

Abstract
Since September 2010, over 10,000 patients have undergone preemptive, panel-based pharmacogenomic testing through the Vanderbilt Pharmacogenomic Resource for Enhanced Decisions in Care and Treatment (PREDICT) program. Analysis of the genetic data from the first 9,589 individuals reveals the frequency of genetic variants is concordant with published allele frequencies. Based on five currently implemented drug-genome interactions, the multiplexed test identified one or more actionable variants in 91% of the genotyped patients and in 96% of African-American patients. Using medication exposure data from electronic medical records, we compared a theoretical “reactive,” prescription-triggered, serial single-gene testing strategy to our preemptive, multiplexed genotyping approach. Reactive genotyping would have generated 14,656 genetic tests. These data highlight three advantages of preemptive genotyping: 1)the vast majority of patients carry at least one pharmacogene variant; 2)data are available at the point of care; and 3)there is a substantial reduction in testing burden compared to a reactive strategy.
Keywords: Pharmacogenomics, Genetic Testing
Introduction
Ten years ago, the development of genome-based approaches to predict drug response was proposed as one of the “grand challenges” in the future of genomics research.(1) Since that time, evidence for implementation in a clinical setting has been established for a number drug-gene interactions (DGIs).(2-8) Implementation of pharmacogenomics into clinical practice, however, has not yet become widely adopted. One reason is the significant challenges associated with implementation, including assessment of the potential benefits for clinical pharmacogenomic testing, definition of the target populations, designation of anticipated scope of pharmacogenomic testing, determination of diagnostic methodologies, development of infrastructure to support reporting, interpretation and use of results, and establishment of reimbursement for testing.(9) A major source of uncertainty surrounding the feasibility of panel-based genotyping programs is whether pharmacogenomic test results will become “actionable” within a patient’s lifetime and provide clinical benefit given the initial investment in genotyping. The opportunities to use pharmacogenomic information and the number of individuals that will have actionable variants are unknown.
The Pharmacogenomic Resource for Enhanced Decisions in Care and Treatment (PREDICT) program at Vanderbilt University Medical Center was created to implement pharmacogenomics into clinical practice and ultimately improve patient care.(9) Genotyping is completed using a panel-based approach, with specific genotypes released into the medical record after review and institutional approval of each target DGI. Patients are selected for genotyping based on anticipated coronary artery stenting via cardiac catheterization, a prognostic risk score that estimates the likelihood of a patients’ exposure to pharmacogenomic medication, or via provider preference. Currently, genotype-guided clinical decision support (CDS) is incorporated into the electronic medical record (EMR) for five well established DGIs: clopidogrel (CYP2C19), simvastatin (SLCO1B1), warfarin (CYP2C9 and VKORC1), thiopurines (TPMT), and tacrolimus (CYP3A5). Specific informative single nucleotide polymorphisms (SNPs) with known drug response associations are interrogated and utilized to determine appropriate CDS.
Since initiation of this program, over 10,000 patients have undergone clinical genotype testing, and CDS for each of the five DGIs has been serially deployed as they were locally approved. Based on this patient cohort, we report here a profile of the genotyped patients including the genotypes identified, the frequency of actionable variants, and the medication exposures among genotyped patients. We also compare the preemptive, multiplexed genotyping model utilized for PREDICT to a “reactive” strategy, where genetic testing would be ordered for individual genes as indicated by medication exposure. Our goal was to begin to quantify benefits of a multiplexed, preemptive approach to pharmacogenomic testing, and to thereby inform evaluation of potential implementation at other centers.
Results
Of the first 10,044 patients genotyped, 455 (4.5%) had one or more “no call” results among the five genes implemented and were excluded from analysis. Table 1 includes demographic and descriptive data for the remaining 9,589 patients with complete genotype data for the five currently implemented DGIs. Supplemental table 1 includes data for all 10,044 individuals. The median age (63 years) and overrepresentation of males (59%) are no different with inclusion of individuals with no-calls. The PREDICT prognostic score represents the estimated likelihood of a patient’s exposure to clopidogrel, warfarin, or a 3-hydroxy-3-methyl-glutaryl (HMG)-CoA reductase inhibitor (statin) within three years. Internally, providers have been encouraged to order PREDICT testing on individuals with scores of at least 40%, although they are free to order the test on other patients as well. We found the median was just higher than this threshold, at 45%. In total, 5,764 (60%) of those genotyped were in the PREDICT preemptive model target population of patients (prognostic scores ≥40% or with a history of coronary artery stent placement), and this subset had more exposures to antiplatelet and cholesterol medications than the genotyped cohort as a whole (Table 1). Among the entire cohort, the race/ethnicity of the majority of the cohort was primarily identified as European American, not Hispanic (EA, N=6,986); 953 African American, Not Hispanic (AA) individuals were also included, among whom women were overrepresented (N=520, 55%). A total of 268 (3%) of individuals were of unknown race, 105 (1%) Asian, and 31 (0.3%) Alaskan, Indian, or Pacific Islander. With respect to ethnicity, 110 (1%) individuals were identified as Hispanic and 1,360 (14%) were unknown.
Table 1.
