

Abstract
Commitment of Runx2-expressing precursor osteoblasts to functional osteoblasts and then osteocytes is triggered by Osterix (Osx), which activates its target genes in those cells during bone formation. It is not yet known whether Osx has a role in remodeling the chromatin architecture of its target genes during the transition from preosteoblast to osteoblast. In testing the hypothesis that Osx is indispensable for active chromatin architecture, we first showed that in Osx-null calvarial cells occupancy of the transcriptional activators including Wdr5, c-Myc and H2A.Z at the Osx-target gene Bsp was very markedly decreased. The levels of methylation of lysines 4 and 36 and acetylation of histone H3, markers for active chromatin, were also reduced at the Bsp gene in these cells. In contrast, occupancy of the transcriptional repressors HP1 and the NO66 histone demethylase, previously identified as an Osx-interacting protein, was increased at the Bsp gene in Osx-null calvarial cells. Furthermore, the Bsp promoter was hypermethylated in embryonic stem (ES) cells and in E9.5 embryos but was markedly hypomethylated in the calvaria of E18.5 embryos, coinciding with robust Bsp expression. In contrast, CpG methylation in the Bsp promoter remained high in Osx-null calvaria compared to Osx-wild type calvaria. Our data also revealed that NO66 interacted with DNMT1A and HDAC1A as well as HP1, which are known to control the histone and DNA methylation. In addition, HP1 stimulated the demethylase activity of NO66 for its substrates H3K4me3 and H3K36me3. Our findings strongly suggest that in the absence of Osx, the chromatin of Osx-target genes is transcriptionally inactive. We propose that Osx is a molecular switch for the formation of an active chromatin state during osteoblast differentiation, whereas NO66 helps gene repression through histone demethylation and/or formation of a repressor complex resulting in multi-layered control of the chromatin architecture of specific osteoblast genes.
Keywords: Osterix, NO66, Histone demethylase, Osteoblasts, Chromatin, DNA methylation
INTRODUCTION
Three transcription factors, Runx2, Osterix (Osx) and β-catenin, have an essential role in osteoblast differentiation and bone formation (1–6). Runx2 is an early, largely osteoblast-specific transcription factor and is required for formation of preosteoblasts, which express early marker genes including Akp and Col1a1. Osx triggers preosteoblasts to differentiate into osteoblasts by activating later markers including Bsp and Oc genes as well as up-regulation of Col1a1. Osx is also needed for the final differentiation of osteoblasts into osteocytes and activates osteocyte-specific markers such as Sost, Dmp1, and Phex (7). In Osx-null embryos, no bone formation takes place and expression of osteoblast-specific genes is severely reduced or absent (6,8). Osx is thus essential for bone formation during embryonic development. Our recent findings strongly suggest that inactivation of the Osx gene postnatally almost completely inhibits bone formation, causes massive accumulation of unresorbed cartilage matrix beneath the growth plate, and produces marked defects in the maturation and function of osteocytes. Hence, Osx continues to play essential roles in bone formation and bone homeostasis in the adult (7,9,10).
Besides the absolute necessity of specific transcription factors for the expression of bone-specific genes, chromatin proteins are also needed in the regulatory network of gene transcription in bones. These proteins regulate the activities of transcription factors as well as histone modifications including acetylation, methylation, phosphorylation, sumoylation, and ubiquitination (11,12). We have shown recently that the JmjC-domain NO66 interacts with Osx and can negatively regulate the transcriptional activity of Osx in reporter assays. NO66 displays histone demethylase activity with dual specificity for H3K4me and H3K36me, two markers of active transcription (13). Knock-down of NO66 in preosteoblast MC3T3 cells accelerates Osx-target gene expression. In addition, during differentiation of MC3T3 cells, occupancy of NO66 at Osx-target genes decreases whereas that of Osx increases. We speculate that the transition of Osx-target genes from inactive to active transcriptional states involves interactions between Osx and NO66 and that NO66 has a role in controlling the levels of H3K4 and H3K36 methylation in the chromatin of these genes (13). NO66 is highly conserved in eukaryotes and is present in both the granular part of the nucleolus and in the nucleoplasm (14).
DNA methylation at CpG dinucleotides is a heritable epigenetic change that occurs throughout the genome and often inversely correlates with gene expression. Repressor protein methyl-CpG-binding protein (MECP2) interacts with methylated DNA and then allows the binding of other repressors such as HP1, histone deacetylases (HDACs), and Co-Rest, thus forming inactive chromatin that is likely to be inaccessible for further interactions with basal and specific transcription factors (15–18). Histone methylation and DNA methylation are interdependent in controlling the chromatin state during gene transcription (19). Other studies have shown that functional interaction between HP1 and DNMT1A leads to increased DNA methylation, resulting in gene silencing (20,21). DNA methylation-induced transcriptional repression is often associated with developmental genes prior to their activation. Subsequent activation of these genes at particular stages of development is needed for proper cellular differentiation and tissue morphogenesis (22). Osteoblast-specific genes are also developmentally expressed, and the function of these genes is required at specific stages such as proliferation, differentiation, and maturation of osteoblasts during bone formation. Whether or not DNA methylation plays an important role in regulation of Osx-target genes during mouse bone development remains elusive. Previous studies indicated that the promoters of genes such as Dlx5, Osx, Oc and Alp as well as osteocyte marker Sost are hypomethylated in osteoblast cells that express these genes but are hypermethylated in non-osteogenic cells, which do not express these genes (23–25).
Even if Osx has an essential role in activation of the genetic program of osteoblast differentiation, we do not currently know whether Osx is needed for chromatin remodeling of osteoblast-specific genes during osteoblast differentiation in order to form a transcriptionally active chromatin environment. In the present study, we showed that in Osx-null mouse embryo calvarial cells, which do not express Bsp and Oc genes despite the expression of early transcription factor Runx2, chromatin of the Osx-target Bsp gene was associated with several repressors including NO66, HP1, and MeCP2 as well as low levels of histone methylation and acetylation at residues that contain high levels of these modifications during active transcription. Our results indicate that the Bsp promoter in mouse calvaria was hypermethylated before activation of this gene or before onset of osteoblast differentiation. Our data strongly suggest that during activation of osteoblast genes at the onset of differentiation, Osx plays a crucial role in remodeling the chromatin into a transcriptionally favorable state. Our findings provide mechanistic insights into the epigenetic transition of repression to activation of Osx-target genes during osteoblast differentiation.
