

Abstract
RelA-mediated NF-κB canonical signaling promotes mesenchymal progenitor cell (MPC) proliferation, but inhibits differentiation of mature osteoblasts (OBs) and thus negatively regulates bone formation. Previous studies suggest that NF-κB RelB may also negatively regulate bone formation through non-canonical signaling, but they involved a complex knockout mouse model and the molecular mechanisms involved were not investigated. Here, we report that RelB−/− mice develop age-related increased trabecular bone mass associated with increased bone formation. RelB−/− bone marrow stromal cells expanded faster in vitro and have enhanced OB differentiation associated with increased expression of the osteoblastogenic transcription factor, Runx2. In addition, RelB directly targeted the Runx2 promoter to inhibit its activation. Importantly, RelB−/− bone-derived MPCs formed bone more rapidly than wild-type cells after they were injected into a murine tibial bone defect model. Our findings indicate that RelB negatively regulates bone mass as mice age and limits bone formation in healing bone defects, suggesting that inhibition of RelB could reduce age-related bone loss and enhance bone repair.
Keywords: RelB, NF-κB, mesenchymal progenitor cells, osteoblasts, bone formation
INTRODUCTION
Pro-inflammatory cytokines, such as RANKL and TNF, mediate bone destruction in common bone diseases, such as postmenopausal osteoporosis, rheumatoid arthritis, and periodontitis (1). In these diseases, unlike in normal bone remodeling, bone formation by osteoblasts (OBs) does not match the bone lost (1), resulting in localized and/or generalized bone loss. A role for the NF-κB family of transcription factors, which includes NF-κB1 (p50 and its precursor p105), NF-κB2 (p52 and its precursor p100), RelA (p65), RelB and c-Rel (2), in bone was first discovered when expression of both NF-κB1 and 2 was found unexpectedly to be required for osteoclast precursor (OCP) differentiation into osteoclasts (OCs) (3, 4). Later it was discovered that they were required for RANKL- and TNF-induced OC formation (5).
RANKL and TNF induce proteasomal processing of p105 to p50 which forms heterodimers typically with RelA to activate canonical NF-κB signaling (2). RANKL and to a much lesser extent TNF activate non-canonical signaling in OCPs leading to NF-κB-inducing kinase (NIK)-mediated processing of p100 to p52 (6) which typically forms heterodimers with RelB to activate non-canonical signaling and transcription of target genes. RelA promotes OC differentiation by blocking a RANKL-inducted apoptotic pathway in OCPs, but it is not involved in terminal OC differentiation (7). RelB in contrast is not required for basal OC formation, but appears to play a role in the enhanced osteoclastogenesis observed in pathologic conditions, such as osteolysis induced by metastatic cancer cells and inflammation (8).
These previous studies have increased understanding of the role for NF-κB in OC formation and functions (3–8), but the role of NF-κB signaling in bone formation is less well understood and the published data are conflicting. For example, several groups have shown that activation of canonical NF-κB signaling inhibits bone formation based on an inhibitory effect of TNF induction of p65 on OB differentiation (9–12). Recently it was reported that inhibition of canonical signaling specifically in mature OBs by genetic manipulation results in a transient increase in bone mass in young mice (13). Furthermore, the NF-κB inhibitor, S1627, promotes murine calvarial defect repair and increased bone mineral density in ovariectomized mice (14), providing additional evidences that canonical NF-κB signaling negatively regulates bone formation. However, several other groups reported that TNF-induced activation of canonical NF-κB signaling in mesenchymal progenitor cells (MPCs) promotes their differentiation into OBs (15–17) through BMP-2-mediated up-regulation of Runx2 and Osterix (Osx) expression (16). These findings indicate that there are complex interactions involving cytokines and canonical NF-κB signaling that can have positive or negative regulatory effects on OBs to influence bone mass depending upon the form of stimulation and the state of osteoblastic cell differentiation.
A role of non-canonical NF-κB signaling in bone formation has also been reported. For example, mice generated to have accumulation of a non-processible form of NF-κB2 p100 have enhanced osteoblastic differentiation (18), and mice with deletion of p100, but retaining a functional p52, have osteopenia due to increased OC activity and impaired OB parameters (19). Interestingly, deletion of both RelB and p100 in these latter mice prevented the osteopenia in the p100−/− mice and actually increased bone mass accompanied by increased OB surfaces (19), suggesting an important role of RelB to inhibit OB differentiation. However, these studies did not include reports of the OB phenotype of single RelB−/− mice and did not examine the molecular mechanisms whereby RelB regulates OB differentiation or function. We have found that bone mass increases in RelB−/− mice as they age due to increased OB precursor proliferation associated with enhanced capacity of their MPCs to differentiate into OBs in vitro and to repair cortical bone defects in mice in vivo.
MATERIALS AMD METHODS
Animals and Reagents
RelB+/− and RelB−/− mice including male and female for all experiments on an inbred C57BL/6 background were obtained from Dr. Mitchell Kronenberg and have been described previously (20, 21). SCID mice were purchased from Jax Labs. The University of Rochester Medical Center Institutional Animal Care and Use Committee approved all animal studies. Labeled antibodies for FACS (APC-CD45, PE-CD105, FITC-Sca-1) were purchased from eBioscience. Ascorbic acid and β-glycerophosphate (β-GP) were purchased from Sigma. The BCIP/NBT alkaline phosphatase substrate was purchased from ScyTek Laboratories. RelB antibody was purchased from Santa Cruz.
OB differentiation
bone marrow (BM) was flushed from long bones of mice with α-MEM containing 20% FBS using a 25G-needle. The cells were filtered with a 40μm cell strainer, and 4×104 cells were cultured in 35mm dishes at 37°C in 5% CO2 for 4 days. Unattached cells were removed and replaced with 10% FBS in α-MEM containing 25μg/ml L-ascorbic acid and 5mM β-GP to induce OB differentiation. After 5–7 days in inducing medium, the cells were stained for alkaline phosphatase (ALP) activity using the ALP substrate, BCIP/NBT. Mineralization typically occurs after 10–14 days in culture, and the cells were stained with the von Kossa method for measurement of mineralized nodule formation. Calvariae from 7 day-old WT and RelB−/− mice were cut into pieces and digested 6 times with a mixture of 0.5% collagenase I and 0.125% trypsin (both from Sigma) for 20 min at 37°C. Cells from the 2nd to 6th digestions were collected for pre-OB cultures and OB differentiation experiments.
Generation of bone-derived mesenchymal progenitor cells (MPCs)
We cut mouse tibiae and femora into small pieces after BM had been flushed out and the cavities had been washed extensively with PBS using a modification of a previously described method (22). The bone fragments were cultured for 4 days with α-MEM containing 20% FBS at 37°C. Bone pieces were transferred into a new dish and cultured for an additional 4–5 days with α-MEM containing 10% FBS. The cells grown on the dish were passaged twice when they were 90% confluent, each time excluding cells tightly attached to the dishes. Third passage cells contained over 99% MPCs, sufficient for our experiments. We named these cells as bone-derived mesenchymal progenitor cells (bMPCs) and they were frozen for use later in OB differentiation and mineralization experiments. mRNA expression levels of OB-related genes were tested using methods we described previously (23).