Demographics and Medication Exposures of Studied Cohort and Subgroups
| All (N=9,589) | European American, not Hispanic (N=6,986) | African American, not Hispanic (N=953) | PREDICT preemptive model target population (N=5,764)ˆ | |
|---|---|---|---|---|
|
| ||||
| Age* | 63 (55-71) | 64 (55-72) | 60 (51-68) | 65 (58-73) |
|
| ||||
| Male Sex | 5,691 (59%) | 4,264 (61%) | 433 (45%) | 3,797 (66%) |
|
| ||||
| Coronary Artery Stent | 2,410 (25%) | 1,741 (25%) | 166 (17%) | 2,410 (42%) |
|
| ||||
| PREDICT Risk Score* | 45 (33-59) | 44 (32-58) | 49 (38-62) | 53 (44-65) |
|
| ||||
| EMR Observation Time (Days)* | 2,090 (599-4561) | 2,213 (743-4627) | 3,457 (1227-5669) | 2,442 (1,017-4,810) |
|
| ||||
| Medication Exposures | ||||
| Clopidogrel | 4,684 (49%) | 3,314 (47%) | 371 (39%) | 3,428 (59%) |
| Clopidogrel or Prasugrel | 4,742 (49%) | 3,364 (48%) | 375 (39%) | 3,472 (60%) |
| Simvastatin | 5,261 (55%) | 3,774 (54%) | 571 (60%) | 3,722 (65%) |
| Any Statin | 7,460 (78%) | 5,478 (78%) | 746 (78%) | 5,052 (88%) |
| Warfarin | 2,069 (22%) | 1,605 (23%) | 199 (21%) | 1,485 (26%) |
| Thiopurine | 129 (1%) | 104 (1%) | 14 (1%) | 84 (1%) |
| Tacrolimus | 256 (3%) | 185 (3%) | 42 (4%) | 176 (3%) |
Median (Interquartile Range)
PREDICT preemptive model target population, including those with history of coronary artery stent and/or with PREDICT risk score > 40, indicating a 40% likelihood of exposure to clopidogrel, warfarin or a statin over 3 years
Table 2 describes the genotype data obtained for actionable SNPs in the genes of interest. The frequency of variant genotypes did not differ between the cohort as a whole and the PREDICT preemptive model target population. The proportion of individuals with variants in genes relevant to simvastatin (SLCO1B1), warfarin (CYP2C9 and VKORC1), and tacrolimus (CYP3A5) differed between EA and AA patients, as expected based on reported differences in minor allele frequencies between these populations (Table 2). Employing the definitions as described in Supplemental table 2, the number of individuals with “actionable” (prompting CDS to suggest a change in dose or medication) and “high risk” (homozygous for variants in CYP2C19, SLCO1B1, CYP2C9, or TPMT known to greatly increase the likelihood of a severe adverse outcome) genotypes for each DGI was determined (Figure 1, Supplemental table 3).(6,8,10-13) For example, identification of either heterozygosity or homozygosity for the SLCO1B1*5 variant leads to genotype-guided advice if a provider orders or prescribes simvastatin, so both genotypes are “actionable,” a finding identified in 26% of all patients. However, individuals homozygous for this variant are at far higher risk than heterozygous or wild-type individuals (20-fold increased risk for homozygotes vs. 4-fold for heterozygotes compared to non-carriers); only homozygosity is considered “high risk,” and was found in 2% of patients. As discussed in the Methods section, the pre-defined high risk alleles are CYP2C19*2/*2, SLCO1B1*5/*5, CYP2C9*3/*3, and homozygosity or compound heterozygosity for TPMT*2 or *3.
Table 2.
Genotypes Identified
| All (N=9,589) | European American, not Hispanic (N=6,986) | African American, not Hispanic (N=953) | PREDICT preemptive model target population (N=5,764)ˆ | |
|---|---|---|---|---|
|
| ||||
| CYP2C19 | ||||
| rs4244285 (*2) heterozygote | 2,398 (25%) | 1,751 (25%) | 235 (25%) | 1,431 (25%) |
| rs4244285 (*2) homozygote* | 238 (2%) | 170 (2%) | 25 (3%) | 123 (2%) |
| rs4986893 (*3) heterozygote | 14 (0%) | 2 (0%) | 1 (0%) | 3 (0%) |
| rs4986893 (*3) homozygote | 2 (0%) | 0 | 0 | 1 (0%) |
| rs28399504 (*4) heterozygote | 46 (0%) | 36 (1%) | 1 (0%) | 26 (0%) |
| rs28399504 (*4) homozygote | 0 | 0 | 0 | 0 |
| rs56337013 (*5) heterozygote | 1 (0%) | 1 (0%) | 0 | 1 (0%) |
| rs56337013 (*5) homozygote | 0 | 0 | 0 | 0 |
| rs72552267 (*6) heterozygote | 6 (0%) | 5 (0%) | 1 (0%) | 4 (0%) |
| rs72552267 (*6) homozygote | 0 | 0 | 0 | 0 |
| rs41291556 (*8) heterozygote | 52 (1%) | 44 (1%) | 1 (0%) | 34 (1%) |
| rs41291556 (*8) homozygote | 1 (0%) | 1 (0%) | 0 | 0 |
|
| ||||
| SLCO1B1 | ||||
| rs4149056 (*5) heterozygote | 2,279 (24%) | 1,805 (26%) | 66 (7%) | 1,346 (23%) |
| rs4149056 (*5) homozygote* | 181 (2%) | 147 (2%) | 3 (0%) | 104 (2%) |
|
| ||||
| CYP2C9 | ||||
| rs1799853 (*2) heterozygote | 1,998 (21%) | 1,622 (23%) | 42 (4%) | 1,229 (21%) |
| rs1799853 (*2) homozygote | 156 (2%) | 129 (2%) | 1 (0%) | 92 (2%) |
| rs1057910 (*3) heterozygote | 1,046 (11%) | 831 (12%) | 31 (3%) | 644 (11%) |
| rs1057910 (*3) homozygote* | 29 (0%) | 23 (0%) | 1 (0%) | 14 (0%) |
|
| ||||
| VKORC1 | ||||
| rs9923231 heterozygote | 4,170 (43%) | 3,305 (47%) | 182 (19%) | 2,507 (43%) |
| rs9923231 homozygote | 1,185 (12%) | 943 (13%) | 10 (1%) | 687 (12%) |
|
| ||||
| TPMT | ||||
| rs1800462 (*2) heterozygote | 52 (1%) | 44 (1%) | 2 (0%) | 32 (1%) |
| rs1800462 (*2) homozygote* | 0 | 0 | 0 | 0 |
| rs1800460 (*3B) heterozygote | 1 (0%) | 0 | 0 | 0 |
| rs1800460 (*3B) homozygote* | 0 | 0 | 0 | 0 |
| rs1142345 (*3C) heterozygote | 171 (2%) | 68 (1%) | 81 (8%) | 107 (2%) |
| rs1142345 (*3C) homozygote* | 2 (0%) | 0 | 2 (0%) | 2 (0%) |
| rs1800460+rs1142545 (*3A, 3D, 3E) heterozygote | 649 (7%) | 514 (7%) | 20 (2%) | 380 (7%) |
| rs1800460+rs1142545 (*3A, 3D, 3E) homozygote* | 9 (0%) | 6 (0%) | 0 | 4 (0%) |
|
| ||||
| CYP3A5 | ||||
| rs776746 (*3) heterozygote | 1,663 (17%) | 918 (13%) | 402 (42%) | 1,003 (17%) |
| rs776746 (*3) homozygote | 7,332 (76%) | 6,022 (86%) | 98 (10%) | 4,357 (76%) |
High risk genotype (not all-inclusive, as compound heterozygosity for TPMT variants also high-risk)
PREDICT preemptive model target population, including those with history of coronary artery stent and/or with PREDICT risk score > 40, indicating a 40% likelihood of exposure to clopidogrel, warfarin or a statin over 3 years
Figure 1. Actionable genotypes in individual and cumulative Drug-Genome Interactions (DGIs).
