

Background: Cellular and γ-herpesvirus Bcl-2 homologs down-regulate autophagy.
Results: A peptide designed to bind to the γ-herpesvirus68 Bcl-2, M11, but not cellular Bcl-2 homologs, abrogates M11-mediated down-regulation of autophagy.
Conclusion: This peptide is a selective M11 inhibitor.
Significance: Such selective inhibitors are important for understanding the role of γ-herpesvirus Bcl-2 homologs in viral reactivation and oncogenic transformation of host cells.
Keywords: Autophagy, Bcl-2 Family Proteins, Crystal Structure, Herpesvirus, Host Defense, Host-Pathogen Interactions, Protein-Protein Interactions
Abstract
γ-herpesviruses (γHVs) are common human pathogens that encode homologs of the anti-apoptotic cellular Bcl-2 proteins, which are critical to viral reactivation and oncogenic transformation. The murine γHV68 provides a tractable in vivo model for understanding general features of these important human pathogens. Bcl-XL, a cellular Bcl-2 homolog, and the murine γHV68 Bcl-2 homolog, M11, both bind to a BH3 domain within the key autophagy effector Beclin 1 with comparable affinities, resulting in the down-regulation of Beclin 1-mediated autophagy. Despite this similarity, differences in residues lining the binding site of M11 and Bcl-XL dictate varying affinities for the different BH3 domain-containing proteins. Here we delineate Beclin 1 differential specificity determinants for binding to M11 or Bcl-XL by quantifying autophagy levels in cells expressing different Beclin 1 mutants and either M11 or Bcl-XL, and we show that a G120E/D121A Beclin 1 mutant selectively prevents down-regulation of Beclin 1-mediated autophagy by Bcl-XL, but not by M11. We use isothermal titration calorimetry to identify a Beclin 1 BH3 domain-derived peptide that selectively binds to M11, but not to Bcl-XL. The x-ray crystal structure of this peptide bound to M11 reveals the mechanism by which the M11 BH3 domain-binding groove accommodates this M11-specific peptide. This information was used to develop a cell-permeable peptide inhibitor that selectively inhibits M11-mediated, but not Bcl-XL-mediated, down-regulation of autophagy.
Introduction
γ-Herpesviruses (γHVs)2 are common human pathogens that infect ∼95% of all adults. Epstein-Barr virus, first isolated from Burkitt's lymphoma, has been detected in several malignant tumors originating in both lymphoid and epithelial tissues (1). Epstein-Barr virus is also the causative agent for infectious mononucleosis and may be responsible for chronic fatigue syndrome. Kaposi's sarcoma-associated herpesvirus (KSHV) is associated with Kaposi sarcoma tumors, which show a high incidence among immunocompromised individuals, such as patients with HIV infection and transplant recipients. Another mammalian γHV, murine γHV68 does not infect humans but provides a tractable model for studying γHV infections in vivo. A murine model has been developed to study the mechanisms and pathogenesis of γHV induction of lympho-proliferative disease (2). All γHVs encode homologs of the anti-apoptotic, cellular Bcl-2 proteins (3, 4), suggesting that these proteins play an important role in the pathogenesis of these viruses.
Bcl-2 was the first cellular protein shown to function as an oncogene by blocking apoptotic cell death rather than by increasing cellular proliferation (5, 6). Bcl-2 family members have now been shown to be multifunctional proteins influencing diverse cellular processes such as autophagy, cell cycle progression, calcineurin signaling, glucose homeostasis, and transcription regulation (7, 8). Members of the Bcl-2 family are identified by the presence of different Bcl-2 homology (BH) domains. This family includes several, pro-apoptotic, BH3-only proteins, such as BIM and BAD; pro-apoptotic homologs with three BH domains (BH3, BH1, and BH2), such as BAX and BAK; and anti-apoptotic homologs with four BH domains (BH4, BH3, BH1, and BH2), such as Bcl-2 and Bcl-XL. Anti-apoptotic Bcl-2 homologs down-regulate apoptosis by binding to the BH3 domain of pro-apoptotic proteins to inhibit their pro-apoptotic function.
The anti-apoptotic γHV Bcl-2 homologs appear to be critical for viral reactivation from latency and replication in immunocompromised hosts (3, 9, 10). Thus, they play important roles in latent and chronic infection. One mechanism by which γHV Bcl-2 homologs may accomplish these physiological functions is by the down-regulation of apoptosis. KSHV Bcl-2 blocks apoptosis stimulated by overexpression of Bax or v-cyclin or by Sindbis virus infection (11, 12); however, in cellular assays, it does not appear to heterodimerize with pro-apoptotic Bcl-2 family members, such as Bax and Bak (13). Epstein-Barr virus encodes a Bcl-2 homolog, BHRF1, which is expressed as an early lytic cycle protein, has anti-apoptotic activity, heterodimerizes with Bax and Bak, and also disrupts the differentiation of epithelial cells (14–17). The γHV68 Bcl-2 homolog, M11, has been shown to down-regulate apoptosis induced by Fas, TNFα, and Sindbis virus infection (18, 19). More recently it has been shown that KSHV Bcl-2 and γHV68 M11 also down-regulate autophagy in cell culture by binding to an essential autophagy effector, Beclin 1 (20–22). M11 is the only γHV Bcl-2 that has been demonstrated to play a role during infection in vivo (3, 19). Thus, the γHV and cellular Bcl-2 homologs are dual regulators of autophagy and apoptosis and serve as a node of cross-talk between these pathways (23–27).
Although human cellular Bcl-2 paralogs share less than 50% sequence identity, known human and murine Bcl-2 ortholog pairs share >85% sequence identity. All anti-apoptotic Bcl-2 homologs have similar three-dimensional structures, consisting of a central hydrophobic α-helix surrounded by six or seven amphipathic helices (28). Previous structural and mutagenic analyses demonstrated that the amphipathic, α-helical BH3 domains of pro-apoptotic proteins bind to a hydrophobic surface groove on Bcl-2 homologs (19, 29–33), with the hydrophobic face of the helix buried in a hydrophobic groove on the surface of the Bcl-2 homolog. Different Bcl-2 homologs have widely varying affinities for BH3 domains from different pro-apoptotic proteins (22, 32–35). The molecular determinants that enforce these varying specificities are not well understood.
It has now been shown that the pro-autophagic effector Beclin 1 also contains a BH3 domain that binds to a hydrophobic surface groove of cellular and γHV Bcl-2 homologs (21, 22, 36–38). Given the differential affinity of Bcl-2 homologs, it is not surprising that although Bcl-2 and Bcl-XL bind to Beclin 1, other cellular Bcl-2 paralogs, Mcl-1, A1, and Bcl-W (20, 39, 40), bind only weakly or not at all. The Beclin 1 BH3 domain is the primary determinant of binding to cellular and γHV Bcl-2 homologs (21, 22, 36–38, 41), binding to different Bcl-2 homologs with affinities in the micromolar range, for example with a Kd of ∼54 μm to KSHV Bcl-2 (22) and ∼9 μm to Bcl-2 (42).
The hypothesis behind this study was that despite the similar general mode of binding of the Beclin 1 BH3 domain to viral M11 and cellular Bcl-XL, the BH3 domain binding grooves of these two homologs are lined by different residues, which results in different atomic details of interaction and different thermodynamic contributions from the interactions of each residue, that would lead to differential affinities for mutant BH3 domain-derived peptides. Further, based on the promiscuity of M11 relative to Bcl-XL for different BH3 domains that has been previously reported (19, 22, 32–35), we expected that a systematic mutagenesis approach would enable us to find a peptide that binds to M11 but does not bind to the cellular Bcl-2 homologs, which have a more stringent binding specificity (22).
Therefore, we used cellular assays to identify Beclin 1 mutations that selectively abrogate down-regulation of autophagy by Bcl-XL, but not M11, then used isothermal titration calorimetry (ITC) to identify a peptide that binds selectively to M11, but not to Bcl-XL. Further, we determined the x-ray crystal structure of this selective peptide bound to M11 to elucidate the mechanism by which it binds to M11. Lastly, we demonstrate that a cell-permeable version of this selective peptide serves as an M11-specific inhibitor that abrogates M11-mediated down-regulation of autophagy in cells. These combined results help explain the atomic bases of the differential specificity of M11 and Bcl-XL and provide a unique tool to target M11-BH3 domain interactions in vivo. This study reports the rational design of an inhibitor that selectively targets M11, which will be valuable in studying its interactions and roles in cell culture and in vivo.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification
cDNA sequences corresponding to the γHV68 M11 and Bcl-XL genes lacking the C-terminal transmembrane helix were cloned and expressed to enable purification of soluble constructs similar to those used for previous structural studies (22, 36). γHV68 M11 residues 1–136 were expressed and purified as previously described (22). The double mutant variant of Bcl-XL (N52D/N66D) was created by two rounds of site-directed mutagenesis using the QuikChange II site-directed mutagenesis kit (Agilent Technologies) and then cloned, along with a C-terminal His6 tag for purification, into the NdeI and NotI restriction sites of pET 29b. The His6-tagged Bcl-XL (residues 1–208, N52D/N66D) was expressed in Escherichia coli BL21(DE3)pLysS cells, and soluble protein in the cell lysate was purified to homogeneity by immobilized metal affinity chromatography using two tandem 5-ml His-Trap HP columns (GE Healthcare) followed by ion exchange chromatography using a Mono Q HR 10/10 column (GE Healthcare) and size exclusion chromatography using a preparative 16/60 Superdex 200 column (GE Healthcare).