FACS analysis
2×106 BM or cultured cells were stained with APC-anti-CD45.2, PE-anti-CD105 and FITC-Sca-1 antibodies. For cell cycle analysis, the collected bMPCs were incubated with 1μM DAPI with PBS containing 2% FBS for 20 min at 37°C. Data were acquired using a FACScanto flow cytometer and analyzed using FlowJo software, as described previously (24).
Micro-CT and bone histomorphometric analysis
Mice were given injections of calcein (10mg/Kg) 5 and 1 day before sacrifice in a standard bone formation double-labeling protocol. Right tibiae were fixed in 10% neutral buffered formalin and micro-CT scanning was performed following guidelines for assessment of bone microstructure in rodents using micro-computed tomography (25). The bones were then processed through graded alcohols and embedded in plastic, and dynamic and static parameters of bone formation were assessed, according to standard methods using OsteoMeasure software (OsteoMetrics). Left tibiae were fixed in 10% neutral buffered formalin for 2 days, decalcified in 10% EDTA for 3 weeks, and the bones were then processed and embedded in paraffin. Sections (4μm-thick) were stained with H&E for analysis of bone volume, OB surface and TRAP+ OCs using OsteoMeasure software.
Tibial Bone Defects
A 2×5 mm full thickness cortical defect was made on the anterior surface of the left and right tibiae of SCID mice. Briefly, a hole was pierced through the cortex ~1mm below the growth plate using a 25-guage needle. Scissors were then inserted into the hole and a 2×5mm defect was created by repeatedly cutting distally through the cortex. The defects were then almost completely filled with decalcified trabecular bone matrix, which had been extracted from bovine femoral necks using the following serial processing: 20% H2O2 for 2 days, 5mmol Sodium Azide (NaN3) overnight, 1mol NaOH containing 1% Triton X-100 overnight, methanol/chloroform (1:1) 24 hours, ether overnight, and 10% EDTA for 2 weeks. 5×105 bMPCs in 5μl of Hank’s solution were then injected into the bone matrix in the defects. The muscle fascia and skin overlying the defects were then sutured closed. Mice were euthanized 2, 4 and 8 weeks post-surgery, and the tibiae were fixed in 10% neutral buffered formalin for 2 days. The volume of new bone formed in the defects was measured using a VivaCT 40 micro-CT scanner (Scanco). The bones were then processed through alcohols, decalcified, and embedded in paraffin. The volume of newly formed bone and fibroblastic tissue was quantified in H&E-stained sections using OsteoMeasure software.
Quantitative Real-Time PCR
Total RNA was extracted from cultured cells using 1 ml TRIzol reagent, and 1 μg was used for synthesis of cDNA using a GeneAmp RNA PCR core kit. Quantitative PCR amplification was performed using an iCycler real-time PCR machine and iQ SYBR Green. Relative mRNA expression levels of target genes were analyzed using the CT value of the gene, normalized to β-actin.
Reporter constructs/Luciferase Assay
To clone the 5′ upstream region of the mouse Runx2 gene, 1997 bp (2kb) fragments in Runx2 promoter region was amplified by PCR from C57Bl6 mouse DNA extracted from bMPCs. The specific forward primer was: 5′-GTATTTCTGTGG-TTTTGTCATTAAAACT-3′, and the reverse primer was: 5′ AGAAAGTTTGCACCGCACTT-3′ overlapping the putative transcriptional start site (Fig. 5B). The 2 kb promoter was found with TFSEARCH software to contain 2 putative NF-κB binding sites. Overhangs containing the Kpn I restriction site were added to the forward primer and an overhang containing the Xho I site was added to the reverse primer at the 5′ end. PCR products were then subcloned into the pCRII TOPO vector (Invitrogen) and sequences were verified before their transfer into the Kpn I/Xho I backbone of pGL3-basic (Promega) driving the firefly Luciferase (Luc) gene. The Runx2 Luc reporter plasmid was co-transfected with either GFP control or RelB plasmid into C2C12 cells using a FuGene6 reagent (Roche). A 0.1μg aliquot of the SV40-Renilla Luc construct (Addgene) was also co-transfected with the above firefly reporters to standardize results for transfection efficiency. Cell lysates were prepared using a reporter lysis buffer (Promega). Luciferase activity was measured using a Microplate Luminometer (PerkinElmer).
Fig. 5. RelB directly targets the Runx2 promoter and inhibits Runx2 expression.

(A) mRNA (left panel) and protein (right panel) expression of Runx2 were tested by Real-time PCR and Western blot from RelB−/− and WT BM stromal cells cultured with OB differentiation medium for 7d. (B) Construction scheme of mouse Runx2 promoter luciferase (Luc) reporter. The 2-kb Runx2 promoter constructs contain 2 putative κB binding sites. (C) The 2-kb Runx2 promoter Luc reporter was co-transfected with RelB plasmid into C2C12 cells and the relative Luc activity was tested. (D) Site-directed mutagenesis of both κB binding sites 1 and 2 in the 2 kb Runx2 promoter was performed by deleting “ATC” and “ACA”. A luciferase activity assay was performed using C2C12 cells that were co-transfected with a RelB plasmid and the mutated Runx2 reporter. (E) ChIP assays were carried out using an anti-RelB or control IgG antibody on sheared chromatin from WT and RelB−/− MPCs. Immunoprecipitated DNA was analyzed by qPCR using primers covering either NF-κB binding site-1 (left panel) or 2 (middle panel) in the Runx2 promoter region or a pair of un-related primers (right panel) designed in the region that is 3 kb apart from the κB binding sites. Results are expressed as fold-enrichment compared with IgG normalized to input. (F) bMPCs from WT and RelB−/− mice were transfected with a scrambled or Runx2 mouse shRNA sequence for 2 days followed by puromycin selection to kill the uninfected cells. The cells were treated with OB differentiation medium for 5d and stained for ALP activity to measure ALP+ cells (left and middle panel), and mRNA expression of Runx2 in these cells was tested by real-time PCR (right panel). All mice in in vitro experiments were 1.5 to 2 months-old. * p<0.05; ** p<0.01 vs. control.
To perform site-directed mutagenesis of both κB binding sites in Runx2 promoter, two pairs of primers: mRunx2-1fw 5′-AATATTTGTAAAGGACCCAGGCTAACACTT, mRunx2-1rv 5′-AAGTGTTAGCCTGGGTCCTTTACAAATATT, and mRunx2-2fw 5′-AGGAGAGAC-AGAGGACCCATAAGTAAAGAG, mRunx2-2rv 5′-CTCTTTACTTATGGGTCCTCTGTC-TCTCCT, was designed using Invitrogen software program and GENEART® Site-Directed Mutagenesis System (Invitrogen) was used to delete “ABC” and “ACA” in the 1 and 2 binding sites following the manual instructions. We followed the above procedure to test the role of RelB on the mutated Runx2 promoter.