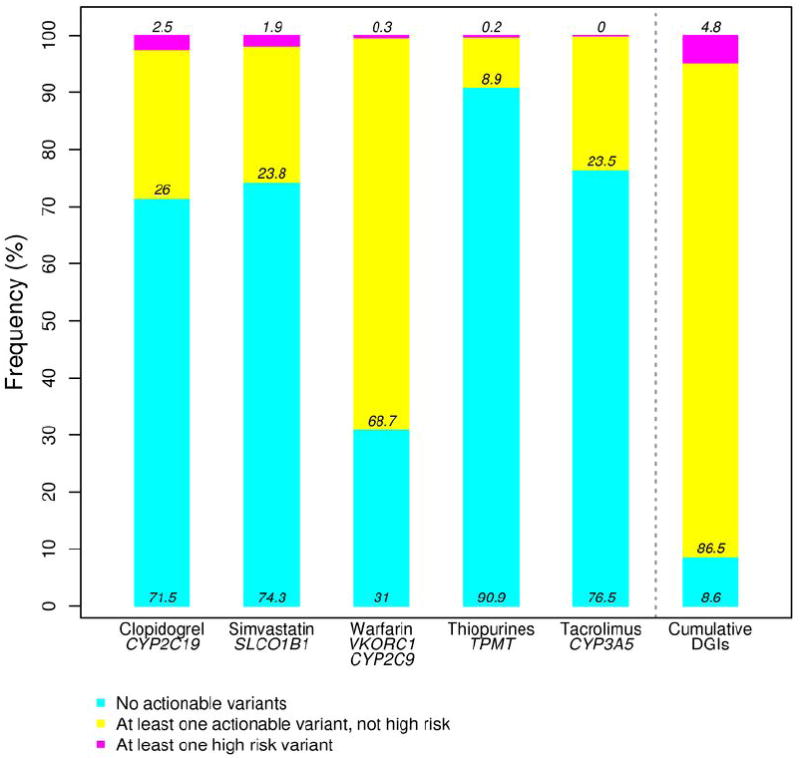
The frequencies of non-actionable, actionable and high risk genotypes for each DGI. The final bar shows frequency of individuals having at least one actionable or high risk genotype among all five DGIs.
The CYP3A5*1 genotype codes for functional CYP3A5 enzyme, and is the most common allele among AAs, but not among EAs. However, standard dosing recommendations for tacrolimus are based on individuals with the CYP3A5*3 (nonfunctional) genotype, the most common genotype in EAs. In our system, actionability is based on CYP3A5*1; having one or no copy of CYP3A5*3, indicating one or two copies of functional CYP3A5*1, respectively, is actionable and was identified in 24% of patients. Analysis of the cumulative frequency of actionable genotypes (e.g., at least one actionable variant identified) revealed that of the 9,589 individuals with complete genotype data for these five DGIs, 8,760 (91%) have at least one actionable genotype (Figure 1), while 5% have at least one high risk genotype (Supplemental table 3).
Each of the genes of interest has been studied in large populations, enabling the comparison of the observed frequency of actionable variants in each population to expected frequency based on published minor allele frequencies for EA and AA populations. The frequency of predicted and observed actionable and high risk genotypes for each of the five DGIs is depicted in Figure 2, panels A-C. For each of the five DGIs, observed frequencies closely approximate expected frequencies.
Figure 2. Predicted and observed actionable genotypes and associated medication exposures.
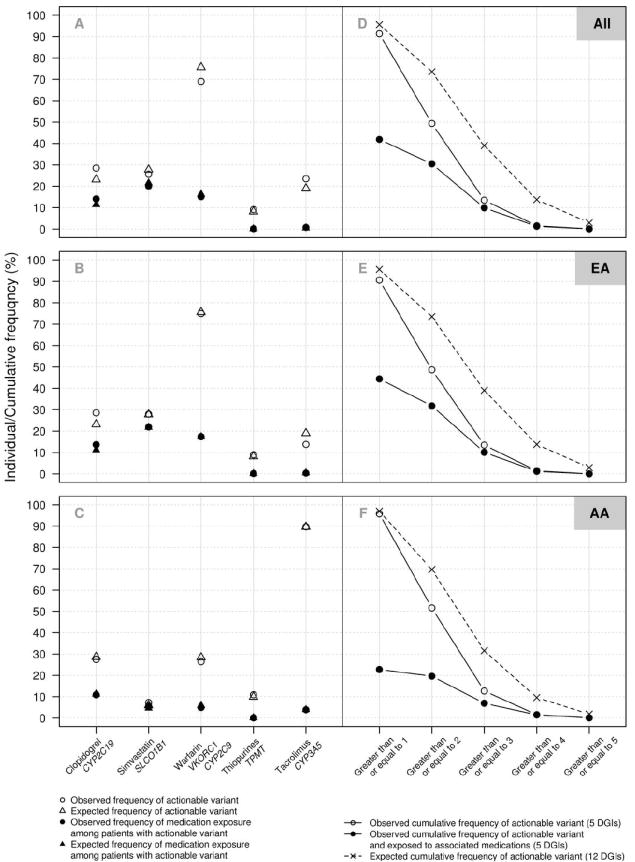
In panels A-C, for each Drug-Genome Interaction (DGI), the frequencies of expected actionable genotypes based on reported minor allele frequencies (open triangles), observed actionable genotypes (open circles), expected frequency of medication exposures among patients with actionable genotypes (filled triangles) and observed actionable genotype with exposure to the associated medication (filled circles) are shown for all 9,589 genotyped patients (A), 6,986 patients of European-American descent (B), and 953 patients of African-American decent (C). Clopidogrel exposure includes clopidogrel and/or prasugrel, the alternative agent. Statin exposure includes simvastatin and/or alternative HMG-CoA reductase inhibitors (atorvastatin, fluvastatin, lovastatin, pravastatin, rosuvastatin). In panels D-F, cumulative frequency of individuals having observed actionable genotypes based on the five currently implemented DGIs (open circles), cumulative frequency of individuals with actionable genotypes exposed to associated medications (filled circles), and predicted cumulative frequency of actionable genotypes based on a total of 12 pharmacogenes (x’s) are shown for all genotyped patients (D), patients of European-American descent (E), and patients of African-American decent (F).