MATERIALS AND METHODS
Isolation and culture of primary osteoblasts from Osx-wild type (wt) and Osx-null embryos
Heterozygous Osx(+/−) mice and homozygous Osx-null(−/ −) mice were generated as previously described (6). Mouse embryos were obtained by timed pregnancy. Calvaria were isolated and treated with collagenase P (1 mg/ml) and dispase (2 mg/ml) in α-MEM for 15 min at 37°C. The first two digests were discarded, and cells obtained after the third digestion were collected, plated, and maintained in complete media supplemented with ascorbate and β-glycerol phosphate whenever needed. Osx-null cells were plated in 12-well plate and transfected with 1μg of Osx expression vector. After transfection and confluency, mock and transfected Osx-null cells and Osx-wt cells (as controls) were cultured in differentiating media for 7 days. Extracellular matrix deposition was analyzed by staining with Alizarin Red S. Cells were viewed under a microscope and photographed with the 20X objective.
Chromatin immunoprecipitation and quantitative real-time PCR
Chromatin immunoprecipitation (ChIP) was performed as previously described (13). After being cross-linked with 1% formaldehyde for 20 min and washed with cold phosphate-buffered saline (PBS), cells were homogenized in hypotonic buffer (25 M HEPES, pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.1% NP-40, 1 mM DTT, 0.5 mM PMSF, and protease inhibitor cocktail). Nuclei were collected by centrifugation at 1500 g for 5 min at 4°C and then resuspended in nuclear lysis buffer (50 mM HEPES pH 7.9, 140 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% Na-deoxycholate, 0.1% SDS, 0.5 mM PMSF, and protease inhibitor cocktail) followed by sonication and then centrifugation. Sonicated chromatin was diluted with lysis buffer and used for immunoprecipitation with antibodies preconjugated to magnetic protein G-coated beads (Invitrogen). After overnight incubation at 4°C with rotation, the beads were washed and reverse cross-linked at 65°C for 5 h, and then DNA was purified with an EZ-ChIP kit (Millipore). Quantitative PCR reactions were performed in triplicate with region-specific primers, purified DNA, and SYBR green I PCR master mix (Applied Biosystems). Results were computed as percentage of antibody bound per input after control IgG values were subtracted.
Genomic DNA isolation, bisulfite approach, and DNA methylation
Mouse tissues were dissolved in lysis buffer containing 50 mM Tris (pH 8.0), 100 mM NaCl, 1% SDS and 50 mg/ml proteinase K by incubation at 55°C overnight. DNA was extracted with phenol-chloroform and once with chloroform and then precipitated with ethanol. Bisulfite treatment of genomic DNA (1 μg) was done with a Methyl Collector DNA kit (Active Motif). Converted DNA was amplified by JumpStart RedTaq (Sigma) with primers designed to anneal to only converted DNA template. PCR products were directly sequenced with the same primers.
Expression of recombinant proteins and histone demethylase activity
Human NO66 (full-length and a fragment of amino acids 168–641) and HP1α cDNAs were cloned into the pET23 vector (Novagen). Proteins were expressed in BL21(DE3) cells upon induction with 0.2 mM IPTG overnight at room temperature and then purified by Ni-NTA agarose (Qiagen). Demethylation reactions were carried out by incubating 2–5 μg of purified protein NO66 and its truncated variant (168–641) with 5–7 μg calf thymus bulk histones (Sigma) in 20 μl demethylase buffer (50 mM HEPES [pH 7.9], 150 mM NaCl, 100 μM ferrous sulphate, 1 mM ketoglutarate, and 2 mM ascorbate). Reactions were carried out at 37°C for 1–3 h, and samples were separated using SDS-PAGE and then immunoblotted.
RESULTS
Osx-null cells remain undifferentiated during in vitro osteoblast differentiation conditions
In Osx-null embryos, Runx2-expressing precursor cells are arrested in their differentiation into osteoblasts, and consequently no expression of Bsp and Oc is observed. Runx2 is expressed in Osx-null mice, but Osx is not expressed in Runx2-null embryos, clearly indicating that Osx is downstream of Runx2 activation at least during embryonic development. In this study, we first asked whether osteoblasts markers can be activated in Osx-null cells upon prolonged culture in osteogenic media. Calvarial cells from Osx-wt and Osx-null embryos at E18.5 were cultured in osteogenic media for 0, 8, and 12 days. Consistent with our previous in vivo results (6), Figure 1A and B indicate that stimulation of alkaline phosphatase (Akp) and Bsp in Osx-wt calvarial cells occurred, but no Bsp expression was observed in Osx-null cells even under prolonged culture of these cells in osteogenic media. Because Osx-null cells are arrested in their differentiation and no expression of Bsp gene was observed, we then overexpressed Osx in these cells to examine whether the osteoblast phenotype could be rescued in Osx-null cells. Our data showed that Osx-null cells transfected with Osx could be differentiated into osteoblasts and produced extracellular matrix, similar to Osx-wt cells, as visualized by Alizarin Red staining (Fig 1C). However, mock transfected Osx-null cells remained osteoblast-arrested and undifferentiated, and no Alizarin Red staining in these cells was observed. Furthermore, levels of endogenous Bsp were also increased in Osx overexpressing Osx-null cells. Expression of Flag-HA-Osx in Osx-null cells and increased level of Bsp in these cells are shown in Suppl. Fig. 1. These observations suggest that Osx-null cells remained undifferentiated even after prolong culture in osteogenic media and thus Osx is required for osteoblast differentiation during in vitro cell culture conditions in which precursor osteoblasts can be differentiated into mature and functional osteoblasts.
Fig. 1.

Osx is required for osteoblasts differentiation in culture upon osteogenic medium. (A–B) Quantitative RT-PCR for Akp and Bsp mRNAs in Osx-wt (Wt) and Osx-null calvarial cells cultured in osteogenic media for the indicated number of days. (C) Alizarin Red S staining for matrix deposition in Osx-wt, Osx-null and overexpressed Osx-null cells which were grown in differentiation media containing ascorbic acid and β-glycerol phosphate for 7 days. After staining, plates were photographed microscopically with a 20X objective and a representative of three independent plates is shown.