Chromatin immunoprecipitation (ChIP)
ChIP was performed using a MAGnify™ ChIP kit (Invitrogen) following the manual instructions. Briefly, sheared chromatin from WT and RelB−/− bMPCs that had been fixed with 1% formaldehyde was immunoprecipitated with 5μg of antibody to RelB (Santa Cruz Biotechnology, Inc), negative control rabbit IgG (Santa Cruz), or positive control H3 histone (Cell Signaling). Immunoprecipitated DNA was then used as a template for quantitative PCR using primers specific for the NF-κB binding sites 1 (forward 5′-tcaactacacagccatgatt and reverse 5′-taagcttggggatctgtaac) or 2 (forward 5′-cttctgaatgccaggaaggc and reverse5′-tgggactgcctaccactgt) of the Runx2 promoter as well as a pair of un-related primers (forward 5′-cactgctgactgaaacaagtc and reverse 5′-agtctgagtgagcttcctgat) designed in the region that is 3 kb apart from the κB binding sites.
ELISA assay
Mouse serum osteocalcin (MyBioSource, San Diego, CA) levels were assessed according to the manufacturer’s instructions.
Statistics
All results are given as mean ± SD. Comparisons between two groups were analyzed using two-tailed unpaired Student’s t-test. One-way ANOVA and Dunnett’s Post-Hoc multiple comparisons were used for comparisons among three or more groups. p values < 0.05 were considered statistically significant.
RESULTs
RelB−/− mice develop increased trabecular bone volume as they age
To fully investigate the role of RelB in bone remodeling, we analyzed the bone phenotype of RelB−/− mice of various ages. Consistent with a previous report that young RelB−/− mice do not appear to have a significant bone phenotype (8), we found that 4-week-old RelB−/− mice have normal trabecular bone volume, as assessed by histomorphometric analysis (Fig. 1A). However, mean diaphyseal trabecular bone volume values in 6–8-week-old RelB−/− mice were double those of WT littermates. Importantly, their mean metaphyseal bone volumes remained within the normal range, similar to those of littermates (Fig. 1A), suggesting that OC function in the RelB−/− mice is normal. Furthermore, when the RelB−/− mice were 10–14 weeks-old, their mean diaphyseal bone volumes had increased further to 4-fold higher than those in WT littermates, while their mean metaphyseal bone volumes still remained within the normal range (Fig. 1A). These histomorphometric findings were confirmed by micro-CT when the mice were 10–14 weeks-old (Fig. 1B), which showed that the increased bone mass was associated with increased trabecular number (Fig. 1B, p<0.01), but not thickness. 10–14-week-old RelB−/− mice also had increased vertebral trabecular bone volume associated with increased trabecular number, but not thickness, as assessed by micro-CT analysis (Fig. 1C), indicating that their increased bone mass was not restricted to diaphyseal bone. Interestingly, however, the RelB−/− mice had normal cortical thickness, and periosteal and endosteal surface areas of the cortical bone in the RelB−/− mice were also similar to that of the WT littermates (Fig. 1D).
Fig. 1. Trabecular bone mass increases in RelB−/− mice as they age.

(A) H&E-stained sections of tibiae from WT and RelB−/− mice and histomorphometric data showing trabecular bone volume (BV/TV) in the tibial metaphyses (area corresponding to the position of the white bar) and proximal diaphysis (vertical green bar) of 4–14-wk-old mice. n=5–8 mice/group. Metaphysic bone (corresponding to ROI-1 of μCT) was defined as a 0.3 mm-region along with the long-axis of tibia beginning at 0.15 mm apart from growth plate. Proximal diaphysis (corresponding to ROI-2 of μCT) was defined as a 0.6 mm-region under ROI-1. (B) Representative μCT scans and bone volume (BV/TV), trabecular number (Tb.N), trabecular thickness (Tb.Th) and trabecular separation (Tb.Sp) in the metaphysis (Region of Interest-1 (ROI-1) and in the proximal part of the diaphysis (ROI-2) of tibiae from 10–14wk-old mice. n=5–8 mice/group. (C) μCT scans and data from 4th lumbar vertebrae from 10–14-wk-old mice. n=10 mice/group. (D) μCT scans and data from cortical bone at the junction between RO-1 and -2 from mice in (B). All groups contained male and female mice. * p<0.05; ** p<0.01 vs. control.
RelB−/− mice have a transient increase in bone formation
The increased diaphyseal and vertebral trabecular bone observed in the RelB−/− mice as they aged could be caused by impaired OC and/or enhanced OB differentiation and function. We confirmed that metaphyseal OC numbers and surfaces are normal in young RelB−/− mice (8) (data not shown) and they remained similar to those in WT control mice in the metaphyseal bone as they aged to 8 weeks-old (data not shown). However, in the proximal diaphyses of RelB−/− mice OC surfaces were increased (15.6±7.3% vs 7.8±3.6 % in WT littermates, p<0.05), while OC numbers were not, presumably reflecting the increased bone mass in the RelB−/− mice (see below). In fact, RelB regulation of OC differentiation is complicated. We found that RelB−/− mice have 3–5 fold increase in monocyte-macrophages in their blood, BM and spleen, and that sorted CD11b−Gr-1− BM cells from RelB−/− mice have significantly enhanced OC differentiation, but their CD11b+Gr-1−/lo+ BM cells, the generally recognized OC precursors (24), did not form OCs in response to RANKL and M-CSF in vitro culture (data not shown). Because the increased trabecular bone volume in RelB−/− mice is not associated with un-resorbed islands of cartilage inside trabeculae, seen typically in osteopetrosis (26,27), we concluded that the increased diaphyseal and vertebral bone mass likely resulted from enhanced OB formation or activity, as has been reported in RelB/p100 double knockout mice (19). Indeed, mean values for OB and mineralizing surfaces and bone formation rates (BFR) in the trabecular bone in the diaphyses of 4-week-old RelB−/− mice are significantly higher than those in WT littermates (Fig. 2A&B). Surprisingly, however, mean values for OB surface and BFRs were reduced significantly in the tibial diaphyseal bone in older (8–9-weeks-old) RelB−/− mice (Fig. 2C), indicating that the increase in bone formation in the RelB−/− mice is transient. Consistent with this, serum osteocalcin levels were significantly increased in younger, but not in older RelB−/− mice (Fig. 2D), and mRNA levels of ALP, Runx2, RANKL and OPG were similar in femoral bone samples from 9-week-old RelB−/− and WT mice, while the osteocalcin mRNA levels were only slightly increased (Supplemental Fig. 1).
Fig 2. RelB−/− mice have a transient increase in bone formation.