EMR data were searched to determine the number of individuals with actionable genotypes who had evidence of exposure to the associated medication or medication class at any time in their history. Due to the recent implementation of the PREDICT program, medication starts may have pre-dated genotyping and incorporation of genotype-guided CDS into the EMR. Over half of the patients with actionable genotypes affecting clopidogrel and simvastatin response have been exposed to these medications at some point, and nearly one-fourth of those with actionable warfarin genotypes have evidence of warfarin exposure (Figure 2, A-C). Among those with high risk genotypes, no individuals with high risk TPMT genotypes (homozygous variant) have been exposed to date to thiopurine medications (N=0/19). Of the 181 individuals homozygous for SLCO1B1 variation, 110 (61%) had evidence of simvastatin exposure, and an additional 32 (18%) have been exposed to a different statin. Of the 110, the last statin mentioned in the EMR was a simvastatin-alternative in 55 (50%). Since the program has only recently started, the frequency with which the data will be used will rise with time.
Analysis of the cumulative frequency of actionable genotypes among African American individuals revealed that all but 40 of the 953 (96%) had at least one actionable genotype (Figure 2, F). Using published minor allele frequencies for variants in six additional genes with known pharmacogenomic associations (CYP2D6, HLA-B, DPYD, G6PD, IL28B, and UGT1A1; Supplemental table 4), we estimate that implementation of these next pharmacogenomic target DGIs will increase the proportion of EA individuals with at least one actionable variant to 96% (Figure 2, E).(2,14-22) EA individuals with two or more actionable genotypes would increase from 49% to 74% with the addition of six more genes, and the proportion with three or more would increase from 14% to 39% (Figure 2, E)
Among the individuals with one or more actionable genotypes, 4,018 (42% of the entire cohort) had evidence of exposure to the risk-associated medication or medication class (Figure 2, D-F). In AA patients, 217 (23% of all AA patients) had actionable genotypes and evidence of an actionable medication exposure. In the PREDICT preemptive model target population, 2,744 (48%) were exposed to one or more medications for which they had actionable genotypes, reflecting the higher medication exposure rates in this subgroup. Medication exposure rates were further elevated by looking at the specific subgroups of patients with PREDICT risk score > 70, where 813/846 (96%) patients were exposed to one or more of the target medications, and in those who had been treated with a coronary artery stent, where all but 12 of the 2,410 patients were exposed (Supplemental table 5). Patients receiving any one of the five target medications had a high likelihood of receiving an additional target medication. Among those who had received antiplatelet therapy with clopidogrel or prasugrel, 93% received a second PREDICT medication. Rates for second target medication exposure for those receiving thiopurine, tacrolimus, warfarin, and statin drugs were 91%, 87%, 84% and 69%, respectively.
The documented exposures to drugs and drug classes with established DGIs provide the opportunity to compare the preemptive, panel-based genetic test model used for PREDICT to a theoretical reactive genotyping model based on serial single-gene testing as indicated by patient prescription for each medication (Figure 3). Compared to the 9,589 panel-based genetic tests performed on patients through PREDICT, determination of genotype using reactive strategy would have resulted in 14,656 tests (1.7-fold more tests). Within the PREDICT preemptive model target population where medication exposures occurred at a higher rate, reactive testing would have required nearly twice as many tests be done (10,269 tests vs. 5,764 panel-based tests completed).
Figure 3. Medication exposures among genotyped individuals.
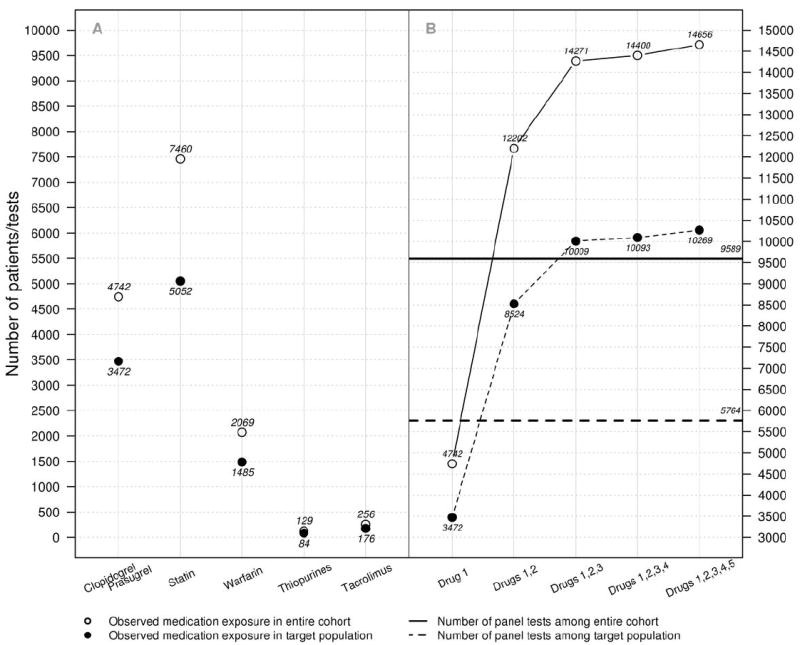
A. Medication exposure among all (open circles) and PREDICT preemptive model target population (filled circles) individuals for each medication currently implemented in the PREDICT program. B. Cumulative number of drug exposures to medications implemented in the PREDICT program, in order of implementation. Drug 1 - clopidogrel and/or prasugrel; 2 - statins; 3 - warfarin; 4 - thiopurines; 5 - tacrolimus. The solid horizontal line indicates the total number of panel tests completed in the entire cohort, and the dotted line indicates the number of panel tests completed in the target population.