Peptide Synthesis
Various Beclin 1 BH3 domain-derived peptides were chemically synthesized and HPLC-purified to >95% purity, with peptide purity confirmed by electrospray mass spectrometry (RSSynthesis/Protein Chemistry Technology Core at the University of Texas Southwestern Medical Center, Dallas, TX).
Isothermal Titration Calorimetry
ITC was performed using a Low Volume Nano ITC (TA Instruments). For all ITC experiments, samples were loaded into separate dialysis cassettes, and co-dialyzed into ITC buffer. The ITC buffer for all experiments comprised of 25 mm HEPES, pH 7.5, 100 mm NaCl, and 2 mm β-mercaptoethanol. ITC was performed at 25 °C with 25 injections of 2 μl each. The data were analyzed using NanoAnalyze Software (TA Instruments) with an independent model. The poor solubility of the D121A peptide in aqueous buffers necessitated a different solubilization and data analysis protocol for ITC. The D121A peptide was mixed into ITC buffer to a concentration of 1 mm and rocked at room temperature overnight. Despite this, a significant fraction of the peptide remained insoluble. This insoluble fraction was pelleted by centrifugation at 13,000 × g for 10 min. The supernatant was collected and co-dialyzed with M11 or Bcl-XL as described above and then used for ITC. Stoichiometry was forced to 1 during data analysis, and the concentration of the D121A peptide was estimated from the fit.
Crystallization
The M11-DS peptide complex was crystallized at 20 °C by hanging drop vapor diffusion from a 1:1 mixture of protein stock (5 mg/ml complex in 20 mm HEPES, pH 7.5, 100 mm NaCl, 1 mm TCEP) and well solution (2.5 m (NH4)2SO4 and 8% v/v 2-propanol). Plate-shaped crystals were harvested and cryoprotected in a cryosolution consisting of 2.5 m (NH4)2SO4 and 25% (v/v) glycerol and then immediately flash-frozen in liquid N2.
Data Collection, Structure Solution, and Refinement
Diffraction intensities from these crystals were recorded at 100 K using 1-s exposures over 0.5° crystal rotation per image, on a 4 × 4 tiled MARmosaic CCD detector (Rayonix) at a crystal to detector distance of 250 mm at Beamline 23ID-D of GMCA@APS (Argonne National Laboratory, Chicago, IL). The data used to solve the structure were collected at an x-ray wavelength of 0.97934 Å in a 360° sweep from a single crystal. The data were processed using HKL2000 (43). The data statistics are summarized in Table 1.
TABLE 1.
Summary of crystallographic data statistics
Values in parentheses pertain to the outermost shell of data.
| Wavelength (Å) | 0.97934 |
| Data range (Å) | 2.1–50.00 (2.1–2.14) |
| Mosaicity | 0.329–0.633 |
| Unique reflections | 24082 |
| Average multiplicity | 3.8 (3.1) |
| Completeness (%) | 99.4 (91.9) |
| Rsym (%)a | 4.9 (44.7) |
| I/σI | 9.2 (2.9) |
a Rsym = Σh,i|Ih,i − <Ih>|/Σh,iIh,i.
Crystals belonged to the space group C21 with unit cell parameters of a = 70.6 Å, b = 140.8 Å, c = 54.0 Å, and β = 127.8°. The crystals contained two copies of the M11-DS peptide complex per asymmetric unit. The positions and orientations of the two M11 (residues 1–136) molecules, monomer A and B, were determined by molecular replacement using HKL3000/MOLREP (44), and a search model extracted from Protein Data Bank code 3DVU, consisting of a single M11 monomer with flexible loop residues 52–73 removed. A helix corresponding to the Beclin 1 BH3 domain (from Protein Data Bank code 3DVU, chain C), with Asp121 mutated to Ala, was manually placed into appropriate density next to monomer A using the program Coot (45) A Glu side chain was built into clear electron density at position 120 after the first cycle of refinement. This defined the structure of the DS peptide. The NCS operator required to superimpose M11 monomer A onto B was used to place a second copy of the DS peptide into appropriate density next to monomer B. The model was refined in the program Refmac5 (46) using imperfect 2-fold NCS restraints (Table 2). The final model is deposited in the Research Collaboratory for Structural Bioinformatics Protein Data Bank with accession code 4MI8. Bcl-2-peptide interactions in different structures were analyzed using PISA (47).
TABLE 2.
Summary of crystallographic refinement statistics
| Model | |
|---|---|
| M11 residues (monomer A) | 135 |
| M11 residues (monomer B) | 136 |
| Beclin 1 DS peptide (chain C) | 20 |
| Beclin 1 DS peptide (chain D) | 22 |
| Water molecules | 133 |
| Sulfate molecules | 4 |
| Data range (Å) | 50–2.1 |
| Rwork (%)a | 16.0 |
| Rfree (%)a | 22.4 |
| Average B-values (Å2) | 34.7 |
| Main chain | 26.7 |
| Side chain | 28.7 |
| Water | 48.0 |
| All atoms | 34.7 |
| B-factor RMSDs between bonded atoms | |
| Main chain | 2.332 |
| Side chain | 4.026 |
| RMSDs from target values | |
| Bond lengths (Å) | 0.020 |
| Bond angles (°) | 1.985 |
| Dihedral angles (°) | 21.33 |
| Improper angles (°) | 1.91 |
| Cross-validated sigma coordinate error (Å) | 0.24 |
| Ramachandran outliers | 0 |
a R factor = Σh, |Fobs − |Fcalc|/Σh|Fobs|. Test set for Rfree consisted of 5.5% of data.
Autophagy Assay
Quantification of fluorescent autophagosomes in MCF7 cells co-transfected with GFP-LC3 (1.6 μg), Beclin 1 (1.2 μg), and either Bcl-XL (1.2 μg) or M11 (1.2 μg) expression plasmids (4 μg of total plasmid) was performed using an inverted Axio Observer (Zeiss). Cells were cultured in DMEM with 10% fetal calf serum (growth medium) in 8-well slides (Millipore) and transfected at 80% confluency with Lipofectamine (Invitrogen). After transfection, cells were either starved overnight in Earle's balanced salt solution (starvation medium) or grown in nutrient-rich media with the addition of 2× essential amino acids and 2× nonessential amino acids. The number of GFP-LC3 puncta per GFP-LC3-positive cell was assessed by counting a minimum of 50 cells via Image ProPlus for duplicate samples per condition in three independent experiments. The significance of alterations in autophagy levels were determined by a two-tailed, heteroscedastic Student's t test, wherein p ≤ 0.05 is considered significant. The effect of a potential inhibitory peptide was investigated by comparing autophagy levels in the absence or presence of 30 μm control or inhibitory peptides.
Western Blot
Expression levels of FLAG-tagged Beclin 1, Bcl-XL, and M11 in MCF7 cells were verified by Western blot analysis using commercial mouse monoclonal anti-FLAG M2-peroxidase antibody (Sigma). As a loading control, the levels of actin in MCF7 cell lysates were detected with mouse anti-actin (Chemicon).
RESULTS
Selection of Beclin 1 Residues Important for Binding to Both M11 and Bcl-XL
Both Beclin 1 and Bcl-XL are highly conserved between human and mice. Human and mouse Beclin 1 orthologs are 99% identical, and their BH3 domains are 100% identical. Similarly, human and murine Bcl-XL orthologs are 97% identical, whereas residues lining the BH3 domain-binding groove are 100% identical (Fig. 1). Therefore, there is no difference in the binding of different BH3 domains, particularly the Beclin 1 BH3 domain, to mouse or human Bcl-XL orthologs.
FIGURE 1.