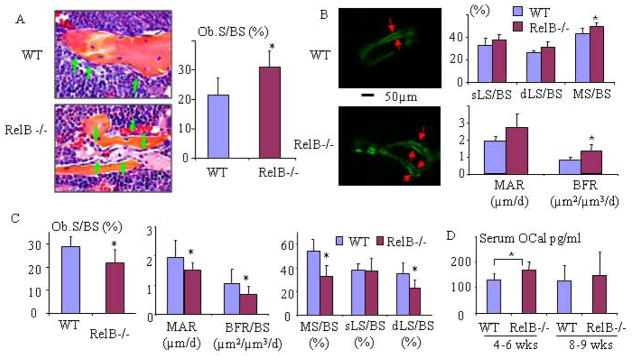
(A) Histology and histomorphometric analysis of OBs (green arrows) and OB surface (OB.S/BS) in the tibiae of 4-wk-old RelB−/− mice and WT littermates (n=7/group). (B) Double calcein labeling (arrows) in trabeculae in proximal tibial diaphyses corresponding to ROI-2 in Fig. 1 and analysis of dynamic parameters of bone formation: single labeled surface (sLS/BS), double labeled surface (dLS/BS), mineralization surface (MS/BS), mineral apposition rate (MAR) and bone formation rate (BFR) in 4-wk-old RelB−/− and WT mice (n=8/group). (C) Analysis of dynamic parameters of bone formation listed in (B) and OB surface in proximal tibial diaphyses of 7–8-week-old RelB−/− and WT mice (n=5–6/group). (D) Serum osteocalcin (OCal) levels from 4–5- (n=9) and 8–9-week-old (n=8) male and female WT and RelB−/− mice tested by ELISA.
Murine MPCs have been defined as CD45−CD105+ cells (28), and we have reported that CD45− cells can be used as mesenchymal stem cell-enriched cells (29). We found that RelB−/− and WT mice have similar numbers of CD45−CD105+ cells in freshly isolated BM from tibiae and femora (Fig. 3A upper panel). However, these BM cells from RelB−/− mice generated 2-fold more CD45−CD105+ MPCs than WT cells 7 days after equal numbers of each were placed in culture dishes (Fig. 3A, lower panel), indicating that RelB−/− MPCs expanded faster in vitro. This was associated with formation of more ALP+ colonies and mineralized nodules (Fig. 3B) from the RelB−/− BM cell cultures and a 3- and 1.5-fold increase in osteocalcin and ALP mRNA expression levels (Fig. 3C) at day 10, respectively, compared to WT cells, consistent with the enhanced OB generation and function in the RelB−/− mice. The enhanced OB proliferation and differentiation persisted in BM cells cultured from 3-month-old RelB−/− mice, despite the reduced BFR in vivo. RelB−/− BM stromal cells formed 3-fold more ALP+ cell colonies than WT cells at day 7, but ALP mRNA expression levels were similar at day 6. This may reflect the fact that the increased numbers of ALP+ cells from RelB−/− stromal cells were mainly due to their faster expansion at an earlier stage of differentiation, as shown in Fig. 3A.
Fig 3. RelB−/− mice have enhanced stromal cell proliferation and OB differentiation in vitro.

(A) FACS analysis showing the % of CD45−CD105+ MPCs in freshly isolated BM cells from 2 month-old WT and RelB−/− mice and BM stromal cells from the mice after being cultured for 7 days with growth-inducing medium. (B) Equal numbers of freshly isolated BM cells from 2 month-old RelB−/− and WT mice were cultured in OB differentiation medium containing 25μg/ml L-ascorbic acid (Vit C) and 5mM β-glycerophosphate (β-GP) for 7 and 12d. ALP+ cells colonies (upper panel) and mineralized nodules (lower panel) were evaluated after ALP and von Kossa staining. (C) Expression of ALP and osteocalcin (OCal) was tested using Real-time PCR in BM stromal cells and in differentiating OBs induced by Vit C and β-GP after 6 and 10 days of culture. (D) BM stromal cells generated from WT and RelB−/− mouse were re-seeded in 12-well plate. After the cells were sub-confluent, they were induced for OB differentiation for 5 days and stained for ALP activity. (E) Calvarial pre-OBs generated from 7-day-old pups were cultured in OB differentiation medium for 5 and 10 days and stained for ALP activity. (F) Calvarial pre-OBs were cultured in OB differentiation medium for the indicated times, total RNA was extracted, and expression of ALP, osteocalcin and Runx2 mRNA was tested by real-time PCR. All mice in in vitro experiments were 1.5–2-months-old males or females except for those used for calvarial pre-osteoblasts in (E&F); 3 wells per group. * p<0.05 vs. control.
To determine if the enhanced differentiation of RelB−/− BM stromal cells contributed to the increased ALP+ colony numbers and mineralized nodule formation, we generated BM stromal cells, re-seeded them onto culture plates, and induced OB differentiation when they were sub-confluent to exclude the influence of proliferation. These re-seeded stromal cells still had increased ALP+ cell formation (Fig. 3D), confirming their enhanced differentiation. Furthermore, we generated calvarial pre-OBs from 7 day-old WT and RelB−/− pups and found that RelB−/− calvarial pre-OBs also have enhanced ALP+ cell differentiation at day 10, but not after 5 day of induction of differentiation (Fig. 3E). Consistent with the findings in BM stromal cells, differentiating RelB−/− OBs from calvariae had significantly increased ALP, osteocalcin and Runx2 mRNA expression, particularly at the later stage of differentiation (day 10, Fig. 3F).
RelB negatively regulates OB differentiation, associated with reduced Runx2 activity
Primary BM stromal cell cultures contain variable numbers of hematopoietic cells, including monocyte-macrophages (30), which could influence stromal cell differentiation. To minimize this possibility, we employed a recently described method (22) in which bone-derived MPCs (bMPCs) are used to study MPC differentiation. We confirmed that almost 100% of these cells from WT and RelB−/− long bones were of mesenchymal origin (i.e. CD45- cells, Fig. 4A left panel), and more than 90% of these cells expressed the stem cell marker, Sca-1 (Fig. 4A right panel). These RelB−/− bMPCs grew faster than WT cells (Fig. 4B). Consistent with this, cell cycle analysis using FACS (Fig. 4C) showed that the % of MPCs undergoing basal proliferation (S and G2/M phase cells) was higher in RelB−/− than in WT cells (36.3% vs. 29.5%, p<0.05) and they formed significantly more ALP+ cells (Fig. 4D), similar to primary BM stromal and calvarial cell cultures. Of note, the ratio of ALP+ cells to total cells was 3-fold higher in RelB−/− bMPCs cultures than in WT cells (Fig. 4D), indicating that RelB−/− bMPCs have enhanced OB differentiation. Importantly and consistently, RelB−/− bMPCs have significantly increased mineralized nodule formation (Fig. 4E) and increased expression of ALP and osteocalcin during their differentiation (Fig. 4F).
Fig. 4. RelB−/− bone-derived mesenchymal progenitor cells (bMPCs) have enhanced proliferation and OB differentiation.