Discussion
In this study of the first ~10,000 individuals clinically genotyped through the PREDICT program, the vast majority of individuals (91%) have at least one actionable genotype among the five DGIs implemented to date, highlighting the utility and potential benefit of panel-based genotyping for pharmacogenomic testing. Although drug dosing regimens are tested and approved based upon population data, significant variation in drug response exists, much of which can be attributed to common genetic variation in genes associated with drug metabolism and response. While it may be expected that testing a large number of pharmacogenes will identify clinically important variants in many patients, the degree of impact may be underestimated, as illustrated by our finding that over nine out of ten patients tested have at least one actionable variant even among the small range of DGIs tested and employing a conservative definition for “actionable.” Importantly, the impact in AA patients is even greater, with nearly all individuals having at least one actionable genotype. Some of this higher estimate is driven by the designation of the ancestral CYP3A5*1 allele as actionable, further emphasizing the need to include diverse ancestries in large genome analyses.
Comparing genotypes from the PREDICT cohort to those reported in previously published cohorts demonstrates that the proportions are similar to predicted minor allele frequencies. This validates the performance of the clinical test, but more importantly suggests that in designing such clinical tests and determining cost and benefit, published minor allele frequencies can be relied upon for modeling the number of clinically significant findings that will be identified through testing. Our estimate of the addition of variants in six additional genes to the PREDICT program shows the impact of these additional genotypes on the frequency of having at least one actionable genotype is minimal, but the frequency of having multiple actionable genotypes increases more substantially.
Critical to the success of a pharmacogenomic testing program is the development of a framework for provider notification and follow-up of actionable genotypes. To improve patient outcomes, identification of risk based on a DGI must be followed by risk modification through dosage adjustment, medication change, or changes in therapeutic monitoring. At the present time, ideal methods have not yet been established for communicating pharmacogenetic test results and their interpretations to the spectrum of providers involved in a patient’s care. In developing such methods, accurate prediction of the number of patients with actionable findings will help determine the requisite resources to optimize patient safety.
The definition of which genotypes are of clinical value has significant impact on the frequency of patients with “valuable” genotypes. The “actionable” genotype definition used here is conservative, requiring the genotype result to trigger a recommendation for change in standard therapy. In practice, all genotypes can be clinically useful; for example, in a patient needing antiplatelet therapy, CYP2C19*1/*1 genotype (homozygous wild type) reassures the provider that clopidogrel is likely efficacious. Using the more stringent criteria of “high risk genotypes,” 5% of patients were identified as at risk. In the panel-based model, these rarer, high risk genotypes are provided along with more common actionable genotypes.
As expected based on published minor allele frequencies, the frequencies of actionable genotypes for specific DGIs are different among EA and AA populations. EAs had a higher frequency of actionable genotypes related to simvastatin than AAs, and participants in pharmacogenomic research studies identifying the SLCO1B1 risk variant have been predominantly of European-American descent. However, individuals of African descent are at higher risk for statin-induced myopathy;(8,23) different SNPs may be present and confer risk for inefficacy or toxicity among AA patients, as well as Asian, Hispanic or other populations. These variants may be incorporated into future clinical tests.(8,24,25) Further, resequencing of large populations is identifying rare variants in pharmacogenes. As these are functionally characterized and incorporated into preemptive testing models, the portion of subjects with no actionable variants will fall further. In this study, AAs have a high frequency of actionable genotypes related to tacrolimus. While clinicians have long been aware of ancestry-based differences in drug response for this medication, the heterogeneity of actionable alleles within each group (e.g. 10% of AA patients are homozygous for CYP3A5*3) suggests that genotyping will provide a more accurate prediction of drug response than race/ethnicity.(26) Using the metric of “actionable genotypes” to determine the value of pharmacogenomic testing, the PREDICT test demonstrates the greatest value for AA patients, where actionable genotypes were identified in 96% of patients. In general, despite a paucity of pharmacogenomic research in minority populations, clinical pharmacogenomic programs such as PREDICT have the potential to improve medication outcomes for non-EA-patients by personalizing therapeutic doses and medication choices, just as incorporation of genetic ancestry has been shown to increase precision in evaluating pulmonary function.(27)
In contrast to indication-based testing, where pharmacogenomic testing is pursued for specific genes as indicated based on medication exposure, the panel-based PREDICT approach tests genotypes for all potential DGIs; this will result in “unnecessary” genetic testing in patients with no exposure to the associated medication. Pharmacogenotypes are most valuable in cases where a patient has an actionable genotype and is exposed to the associated medication. Given the relatively recent initiation of the PREDICT program and the serial implementation of DGIs, there are not sufficient accumulated data to specifically quantify drug exposures occurring after genotype and CDS data are in the EMR. However, using lifetime exposures (to date) to the associated medications as an approximation, the majority of patients genotyped were prescribed one of the medications, and more importantly, among those with actionable genotypes, over 40% of patients had evidence of exposure to the specific medication associated with that genotype. Moving forward, each of these cases represents an opportunity to improve therapeutic outcomes.
Our comparison of preemptive panel-based testing with a reactive testing strategy demonstrates that preemptive testing results in far fewer tests being performed. A formal pharmacoeconomic evaluation is underway, but given that the panel-based test is comparable in cost to single-gene assays, it is reasonable to expect that the panel-based test will prove to be more cost effective than individual assays. Preemptive panel-based testing has the added benefit of timeliness. If the panel is completed prior to medication exposure or at the point of first medication prescription, genotypes are available at the time of medication order/prescription for all subsequent medications. Our prior work demonstrated that the opportunities to use genetic test results to guide care are common, with 65% of regular clinic patients receiving a medication with pharmacogenetic indications.(28) Additional genotypes present on the panel or new DGIs for existing genotypes may also prove clinically useful in the future, increasing utility without additional testing cost.
The retrospective nature of this study and the selection process for inclusion of patients in the PREDICT program, either by indication, identification via predictive modeling, patient request, or physician preference, may limit the applicability of these findings to other cohorts. However, the finding that this selected patient population closely mirrors expected results based on population data is encouraging that accurate predictions can be made for other clinical settings. Provided there are reported genotype-frequency data for appropriate race/ethnicities in a population, accurate predictions can be made for other clinical settings.
Taken together, these data highlight the potential of panel-based pharmacogenotyping to identify actionable variants. The frequency with which patients harboring actionable variants are exposed to the medications of interest, the degree to which providers make use of genotype-guided CDS, and the extent that adverse therapeutic outcomes will be reduced remain to be determined.