Sequence alignment of γHV Bcl-2 and Bcl-XL homologs. The top sequence is γHV68 M11, and the bottom sequences are human and mouse Bcl-XL as indicated. The numbers after each alignment block indicate the last residue of each sequence in that block. Green or red backgrounds indicate residues lining the BH3 binding groove in either M11 or Bcl-XL, respectively. Sequence conservation between all three homologs is indicated above the sequence alignment, whereas that between human and mouse Bcl-XL is indicated below the alignment. Asterisks denote invariant residues, double dots indicate highly conserved residues, and single dots indicate similar residues.
In contrast, M11 and Bcl-XL share only 16.2% sequence identity (Fig. 1), although they also have similar functions, three-dimensional structures, and modes of binding. A comparison of complex structures of the Beclin 1 BH3 domain bound to M11 (21, 22) or Bcl-XL (36, 37) demonstrates that each interaction involves the same 12 Beclin 1 residues (Fig. 2) and buries 978 and 1052 Å2 respectively, of surface area from each molecule at the interface, as calculated using PISA (47). Among these 12 Beclin 1 BH3 domain residues, the six that have the most extensive interactions (Fig. 2) are also highly conserved among other BH3 domains (38). Therefore these six residues were selected for mutagenesis. Of these residues, Leu112, Leu116, and Gly120 are completely buried; Phe123 is partially buried; the aliphatic part of the Lys117 side chain hydrophobically packs against M11 residues Asp81 and Arg87, whereas the amino group makes polar and charged interactions; and Gly120 and Asp121 interact with a Gly-Arg pair conserved in most Bcl-2 homologs, including Bcl-XL and M11 (21, 22, 36–38). This Gly-Arg pair, especially the Arg, has been shown to contribute significantly to the interaction of many Bcl-2 homologs with diverse BH3 domains (19, 48). Gly120 is packed against the conserved Bcl-2 Gly-Arg main chain, while the BH3 domain Asp121 makes a bidentate salt bridge with the conserved Bcl-2 Arg. Therefore, Leu112, Leu116, Lys117, Gly120, Asp121, and Phe123 were selected for further investigation in this study.
FIGURE 2.
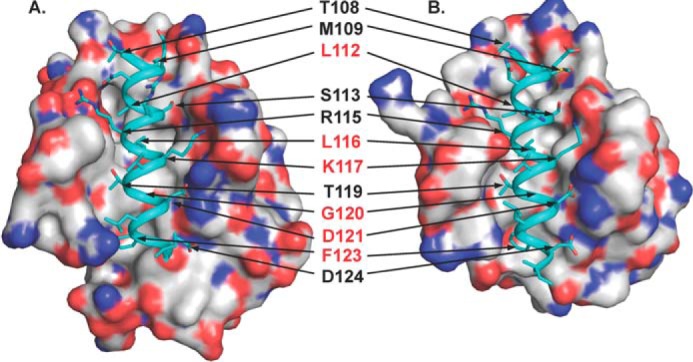
Comparison of the WT Beclin 1 BH3 domain bound to Bcl-XL (Protein Data Bank code 2PIL, A) and M11 (Protein Data Bank code 3DVU, B). Each complex is shown in a superimposable view with Bcl-XL and M11 shown as molecular surface colored by atom type: oxygen, red; nitrogen, blue; sulfur, yellow; and carbon, light gray. In each complex structure, the WT BH3 domain is rendered as teal ribbon, and residues are displayed in stick. The 12 BH3 domain residues involved in binding to both Bcl-XL and M11 are labeled, with the residues selected for mutagenesis highlighted in red. This and all other molecular figures were prepared with the program PyMOL.
The Beclin 1 BH3 domain binds with a very similar, moderate binding affinity of ∼1.5 μm to both M11 and Bcl-XL (see Table 3). Further, for both interactions, the favorable free energy of association (ΔG) is due to enthalpic contributions (ΔHapp) rather than due to entropic contributions (ΔSapp), which are negative in each case (see Table 3). We have recently shown that the Beclin 1 BH3 domain is disordered in solution and that BH3 domain residues 116–127 appear to serve as an “anchor” that nucleates concomitant folding and binding of the Beclin 1 BH3 domain to Bcl-2 and includes most of the residues important for binding to Bcl-2 (42). Therefore, the negative ΔSapp likely reflects BH3 domain desolvation and increased structure upon binding, which proceeds despite the negative ΔSapp, because of enthalpic compensation.
TABLE 3.
Thermodynamic parameters for binding of various Beclin 1 BH3 domain-derived peptides to M11 and Bcl-XL
| Peptide | M11 |
Bcl-XL |
||||||
|---|---|---|---|---|---|---|---|---|
| Kd | ΔH | ΔG | ΔS | Kd | ΔH | ΔG | ΔS | |
| μm | kJ/mol | kJ/mol | J/K·mol | μm | kJ/mol | kJ/mol | J/K·mol | |
| WT | 1.38 ± 0.41 | −70.97 ± 6.39 | −33.51 ± 0.71 | −125.72 ± 22.53 | 1.95 ± 0.19 | −43.57 ± 1.44 | −32.58 ± 0.23 | −36.89 ± 4.53 |
| L112A | 4.33 ± 0.86 | −47.36 ± 1.53 | −30.63 ± 0.53 | −56.14 ± 4.10 | 109.71 ± 3.10 | −42.36 ± 1.07 | −22.59 ± 0.07 | −66.34 ± 3.84 |
| L116A | 177.62 ± 16.95 | −36.40 ± 4.64 | −21.40 ± 0.24 | −50.33 ± 16.36 | No binding | |||
| K117A | 0.66 ± 0.17 | −69.08 ± 3.05 | −35.32 ± 0.67 | −113.29 ± 11.74 | 18.93 ± 4.15 | −37.66 ± 3.72 | −26.99 ± 0.58 | −35.83 ± 11.52 |
| G120E | 36.49 ± 8.10 | −52.17 ± 5.81 | −25.36 ± 0.52 | −89.97 ± 20.91 | No binding | |||
| D121A | 1.33 ± 0.72 | −58.54 ± 12.21 | −33.74 ± 1.23 | −83.13 ± 45.04 | No binding | |||
| F123A | 5.20 ± 1.53 | −59.36 ± 2.72 | −30.20 ± 0.74 | −97.84 ± 11.61 | 407.56 ± 75.68 | −30.70 ± 8.28 | −19.36 ± 0.46 | −38.04 ± 29.34 |
| G120E/D121A | 6.43 ± 0.15 | −62.34 ± 2.72 | −29.62 ± 0.06 | −109.81 ± 9.28 | No binding | |||
Despite the general overall similarity in the interaction of the Beclin 1 BH3 domain with M11 and Bcl-XL described above, the specific interactions of each BH3 domain residue with residues in the two homologs are different, suggesting that each BH3 domain residue will have a different thermodynamic contribution to the overall binding. Indeed, despite the similar ΔG of binding to M11 and Bcl-XL, the magnitudes of the entropic and enthalpic contributions to binding are different, with ΔHapp for binding to M11 being ∼2-fold higher, and TΔSapp being ∼4-fold lower than that for binding to Bcl-XL (see Table 3). This supports the hypothesis that the different binding interactions of each BH3 domain residue dictate a different thermodynamic contribution to the overall similar affinity of binding in the interaction with M11 and Bcl-XL. Thus, the similar interactions of the Beclin 1 BH3 domain with M11 and Bcl-XL provide a good model system for a detailed mutational and thermodynamic analysis elucidating how differences in the binding determinants of M11 and Bcl-XL can translate to differential affinities for various BH3 domain-containing proteins, despite a similar overall mode of binding.
Specific Beclin 1 Mutations Abrogate Autophagy Down-regulation by Bcl-XL but Not by M11
Based on the analysis above, we created the following single mutant Beclin 1 constructs: L112A, L116A, K117A, G120E, and F123A. The D121A mutant was not investigated using cellular assays because our previously published results show that M11 binds to a Beclin 1 G120A/D121A mutant (22). Expression of all Beclin 1 mutants was comparable to that of WT Beclin 1 in both starvation and nutrient-rich conditions (Fig. 3A). Bcl-XL and M11 also had comparable expression in both starvation and nutrient-rich conditions.
FIGURE 3.

Effect of different Beclin 1 mutations on down-regulation of autophagy by Bcl-XL or M11. A, Western blots of MCF7 cell extracts indicating comparable expression levels of WT and mutant FLAG-tagged Beclin 1 constructs and of Bcl-XL and M11 in starvation and nutrient-rich conditions, with actin as a loading control. B–D, bar graphs representing light microscopy quantification of the number of discrete GFP-LC3 puncta per cell in GFP-positive MCF7 cells co-transfected with GFP-LC3, WT, or mutant Beclin 1 as indicated below the x axis and either no Bcl-2 homolog (B), Bcl-XL (C), or M11 (D).