Repression of Bsp in Osx-null cells is associated with increased occupancy of NO66 histone demethylase
The JmjC-domain-containing NO66 is an Osx-interacting protein and a histone demethylase specific for H3K4me3 and H3K36me3. NO66 inhibits the Osx-dependent activation of Osx-target promoters in reporter assays and inhibits expression of these target genes in osteoblasts. In differentiating MC3T3 cells induced by BMP2, interaction of NO66 with the Bsp and Oc genes decreases while that of Osx increases with concomitant expression of these genes (13). Those studies suggest the hypothesis that activation by Osx might cause the removal of NO66 from Osx-target genes. One prediction from this hypothesis would be that in Osx-null cells, the occupancy of NO66 would be maintained at a high level. We tested this prediction by comparing the occupancy of NO66 in Osx-wt and Osx-null calvarial cells. ChIP experiments showed that the occupancy of Osx at the Bsp gene was observed only in Osx-wt cells, but not in Osx-null cells (Fig. 2A, Left). Interestingly, the NO66 occupancy was markedly higher at the Bsp gene in Osx-null cells than in Osx-wt cells, indicating that NO66 remains present at the Osx-target Bsp in the absence of Osx when this gene is repressed (Fig 2B, Left). In addition, there was no apparent change in levels of NO66 in Osx-null cells as shown by western blot (Fig 2C). The Osx occupancy at control genes including βactin, cyclin B1(ccn b1), and grp as well as that of NO66 at βactin was observed at background level, indicating the specificity of our ChIP experiments (Fig 2A and B, Right). Furthermore, when Osx-wt calvarial cells were cultured in osteogenic media for 8 days and Bsp expression was induced (Fig. 1B), occupancy of Osx at the Bsp gene increased particularly at the P2 site, but that of NO66 remarkably decreased in the Bsp chromatin (Fig. 2D). These results suggest that 1) interactions of NO66 with Osx-target Bsp were associated with osteoblast-arrested precursor cells when Bsp expression was repressed, 2) depletion of NO66 from the Bsp occurred during expression of this gene and osteoblast differentiation, and 3) when Osx occupancy increased, the levels of NO66 occupancy at the target sites decreased. We speculate that Osx-mediated activation is needed to displace NO66 from Bsp gene and most likely from other Osx-target osteoblast genes, which might facilitate activation of these genes in osteoblasts.
Fig. 2.

Higher levels of NO66 occupancy in the chromatin of the BSP gene in Osx-null calvarial cells compared to Osx-wt cells. (A) ChIP assays for the occupancy of Osx and (B) for NO66 occupancy in chromatin fragments of the Bsp gene in Osx-wt and Osx-null calvarial cells. Osx occupancy at the Bsp gene in Osx-null cells or at promoters of the control genes is above the background owing to non-specific binding of the Osx antibody with genomic material. ChIPed DNA samples derived from Osx and NO66 antibodies were amplified within the promoter regions of βactin, cyclin B1 (ccn b1), and grp genes to indicate the specificity of the ChIP. (C) Western blots for Osx and NO66 levels in Osx-wt and Osx-null calvarial cells. β-actin served as loading control. (D) ChIP assays for Osx and NO66 after Osx-wt cells were cultured in the presence or absence of osteogenic media for 8 days (in - induced, un - uninduced). The ChIP data are presented as a percentage of input after subtracting control IgG values. Error bars represents standard error of the mean of triplicate qPCR of representative ChIP assays. Schematic of the Bsp gene is shown with primers (indicated as P1 –P5) used in this study for PCR amplification.
Inactivation of Osx prevents active histone methylation and acetylation to take place in the chromatin of the Osx-target Bsp gene
Runx2 and Osx are both essential for expression of Bsp and Oc during mouse embryonic development. In Runx2-expressing Osx-null embryos, no expression of Bsp and Oc occurs (6), thus raising an important question of whether chromatin architecture of these genes is transcriptionally favorable. To address this, we examined the occupancy of chromatin modifying activities at chromatin segments of the Bsp gene in Osx-wt and Osx-null calvarial cells. ChIP assays showed that the occupancy of lysine 4 methyl transferase (Wdr5) at the two chromatin fragments of the Bsp promoter was significantly decreased in the Osx-null cells compared to Osx-wt cells (Fig. 3A). Wdr5 is a BMP-2-inducible gene and its protein product is a positive regulator of osteoblast differentiation (28). The levels of trimethylated lysine 4 (H3K4me3) were also decreased at the Bsp gene in Osx-null cells compared to Osx-wt cells (Fig. 3B). As expected, H3K4me3 levels were enriched within 1 kb of the Bsp-proximal promoter in Osx-wt cells compared to the rest of the gene. Furthermore, the levels of H3K36me3 within the coding region of the Bsp gene were significantly decreased in Osx-null cells (Fig. 3C). Consistent with our previous studies, increased occupancy of NO66 within the coding region of the Bsp gene is inversely correlated with that of H3K36me3. This indicates that lower levels of H3K36me3 might be due to the presence of NO66 within the coding region of the Bsp gene. Moreover, levels of H3 acetylation at lysine 9/16, which remain high in active chromatin, were almost undetectable at the Bsp promoter in Osx-null cells and were significantly enriched in Osx-wt cells (Fig. 3D). Levels of H3K27me3 in the promoter and coding regions of the Bsp gene were not changed in Osx-null cells compared to Osx-wt cells (Fig 3E). These results of ChIP experiments are based on limited intragenic regions; further genome-wide analysis of the distribution of histone methylation in osteoblast genes during bone formation will elucidate the role of histone methylation as well as that of Runx2 and Osx in the chromatin architecture of osteoblast genes. Nevertheless, our results clearly indicate that Bsp remains repressed in the absence of active histone methylation and acetylation together in parallel to increased occupancy of NO66, which demethylates active marks lysine 4 and 36 (13), within promoter and coding regions of the chromatin of Bsp in Osx-null cells.
Fig. 3.