Methods
Institutional Pharmacogenotyping Program
At Vanderbilt, the PREDICT program began pharmacogenomic testing in September 2010. This program is unique in its approach to pre-prescription genotyping, leveraging both a predictive model to identify patients at risk of future exposure to target medications and indication-triggered testing to obtain a multiplexed genotype test.(9,28) This preemptive approach enables genotype-guided CDS for providers at the time of medication initiation by prescription or order. Genotyping is completed using a panel-based test, currently with the Illumina VeraCode ADME Core panel, through which genotypes for a 184 variants in 34 genes are determined. Testing is performed in a Clinical Laboratory Improvement Amendments (CLIA)-certified laboratory that participates in the College of American Pathologists (CAP) biannual pharmacogenetics (PGX) proficiency exchange. Prior to reporting clinical results for the initial gene, CYP2C19, the genotypes of 56 commercially purchased control samples were tested and 100% concordance for CYP2C19 was obtained. Subsequent clinically actionable SNPs were monitored for accuracy before reporting into patient medical records by measuring concordance rates. While 100% concordance rates would be ideal, for most, concordance rates of >99% was achieved. After the launching of an actionable SNP, the performance of each is monitored monthly and documented as part of lab quality control. Specific genotype results are released into the EMR after review of the relevant evidence, development of genotype-specific CDS, and approval by institutional oversight.
In September 2010, PREDICT testing was initiated for patients undergoing cardiac catheterization since ~40% of these patients receive clopidogrel. CDS for clopidogrel prescription based on patient genotypes for CYP2C19 represented the first targeted patient genotyping and clinically-implemented DGI at Vanderbilt. Since then, the program has expanded to provide genotyping to patients presenting for both inpatient and outpatient care, and accompanying prompts to providers to consider testing in patients presenting for cardiac catheterization, treatment of acute lymphocytic leukemia (due to exposure to thiopurine therapy), and for those with 40% or higher likelihood of exposure to clopidogrel, warfarin or a statin in the next three years by the PREDICT statistical model.(28) Providers may also order the test outside of these scenarios as they would any other laboratory test.
Patient Population
Patient data collected in the course of patient care and recorded in the EMR, including routine laboratory, prescription, and administrative records for patients affected by the PREDICT program, were included in an IRB-approved data repository. For this study, use of de-identified data from the repository was reviewed by the IRB and granted exempt status. Data for all patients age 18 and older with PREDICT genetic testing prior to 9/30/2012 were extracted from the repository for analysis. Demographic data and clinical data were determined from the EMR. Medication exposure data were determined using a combination of structured medication entries and MedEx, a natural language processing system that extracts medication entries from free text.(29) Genotype data were made available from the PREDICT repository.
Analysis
Summary statistics and calculation of observed vs. expected genotypes were performed using R version 3.0.1 (Vienna, Austria). For all analyses, individuals with a “no call” for one or more genotypes of interest were excluded. Actionable and deterministic genotypes were defined as shown in Supplemental table 2. These definitions are based upon institutionally-approved clinical decision support advisors that prompt providers to alter dosing or medication choice based on genotype for clopidogrel, simvastatin, thiopurines and tacrolimus. For warfarin, clinical decision support includes a dosing advisor which incorporates CYP2C9 and VKORC1 genotype, whether wild type or variant. Actionable genotypes were defined as having one or more CYP2C9*2 or *3 variant, or VKORC1 rs9923231.(3) High risk genotypes were also defined a priori as those genotypes most strongly associated with risk of adverse outcomes, including homozygosity for CYP2C19*2 (clopidogrel resistance), SLCO1B1*5 (myopathy due to simvastatin), CYP2C9*3 (warfarin sensitivity/unstable dosing), and homozygosity or compound heterozygosity for TPMT*2 or *3 (bone marrow toxicity due to thiopurines). Expected frequencies for actionable genotypes were calculated using published minor allele frequencies.(3,4,6,8) To calculate expected rates of actionable genotypes without available guidelines, reported genotype frequencies were determined from the literature (Supplemental table 4).(20,30-36) For medication exposure analyses, individuals exposed to either clopidogrel or alternative medication (prasugrel for the time frame of data collection) were included as clopidogrel-exposed, as the prasugrel prescription may have been prompted by the PREDICT genotyping result. Similarly, all HMG-CoA reductase inhibitors were included when determining statin exposure rates, as genotype data may or may not have been available to providers at the time of prescription, prompting the choice of an alternate to simvastatin.
Supplementary Material
Study Highlights.
What is the current knowledge on the topic?
Genotype-guided pharmacotherapy has been clinically implemented in limited settings. At Vanderbilt, the PREDICT program has preemptively tested pharmacogenomic variants for the clinical care of over 10,000 patients to date.
What question this study addressed?
We determined the frequencies of actionable genotypes and medication exposures for five drug-genome interactions employed in PREDICT, and compared the number of tests obtained under panel-based preemptive testing vs. indication-based testing.
What this study adds to our knowledge?
One or more actionable variants are identified in 91% of patients. Most genotyped patients had at least one drug exposure, and those with one drug exposure were likely to be exposed to a second target drug. Fewer genetic tests are performed with preemptive genotyping than a reactive strategy.
How this might change clinical pharmacology and therapeutics?
These data inform potential implementation of pharmacogenomic programs and may lead to increased adoption of clinical pharmacogenomic programs.
Acknowledgments
The authors would like to acknowledge the contributions of Marc Bellar, Julie Field, Jennifer Mitchell and Cindy Vnencak-Jones in developing and implementing the PREDICT program. Portions of this study were supported by the Vanderbilt Institute for Clinical and Translational Research (VICTR), NCATS/NIH grant UL1 TR000445; and NIH/NIGMS Clinical Pharmacology Training Program 5T32 GM007569-33 (SLV).
Funding/Research Support
All Authors: Portions of this study were supported by NCATS/NIH grant UL1 TR000445, supporting the Vanderbilt Institute for Clinical and Translational Research (VICTR). SLV: Portions of this study supported by NIH/NIGMS Clinical Pharmacology Training Program 5T32 GM007569-33.