(A) bMPCs generated from 8 week-old RelB−/− mice and WT littermates were analyzed by FACS using CD45, CD105 and Sca-1 antibodies. (B) 2×104 bMPCs from WT and RelB−/− mice were seeded in 12-well plates for indicated times. The cells were fixed with 10% formalin followed by H&E staining to count cell numbers (4 wells/time-point). (C) Cultured bMPCs at 60–70% confluent were collected and incubated with DAPI (1μg/ml). The cell cycle was analyzed by FACS, and the data presented were from 3 pair of mice. (D) bMPCs from RelB−/− and WT littermates were cultured in OB differentiation medium for 5d and stained for ALP activity followed by eosin counterstaining. ALP+ and total cell numbers were counted and the ALP+/total cell ratio was calculated. (E) 1×104 bMPCs were cultured in 12-well plates for 5 days to sub-confluence and OB differentiation medium was added for 21 days when nodules had formed. Von Kossa staining was performed to quantify nodule area. (F) bMPCs cultured in 60 mm dish were induced for OB differentiation for the indicated times. Expression levels of ALP and osteocalcin (OCal) were tested using Real-time PCR. All mice in in vitro experiments were 1.5–2-months-old males or females; 3–4 wells per group. * p<0.05; ** p<0.01 vs. control.
Runx2 and Osx are two critical transcription factors for OB differentiation and maturation. Osx is a downstream target of Runx2 and regulates expansion of an early osteoblastic pool derived from MPCs (31,32). Runx2 is required for commitment of mesenchymal osteochondroprogenitors to the osteoblastic lineage and OB differentiation during both endochondral and intramembranous ossification (33). We found that differentiated RelB−/− stromal cells have 3-fold higher Runx2 (Fig. 5A left panel) and 4-fold higher Osx (data not shown) mRNA expression levels than WT cells. Although the endogenous Runx2 protein level is low in osteoblastic cells (23), we observed an increased Runx2 protein in the differentiating RelB−/− stromal cells compared to the WT cells tested by Western blot (Fig. 5A right panel). Sequence analysis with TFSEARCH software showed that the mouse Runx2 promoter contains two putative NF-κB binding sites, at −653/−644 and −1262/−1253 (Fig. 5B). We constructed a 2-kb mouse Runx2 promoter Luc reporter, which contains 2 putative NF-κB binding sites (Fig. 5B). We then co-transfected the Runx2 Luc reporter with RelB plasmids into C2C12 pre-osteoblastic cells, and found that RelB dose-dependently inhibited Runx2 Luc activity (Fig. 5C). We performed the site-directed mutagenesis of both κB binding sites in the promoter by deleting “ATC” and “ACA” in binding sites 1 and 2, which was confirmed by sequencing. Then, using the mutated Runx2 reporter we found that RelB did not affect the Luc activity of the mutated Runx2 promoter reporter (Fig. 5D), confirming that RelB does indeed regulate Runx2 activity via κ B binding sites.
To further test if RelB directly binds to the putative κB binding sites of the Runx2 promoter, we performed ChIP assays using WT and RelB−/− MPCs. As shown in Fig. 5E, immunoprecipitation of both binding sites with anti-RelB antibody followed by qPCR with specific primers for each of the putative binding sites yielded distinct enrichment in WT MPCs over the input chromatin compared to that of an IgG negative control antibody (Fig. 5E). No chromatin enrichment by the RelB antibody was observed in RelB−/− MPCs, indicating specific binding of RelB protein to the Runx2 promoter. Importantly, a Runx2 shRNA abolished the enhanced OB differentiation by RelB−/− MPCs (Fig 5F).
RelB−/− MPCs induce bone formation and repair in tibial bone defects more rapidly than WT MPCs
To test if RelB−/− MPCs have enhanced bone forming potential in vivo, we made 5×2mm cortical defects in the anterior tibiae of SCID mice. We filled the defects with decalcified bovine bone matrix (DBM), which is used commonly to fill bone defects and stimulate bone formation. bMPCs from RelB−/− and WT littermates were then injected into the DBM in the left and right tibiae, respectively. We found that the defects injected with RelB−/− bMPCs had significantly more new bone formation than those injected with WT cells, as assessed by micro-CT and histologic analysis, 4 weeks post-transplantation (Fig. 6A&B). The volume of new bone formed from transplanted WT MPCs almost matched that formed by the RelB−/− cells at 8 weeks post-surgery. However, at 8 weeks post-surgery almost all the area around the new bone and surviving fragments of bone matrix implanted before the injection of RelB−/− MPCs had become filled with hematopoietic marrow, while this space in the defects injected with WT MPCs was still largely filled with immature fibroblastic tissue and unresorbed DBM (Fig. 6B), indicating that the transplanted RelB−/− MPCs induced accelerated bone repair and resorption of the DBM. In addition, by 8 weeks post-surgery, new bone containing live osteocytes and partly covered with a periosteum-like membrane extended along ~70% of the surface of the DBM particles injected with RelB−/− MPCs. In contrast, only ~20% of the surface of DBM injected with WT MPCs was covered by viable new bone and the remainder of the surface was covered by fibrotic tissue, indicating a marked delay in the healing process (Fig. 6C).
Fig. 6. RelB−/− MPCs induce bone formation and repair in tibial bone defects more rapidly than WT MPCs.

Bilateral 2×5mm cortical defects were made in the anterior proximal tibiae of SCID mice and filled with decalcified bovine bone matrix. 5×105 bMPCs from 7-wk-old RelB−/− and WT mice were injected into the bone matrix in the left and right tibial defects, respectively. The mice were sacrificed 4 and 8 weeks post-surgery and the volume of new bone (BV/TV, expressed as a percentage of the total defect volume) formed in the defects was measured by μCT (A) followed by histomorphometric analysis of the area of newly formed trabecular bone (Trab) and fibrosis tissue (Fib, black arrows) containing spindle-shaped fibroblast-like cells observed in decalcified H&E-stained sections of the bones (B). (C) Representative images of bone in the defect sites 8 weeks post-surgery, and the extent of new bone (with viable osteocytes and covered with a periosteum-like membrane, green arrow) formed on the DBM was quantified and expressed as a % of the total length of implanted DBM. The black arrow shows fibrous tissue covering the acellular DBM. n=5/group. * p<0.05 vs. control.
DISCUSSION
Here, we report that RelB−/− mice have increased trabecular bone volume in the diaphyses of their long bones and in their vertebral bodies as they age. The mice have relatively normal OC numbers and functions in vivo, but impaired RANKL-induced OC formation in vitro, similar to findings reported in RelB−/− mice by other investigators (8). Importantly, the metaphyseal bone in the −/− mice does not have the typical histologic features of osteopetrosis, such as increased trabecular bone volume with un-resorbed islands of cartilage inside trabeculae (26,27). In contrast, young RelB−/− mice have significantly increased OB surfaces and BFRs in tibial and vertebral bone sections, consistent with an enhanced OB phenotype being responsible for the increased bone mass observed when the mice are older. The RelB−/− mice have normal numbers of bone marrow MPCs, indicating that RelB does not have a role in the maintenance of these progenitor cells or in their differentiation from mesenchymal stem cells, as it does along with p52 in the maintenance of hematopoietic stem cells (34). However, RelB−/− MPCs derived from either BM or bone have enhanced commitment of pre-OBs and their subsequent proliferation, OB differentiation and mineralization potential in vitro accompanied by increased expression of Runx2, suggesting that RelB negatively regulates their differentiation in part at least by limiting expression of this essential osteoblastogenic transcription factor. These findings suggest that RelB plays an important role to limit bone formation in the diaphysis, which could potentially reduce the strength of bone near the distal ends of long bones as mice and other mammals age and increase the risk of fracture through them.