Assays to monitor autophagy levels were performed using MCF7 cells, which express low levels of Beclin 1 and do not show starvation-induced increases in autophagy unless Beclin 1 is ectopically expressed (20, 49–51) (Fig. 3). This allows the effect of Beclin 1 mutants to be assayed in the absence of endogenous Beclin 1. Earlier studies have utilized multiple diverse methods to conclusively demonstrate that in starvation conditions cellular and γHV Bcl-2 homologs, including Bcl-XL and γHV68 M11, reduce autophagic flux by binding to Beclin 1 (20–22, 40, 41, 52–54). Therefore, here we monitored autophagy levels simply by quantifying the change in cellular localization of a GFP-tagged, transiently expressed mammalian autophagy-specific marker, LC3 (GFP-LC3) from a diffuse cytoplasmic distribution to localized punctae corresponding to autophagosomal structures (Fig. 3). Transient expression of Beclin 1 in MCF7 cells led to a marked increase in autophagy upon starvation (p = 0.00060 for starved versus nutrient-rich cells; Fig. 3, B–D). Basal autophagy levels in nutrient-rich media are typically much lower and less consistent than in starvation conditions; therefore here we focus on the autophagy levels observed in starvation conditions. The levels of autophagy mediated by each Beclin 1 mutant tested was comparable to that mediated by WT Beclin 1 (ranging between p = 0.10915 and 0.93428 for mutants versus WT Beclin 1; Fig. 3B).
The transient co-expression of either Bcl-XL or M11 was used to assay the ability of these homologs to down-regulate autophagy upon expression of each Beclin 1 single mutant (Fig. 3). Starvation-induced, Beclin 1-dependent autophagy is significantly down-regulated by expression of either Bcl-XL (p = 0.00033 for Bcl-XL versus empty vector; Fig. 3C) or M11 (p = 0.00434 for M11 versus empty vector; Fig. 3D), as has been previously shown (20–22). We find that M11 down-regulates starvation-induced autophagy at least as potently as Bcl-XL (Fig. 3, C and D), and in general, Beclin 1 BH3 domain mutations are less deleterious for the M11-mediated down-regulation of Beclin 1-dependent autophagy.
Under starvation conditions, Bcl-XL down-regulates autophagy mediated by the K117A Beclin 1 mutant as effectively as that mediated by WT Beclin 1 (p = 0.50430 for mutant versus WT Beclin 1; Fig. 3C). However, Bcl-XL-mediated down-regulation of autophagy is less pronounced upon expression of L112A (p = 0.06209 for mutant versus WT Beclin 1) or G120E (p = 0.01190 for mutant versus WT Beclin 1) Beclin 1 mutants (Fig. 3C). Among the Beclin 1 single mutants, the most substantial abrogation of Bcl-XL-mediated down-regulation of autophagy was observed upon expression of the mutants F123A (p = 0.00246 for mutant versus WT Beclin 1) and L116A (p = 0.00212 for mutant versus WT Beclin 1; Fig. 3C).
Similar to Bcl-XL, expression of the Beclin 1 K117A mutant (p = 0.15725 for mutant versus WT Beclin 1) did not affect M11-mediated down-regulation of autophagy (Fig. 3D). M11-mediated autophagy down-regulation is significantly weaker upon expression of the mutants F123A (p = 0.01070 for mutant versus WT Beclin 1) and L112A (p = 0.00065 for mutant versus WT Beclin 1). The most substantial abrogation of M11-mediated autophagy down-regulation is observed when L116A mutant Beclin 1 was expressed (p = 0.04316 for mutant versus WT Beclin 1).
Surprisingly however, and contrary to expectations from structural analysis, M11 effectively down-regulates autophagy upon expression of the G120E single mutant (p = 0.03131 for mutant versus WT Beclin 1). Despite the previous cellular co-immunoprecipitation assays showing that a Beclin 1 G120A/D121A mutant binds to M11 (22), we expected that the mutation of G120 to the large and negatively charged Glu residue would disrupt binding to both Bcl-XL and M11, consequently abrogating the down-regulation of autophagy by these Bcl-2 homologs. However, our data indicate that unlike Bcl-XL (Fig. 3C), the M11 binding site accommodates the Glu side chain, allowing M11 to effectively down-regulate autophagy mediated by G120E Beclin 1 (Fig. 3D).
Therefore, we further examined the role of Asp121 in the context of the G120E mutation by assaying the ability of Bcl-XL and M11 to down-regulate autophagy mediated by a G120E/D121A Beclin 1 double mutant. As expected, expression of the G120E/D121A double mutant resulted in abrogation of Bcl-XL-mediated autophagy down-regulation (p = 0.00079 for double mutant versus WT Beclin 1), comparable to the effect seen upon expression of the L116A mutant Beclin 1 (Fig. 3C). However, in complete contrast to Bcl-XL, M11 effectively down-regulates autophagy mediated by the G120E/D121A Beclin 1 double mutant (p = 0.22842 for double mutant versus WT Beclin 1; Fig. 3D). Thus, the G120E mutation enables selective inhibition of autophagy by M11.
Identification of Peptides That Bind to M11, but Not to Bcl-XL.
We used ITC to quantify and compare binding of a systematic set of Beclin 1 BH3 domain-derived peptides whose residues are numbered according to the Beclin 1 sequence. In general, each residue substitution impacted binding to Bcl-XL more than to M11, with the different substitutions having very diverse thermodynamic effects on binding to either M11 or Bcl-XL (Table 3). All substitutions weakened binding to Bcl-XL, but not to M11, and the binding affinity was generally consistent with results monitoring the effect of these mutations on down-regulation of autophagy by Bcl-XL and M11. The L112A substitution weakened binding to Bcl-XL to barely detectable levels but reduced binding to M11 by only ∼3-fold. Similarly, the F123A substitution weakened binding to Bcl-XL by ∼200-fold and to M11 by ∼4-fold, although the equivalent mutation in Beclin 1 had a more dramatic impact on the down-regulation of cellular autophagy by M11, suggesting that this mutation is more deleterious in the context of full-length Beclin 1 interactions in the cell (Fig. 3D). Interestingly, although relative to the WT BH3 domain, the K117A peptide bound with ∼10-fold weaker affinity to Bcl-XL, it actually bound with ∼2-fold tighter affinity to M11 (Table 3). Lastly, no single substitution abolished binding to M11, but three single substitutions, L116A, G120E, and D121A, abrogated binding to Bcl-XL. The L116A and G120E substitutions were also the most deleterious for binding to M11, reducing binding affinity for M11 more than 120- and 26-fold, respectively (Table 3), but binding to M11 seems unaffected by the D121A substitution.
Contrary to initial expectations based on the structure of the WT BH3 domain bound to M11, but consistent with the cellular autophagy assays, the G120E and D121A peptides are still able to bind to M11 (Table 3). Therefore, we quantified and compared the ability of a G120E/D121A double-substituted peptide (DS peptide) to bind to Bcl-XL and M11. Consistent with our cellular experiments in the previous section, the DS peptide does not bind to Bcl-XL, but importantly, it binds to M11 with ∼5.7-fold better affinity compared with the G120E peptide and only ∼4.7-fold weaker affinity compared with the WT BH3 domain. Thus, in the context of the G120E substitution, the removal of the carboxylate group at the 121 position improves binding.
Structure of the DS Peptide Bound to M11
To elucidate the mechanism by which M11 is able to bind the DS peptide, we determined the x-ray crystal structure of the M11-DS peptide complex to 2.1 Å resolution. Residues altered in the DS peptide, Glu120 and Ala121, have very well defined electron density (Fig. 4A). Both the DS peptide (Fig. 4A) and the WT BH3 domain (Fig. 4B) bind by a similar mode within the M11 hydrophobic surface groove. The two complexes superimpose with an RMSD of 0.451 Å over 148 Cα atoms, indicating that they are fairly similar, although the superposition is somewhat worse than that of the two complexes within the asymmetric units of structures of either the M11-DS peptide complex (0.162 Å) or the WT BH3 domain complex (0.031 Å). Despite this similarity of interaction, the surface area of each molecule buried in the interaction interface is significantly reduced in the M11-DS peptide complex, to 868 Å2, compared with 978 Å2 in the M11-WT BH3 domain complex. This reduced buried surface area likely accounts for the reduced binding affinity of the DS peptide and is the result of the substantial main chain shifts and side chain movements in the bound DS peptide relative to the WT BH3 domain, as well as subtle compensatory side chain changes in M11 that facilitate binding of the DS peptide.