Abbreviations
- AA
African American, Non-Hispanic
- CAP
College of American Pathologists
- CLIA
Clinical Laboratory Improvement Amendments
- CDS
Clinical Decision Support
- DGI
Drug-Genome Interaction
- EA
European American, Non-Hispanic
- EMR
Electronic Medical Record
- HMG
3-hydroxy-3-methyl-glutaryl
- PGX
Pharmacogenetics
- PREDICT
Pharmacogenomic Resource for Enhanced Decisions in Care and Treatment
- SNP
Single Nucleotide Polymorphism
Footnotes
Author Contributions:
Van Driest, Shi, Bowton, Schildcrout, Peterson, Pulley, Denny, and Roden wrote the manuscript
Van Driest, Shi, Bowton, Schildcrout, Peterson, Pulley, and Roden designed the research
Van Driest, Bowton, Pulley, Denny, and Roden performed the research
Shi and Schildcrout analyzed the data
Conflict of Interest/Disclosure: The authors declare no relevant financial interests in this manuscript. The authors declare no other relationships/conditions/circumstances that present a potential conflict of interest that have influenced or give the appearance of potentially influencing the work submitted in this manuscript, other than the funding and research support for the institution and individual authors as described below.
References
- 1.Collins FS, Green ED, Guttmacher AE, Guyer MS. US National Human Genome Research Institute. A vision for the future of genomics research. Nature. 2003 Apr 24;422(6934):835–47. doi: 10.1038/nature01626. [DOI] [PubMed] [Google Scholar]
- 2.Crews KR, Gaedigk A, Dunnenberger HM, Klein TE, Shen DD, Callaghan JT, et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) guidelines for codeine therapy in the context of cytochrome P450 2D6 (CYP2D6) genotype. Clin Pharmacol Ther. 2012 Feb;91(2):321–6. doi: 10.1038/clpt.2011.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Johnson JA, Gong L, Whirl-Carrillo M, Gage BF, Scott SA, Stein CM, et al. Clinical Pharmacogenetics Implementation Consortium Guidelines for CYP2C9 and VKORC1 genotypes and warfarin dosing. Clin Pharmacol Ther. 2011 Oct;90(4):625–9. doi: 10.1038/clpt.2011.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scott SA, Sangkuhl K, Gardner EE, Stein CM, Hulot J-S, Johnson JA, et al. Clinical Pharmacogenetics Implementation Consortium guidelines for cytochrome P450-2C19 (CYP2C19) genotype and clopidogrel therapy. Clin Pharmacol Ther. 2011 Aug;90(2):328–32. doi: 10.1038/clpt.2011.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scott SA, Sangkuhl K, Stein CM, Hulot J-S, Mega JL, Roden DM, et al. Clinical pharmacogenetics implementation consortium guidelines for CYP2C19 genotype and clopidogrel therapy: 2013 update. Clin Pharmacol Ther. 2013 Sep;94(3):317–23. doi: 10.1038/clpt.2013.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Relling MV, Gardner EE, Sandborn WJ, Schmiegelow K, Pui C-H, Yee SW, et al. Clinical Pharmacogenetics Implementation Consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing. Clin Pharmacol Ther. 2011 Mar;89(3):387–91. doi: 10.1038/clpt.2010.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Relling MV, Gardner EE, Sandborn WJ, Schmiegelow K, Pui C-H, Yee SW, et al. Clinical pharmacogenetics implementation consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing: 2013 update. Clin Pharmacol Ther. 2013 Apr;93(4):324–5. doi: 10.1038/clpt.2013.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wilke RA, Ramsey LB, Johnson SG, Maxwell WD, McLeod HL, Voora D, et al. The clinical pharmacogenomics implementation consortium: CPIC guideline for SLCO1B1 and simvastatin-induced myopathy. Clin Pharmacol Ther. 2012 Jul;92(1):112–7. doi: 10.1038/clpt.2012.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pulley JM, Denny JC, Peterson JF, Bernard GR, Vnencak-Jones CL, Ramirez AH, et al. Operational Implementation of Prospective Genotyping for Personalized Medicine: The Design of the Vanderbilt PREDICT Project. Clin Pharmacol Ther 2012. 92(1):87–95. doi: 10.1038/clpt.2011.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mega JL, Hochholzer W, Frelinger AL, 3rd, Kluk MJ, Angiolillo DJ, Kereiakes DJ, et al. Dosing clopidogrel based on CYP2C19 genotype and the effect on platelet reactivity in patients with stable cardiovascular disease. J Am Med Assoc. 2011 Nov 23;306(20):2221–8. doi: 10.1001/jama.2011.1703. [DOI] [PubMed] [Google Scholar]
- 11.Ogg MS, Brennan P, Meade T, Humphries SE. CYP2C9*3 allelic variant and bleeding complications. Lancet. 1999 Sep 25;354(9184):1124. doi: 10.1016/S0140-6736(05)76918-X. [DOI] [PubMed] [Google Scholar]
- 12.Ablin J, Cabili S, Lagziel A, Peretz H. Warfarin therapy in a patient homozygous for the CYP2C9 3 allele. Isr Med Assoc J. 2002 Feb;4(2):139–41. [PubMed] [Google Scholar]
- 13.Steward DJ, Haining RL, Henne KR, Davis G, Rushmore TH, Trager WF, et al. Genetic association between sensitivity to warfarin and expression of CYP2C9*3. Pharmacogenetics. 1997 Oct;7(5):361–7. doi: 10.1097/00008571-199710000-00004. [DOI] [PubMed] [Google Scholar]