Chromatin of the Bsp gene lacks the active marks of histone methylation and acetylation in absence of Osx. (A–E) ChIP assays for the occupancy of Wdr5, H3K4me3, H3K36me3, H3K9/16Ac, and H3K27me3 at chromatin segments of the Bsp gene in Osx-wt (Wt) and Osx-null (mt) calvarial cells as indicated in the panels. ChIP data for Wdr5 occupancy are presented as fold enrichment over control rabbit IgG values in the promoter of the Bsp gene. ChIP data for methylation and acetylation are normalized with occupancy of total histone H3 at the respective sites after subtracting control rabbit IgG values. Site-specific primers used in this study are described above.
c-Myc induces osteoblast differentiation through positive regulation of Osx activity
Transcription factor c-Myc induces osteoblast differentiation by first targeting Runx2 expression and then other osteoblast markers including Akp and Oc (29). However, the regulatory role of c-Myc on Osx activity in the transcriptional program of Osx-target genes has not been tested. To address this, we co-transfected c-Myc and Osx and showed in reporter assays that co-transfection enhanced the Osx-dependent activity of the Bsp and Oc promoters (Fig. 4A and B). c-Myc alone, however, had little effect on the basal activity level of these promoters. Consistent with our previous studies, NO66 inhibited Osx-dependent stimulation as well as c-Myc-stimulated Osx activity of the Bsp promoter (Fig. 4A). ChIP assays showed that c-Myc interacted with the chromatin of the Bsp promoter only in Osx-wt osteoblasts, but not in Osx-null cells (Fig. 4C). Interactions of c-Myc at the proximal promoter region (P4) appeared to be very specific when compared to that at other regions. Note that c-Myc interacted with the same chromatin segments of the Bsp gene (P4) with which Osx also interacted (see Fig. 2A and D). As was the case for Osx-null cells, we found that despite high levels of c-myc expression in osteoblast-arrested Osx-null embryos (30), c-Myc did not interact with the Bsp gene in these cells. Taken together, we conclude that this failure of c-Myc to interact with the Bsp gene in Osx-null cells could be due to the absence of Osx, which causes an inactive chromatin structure. Furthermore, our results identified c-Myc as a positive modulator of Osx-dependent gene activation.
Fig. 4.

Transcription factor c-Myc potentiates Osx-dependent Bsp activation through interactions with the Bsp gene. (A) Reporter assay using the 2-kb mouse Bsp promoter after transfection with Osx and c-Myc expression vectors in HEK293T cells in dose-dependent manner as indicated (100ng for Osx and 1x=100ng for c-Myc). Co-transfection of NO66 (200ng plasmid) inhibited Osx-dependent Bsp promoter activity. (B) Reporter assay using the 1.1kb mouse osteocalcin promoter after transfection with Osx and c-Myc expression vectors in HEK293T cells (1x=25ng for Osx, and 200ng for c-Myc). The error bars represent standard error of the mean of at least triplicate experiments. The P value (<0.05) for statistical difference was calculated using one way ANOVA test and was indicated by asterisks. (C) Interaction of c-Myc with Bsp gene occurred only in the presence of Osx. ChIP assays for c-Myc occupancy at the chromatin segments of the Bsp gene in Osx-wt (wt) and Osx-null calvarial cells. Data were analyzed as described above.
Hypermethylation of the Bsp promoter inversely correlates with Bsp expression in developing mouse calvarial bone
DNA methylation plays unequivocally important roles in the regulation of developmentally expressed genes by regulating chromatin structure and function. To test the likelihood that DNA methylation also regulates the expression of osteoblast genes, we first identified the sites of DNA methylation in the Bsp promoter and then analyze CpG methylation during mouse embryonic development. Analysis of the genomic sequence of the mouse Bsp gene using the Methyl Express software (ABI) identified a cluster of 19 CpG sites that formed a potential CpG island within the DNA segment located between −2.4 and −1.5 kb in the region surrounding Bsp and 12 other CpG sites between the gene’s proximal promoter and first intron (−1.5 to +0.2 kb). Figure 5A illustrates these methylatable CpG sites. No information is available yet on whether these CpG sites are methylated and contribute to the regulation of Bsp expression.
Fig. 5.

DNA methylation in the Bsp promoter is significantly lost preceding the Bsp expression in Osx-wt calvaria but not in Osx-null calvaria. (A) Schematic of the Bsp promoter showing CpG sites (indicated with diamond) within the −2.4 kb to +0.2 kb region. PCR products for the region I–IV were generated after bisulfite-treated DNA template and sequenced. (B) Bisulfite approach showing conversion of unmethylated cytosine to thymidine but not methylated cytosine. (C–G) Sequencing chromatogram of PCR products of region I amplified from bisulfite-treated DNA isolated at developmental stages of Osx-wt and Osx-null calvaria, as indicated in the Panel. Arrow indicates methylated cytosine undergoes DNA demethylation. (H) No DNA demethylation occurs in non-osseous brain and liver tissues of E18.5 embryos, which lacked Bsp expression, as shown by sequencing (Region I, site 7).
Genomic DNA was prepared from mouse ES cells, E9.5 embryo heads, and E13.5 and E18.5 calvaria. We also prepared DNA from E18.5 calvaria of osteoblast-arrested Osx-null embryos. These stages correspond to early embryogenesis (ES cells), osteoblast progenitor cells (E9.5), preosteoblasts (E13.5), and mature osteoblasts in calvarial bone (E18.5). Using several primer sets covering −2.4 to + 0.2 kb (from I to IV, Figure 5A), we amplified bisulfite-treated genomic DNA fragments and then sequenced the products. Conversion of non-CpG cytosine to thymidine after PCR amplification (indicated with red label T, Panel B) indicated that bisulfite treatment was very efficient to identify methylated cytosines (arrows in Fig. 5C–H). Using this approach, we found that 31CpG sites in the Bsp promoter were methylated in mouse ES cells. Representative chromatograms of cytosine methylation in region IV are shown in Figure 5C–H.
Analyses of DNA methylation specifically in promoter segment IV, which harbors essential cis-regulatory elements for Bsp expression, revealed that five CpG sites (Fig. 5C and D, indicated with arrow) in region IV were completely methylated in ES cells and in whole-head embryos (E9.5). In contrast, these CpG sites were noticeably hypomethylated in E13.5 and E18.5 mouse calvaria because both cytosine and thymidine peaks were present at these CpG sites (compare C peaks of sites 1–7 in ES cells and in E9.5 embryos with those of E18.5 embryos). These data indicate that inheritable methylated cytosines in Bsp gene from early embryogenesis had undergone DNA demethylation during calvarial bone development. For instance, CpG methylation at site 7 was completely lost in calvaria of E18.5 embryos (Panel F), when Bsp is robustly expressed. Our results suggest that demethylation of the Bsp promoter begins in osteo-chondro progenitors during mesenchyme condensation (around E11) and continues until the chromatin of the Bsp gene becomes amenable to induction of expression in differentiating osteoblasts. The promoter hypomethylation was much more apparent at embryonic stage E18.5 than at E13.5, further indicating that DNA methylation restricts Bsp expression in precursor osteoblasts or prior to osteoblast differentiation. Figure 5H shows that the Bsp promoter is still hypermethylated in adult brain and liver tissues, which lack Bsp expression.