FIGURE 4.

Stereo view of complexes of M11 bound to DS peptide (A) and WT BH3 domain (B) (Protein Data Bank code 3DVU). Atoms are colored by type as in Fig. 2, with M11 shown as molecular surface, whereas the DS peptide (magenta carbons) and WT BH3D (teal carbons) are displayed in atomic detail. The blue mesh represents the electron density contoured at 1 σ above the mean for a 1.6 Å radius around the peptide atoms from the 2Fo − Fc map at 2.1 Å for the DS peptide and at 2.5 Å for the WT BH3 domain complexes. Labels indicate residues substituted in the DS peptide.
Separate superpositions of the M11 molecule in each complex indicate that there is limited conformational change in the M11 structure (Fig. 5A), with RMSDs of 0.38 Å over 130 Cα atoms. This superposition is slightly worse than superposition of two M11 subunits within the asymmetric unit of either the M11-DS peptide complex (0.17 Å) or the WT BH3 domain complex (0.03 Å) but lies within experimental error for these structures. Maximal M11 conformational change is seen not at the BH3 domain binding groove but rather at the flexible α1-α2 loop, which is distant from the binding site; however, this change is similar to conformational variation in this loop between different copies of the same complex present in the asymmetric unit of each crystal. Therefore, the conformational changes in this flexible loop do not relate to the binding of different peptides to M11.
FIGURE 5.
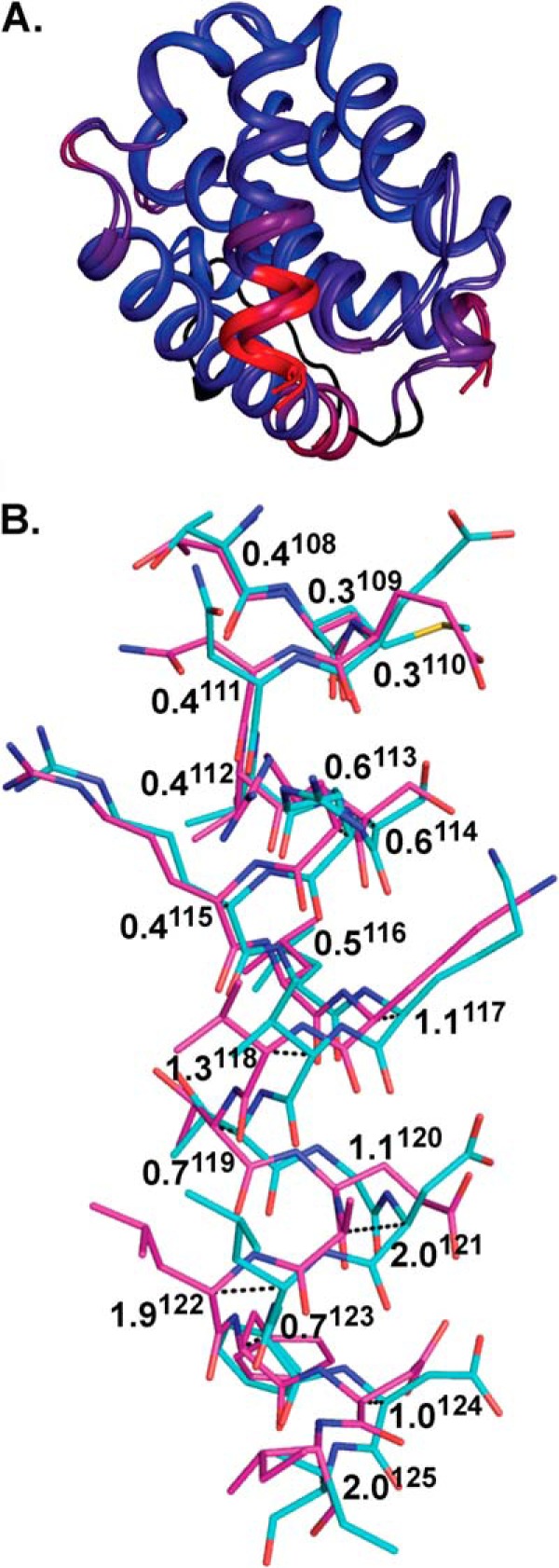
Superposition of complexes of M11 bound to DS peptide and WT BH3D. A, M11-DS peptide and M11-WT BH3 domain (Protein Data Bank code 3DVU) are aligned and colored by RMSD, with colors from blue to red corresponding to the range of pairwise RMSDs from a minimum of 0.07 to a maximum of 4.30. The M11 flexible loop located on the M11 face opposite the BH3 domain-binding groove was not included in these calculations and is colored black. Besides this loop, the most significant shift between the two complexes is observed in the C-terminal half of the peptide, as indicated by the red color. B, pairwise Cα shifts between DS peptide (magenta) and WT BH3 domain (teal). Atoms are color-coded as in Fig. 4. Pairwise shifts between the WT BH3 domain and DS peptide are displayed in Å, with superscript numbers denoting residue numbers relative to the Beclin 1 BH3 domain.
In contrast to M11, significant changes are seen between the bound DS peptide and WT BH3 domain conformations (Fig. 5B). The bound WT and DS peptides superimpose with an RMSD of 0.99 Å over 18 Cα atoms, with the comparatively poorer alignment chiefly attributable to the shifted positions of residues 117–125. The identical N-terminal halves of the two peptides superimpose fairly well between the WT BH3 domain and DS peptide structures, with an RMSD of 0.38 Å over 9 Cα atoms. However, superposition of the C-terminal half is poorer, with an RMSD of 1.35 Å over 9 Cα atoms. Thus, binding to M11 is enabled by significant shifts of the DS peptide main chain, especially of its C-terminal half (Fig. 5), relative to WT BH3 domain.
Differences in the Interactions of M11 with the DS Peptide or the WT BH3 Domain
Peptide amino acids corresponding to BH3 domain residues Leu112 and Leu116 bind in similar locations in the DS peptide and WT BH3 domain complexes, with pairwise differences in the Cα positions being 0.4 and 0.5 Å, respectively (Fig. 5B). The packing of Leu112 is virtually identical in each complex, with Leu112 being sandwiched between Met109 and Leu116, which are approximately one helical turn away on each side within the peptide, and surrounded by a hydrophobic pocket lined by M11 residues Tyr60, Ala63, and Leu74. Similarly, in each complex Leu116 is packed into a hydrophobic pocket lined by M11 residues Phe48, Tyr60, Leu78, and Val94, although there are some subtle differences in the atomic details of the interaction (Fig. 6).
FIGURE 6.

Stereo view showing details of M11 residue interactions with the DS peptide (A) and the WT BH3 domain (B) (Protein Data Bank code 3DVU). Bond lengths are shown for polar interactions. Atoms are color-coded as in Fig. 5, and residue labels are color-coded by molecule.
Starting at Lys117, there are incrementally increasing shifts in the DS peptide residue positions relative to those in the WT BH3 domain. The pairwise shift at Lys117 Cα is 1.1 Å (Fig. 5B), which enables additional interactions between Lys117 and M11 in the DS peptide complex. The aliphatic part of Lys117 packs against the aliphatic parts of M11 Asp81 and Arg87 in both complexes (Fig. 6) but in the DS peptide complex also interacts with M11 Leu78 and Ser77 (Fig. 6A). Further, although the Lys117 amino group does not make any interactions in the WT complex (Fig. 6B), in the DS complex it electrostatically bonds with the M11 Asp81 carboxylate and hydrogen bonds the Ser77 hydroxyl (Fig. 6A). Similarly, the next peptide residue, Val118, is solvent-exposed and does not interact with M11 in the WT BH3 domain complex, but a 1.3 Å Cα shift (Fig. 5B) at this position in the DS peptide complex results in packing against M11 Tyr56 (Fig. 6A). The following residue, Thr119, has a smaller Cα shift between the WT BH3 domain and DS peptide (Fig. 5), and maintains similar, but slightly different, interactions in both complexes, with the aliphatic parts of the side chain packed against M11 residues Phe48, Tyr52, and the His51 main chain (Fig. 6).