- 14.Hicks JK, Swen JJ, Thorn CF, Sangkuhl K, Kharasch ED, Ellingrod VL, et al. Clinical Pharmacogenetics Implementation Consortium guideline for CYP2D6 and CYP2C19 genotypes and dosing of tricyclic antidepressants. Clin Pharmacol Ther. 2013 May;93(5):402–8. doi: 10.1038/clpt.2013.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martin MA, Klein TE, Dong BJ, Pirmohamed M, Haas DW, Kroetz DL, et al. Clinical pharmacogenetics implementation consortium guidelines for HLA-B genotype and abacavir dosing. Clin Pharmacol Ther. 2012 Apr;91(4):734–8. doi: 10.1038/clpt.2011.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hershfield MS, Callaghan JT, Tassaneeyakul W, Mushiroda T, Thorn CF, Klein TE, et al. Clinical Pharmacogenetics Implementation Consortium guidelines for human leukocyte antigen-B genotype and allopurinol dosing. Clin Pharmacol Ther. 2013 Feb;93(2):153–8. doi: 10.1038/clpt.2012.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Caudle KE, Thorn CF, Klein TE, Swen JJ, McLeod HL, Diasio RB, et al. Clinical Pharmacogenetics Implementation Consortium Guidelines for Dihydropyrimidine Dehydrogenase Genotype and Fluoropyrimidine Dosing. Clin Pharmacol Ther. 2013 Aug 29; doi: 10.1038/clpt.2013.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McDonagh EM, Thorn CF, Bautista JM, Youngster I, Altman RB, Klein TE. PharmGKB summary: very important pharmacogene information for G6PD. Pharmacogenet Genomics. 2012 Mar;22(3):219–28. doi: 10.1097/FPC.0b013e32834eb313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Muir AJ, Gong L, Johnson SG, Michael Lee MT, Williams MS, Klein TE, et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) guidelines for IFNL3 (IL28B) genotype and peginterferon alpha based regimens. Clin Pharmacol Ther. 2013 Oct 4; doi: 10.1038/clpt.2013.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu X, Cheng D, Kuang Q, Liu G, Xu W. Association of UGT1A1*28 polymorphisms with irinotecan-induced toxicities in colorectal cancer: a meta-analysis in Caucasians. Pharmacogenomics J. 2013 Mar 26; doi: 10.1038/tpj.2013.10. [epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ramsey BW, Davies J, McElvaney NG, Tullis E, Bell SC, Dřevínek P, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med. 2011 Nov 3;365(18):1663–72. doi: 10.1056/NEJMoa1105185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leckband SG, Kelsoe JR, Dunnenberger HM, George AL, Jr, Tran E, Berger R, et al. Clinical pharmacogenetics implementation consortium guidelines for HLA-B genotype and carbamazepine dosing. Clin Pharmacol Ther. 2013 Sep;94(3):324–8. doi: 10.1038/clpt.2013.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hippisley-Cox J, Coupland C. Individualising the risks of statins in men and women in England and Wales: population-based cohort study. Heart. 2010 Jun 1;96(12):939–47. doi: 10.1136/hrt.2010.199034. [DOI] [PubMed] [Google Scholar]
- 24.Chung J-Y, Cho J-Y, Yu K-S, Kim J-R, Oh D-S, Jung H-R, et al. Effect of OATP1B1 (SLCO1B1) variant alleles on the pharmacokinetics of pitavastatin in healthy volunteers. Clin Pharmacol Ther. 2005 Oct;78(4):342–50. doi: 10.1016/j.clpt.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 25.Lee E, Ryan S, Birmingham B, Zalikowski J, March R, Ambrose H, et al. Rosuvastatin pharmacokinetics and pharmacogenetics in white and Asian subjects residing in the same environment. Clin Pharmacol Ther. 2005 Oct;78(4):330–41. doi: 10.1016/j.clpt.2005.06.013. [DOI] [PubMed] [Google Scholar]
- 26.Ramirez AH, Shi Y, Schildcrout JS, Delaney JT, Xu H, Oetjens MT, et al. Predicting warfarin dosage in European-Americans and African-Americans using DNA samples linked to an electronic health record. Pharmacogenomics. 2012 Mar;13(4):407–18. doi: 10.2217/pgs.11.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kumar R, Seibold MA, Aldrich MC, Williams LK, Reiner AP, Colangelo L, et al. Genetic ancestry in lung-function predictions. N Engl J Med. 2010 Jul 22;363(4):321–30. doi: 10.1056/NEJMoa0907897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schildcrout JS, Denny JC, Bowton E, Gregg W, Pulley JM, Basford MA, et al. Optimizing drug outcomes through pharmacogenetics: a case for preemptive genotyping. Clin Pharmacol Ther. 2012 Aug;92(2):235–42. doi: 10.1038/clpt.2012.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu H, Stenner SP, Doan S, Johnson KB, Waitman LR, Denny JC. MedEx: a medication information extraction system for clinical narratives. J Am Med Informatics Assoc. 2010 Feb;17(1):19–24. doi: 10.1197/jamia.M3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chinevere TD, Murray CK, Grant E, Jr, Johnson GA, Duelm F, Hospenthal DR. Prevalence of glucose-6-phosphate dehydrogenase deficiency in U.S. Army personnel Mil Med. 2006 Sep;171(9):905–7. doi: 10.7205/milmed.171.9.905. [DOI] [PubMed] [Google Scholar]
- 31.Ge D, Fellay J, Thompson AJ, Simon JS, Shianna KV, Urban TJ, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature. 2009 Sep 17;461(7262):399–401. doi: 10.1038/nature08309. [DOI] [PubMed] [Google Scholar]
- 32.Girard H, Butler LM, Villeneuve L, Millikan RC, Sinha R, Sandler RS, et al. UGT1A1 and UGT1A9 functional variants, meat intake, and colon cancer, among Caucasians and African-Americans. Mutat Res. 2008 Sep 26;644(1-2):56–63. doi: 10.1016/j.mrfmmm.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mattison LK, Fourie J, Desmond RA, Modak A, Saif MW, Diasio RB. Increased Prevalence of Dihydropyrimidine Dehydrogenase Deficiency in African-Americans Compared with Caucasians. Clin Cancer Res. 2006 Sep 15;12(18):5491–5. doi: 10.1158/1078-0432.CCR-06-0747. [DOI] [PubMed] [Google Scholar]
- 34.Monaghan KG, Bluhm D, Phillips M, Feldman GL. Preconception and prenatal cystic fibrosis carrier screening of African Americans reveals unanticipated frequencies for specific mutations. Genet Med. 2004 Jun;6(3):141–4. doi: 10.1097/01.gim.0000127269.42279.83. [DOI] [PubMed] [Google Scholar]
- 35.Saif MW. Dihydropyrimidine Dehydrogenase Gene (DPYD) Polymorphism among Caucasian and non-Caucasian Patients with 5-FU- and Capecitabine-related Toxicity Using Full Sequencing of DPYD. Cancer Genomics Proteomics. 2013 Mar 1;10(2):89–92. [PubMed] [Google Scholar]
- 36.Wei X, Elizondo G, Sapone A, McLeod HL, Raunio H, Fernandez-Salguero P, et al. Characterization of the Human Dihydropyrimidine Dehydrogenase Gene. Genomics. 1998 Aug 1;51(3):391–400. doi: 10.1006/geno.1998.5379. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.