The role of Runx2 in the regulation of bone formation is complex. For example, it is required for commitment of progenitors to the OB lineage and for OB differentiation, but it also inhibits proliferation of MPCs and of mature OBs (33, 35, 36); thus its presence reflects induction of differentiation of osteoblastic cells. We found that MPCs and neonatal calvarial cells from RelB−/− mice have increased OB differentiation capacity and Runx2 mRNA expression levels (Fig. 3F & Fig. 5A) and that RelB inhibits Runx2 expression in osteoblastic cells by directly binding to κB binding sites in the Runx2 promoter. These findings are consistent with RelB inhibition of Runx2 expression playing an important contributory role to insure that the expansion of MPCs in WT mice is appropriate prior to induction of osteoblast differentiated activities. In this way, RelB could limit trabecular bone formation in mice as they age. However, there are other possible mechanisms, including RelB regulation of expression of other genes that control proliferation, including cyclin D1, p53 etc, and these will require further study.
The increase in bone formation rate and diaphyseal OB numbers in the RelB−/− mice is transient, indicating that the role of RelB in the regulation of OB differentiation and function is complex. This could reflect the fact that diaphyseal bone volume is not comparable between RelB−/− and WT mice as they age since very little trabecular bone remains in this region in WT mice. However, another possibility is that because RelB−/− mice develop multi-organ inflammation (21) and increased production of cytokines, such as TNF (37), these could inhibit bone formation as the mice age. Importantly, BM cells from 3-month-old RelB−/− mice (which have reduced bone formation in vivo) have very significantly enhanced capacity to differentiate into ALP+ cells in vitro, suggesting that the reduced bone formation in older RelB−/− mice results from the effects of secondary factors. Further studies using mice with tissue-specific deletion of RelB will be necessary to address this issue definitively.
Age-related bone loss is a major health problem associated with increased risk of fracture and morbidity and it will become more prevalent as the aging population increases. Thus, it is important to better understand the mechanisms that lead to loss of trabecular and cortical bone with aging so that novel therapeutic strategies can be developed to prevent it. Age-related osteoporosis is associated with so-called low-grade molecular inflammation induced by increased production of cytokines, including TNF, by immune and other cells as mice and humans age (38–40). RelB could also play a role in this cytokine-associated bone loss with aging since it negatively regulates TNF expression (37). Increased TNF production can lead to bone loss by a variety of mechanism, including increased OC formation and activation, both directly (6) and indirectly (41), and inhibition of OB differentiation and function (9–11). Thus, increasing levels of cytokines, such as TNF, in blood and BM of RelB−/− mice could secondarily mediate inhibition of bone formation in the mice as they age and account for the decreased bone formation rate we observed in older mice. The enhanced differentiation of RelB−/− MPCs occurred in vitro in the absence of added TNF, and importantly, RelB−/− MPCs injected into tibial cortical bone defects experienced the same cytokine micro-environment as WT MPCs in recipient SCID mice, but they induced more bone formation in the defects, suggesting they have intrinsically enhanced OB differentiation.
Transplanted DBM, similar to allograft bone, is osteoinductive (42), and likely undergoes creeping substitution with new bone forming on it as bone defects are repaired. Once formed, new bone and some of the DBM to which it attaches is continually remodeled, but DBM typically is not fully resorbed even at 42 weeks after surgery (43). Thus, although it is hard to predict when transplanted DBM will be resorbed completely to allow restoration of normal cortical structure in tibial cortical defects, the presence of new bone forming on and around DBM would be indicative of healing. Our observation that by 8 weeks after transplantation, 70% of the surface of DBM in defects injected with RelB−/− MPCs was covered by newly-formed viable bone compared with only ~20 % of the surface of DBM injected with WT MPCs further supports an important negative regulatory role for RelB in bone defect repair. In addition to directly differentiating into bone tissue, donor MPCs injected into bone defects also produce cytokines and chemokines, which can recruit host MPCs, promote angiogenesis and cellular migration, and inhibit apoptosis (44–46) to enhance tissue repair. Further studies will be required to determine how much of the new bone formed in the tibial bone defects in the SCID mice was derived directly from RelB−/− MSCs and if and how these injected MPCs recruited host MPCs or promoted angiogenesis and callus remodeling to enhance bone repair.
Interestingly, cortical thickness as well as the periosteal and endosteal surface area did not increase in the RelB−/− mice as they aged. This may explain why vertebral bodies from 10–14-month-old mice did not have increased biomechanical properties when we tested them ex vivo (data not shown). We do not have an explanation for this differential role for RelB in trabecular versus cortical bone, but we did not observe an increase in BFRs on the endosteal bone surfaces of the RelB−/− mice (data not shown) as we did on their trabecular surfaces. That OBs on trabecular and cortical bone surfaces may be regulated differentially is supported by a recent study reporting the in vivo effects of a peptide that inhibits OC formation by binding to RANKL (47). It increased cortical, but not trabecular bone formation and mass in mice by activating Smad1/5/8 and working synergistically with BMP-2 signaling in osteoblasts to enhance OB differentiation and matrix mineralization (47). Another study reported that TRAF family-member-associated NF-κB activator (TANK)-deficient mice have increased cortical bone mineral density and thickness, but trabecular osteopenia (48), providing further support that signaling in trabecular and cortical OBs can be regulated differentially.
In summary, our findings indicate that RelB inhibits OB differentiation and bone formation directly, associated with reduced Runx2 activation, and contributes to age-related bone loss in mice. The enhanced bone formation in RelB−/− mice and from RelB−/− MPCs transplanted into SCID mice suggests that strategies to reduce or inhibit RelB functions in MPCs should increase bone mass and enhance repair of bone defects and possibly also of fractures.
Supplementary Material
Supplemental Fig. 1. Total RNA was extracted from femora of 2 RelB−/− and 2 WT littermate (1 and 2, representing each mouse) 9-week-old mice using Trizol reagents. mRNA expression levels of ALP, osteocalcin (OCal), Runx2, RANKL, OPG were tested by real-time PCR and the RANKL/OPG expression ratio was calculated.
Acknowledgments
We thank Michael Thullen for help with μCT scanning and analysis. Research reported in this publication was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health under Award Number AR43510 to B.F. Boyce, AR48697 to L. Xing, P30AR061307 to E. Schwarz (and a Pilot grant to Z. Yao from P30AR061307) and grant number 1S10RR027340 to B.F. Boyce from the NIH. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
The authors have declared that no conflict of interest exists.
Authors’ roles: Study design: ZY, BFB. Study conduct and data collection: ZY, YL, XY, YD. Data analysis and interpretation: ZY, LX and BFB. Manuscript preparation: ZY, LX and BFB. All authors approved final version of manuscript: ZY and BFB take responsibility for the integrity of the data analysis.