The next two residues are altered in the DS peptide: Glu120 and Ala121, compared with Gly120 and Asp121 in the WT BH3 domain. The incremental shifts preceding these residues result in a shift of 1.1 Å at the Glu120 Cα from the WT BH3 domain Gly120 Cα position, and of a maximal shift of 2.0 Å at the Cα of residue 121. The Cα shift at residue 121 corresponds to approximately half a helical turn relative to the WT BH3 domain-M11 complex (Fig. 5B). In the M11-WT BH3 domain complex (Fig. 6B), the Gly120–Asp121 main chain packs in an anti-parallel manner against the main chain of two conserved M11 residues: Gly86 and Arg87. In contrast, in the M11-DS peptide complex (Fig. 6A), Glu120 extends across the M11 hydrophobic groove, with the aliphatic part of the side chain packed against the M11 Gly86 main chain and the aliphatic parts of Arg87 and Phe48, to make one salt bridge with M11 Arg87. Thus, the M11 binding groove accommodates the larger Glu side chain and stabilizes the altered Glu120 by electrostatic interactions with the conserved M11 Arg87. Further, although in the WT BH3 domain complex (Fig. 6B) the Asp121 side chain is stabilized by packing against the aliphatic part of Arg87 and a bidentate salt bridge to M11 Arg87, in the DS peptide complex (Fig. 6A) Ala121 makes no contacts with M11 and is completely solvent-exposed as a consequence of the main chain shifts.
Leu122, the peptide residue that follows the two altered residues, is also significantly shifted and has a completely different environment in the WT BH3 domain and DS peptide structures (Fig. 6). In the WT peptide, Leu122 is solvent-exposed and makes no contacts with M11 (Fig. 6B), whereas in the DS peptide complex, it is shifted to pack against M11 His51 and Val55 (Fig. 6A) with this interaction being accommodated by a rotation of the His51 imidazole.
Pairwise Cα shifts between the M11-bound DS peptide and WT BH3 domain decrease to 0.7 Å at Phe123 (Fig. 5), which allows the side chain to bind in equivalent M11 hydrophobic surface pockets comprised of residues Leu44, Glu47, Phe48, His51, Gly86, and Val89 in each complex, but with an altered orientation of the Phe123 aromatic ring and subtly different interactions with M11 (Figs. 4 and 6). The relative shifts between the WT BH3 domain and DS peptide are retained at the Asp124 Cα position (Fig. 5). This allows the aliphatic part of the Asp124 side chain to pack against the M11 G86 Cα in both complexes. In addition, it packs against the aliphatic parts of M11 Asn84 in the WT BH3 domain complex and against the peptide Glu120 in the DS peptide complex. Further, the Asp124 carboxylate group of the WT BH3 domain (Fig. 6B) hydrogen bonds to the M11 Gly86 amide, but a similar interaction is not seen in the DS peptide complex (Fig. 6A).
Thus, the main chain shifts of the DS peptide enable the Glu120-M11 Arg87 interaction and remove Ala121 from M11 interactions. It is likely that the improved binding of M11 to the DS peptide, relative to the G120E peptide, is due to the elimination of the competition between the G120E and Asp121 carboxylates for the M11 Arg87 interaction and the helix strain associated with the binding of the latter peptide.
A Cell-permeable DS Peptide Selectively Abrogates Down-regulation of Autophagy by M11, but Not by Bcl-XL
Lastly, we investigated whether the DS peptide would specifically prevent M11-mediated, but not Bcl-XL-mediated, down-regulation of Beclin 1-dependent autophagy. To make the peptide cell-permeable, the transactivating HIV-1 transcriptional activator protein transduction domain (TAT), which is a cell-penetrating peptide previously shown to facilitate entry of extended polypeptides into mammalian cells under the conditions used here (55), was attached via a diglycine linker to the N terminus of the DS peptide (TAT-DS peptide). As a control we also assayed the effect of a TAT-BH3 domain fused peptide (TAT-WT peptide) on M11-and Bcl-XL-mediated down-regulation of Beclin 1-dependent autophagy.
Treatment of Beclin 1-transfected MCF7 cells with the TAT-WT peptides did not significantly increase levels of autophagy in either nutrient-rich or starvation conditions (Fig. 7). As expected, compared with untreated cells, TAT-WT peptide treatment markedly increases autophagy levels in cells that are transiently transfected with either M11 (p = 0.00194 for treated versus untreated cells; Fig. 7) or Bcl-XL (p = 0.00007 for treated versus untreated cells; Fig. 7). This suggests that the TAT-WT peptide binds to both M11 and Bcl-XL, preventing both Bcl-2 homologs from down-regulating Beclin 1-mediated autophagy.
FIGURE 7.
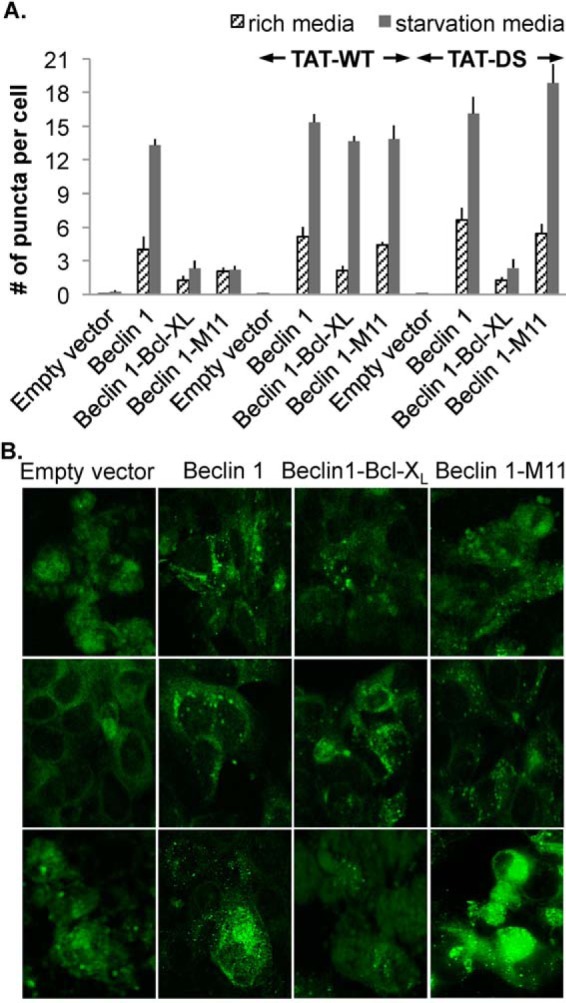
Effect of TAT-DS peptide treatment on down-regulation of autophagy by Bcl-XL and M11. A, light microscopy quantification of the number of discrete GFP-LC3 puncta per cell in GFP-positive MCF7 cells are co-transfected with GFP-LC3, WT Beclin 1, and either WT M11 or Bcl-XL and then treated with either no peptide, TAT-WT, or TAT-DS peptide. B, representative images of GFP-LC3 staining in these cells corresponding to cells that were untreated (top row), TAT-WT treated (middle row), and TAT-DS treated (bottom row).
TAT-DS peptide treatment of MCF7 cells that express Beclin 1, but not Bcl-XL or M11, causes an insignificant elevation in autophagy levels relative to untreated cells (p = 0.20340 for treated versus untreated cells; Fig. 7). TAT-DS peptide treatment of cells that transiently express Bcl-XL in addition to Beclin 1 had an insignificant effect compared with untreated cells (p = 0.92294 for treated versus untreated cells; Fig. 7), presumably because the TAT-DS peptide does not bind to Bcl-XL and so does not prevent Bcl-XL from down-regulating autophagy. Strikingly, however, TAT-DS peptide treatment of cells that transiently express M11 in addition to Beclin 1 markedly increases autophagy levels compared with untreated cells (p = 0.042479 for treated versus untreated cells; Fig. 7), indicating that the TAT-DS peptide binds to M11, preventing M11 from down-regulating Beclin 1-mediated autophagy. Thus, the TAT-DS peptide inhibits M11-mediated down-regulation of autophagy, but not Bcl-XL-mediated down-regulation of autophagy.
DISCUSSION
In this study we have shown that despite the similar general mode of binding of the Beclin 1 BH3 domain to M11 and Bcl-XL, the different residues lining the binding grooves of each homolog dictate differences in the atomic details and the thermodynamics of each interaction and consequently affect biological function. We detailed the effect of mutations on binding of the BH3 domain to two different Bcl-2 homologs, which combined with our structural information, helps illuminate the atomic bases of the differential specificity of these homologs for different BH3 domain-containing binding partners. We found that, consistent with the promiscuity of M11 and specificity of Bcl-XL for diverse BH3 domains, the M11 binding site accommodates more peptide residue substitutions than the binding sites of Bcl-XL or Bcl-2 (42). These changes are accommodated chiefly by subtle M11 side chain conformational changes that allow for more dramatic changes in the bound peptide, which enable alternate interactions with M11. Either the G120E or D121A mutation is sufficient for preventing binding to Bcl-XL or Bcl-2 (42), suggesting that unlike for M11, Bcl-2 and Bcl-XL cannot accommodate a large side chain at the Gly120 position and that the salt bridge formed with Arg87 of Bcl-XL is critical for binding to Bcl-XL and Bcl-2, but not to M11. Based on superpositions of the complexes of M11-DS peptide, M11-WT BH3 domain, and Bcl-XL-WT BH3 domain, it appears that the peptide shifts observed in the M11-DS peptide complex, which enable the G120E to interact with Arg87, would not be easily accommodated in Bcl-XL. For instance, an obvious steric clash would occur between the Leu122 main chain of the shifted DS peptide and the Bcl-XL R100, whose conformation is stabilized by salt bridge networks (56). This information adds significantly to our understanding of interactions between Bcl-2 homologs and BH3 domain-containing proteins, explaining how mutated or diverse BH3 domains may be bound by one homolog and not another, thus also adding to our repertoire of information on protein-protein interactions in general.