Promoter methylation persists in Osx-null cells that do not express Bsp
Although our approach for detecting cytosine methylation does not permit rigorous quantitative analysis, the peak height ratio of C to T at a specific CpG site could be considered an indirect measurement of the methylation level. The presence of both cytosine and thymidine at a CpG site indicates that methylated cyosine became progressively demethylated to cytosine which gets converted to thymidine after bisulfite treatment (Fig 5, compare mosaics of C/T at site 7 in ES cells, E9.5 embryos, E13.5, E18.5 embryos and E18.5 Osx-null embryo). Further DNA demethylation of these CpG sites was relatively slower in E18.5 Osx-null calvaria than in E18.5 Osx-wt calvaria. The levels of methylation in some areas of E18.5 calvaria of Osx-null embryos were similar to those in E13.5 calvaria of Osx-wt embryos. These observations suggest that DNA methylation in the Bsp promoter persists in Osx-null calvaria (compare panels E, F and G, and Suppl Fig. 2 and 3). Osx begins to be expressed around E13.5 and is followed at E15.5 by expression of Bsp in osteoblasts of developing calvaria (31). Moreover, several CpG sites (for example 8–10,12–14; Suppl Fig. 2 and 3) were noticeably demethylated in Osx-wt calvaria when compared to those in ES cells and Osx-null calvaria at E.18.5. These findings further suggest that from ES cell stage to E18.5 embryo calvaria, DNA demethylation of the Bsp gene has progressed more in Osx-wt than in Osx-null calvaria. Our data indeed suggest that although DNA demethylation of the Bsp promoter is Osx-independent event and normally continues prior to Osx expression when Bsp is repressed; further DNA demethylation is likely to continue during development coinciding with Bsp expression during osteoblast differentiation. Continued DNA demethylation of the Bsp promoter in Osx-wt calvaria might be a developmental process coupled with recruitment of transcription factors including Osx to the target sites of the chromatin during initiation of Bsp expression. We speculate that in Osx-null calvaria DNA methylation in the Bsp gene may contribute to establishing an inactive chromatin state.
Association of epigenetic regulators MeCP2, HP1, and H2A.Z regulates Bsp expression
DNA methylation mediates gene repression through a sequential process, first by interactions of methyl CpG-binding proteins (MeCP2) with methylated DNA and then by formation of a repressor complex including HP1 and HDACs at the target gene. These steps lead to chromatin compaction and gene silencing (32). To gain further insights into the epigenetic control of Bsp expression by DNA methylation, we examined the occupancies of MeCP2, HP1 and H2A.Z in the chromatin of the Bsp gene in Osx-wt and Osx-null calvarial cells. Figure 6 shows that the occupancy of MeCP2 and HP1 at the Bsp gene was higher in Osx-null cells than Osx-wt cells (Fig. 6A and B). In contrast, occupancy of H2A.Z at the proximal promoter (core) and in the coding region (P5+) of the Bsp gene was significantly decreased in Osx-null cells (Fig. 6C). Note that H2A.Z occupancy inversely correlates with DNA methylation because interaction of H2A.Z with the promoter protects DNA from further methylation during transcription initiation (33,34). Together, these data are consistent with previous reports and our hypothesis that the higher levels of MeCP2 and HP1 and the lower levels of H2A.Z at the Bsp gene in Osx-null calvarial cells were most likely due to higher DNA methylation of this gene in these cells than in Osx-wt cells.
Fig. 6.

Increased occupancy of MeCP2 and HP1 but not H2A.Z at the Bsp gene in Osx-null calvarial cells. (A–C) ChIP assays for occupancy of MeCP2, HP1, and H2A.Z at chromatin segments of the Bsp gene in Osx-wt (Wt) and Osx-null calvarial cells. ChIP data for MeCP2 and HP1α occupancy are presented as fold enrichment over control rabbit IgG values and for H2A.Z as % bound/input after subtracting control rabbit IgG values. Site-specific primers P2 and P4 are within the promoter and P5+ within the coding region of the Bsp gene. (D) Panel 1, Coimmunoprecipitation of endogenous HP1α in the lysates of Osx-wt calvarial cells with NO66 antibody followed by western blot with HP1α antibody. Panel 2–4, Purification of Flag-NO66 from nuclear extract of Flag-NO66 expressing HEK293 cells with anti-Flag M2 agarose (Sigma) and then elution with 3X Flag peptides, followed by separation of eluate on SDS-PAGE and western blot with antibodies as indicated in the panel. (E) HP1α stimulates H3K4me3 and H3K36me3 demethylation by the NO66 demethylase. The demethylation reaction was carried out with E. coli expressed NO66 (full-length) and HP1α using calf-thymus bulk histones as substrates. Lower amount of NO66 that showed no demethylase activity was used in this reaction (1x equivalent to 1–2 μg). (F) Osx inhibits the NO66-dependent demethylation of H3K4me3 and H3K36me3 in a reaction that contained E. coli-expressed His-NO66 (168–641) and His-Osx (1–225). His-NO66 (168–641) that contained an intact JmjC domain also exhibited demethylase activity. Samples were run on the same gel and relevant lanes were shown (Panel E and F).
It is clear from the above data that repression of the Bsp gene in Osx-null cells was correlated with DNA methylation and increased interactions of the repressors NO66, HP1, and MeCP2 with the Bsp gene. We also performed a co-immunoprecipitation to demonstrate interactions between NO66 and HP1α in Osx-wt calvarial cells (Fig. 6D, Top), consistent with an earlier report indicating the co-localization of NO66 and HP1α at heterochromatic regions within the cell (14). In addition, in HEK293 cells stably transfected with a Flag-NO66-encoding expression vector, NO66 also interacted with endogenous DNMT1a and HDAC1 (Fig. 6D), suggesting the hypothesis that NO66 could form a repressor complex with these epigenetic regulators at Osx-target genes. HP1α was reported to interact with the drosophila KDM4 histone demethylase and to stimulate its activity for lysine 36 (35). Similar to this our data also indicate that HP1α stimulated the NO66 demethylase activity for H3K4me3 and H3K36me3 as substrates (Fig. 6E, lane 4 and 5). Further NO66 variants (168–641) that have an intact JmjC domain also exhibited demethylase activity toward lysine 4 and 36 (Fig. 6F, lane 1–3), and this activity was inhibited in the presence of Osx. Altogether, NO66 and HP1α might act as co-repressors for Osx-target genes in preosteoblasts to demethylate H3K4me and H3K36me. We also speculate that interactions of Osx at its target genes in expression of those genes during osteoblast differentiation might trigger the local removal of NO66 from chromatin and/or inhibit the demethylase activity of NO66.