References
- 1.Mundy GR. Osteoporosis and inflammation. Nutr Rev. 2007;65(12 Pt 2):S147–51. doi: 10.1111/j.1753-4887.2007.tb00353.x. [DOI] [PubMed] [Google Scholar]
- 2.Vallabhapurapu S, Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol. 2009;27:693–733. doi: 10.1146/annurev.immunol.021908.132641. [DOI] [PubMed] [Google Scholar]
- 3.Franzoso G, Carlson L, Xing L, et al. Requirement for NF-kappaB in osteoclast and B-cell development. Genes Dev. 1997;11(24):3482–96. doi: 10.1101/gad.11.24.3482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Iotsova V, Caamaño J, Loy J, Yang Y, Lewin A, Bravo R. Osteopetrosis in mice lacking NF-kappaB1 and NF-kappaB2. Nat Med. 1997;3(11):1285–9. doi: 10.1038/nm1197-1285. [DOI] [PubMed] [Google Scholar]
- 5.Yamashita T, Yao Z, Li F, et al. NF-kappaB p50 and p52 regulate receptor activator of NF-kappaB ligand (RANKL) and tumor necrosis factor-induced osteoclast precursor differentiation by activating c-Fos and NFATc1. J Biol Chem. 2007;282(25):18245–53. doi: 10.1074/jbc.M610701200. [DOI] [PubMed] [Google Scholar]
- 6.Yao Z, Xing L, Boyce BF. NF-kappaB p100 limits TNF-induced bone resorption in mice by a TRAF3-dependent mechanism. J Clin Invest. 2009;119(10):3024–34. doi: 10.1172/JCI38716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vaira S, Alhawagri M, Anwisye I, et al. RelA/p65 promotes osteoclast differentiation by blocking a RANKL-induced apoptotic JNK pathway in mice. J Clin Invest. 2008;118(6):2088–97. doi: 10.1172/JCI33392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vaira S, Johnson T, Hirbe AC, et al. RelB is the NF-kappaB subunit downstream of NIK responsible for osteoclast differentiation. Proc Natl Acad Sci U S A. 2008;105(10):3897–902. doi: 10.1073/pnas.0708576105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li Y, Li A, Strait K, et al. Endogenous TNF alpha lowers maximum peak bone mass and inhibits osteoblastic Smad activation through NF-kappaB. J Bone Miner Res. 2007;22 (5):646–655. doi: 10.1359/jbmr.070121. [DOI] [PubMed] [Google Scholar]
- 10.Gilbert LC, Rubin J, Nanes MS. The p55 TNF receptor mediates TNF inhibition of osteoblast differentiation independently of apoptosis. Am J Physiol Endocrinol Metab. 2005;288(5):E1011–8. doi: 10.1152/ajpendo.00534.2004. [DOI] [PubMed] [Google Scholar]
- 11.Yamazaki M, Fukushima H, Shin M, et al. Tumor necrosis factor alpha represses bone morphogenetic protein (BMP) signaling by interfering with the DNA binding of Smads through the activation of NF-kappaB. J Biol Chem. 2009;284(51):35987–95. doi: 10.1074/jbc.M109.070540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gilbert L, He X, Farmer P, et al. Expression of the osteoblast differentiation factor RUNX2 (Cbfa1/AML3/Pebp2alpha A) is inhibited by tumor necrosis factor-alpha. J Biol Chem. 2002;277(4):2695–701. doi: 10.1074/jbc.M106339200. [DOI] [PubMed] [Google Scholar]
- 13.Chang J, Wang Z, Tang E, et al. Inhibition of osteoblastic bone formation by nuclear factor-kappaB. Nat Med. 2009;15(6):682–9. doi: 10.1038/nm.1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alles N, Soysa NS, Hayashi J, Khan M, Shimoda A, Shimokawa H, Ritzeler O, Akiyoshi K, Aoki K, Ohya K. Suppression of NF-κB increases bone formation and ameliorates osteopenia in ovariectomized mice. Endocrinology. 2010;151(10):4626–34. doi: 10.1210/en.2010-0399. [DOI] [PubMed] [Google Scholar]
- 15.Cho HH, Shin KK, Kim YJ, et al. NF-kappaB activation stimulates osteogenic differentiation of mesenchymal stem cells derived from human adipose tissue by increasing TAZ expression. J Cell Physiol. 2010;223(1):168–77. doi: 10.1002/jcp.22024. [DOI] [PubMed] [Google Scholar]
- 16.Hess K, Ushmorov A, Fiedler J, et al. TNF alpha promotes osteogenic differentiation of human mesenchymal stem cells by triggering the NF-kappaB signaling pathway. Bone. 2009;45(2):367–76. doi: 10.1016/j.bone.2009.04.252. [DOI] [PubMed] [Google Scholar]
- 17.Lencel P, Delplace S, Hardouin P, Magne D. TNF-α stimulates alkaline phosphatase and mineralization through PPARγ inhibition in human osteoblasts. Bone. 2011;48(2):242–9. doi: 10.1016/j.bone.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 18.Seo Y, Fukushima H, Maruyama T, et al. Accumulation of p100, a precursor of NF-κB2, enhances osteoblastic differentiation in vitro and bone formation in vivo in aly/aly mice. Mol Endocrinol. 2012;26(3):414–22. doi: 10.1210/me.2011-1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Soysa NS, Alles N, Weih D, et al. The pivotal role of the alternative NF-kappaB pathway in maintenance of basal bone homeostasis and osteoclastogenesis. J Bone Miner Res. 2010;25(4):809–18. doi: 10.1359/jbmr.091030. [DOI] [PubMed] [Google Scholar]
- 20.Elewaut D, Shaikh RB, Hammond KJ, et al. NIK-dependent RelB activation defines a unique signaling pathway for the development of V alpha 14i NKT cells. J Exp Med. 2003;197(12):1623–33. doi: 10.1084/jem.20030141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Burkly L, Hession C, Ogata L, et al. Expression of relB is required for the development of thymic medulla and dendritic cells. Nature. 1995;373(6514):531–6. doi: 10.1038/373531a0. [DOI] [PubMed] [Google Scholar]
- 22.Zhu H, Guo ZK, Jiang XX, et al. A protocol for isolation and culture of mesenchymal stem cells from mouse compact bone. Nat Protoc. 2010;5(3):550–60. doi: 10.1038/nprot.2009.238. [DOI] [PubMed] [Google Scholar]
- 23.Kaneki H, Guo R, Chen D, et al. Tumor necrosis factor promotes Runx2 degradation through up-regulation of Smurf1 and Smurf2 in osteoblasts. J Biol Chem. 2006;281(7):4326–33. doi: 10.1074/jbc.M509430200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yao Z, Li P, Zhang Q, et al. Tumor necrosis factor-alpha increases circulating osteoclast precursor numbers by promoting their proliferation and differentiation in the bone marrow through up-regulation of c-Fms expression. J Biol Chem. 2006;281 (17):11846–55. doi: 10.1074/jbc.M512624200. [DOI] [PubMed] [Google Scholar]