Consistent with our expectations, we were able to exploit the subtle differences in binding by M11 and Bcl-XL to design specific inhibitors of M11 that abrogate M11 function in cells. Our results provide a tool to directly target M11-BH3 domain interactions in cellular studies. The selective peptide inhibitor identified in this study will be useful in studying the role of M11 at different stages of the γHV68 life-cycle, by treating virus-infected cells with the peptide at different time points and assaying the effects on viral infection. In vivo studies in mice may require the development of more bio-stable small molecules. The structural information presented here will be invaluable for the future rational design of such small molecules that can selectively inhibit M11, but do not affect cellular Bcl-2 homologs. Although a general M11-specific inhibitor would help elucidate the general function of M11, a specific inhibitor that selectively inhibits only the γHV Bcl-2-Beclin 1 interaction would remove only the γHV blockade of autophagy and therefore would be an extremely useful tool to study the role of autophagy in regulating γHV infections.
Lastly, this study also elucidates methods that may allow us to identify determinants specific for binding to other Bcl-2 homologs, especially those from other γHVs such as KSHV and Epstein-Barr virus. Such studies will provide basic mechanistic explanations about their ability to bind to diverse BH3 domain-containing proteins, and consequently their ability to differentially regulate various pathways, and may further enable us to design inhibitors that specifically target these proteins. Thus, this research will substantially assist and inform future research on the pathogenesis of infections caused by γHVs. Ultimately, such small molecule inhibitors may even form the basis of novel therapeutics to treat γHV infection, which currently cannot be cured, by promoting the autophagic degradation of viruses, apoptotic destruction of infected host cells, and/or restoration of the tumor suppressor activity of Beclin 1.
Acknowledgments
We acknowledge usage of the North Dakota State University Advanced Imaging and Microscopy Core Laboratory and usage of the Low Volume Nano ITC (courtesy of Dr. Sanku Mallik). We also thank Dr. Dominika Borek (University of Texas Southwestern Medical Center) for assistance with structure solution during the CCP4/APS summer school held at the Advanced Photon Source.
This work was supported, in whole or in part, by National Institutes of Health Grants P20 RR015566 and P30 GM103332-01 (to S. S. and C. L. C.), R21 AI078198 (to S. S.), and RO1 CA109618 and CPRIT PR120718-P1 (to B. L.). This work was also supported by National Science Foundation Grants EPS-0814442 (to S. S. and C. L. C.), HRD-0811239 (to S. S.).
- HV
- herpesvirus
- RMSD
- root mean square deviation
- ITC
- isothermal titration calorimetry
- KSHV
- Kaposi's sarcoma-associated herpesvirus
- BH
- Bcl-2 homology
- TAT
- transactivating HIV-1 transcriptional activator protein transduction domain.
REFERENCES
- 1. Roizman B., Pellet P. E. (2001) The family herpesviridae. A brief introduction. In Field's Virology (Knipe D. M., Howley P. M., eds) pp. 2381–2398, Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 2. Tarakanova V. L., Suarez F., Tibbetts S. A., Jacoby M. A., Weck K. E., Hess J. L., Speck S. H., Virgin H. W. (2005) Murine gammaherpesvirus 68 infection is associated with lymphoproliferative disease and lymphoma in BALB beta2 microglobulin-deficient mice. J. Virol. 79, 14668–14679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gangappa S., van Dyk L. F., Jewett T. J., Speck S. H., Virgin H. W. (2002) Identification of the in vivo role of a viral Bcl-2. J. Exp. Med. 195, 931–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cuconati A., White E. (2002) Viral homologs of Bcl-2. Role of apoptosis in the regulation of virus infection. Genes Dev. 16, 2465–2478 [DOI] [PubMed] [Google Scholar]
- 5. Vaux D. L., Cory S., Adams J. M. (1988) Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature 335, 440–442 [DOI] [PubMed] [Google Scholar]
- 6. McDonnell T. J., Deane N., Platt F. M., Nunez G., Jaeger U., McKearn J. P., Korsmeyer S. J. (1989) bcl-2-immunoglobulin transgenic mice demonstrate extended B cell survival and follicular lymphoproliferation. Cell 57, 79–88 [DOI] [PubMed] [Google Scholar]
- 7. Reed J. C. (1998) Bcl-2 family proteins. Oncogene 17, 3225–3236 [DOI] [PubMed] [Google Scholar]
- 8. Danial N. N., Korsmeyer S. J. (2004) Cell death. Critical control points. Cell 116, 205–219 [DOI] [PubMed] [Google Scholar]
- 9. Hardwick J. M. (1998) Viral interference with apoptosis. Semin. Cell. Dev. Biol. 9, 339–349 [DOI] [PubMed] [Google Scholar]
- 10. Benedict C. A., Norris P. S., Ware C. F. (2002) To kill or be killed. Viral evasion of apoptosis. Nat. Immunol. 3, 1013–1018 [DOI] [PubMed] [Google Scholar]
- 11. Sarid R., Sato T., Bohenzky R. A., Russo J. J., Chang Y. (1997) Kaposi's sarcoma-associated herpesvirus encodes a functional bcl-2 homologue. Nat. Med. 3, 293–298 [DOI] [PubMed] [Google Scholar]
- 12. Ojala P. M., Tiainen M., Salven P., Veikkola T., Castaños-Vélez E., Sarid R., Biberfeld P., Mäkelä T. P. (1999) Kaposi's sarcoma-associated herpesvirus-encoded v-cyclin triggers apoptosis in cells with high levels of cyclin-dependent kinase 6. Cancer Res. 59, 4984–4989 [PubMed] [Google Scholar]
- 13. Cheng E. H., Nicholas J., Bellows D. S., Hayward G. S., Guo H.-G., Reitz M. S., Hardwick J. M. (1997) A Bcl-2 homolog encoded by Kaposi's sarcoma-associated virus, human herpesvirus 8, inhibits apoptosis but does not heterodimerize with Bax or Bak. Proc. Natl. Acad. Sci. U.S.A. 94, 690–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Henderson S., Huen D., Rowe M., Dawson C., Johnson G., Rickinson A. (1993) Epstein-Barr virus-encoded BHRF1 protein, a viral homologue of Bcl-2, protects human B-cells from programmed cell death. Proc. Natl. Acad. Sci. U.S.A. 90, 8479–8483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Foghsgaard L., Jäättelä M. (1997) The ability of BHRF1 to inhibit apoptosis is dependent on stimulus and cell type. J. Virol. 71, 7509–7517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Marshall W. L., Yim C., Gustafson E., Graf T., Sage D. R., Hanify K., Williams L., Fingeroth J., Finberg R. W. (1999) Epstein-Barr virus encodes a novel homolog of the Bcl-2 oncogene that inhibits apoptosis and associates with Bax and Bak. J. Virol. 73, 5181–5185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Theodorakis P., D'Sa-Eipper C., Subramanian T., Chinnadurai G. (1996) Unmasking of a proliferation-restraining activity of the anti-apoptosis protein EBV BHRF1. Oncogene 12, 1707–1713 [PubMed] [Google Scholar]
- 18. Wang G.-H., Garvey T. L., Cohen J. I. (1999) The murine gammaherpesvirus-68 M11 protein inhibits Fas- and TNF- induced apoptosis. J. Gen. Virol. 80, 2737–2740 [DOI] [PubMed] [Google Scholar]
- 19. Loh J., Huang Q., Petros A. M., Nettesheim D., van Dyk L. F., Labrada L., Speck S. H., Levine B., Olejniczak E. T., Virgin H. W. (2005) A surface groove essential for viral Bcl-2 function during chronic infection in vivo. PLoS Pathog. 1, e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pattingre S., Tassa A., Qu X., Garuti R., Liang X. H., Mizushima N., Packer M., Schneider M. D., Levine B. (2005) Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 122, 927–939 [DOI] [PubMed] [Google Scholar]