Collectively, these findings support our hypothesis that both DNA methylation and histone demethylation of lysines 4 and 36 contribute to the transcriptionally inactive chromatin state of the Bsp gene in Osx-null calvarial cells. Our data provide evidence that NO66 plays a very significant role in epigenetic control of Osx-target genes.
Discussion
We present evidence that Osx, a downstream target of Runx2, is required for the formation of transcriptionally active chromatin at the Bsp gene during the differentiation of preosteoblasts to osteoblasts. We have shown previously that Osx is required for activation of osteoblast markers including Col1a1, Bsp, OC, and osteocyte genes Sost, Dmp1, and Dkk1. Expression of these genes in Osx-null embryos is either absent or markedly reduced. In this study, we examined the Osx-mediated mechanisms in gene activation during the transition of precursor osteoblasts to differentiating osteoblasts. We emphasize that expression of the Bsp is epigenetically controlled by histone and DNA methylation. The mechanism underlying the transition from repression to activation of Bsp and likely other Osx-target genes during osteoblast differentiation is based on our observations that include DNA demethylation, removal of a repressor complex from chromatin, recruitment of activators, and chromatin-modifying activities.
Several lines of evidence support the hypothesis that in Osx-null calvarial cells, the chromatin state of the Osx-target genes is markedly different from that of Osx-wt calvarial cells. In absence of Osx, the occupancy of the transcriptional activators and active chromatin modifiers including histone methylation and acetylation at specific sites in the Bsp gene, tested in this study, is abolished in Osx-null cells but not in Osx-wt cells. In contrast, occupancy of inactive chromatin modifiers including NO66, HP1, and MeCP2 in the Bsp gene is relatively high in osteoblasts-arrested Osx-null compared to Osx-wt cells. Further genome-wide analysis using a ChIPSeq approach to reveal other interactions sites of Osx as well as histone methylation in their target osteoblast genes will help us better understand the regulation of those target genes in osteoblasts.
We also demonstrated the reduced levels of histone H3K9/16 acetylation and of active histone methylation marks H3K4me3 and H3K36me3 within promoter and coding regions in the chromatin of the Bsp gene. We further observed a markedly lower occupancy of the transcriptional activators c-Myc and Wdr5 as well as that of H2A.Z in Osx-null calvarial cells. In contrast, a much higher occupancy of the repressor proteins NO66, HP1 and MeCP2 was observed in the chromatin of the Bsp gene. Moreover, in Osx-null calvaria, DNA demethylation at specific CpG sites in the Bsp promoter was delayed compared to that normally observed during Bsp expression in Osx-wt calvarial bone. Taken together, our results strongly suggest the hypothesis that in the absence of Osx, osteoblast target Bsp and most likely other genes are repressed because of an inability of Wdr5 and c-Myc to maintain stable interactions with chromatin and because the histone-modifying enzymes for acetylation and methylation cannot get recruited to these sites for transcription activation. Therefore, Osx acts as a molecular switch to trigger chromatin remodeling from an inactive to an active transcriptional state in osteoblasts (see Fig 7 for a proposed model). This chromatin remodeling also likely promotes localized DNA demethylation. It is also possible that additional mechanisms contribute to the control of chromatin remodeling of Osx-target genes. Our experiments were performed with ex vivo calvarial cells isolated from Osx-null E18.5 embryos (representing Runx2-expressing Osx-null preosteoblasts), and from Osx-wt E18.5 embryos, (representing Runx2- and Osx-expressing osteoblasts). Although Runx2 acts as upstream of Osx and is expressed in Osx-null calvarial cells but Runx2 cannot compensate for the functional loss of Osx. Indeed, interactions of both Runx2 and Osx with the chromatin of the Bsp and Oc genes in Osx-wt osteoblasts are likely needed for the optimal expression of these genes in osteoblasts.
Fig. 7.
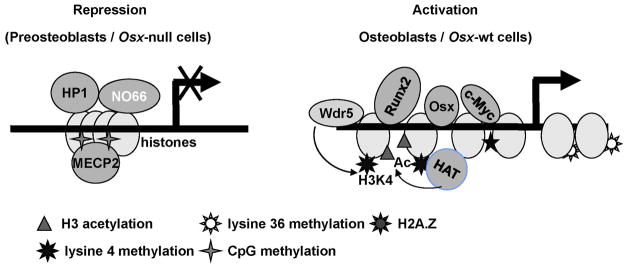
Osx is required for chromatin relaxation and stable interactions of transcriptional regulators during activation of its target genes in osteoblasts. A proposed model for the molecular role of Osx in chromatin remodeling of its target Bsp gene is depicted based on our results presented in this study.
We have shown here that the process that prevents Bsp activation in the absence of Osx includes the maintenance of DNA methylation and that of demethylated H3K4me3 and K36me3 as well as interactions of repressors with the Bsp gene. Because HP1 stimulated the demethylase activity of NO66, the interactions of NO66 and HP1 at the Bsp gene in Osx-null cells may help maintain the demethylated state of H3K4 and H3K36. Our data further suggest the hypothesis that Osx expression in osteoblasts could trigger the removal of NO66 as well as that of HP1and that of other potential repressors (including HDAC1) from the Bsp gene, thereby allowing interactions of c-Myc, Wdr5, and H2A.Z as well as other unknown factors with the chromatin of this gene during transcription. Several HDACs have been reported to negatively regulate Runx2 activity and osteoblast differentiation (36). Notably, c-Myc has the ability to potentiate Osx activity and its interactions with the Osx-target Bsp gene in osteoblasts appear to depend largely on Osx, because no interactions of c-Myc occurred at the Bsp gene in Osx-null cells.
Hypermethylation of the Bsp promoter was observed in ES cells and is epigenetically inherited in subsequent developmental stages up to a point that, however, largely precedes Bsp expression. Histone and DNA methylation produce epigenetic repressive marks and are interlinked in gene repression. Previous studies have shown that HP1 interacts with DNMT1 in vitro and in vivo and induces DNMT1 activity, resulting in increased DNA methylation (20). Also DNMT1A interacts with histone methyltransferase (G9a) or SUV39H1, and this interaction coordinates trimethylation of lysine 9, through which HP1 interacts with chromatin (37). In Osx-null calvaria, increased DNA methylation in the Bsp promoter compared to that in Osx-wt, coupled with increased occupancies of MeCP2 and HP1, supports the view that DNA methylation might further potentiate the restriction in Bsp expression in these Osx-null cells. Increased levels of H2A.Z in the chromatin of the Bsp gene in Osx-wt cells maintain active transcription of this gene. Based on these observations, we speculate that upon Osx expression in Osx-wt cells, although not an Osx-dependent event, erasure of DNA methylation in other Osx-target genes might continue, which should allow formation of a transcriptionally active chromatin state. This is also seen in the case of the Sost promoter which is methylated in osteoblasts but is demethylated and actively expressed in osteocytes (25).
