholar]
- 97. Schultz D, Friedman J, Rauscher F (2001) Targeting histone deacetylase complexes via KRAB-zinc finger proteins: the PHD and bromodomains of KAP-1 form a cooperative unit that recruits a novel isoform of the Mi-2alpha subunit of NuRD . Genes Dev 15: 428–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Ni Y, Wang X, Li D, Wu Y, Xu W, et al. (2008) Novel cotton homeobox gene and its expression profiling in root development and in response to stresses and phytohormones. Acta Biochim Biophys Sin (Shanghai) 40: 78–84. [DOI] [PubMed] [Google Scholar]
- 99. Zahur M, Asif MA, Zeeshan N, Mehmood S, Malik MF, et al. (2013) Homeobox leucine zipper proteins and cotton improvement. Adv Biosci Biotechnol 4: 15.24558636 [Google Scholar]
- 100. Sun J, Jiang H, Xu Y, Li H, Wu X, et al. (2007) The CCCH-type zinc finger proteins AtSZF1 and AtSZF2 regulate salt stress responses in Arabidopsis. Plant Cell Physiol 48: 1148–1158. [DOI] [PubMed] [Google Scholar]
- 101. Saibo NJ, Lourenco T, Oliveira MM (2009) Transcription factors and regulation of photosynthetic and related metabolism under environmental stresses. Ann Bot 103: 609–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Ciftci-Yilmaz S, Morsy MR, Song L, Coutu A, Krizek BA, et al. (2007) The EAR-motif of the Cys2/His2-type zinc finger protein Zat7 plays a key role in the defense response of Arabidopsis to salinity stress. J Biol Chem 282: 9260–9268. [DOI] [PubMed] [Google Scholar]
- 103. Scharf KD, Berberich T, Ebersberger I, Nover L (2012) The plant heat stress transcription factor (Hsf) family: structure, function and evolution. Biochim Biophys Acta 1819: 104–119. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Random distribution of clean reads. The x-axis describes the number of reads and the y-axis indicates the number of clean reads mapped to relative positions in unigenes for the six subtranscriptomes. The orientations of the genes are in 5′ to 3′direction and the gene lengths are normalized.
(TIF)
GO annotations of all-unigenes. The annotations were performed with Blast2GO software. The length of each bar indicated the percentage of all unigenes falls under each GO terms. The x-axis is in logarithmic scale.
(TIF)
COG function prediction of all-unigenes. The possible functions of all-unigenes were predicted by alignment to COG database. Each letter in the x-axis represented the COG categories listed on the right of the graph.
(TIFF)
List of selected all-unigenes for comparison of qRT-PCR and RNA-seq analysis.
(XLSX)
List of primers to amplify the selected all-unigenes for qRT-PCR analysis.
(XLSX)
KEGG pathway annotations for all-unigenes.
(XLSX)
Differentially expressed unigenes in leaf tissues.
(XLSX)
Differentially expressed unigenes in root tissues.
(XLSX)
Gene Ontology funtional enrichment analysis of leaf DEGs. (FDR-value <0.05).
(XLSX)
Gene Ontology functional enrichment analysis of root DEGs. (FDR-value <0.05).
(XLSX)
Pathway enrichment analysis of leaf DEGs. (Adjusted-P-value <0.05).
(XLSX)
Pathway enrichment analysis of root DEGs. (Adjusted-P-value <0.05).
(XLSX)
The number of genes in TF families.
(XLSX)