- 25.Bouxsein ML, Boyd SK, Christiansen BA, et al. Guidelines for assessment of bone microstructure in rodents using micro–computed tomography. J Bone Miner Res. 2010;25 (7):1468–86. doi: 10.1002/jbmr.141. [DOI] [PubMed] [Google Scholar]
- 26.Dougall WC, Glaccum M, Charrier K, et al. RANK is essential for osteoclast and lymph node development. Genes Dev. 1999;13(18):2412–24. doi: 10.1101/gad.13.18.2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lowe C, Yoneda T, Boyce BF, Chen H, Mundy GR, Soriano P. Osteopetrosis in Src-deficient mice is due to an autonomous defect of osteoclasts. Proc Natl Acad Sci U S A. 1993;90(10):4485–9. doi: 10.1073/pnas.90.10.4485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mukherjee S, Raje N, Schoonmaker JA, et al. Pharmacologic targeting of a stem/progenitor population in vivo is associated with enhanced bone regeneration in mice. J Clin Invest. 2008;118(2):491–504. doi: 10.1172/JCI33102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao L, Huang J, Zhang H, et al. Tumor necrosis factor inhibits mesenchymal stem cel differentiation into osteoblasts via the ubiquitin E3 ligase Wwp1. Stem Cells. 2011;29(10):1601–10. doi: 10.1002/stem.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chang MK, Raggatt LJ, Alexander KA, et al. Osteal tissue macrophages are intercalated throughout human and mouse bone lining tissues and regulate osteoblast function in vitro and in vivo. J Immunol. 2008;181(2):1232–44. doi: 10.4049/jimmunol.181.2.1232. [DOI] [PubMed] [Google Scholar]
- 31.Nishio Y, Dong Y, Paris M, et al. Runx2-mediated regulation of the zinc finger Osterix/Sp7 gene. Gene. 2006;372:62–70. doi: 10.1016/j.gene.2005.12.022. [DOI] [PubMed] [Google Scholar]
- 32.Nakashima K, et al. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell. 2002;108(1):17–29. doi: 10.1016/s0092-8674(01)00622-5. [DOI] [PubMed] [Google Scholar]
- 33.Komori T. Signaling networks in RUNX2-dependent bone development. J Cell Biochem. 2011;112(3):750–5. doi: 10.1002/jcb.22994. [DOI] [PubMed] [Google Scholar]
- 34.Zhao C, Xiu Y, Ashton J, Xing L, Morita Y, Jordan CT, Boyce BF. Noncanonical NF-κB signaling regulates hematopoietic stem cell self-renewal and microenvironment interactions. Stem Cells. 2012;30(4):709–18. doi: 10.1002/stem.1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pratap J, Galindo M, Zaidi SK, et al. Cell growth regulatory role of Runx2 during proliferative expansion of preosteoblasts. Cancer Res. 2003;63(17):5357–62. [PubMed] [Google Scholar]
- 36.Zaidi SK, Pande S, Pratap J, et al. Runx2 deficiency and defective subnuclear targeting bypass senescence to promote immortalization and tumorigenic potential. Proc Natl Acad Sci U S A. 2007;104(50):19861–6. doi: 10.1073/pnas.0709650104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kiebala M, Polesskaya O, Yao Z, et al. Nuclear factor-kappa B family member RelB inhibits human immunodeficiency virus-1 Tat-induced tumor necrosis factor-alpha production. PLoS One. 2010;5(7):e11875. doi: 10.1371/journal.pone.0011875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kitaura H, Zhou P, Kim HJ, Novack DV, Ross FP, Teitelbaum SL. M-CSF mediates TNF-induced inflammatory osteolysis. J Clin Invest. 2005;115(12):3418–27. doi: 10.1172/JCI26132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Clowes JA, Riggs BL, Khosla S. The role of the immune system in the pathophysiology of osteoporosis. Immunol Rev. 2005;208:207–27. doi: 10.1111/j.0105-2896.2005.00334.x. [DOI] [PubMed] [Google Scholar]
- 40.Cao JJ, Wronski TJ, Iwaniec U, et al. Aging increases stromal/osteoblastic cell-induced osteoclastogenesis and alters the osteoclast precursor pool in the mouse. J Bone Miner Res. 2005;20(9):1659–68. doi: 10.1359/JBMR.050503. [DOI] [PubMed] [Google Scholar]
- 41.Lam J, Takeshita S, Barker JE, et al. TNF-alpha induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. J Clin Invest. 2000;106(12):1481–8. doi: 10.1172/JCI11176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hosny M, Sharawy M. Osteoinduction in rhesus monkeys using demineralized bone powder allografts. J Oral Maxillofac Surg. 1985;43(11):837–44. doi: 10.1016/0278-2391(85)90219-8. [DOI] [PubMed] [Google Scholar]
- 43.Tuli SM, Singh AD. The osteoninductive property of decalcified bone matrix. J Bone Joint Surg Br. 1978;60(1):116–23. doi: 10.1302/0301-620X.60B1.342532. [DOI] [PubMed] [Google Scholar]
- 44.Boomsma RA, Geenen DL. Mesenchymal stem cells secrete multiple cytokines that promote angiogenesis and have contrasting effects on chemotaxis and apoptosis. PLoS One. 2012;7(4):e35685. doi: 10.1371/journal.pone.0035685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schinköthe T, Bloch W, Schmidt A. In vitro secreting profile of human mesenchymal stem cells. Stem Cells Dev. 2008;17(1):199–206. doi: 10.1089/scd.2007.0175. [DOI] [PubMed] [Google Scholar]
- 46.Oh JY, Kim MK, Shin MS, Wee WR, Lee JH. Cytokine secretion by human mesenchymal stem cells cocultured with damaged corneal epithelial cells. Cytokine. 2009;46(1):100–3. doi: 10.1016/j.cyto.2008.12.011. [DOI] [PubMed] [Google Scholar]
- 47.Furuya Y, Inagaki A, Khan M, et al. Stimulation of bone formation in cortical bone of mice treated with a receptor activator of nuclear factor-κB ligand (RANKL)-binding peptide that possesses osteoclastogenesis inhibitory activity. J Biol Chem. 2013;288(8):5562–71. doi: 10.1074/jbc.M112.426080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maruyama K, Kawagoe T, Kondo T, Akira S, Takeuchi O. TRAF family member-associated NF-κB activator (TANK) is a negative regulator of osteoclastogenesis and bone formation. J Biol Chem. 2012;287(34):29114–24. doi: 10.1074/jbc.M112.347799. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Fig. 1. Total RNA was extracted from femora of 2 RelB−/− and 2 WT littermate (1 and 2, representing each mouse) 9-week-old mice using Trizol reagents. mRNA expression levels of ALP, osteocalcin (OCal), Runx2, RANKL, OPG were tested by real-time PCR and the RANKL/OPG expression ratio was calculated.