In summary, we have presented three important observations regarding the epigenetic control of expression of the Osx-target Bsp gene during osteoblast differentiation. First, Osx is needed for osteoblasts differentiation in vitro and is crucial for chromatin remodeling during gene activation; second, the osteoblast-specific Bsp gene is methylated prior to its activation in osteoblasts; and third, in addition to demethylation of the active lysines 4 and 36 marks, NO66 likely forms a strong repressor complex, through interactions with HP1, DNMT1, and HDAC1, at Osx-target genes in osteoblasts, causing further repression of these genes. Although our analysis of transcriptional regulators was limited to a few sites within the chromatin of the Bsp gene, we speculate that similar mechanisms may control chromatin remodeling of other Osx-target genes in the transition from gene repression to activation during osteoblast differentiation.
Supplementary Material
Acknowledgments
The study was supported by Grant AR49072 (BdC), AR061590 (KS) and CA16672 (DNA Sequencing Core Facility) from NIAMS/NIH, and its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIAMS/NIH. KS is a recipient of the Rolanette and Berdon Lawrence Research Award of the Bone Disease Program of Texas.
Footnotes
Additional Supplemental information may be found in the online version of this aticle.
Authors’ role: Study design: KS, HY, and BdC. Study conduct: KS. Data interpretation: KS and BdC. HY provided technical help and discussion. XZ provided Osx KO mice. Manuscript preparation: KS and BdC.
References
- 1.Bodine PV, Komm BS. Wnt signaling and osteoblastogenesis. Rev Endocr Metab Disord. 2006;7(1–2):33–9. doi: 10.1007/s11154-006-9002-4. [DOI] [PubMed] [Google Scholar]
- 2.Day TF, Guo X, Garrett-Beal L, Yang Y. Wnt/beta-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Developmental cell. 2005;8(5):739–50. doi: 10.1016/j.devcel.2005.03.016. [DOI] [PubMed] [Google Scholar]
- 3.Ducy P, Zhang R, Geoffroy V, Ridall AL, Karsenty G. Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell. 1997;89(5):747–54. doi: 10.1016/s0092-8674(00)80257-3. [DOI] [PubMed] [Google Scholar]
- 4.Hill TP, Spater D, Taketo MM, Birchmeier W, Hartmann C. Canonical Wnt/beta-catenin signaling prevents osteoblasts from differentiating into chondrocytes. Dev Cell. 2005;8(5):727–38. doi: 10.1016/j.devcel.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 5.Rodda SJ, McMahon AP. Distinct roles for Hedgehog and canonical Wnt signaling in specification, differentiation and maintenance of osteoblast progenitors. Development. 2006;133(16):3231–44. doi: 10.1242/dev.02480. [DOI] [PubMed] [Google Scholar]
- 6.Nakashima K, Zhou X, Kunkel G, Zhang Z, Deng JM, Behringer RR, de Crombrugghe B. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell. 2002;108(1):17–29. doi: 10.1016/s0092-8674(01)00622-5. [DOI] [PubMed] [Google Scholar]
- 7.Zhou X, Zhang Z, Feng JQ, Dusevich VM, Sinha K, Zhang H, Darnay BG, de Crombrugghe B. Multiple functions of Osterix are required for bone growth and homeostasis in postnatal mice. Proc Natl Acad Sci U S A. 2010;107(29):12919–24. doi: 10.1073/pnas.0912855107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nakashima K, de Crombrugghe B. Transcriptional mechanisms in osteoblast differentiation and bone formation. Trends Genet. 2003;19(8):458–66. doi: 10.1016/S0168-9525(03)00176-8. [DOI] [PubMed] [Google Scholar]
- 9.Baek WY, Lee MA, Jung JW, Kim SY, Akiyama H, de Crombrugghe B, Kim JE. Positive regulation of adult bone formation by osteoblast-specific transcription factor osterix. J Bone Miner Res. 2009;24(6):1055–65. doi: 10.1359/jbmr.081248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baek WY, de Crombrugghe B, Kim JE. Postnatally induced inactivation of Osterix in osteoblasts results in the reduction of bone formation and maintenance. Bone. 2010;46(4):920–8. doi: 10.1016/j.bone.2009.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Margueron R, Trojer P, Reinberg D. The key to development: interpreting the histone code? Curr Opin Genet Dev. 2005;15(2):163–76. doi: 10.1016/j.gde.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 12.Martin C, Zhang Y. The diverse functions of histone lysine methylation. Nat Rev Mol Cell Biol. 2005;6(11):838–49. doi: 10.1038/nrm1761. [DOI] [PubMed] [Google Scholar]
- 13.Sinha KM, Yasuda H, Coombes MM, Dent SY, de Crombrugghe B. Regulation of the osteoblast-specific transcription factor Osterix by NO66, a Jumonji family histone demethylase. EMBO J. 2010;29(1):68–79. doi: 10.1038/emboj.2009.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eilbracht J, Reichenzeller M, Hergt M, Schnolzer M, Heid H, Stohr M, Franke WW, Schmidt-Zachmann MS. NO66, a highly conserved dual location protein in the nucleolus and in a special type of synchronously replicating chromatin. Mol Biol Cell. 2004;15(4):1816–32. doi: 10.1091/mbc.E03-08-0623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fujita N, Watanabe S, Ichimura T, Tsuruzoe S, Shinkai Y, Tachibana M, Chiba T, Nakao M. Methyl-CpG binding domain 1 (MBD1) interacts with the Suv39h1-HP1 heterochromatic complex for DNA methylation-based transcriptional repression. J Biol Chem. 2003;278(26):24132–8. doi: 10.1074/jbc.M302283200. [DOI] [PubMed] [Google Scholar]
- 16.Geiman TM, Sankpal UT, Robertson AK, Zhao Y, Robertson KD. DNMT3B interacts with hSNF2H chromatin remodeling enzyme, HDACs 1 and 2, and components of the histone methylation system. Biochem Biophys Res Commun. 2004;318(2):544–55. doi: 10.1016/j.bbrc.2004.04.058. [DOI] [PubMed] [Google Scholar]