- 21. Ku B., Woo J.-S., Liang C., Lee K.-H., Hong H.-S., Xiaofei E., Kim K.-S., Jung J. U., Oh B.-H. (2008) Structural and biochemical bases for the inhibition of autophagy and apoptosis by viral Bcl-2 of murine γ-Herpesvirus 68. PLoS Pathog. 4, e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sinha S., Colbert C. L., Becker N., Wei Y., Levine B. (2008) Molecular basis of the regulation of Beclin 1-dependent autophagy by the γ-herpesvirus 68 Bcl-2 homolog M11. Autophagy 4, 989–997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wei Y., Sinha S., Levine B. (2008) Dual role of JNK1-mediated phosphorylation of Bcl-2 in autophagy and apoptosis regulation. Autophagy 4, 949–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Levine B., Sinha S., Kroemer G. (2008) Bcl-2 family members. Dual regulators of apoptosis and autophagy. Autophagy 4, 600–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Maiuri M. C., Criollo A., Kroemer G. (2010) Crosstalk between apoptosis and autophagy within the Beclin 1 interactome. EMBO J. 29, 515–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kang R., Zeh H. J., Lotze M. T., Tang D. (2011) The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 18, 571–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Su M., Mei Y., Sinha S. (2013) Role of the crosstalk between autophagy and apoptosis in cancer. J. Oncol. 2013, 102735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Muchmore S. W., Sattler M., Liang H., Meadows R. P., Harlan J. E., Yoon H. S., Nettesheim D., Chang B. S., Thompson C. B., Wong S. L., Ng S. L., Fesik S. W. (1996) X-ray and NMR structure of human Bcl-XL, an inhibitor of programmed cell death. Nature 381, 335–341 [DOI] [PubMed] [Google Scholar]
- 29. Liu X., Dai S., Zhu Y., Marrack P., Kappler J. W. (2003) The structure of a Bcl-xL/Bim fragment complex. Implications for Bim function. Immunity 19, 341–352 [DOI] [PubMed] [Google Scholar]
- 30. Petros A. M., Nettesheim D. G., Wang Y., Olejniczak E. T., Meadows R. P., Mack J., Swift K., Matayoshi E. D., Zhang H., Thompson C. B., Fesik S. W. (2000) Rationale for Bcl-xL/BAD peptide complex formation from structure, mutagenesis, and biophysical methods. Protein Sci. 9, 2528–2534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sattler M., Liang H., Nettesheim D., Meadows R. P., Harlan J. E., Eberstadt M., Yoon H. S., Shuker S. B., Chang B. S., Minn A. J., Thompson C. B., Fesik S. W. (1997) Structure of the Bcl-XL-Bak peptide complex. Recognition between regulators of apoptosis. Science 275, 983–986 [DOI] [PubMed] [Google Scholar]
- 32. Huang Q., Petros A. M., Virgin H. W., Fesik S. W., Olejniczak E. T. (2002) Solution structure of a Bcl-2 homolog from Kaposi sarcoma virus. Proc. Natl. Acad. Sci. U.S.A. 99, 3428–3433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Huang Q., Petros A. M., Virgin H. W., Fesik S. W., Olejniczak E. T. (2003) Solution structure of the BHRF1 protein from Epstein-Barr virus, a homolog of human Bcl-2. J. Mol. Biol. 332, 1123–1130 [DOI] [PubMed] [Google Scholar]
- 34. Chen L., Willis S. N., Wei A., Smith B. J., Fletcher J. I., Hinds M. G., Colman P. M., Day C. L., Adams J. M., Huang D. C. (2005) Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic funciton. Mol. Cell 17, 393–403 [DOI] [PubMed] [Google Scholar]
- 35. DeBartolo J., Dutta S., Reich L., Keating A. E. (2012) Predictive Bcl-2 family binding models rooted in experiment or structure. J. Mol. Biol. 422, 124–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Oberstein A., Jeffrey P. D., Shi Y. (2007) Crystal structure of the Bcl-XL-Beclin 1 peptide complex. Beclin 1 is a novel BH3-only protein. J. Biol. Chem. 282, 13123–13132 [DOI] [PubMed] [Google Scholar]
- 37. Feng W., Huang S., Wu H., Zhang M. (2007) Molecular basis of Bcl-XL's target recognition versatility revealed by the structure of Bcl-XL in complex with the BH3 domain of Beclin-1. J. Mol. Biol. 372, 223–235 [DOI] [PubMed] [Google Scholar]
- 38. Sinha S., Levine B. (2008) The autophagy effector Beclin 1. A novel BH3-only protein. Oncogene 27, S137–S148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liang X. H., Kleeman L. K., Jiang H. H., Gordon G., Goldman J. E., Berry G., Herman B., Levine B. (1998) Protection against fatal Sindbis virus encephalitis by Beclin 1, a novel Bcl-2-interacting protein. J. Virol. 72, 8586–8596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Erlich S., Mizrachy L., Segev O., Lindenboim L., Zmira O., Adi-Harel S., Hirsch J. A., Stein R., Pinkas-Kramarski R. (2007) Differential interactions between Beclin 1 and Bcl-2 family members. Autophagy 3, 561–568 [DOI] [PubMed] [Google Scholar]
- 41. Maiuri M. C., Le Toumelin G., Criollo A., Rain J. C., Gautier F., Juin P., Tasdemir E., Pierron G., Troulinaki K., Tavernarakis N., Hickman J. A., Geneste O., Kroemer G. (2007) Functional and physical interaction between Bcl-XL and a BH3-like domain in Beclin-1. EMBO J. 26, 2527–2539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mei Y., Su M., Soni G., Salem S., Colbert C. L., Sinha S. C. (2013) Intrinsically disordered regions in autophagy proteins. Proteins 10.1002/prot.24424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 44. Kissinger C. R., Gehlhaar D. K., Fogel D. B. (1999) Rapid automated molecular replacement by evolutionary search. Acta Crystallogr. D Biol. Crystallogr. 55, 484–491 [DOI] [PubMed] [Google Scholar]
- 45. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Brünger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., Warren G. L. (1998) Crystallography & NMR system (CNS). A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54, 905–921 [DOI] [PubMed] [Google Scholar]
- 47. Krissinel E., Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 48. Moroy G., Martin E., Dejaegere A., Stote R. H. (2009) Molecular basis for Bcl-2 homology 3 domain recognition in the Bcl-2 protein family. Identification of conserved hot spot interactions. J. Biol. Chem. 284, 17499–17511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Liang X. H., Jackson S., Seaman M., Brown K., Kempkes B., Hibshoosh H., Levine B. (1999) Induction of autophagy and inhibition of tumorigenesis by Beclin 1. Nature 402, 672–676 [DOI] [PubMed] [Google Scholar]
- 50. Liang X. H., Yu J., Brown K., Levine B. (2001) Beclin 1 contains a leucine-rich nuclear export signal that is required for its autophagy and tumor suppressor function. Cancer Res. 61, 3443–3449 [PubMed] [Google Scholar]
- 51. Furuya N., Yu J., Byfield M., Pattingre S., Levine B. (2005) The evolutionarily conserved domain of Beclin 1 is required for Vps34 binding, autophagy and tumor suppressor function. Autophagy 1, 46–52 [DOI] [PubMed] [Google Scholar]
- 52. Wei Y., Pattingre S., Sinha S., Bassik M., Levine B. (2008) JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol. Cell. 30, 678–688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ku B., Woo J.-S., Liang C., Lee K.-H., Jung J. U., Oh B.-H. (2008) An insight into the mechanistic role of Beclin 1 and its inhibition by prosurvival Bcl-2 family proteins. Autophagy 4, 519–520 [DOI] [PubMed] [Google Scholar]
- 54. Pattingre S., Bauvy C., Levade T., Levine B., Codogno P. (2009) Ceramide-induced autophagy. To junk or to protect cells? Autophagy 5, 558–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Shoji-Kawata S., Sumpter R., Leveno M., Campbell G. R., Zou Z., Kinch L., Wilkins A. D., Sun Q., Pallauf K., MacDuff D., Huerta C., Virgin H. W., Helms J. B., Eerland R., Tooze S. A., Xavier R., Lenschow D. J., Yamamoto A., King D., Lichtarge O., Grishin N. V., Spector S. A., Kaloyanova D. V., Levine B. (2013) Identification of a candidate therapeutic autophagy-inducing peptide. Nature 494, 201–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Maity A., Yadav S., Verma C. S., GhoshDastidar S. (2013) Dynamics of Bcl-xL in water and membrane. Molecular simulations. PLoS One 8, e76837. [DOI] [PMC free article] [PubMed] [Google Scholar]