- 17.Fuks F. DNA methyltransferases: from chromatin remodeling to cancer. Med Sci (Paris) 2003;19(4):477–80. doi: 10.1051/medsci/2003194477. [DOI] [PubMed] [Google Scholar]
- 18.Robertson KD, Keyomarsi K, Gonzales FA, Velicescu M, Jones PA. Differential mRNA expression of the human DNA methyltransferases (DNMTs) 1, 3a and 3b during the G(0)/G(1) to S phase transition in normal and tumor cells. Nucleic Acids Res. 2000;28(10):2108–13. doi: 10.1093/nar/28.10.2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ciccone DN, Su H, Hevi S, Gay F, Lei H, Bajko J, Xu G, Li E, Chen T. KDM1B is a histone H3K4 demethylase required to establish maternal genomic imprints. Nature. 2009;461(7262):415–8. doi: 10.1038/nature08315. [DOI] [PubMed] [Google Scholar]
- 20.Smallwood A, Esteve PO, Pradhan S, Carey M. Functional cooperation between HP1 and DNMT1 mediates gene silencing. Genes & development. 2007;21(10):1169–78. doi: 10.1101/gad.1536807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Honda S, Selker EU. Direct interaction between DNA methyltransferase DIM-2 and HP1 is required for DNA methylation in Neurospora crassa. Mol Cell Biol. 2008;28(19):6044–55. doi: 10.1128/MCB.00823-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mabaera R, Richardson CA, Johnson K, Hsu M, Fiering S, Lowrey CH. Developmental- and differentiation-specific patterns of human gamma- and beta-globin promoter DNA methylation. Blood. 2007;110(4):1343–52. doi: 10.1182/blood-2007-01-068635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee JY, Lee YM, Kim MJ, Choi JY, Park EK, Kim SY, Lee SP, Yang JS, Kim DS. Methylation of the mouse DIx5 and Osx gene promoters regulates cell type-specific gene expression. Mol Cells. 2006;22(2):182–8. [PubMed] [Google Scholar]
- 24.Hupkes M, van Someren EP, Middelkamp SH, Piek E, van Zoelen EJ, Dechering KJ. DNA methylation restricts spontaneous multi-lineage differentiation of mesenchymal progenitor cells, but is stable during growth factor-induced terminal differentiation. Biochim Biophys Acta. 2011;1813(5):839–49. doi: 10.1016/j.bbamcr.2011.01.022. [DOI] [PubMed] [Google Scholar]
- 25.Delgado-Calle J, Sanudo C, Bolado A, Fernandez AF, Arozamena J, Pascual-Carra MA, Rodriguez-Rey JC, Fraga MF, Bonewald L, Riancho JA. DNA methylation contributes to the regulation of sclerostin expression in human osteocytes. J Bone Miner Res. 2012;27(4):926–37. doi: 10.1002/jbmr.1491. [DOI] [PubMed] [Google Scholar]
- 26.Roca H, Phimphilai M, Gopalakrishnan R, Xiao G, Franceschi RT. Cooperative interactions between RUNX2 and homeodomain protein-binding sites are critical for the osteoblast-specific expression of the bone sialoprotein gene. J Biol Chem. 2005;280(35):30845–55. doi: 10.1074/jbc.M503942200. [DOI] [PubMed] [Google Scholar]
- 27.Roca H, Franceschi RT. Analysis of transcription factor interactions in osteoblasts using competitive chromatin immunoprecipitation. Nucleic Acids Res. 2008;36(5):1723–30. doi: 10.1093/nar/gkn022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhu ED, Demay MB, Gori F. Wdr5 is essential for osteoblast differentiation. J Biol Chem. 2008;283(12):7361–7. doi: 10.1074/jbc.M703304200. [DOI] [PubMed] [Google Scholar]
- 29.Zhang Y, Hassan MQ, Li ZY, Stein JL, Lian JB, van Wijnen AJ, Stein GS. Intricate gene regulatory networks of helix-loop-helix (HLH) proteins support regulation of bone-tissue related genes during osteoblast differentiation. J Cell Biochem. 2008;105(2):487–96. doi: 10.1002/jcb.21844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang C, Cho K, Huang Y, Lyons JP, Zhou X, Sinha K, McCrea PD, de Crombrugghe B. Inhibition of Wnt signaling by the osteoblast-specific transcription factor Osterix. Proc Natl Acad Sci U S A. 2008;105(19):6936–41. doi: 10.1073/pnas.0710831105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mikura A, Okuhara S, Saito M, Ota M, Ueda K, Iseki S. Association of tenascin-W expression with mineralization in mouse calvarial development. Congenital anomalies. 2009;49(2):77–84. doi: 10.1111/j.1741-4520.2009.00227.x. [DOI] [PubMed] [Google Scholar]
- 32.Fuks F, Hurd PJ, Deplus R, Kouzarides T. The DNA methyltransferases associate with HP1 and the SUV39H1 histone methyltransferase. Nucleic Acids Res. 2003;31(9):2305–12. doi: 10.1093/nar/gkg332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kobor MS, Lorincz MC. H2A.Z and DNA methylation: irreconcilable differences. Trends Biochem Sci. 2009;34(4):158–61. doi: 10.1016/j.tibs.2008.12.006. [DOI] [PubMed] [Google Scholar]
- 34.Zilberman D, Coleman-Derr D, Ballinger T, Henikoff S. Histone H2A.Z and DNA methylation are mutually antagonistic chromatin marks. Nature. 2008;456(7218):125–9. doi: 10.1038/nature07324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lin CH, Li B, Swanson S, Zhang Y, Florens L, Washburn MP, Abmayr SM, Workman JL. Heterochromatin protein 1a stimulates histone H3 lysine 36 demethylation by the Drosophila KDM4A demethylase. Mol Cell. 2008;32(5):696–706. doi: 10.1016/j.molcel.2008.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jensen ED, Nair AK, Westendorf JJ. Histone deacetylase co-repressor complex control of Runx2 and bone formation. Crit Rev Eukaryot Gene Expr. 2007;17(3):187–96. doi: 10.1615/critreveukargeneexpr.v17.i3.20. [DOI] [PubMed] [Google Scholar]
- 37.Esteve PO, Chin HG, Smallwood A, Feehery GR, Gangisetty O, Karpf AR, Carey MF, Pradhan S. Direct interaction between DNMT1 and G9a coordinates DNA and histone methylation during replication. Genes & development. 2006;20(22):3089–103. doi: 10.1101/gad.1463706. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.