

Background: Neurodegenerative diseases are associated with inflammation in the brain and other organs.
Results: In murine Niemann-Pick Type C, cathepsin S was elevated and a plasma marker of liver inflammation. Lysozyme was an inflammatory plasma marker derived from both liver and brain.
Conclusion: Their dual analysis suggested four distinct severity states of neurodegeneration.
Significance: Cerebral inflammatory disease may be detectable in plasma.
Keywords: Biomarkers, Genetic Diseases, Inflammation, Neurodegeneration, Neuroinflammation, Cathepsin S, Lysozyme
Abstract
Early diagnosis of neurological disorders would greatly improve their management and treatment. A major hurdle is that inflammatory products of cerebral disease are not easily detected in blood. Inflammation in multiple organs and heterogeneity in disease present additional challenges in distinguishing the extent to which a blood-based marker reflects disease in brain or other afflicted organs. Murine models of the monogenetic disorder Niemann-Pick Type C present aggressive forms of cerebral and liver inflammatory disease. Microarray analyses previously revealed age-dependent changes in innate immunity transcripts in the mouse brain. We have now validated four putative secretory inflammatory markers that are also elevated in mouse liver. We include limited, first time analysis of human Niemann-Pick Type C liver and cerebellum. Furthermore, we utilized 2-hydroxypropyl-β-cyclodextrin (HPβCD, an emerging therapeutic) administered intraperitoneally in mice, which abrogates inflammatory pathology in the liver but has limited effect on the brain. By analyzing the corresponding effects on inflammatory plasma proteins, we identified cathepsin S as a lead indicator of liver disease. In contrast, lysozyme was a marker of both brain and liver disease. 2-Hydroxypropyl-β-cyclodextrin had no effect on transcripts of neuron-specific 24-hydroxylase, and its product 24(S)-hydroxycholesterol was not a useful indicator in mouse plasma. Our data suggest that dual analysis of levels of the inflammatory markers lysozyme and cathepsin S may enable detection of multiple distinct states of neurodegeneration in plasma.
Introduction
Inflammatory proteins, especially those of innate immunity, are under investigation as biomarkers to monitor disease onset and progression in a wide range of neurodegenerative and metabolic disorders (1–3). Heterogeneity in the progression of these diseases underscores the critical need for biomarkers. This is particularly so for inherited lysosomal disorders because they are rare, which increases the challenges of detection and treatment. Multiple organs may be affected, raising the question of whether markers reflect change in one or more organ systems. Plasma markers for neurological disease have been particularly elusive in both rare and more prevalent neurodegenerative disorders (such as Alzheimer and Parkinson diseases).
Niemann-Pick Type C (NPC)2 is an autosomal recessive neurodegenerative, lysosomal disorder caused by defects in function of either the NPC1 or NPC2 gene, although in ∼95% of patients, disease is caused by a defect in NPC1 (4). Progressive neurodegeneration is a prominent feature. In addition, NPC is also recognized as a significant cause of liver disease in early life (5–7). A mouse model, BALB/c Npc1−/− also known as Npc1nih, where the Npc1 gene is truncated (8) enables the study of aggressive forms of brain and liver disease. Furthermore, because terminal stage disease manifests in less than 90 days, it provides a relatively short model to monitor both neurodegenerative and liver disease.
Multiple inflammatory, innate immune changes have been reported by transcriptional and protein analyses in the liver, spleen, and brain of NPC animals (9–12). At the cellular level, there is prominent accumulation of foamy macrophages in liver (9, 10, 13) and activation of microglia in brain (14). Impaired development and reduced natural killer T cells in spleen and thymus have been found in NPC null mice (15, 16). In addition, expression arrays suggest transcriptional changes in NPC cells grown in in vitro cultures (17, 18).
We investigated conserved transcriptional changes seen in the brain throughout the life span of the Npc1nih mouse by examining animals at six different ages from weaning to late neurodegeneration (19). These analyses revealed innate immunity trends that could not be obtained from isolated (or a few) time points. We compared them with changes in the liver to identify age-dependent elevation of eight genes of lysosomal innate immunity proteins in the brain and liver, and results suggested that they may be potentially suitable as biomarkers for disease in both organs and secreted into plasma. The top candidate, lysozyme, was validated in plasma of Npc1nih and Npc1nmf164 (Npc1nmf; a BALB/c strain with a point mutation (D1005G) in the NPC1 protein). Our analyses also revealed that neutrophils accumulate in the NPC liver, suggesting a new cellular component that contributes to inflammatory damage there. In independent studies, Cluzeau et al. (20) correlated age-dependent gene expression in mouse liver to identify two plasma markers validated in mice and humans, but their link to molecular changes in the brain was not investigated.
Our interest is also to understand how potential biomarkers and inflammatory changes will serve to assess therapies and their differential effects on disease in brain. To do this, we expanded validation of candidate genes using multiple members of the cathepsin family in brain and liver of murine models. We also extended findings in mice in a limited, first time molecular analysis of human cerebellum and liver. Furthermore, we monitored changes in cathepsins as well as previously identified lysozyme in mice treated with 2-hydroxypropyl-β-cyclodextrin (HPβCD; commonly known as cyclodextrin), an emerging therapeutic known to improve disease outcomes in mice (21–24) and being expanded for use in humans. Cathepsins are cysteine and aspartic proteases that are secreted into the body fluid including blood, and several cathepsins have been identified as blood-based markers for several cancers and inflammatory diseases (25–27). However, use of cathepsins as plasma biomarkers in neurodegenerative lysosomal disorders has been poorly explored. Lysozyme transcripts are the most highly elevated in the brain, and their elevation in mouse plasma has been reported (19), but how the contribution from the liver could be distinguished from that in the brain remained unknown.
EXPERIMENTAL PROCEDURES
Materials
All fine chemicals were obtained from Sigma unless otherwise indicated. For immunohistochemistry, rat anti-mouse Ly-6G (clone 1A8, BioXcell) was used to detect neutrophils, and monoclonal anti-calbindin (C9848, Sigma) antibody was used for Purkinje neurons. Rabbit anti-cathepsin S (CTSS) (H-50) antibody was from Santa Cruz Biotechnology (Dallas, TX). Antibodies to lysozyme (28) were a kind gift from Professor Tomas Ganz (University of California at Los Angeles). Oligonucleotides for quantitative PCR (qPCR) were purchased from Invitrogen.
Production of Npc1nih and Npc1nmf164 Mutant Mice
A breeding pair of Npc1nih (BALB/c Nctr-Npc1m1N/J) mice was purchased from The Jackson Laboratory (Bar Harbor, ME). This strain is a widely used NPC BALB/c strain (8) carrying a truncation and premature translation of NPC1 protein that was originally established by Peter Pentchev at the National Institutes of Health (Bethesda, MD). Npc1nmf164 is a BALB/c strain derived from the recently described Npc1nmf164 in C57BL/6J (29) that contains an ethylnitrosourea-induced point mutation in the Npc1 gene. The mutation is a single nucleotide change (A to G at cDNA bp 3163) causing an aspartate to glycine change at position 1005 (D1005G) that results in slower disease progression due to partial loss of NPC1 function. The mutation was transferred from C57BL/6J to the BALB/c strain by Robert P. Erickson, University of Arizona Health Sciences Center (Tucson, AZ). Homozygous mutants of both strains (Npc1−/−) along with wild type littermates (Npc1+/+) were generated by crossing heterozygous mutant (Npc1+/−) males and females in house. Npc1nih mouse pups were genotyped according to published protocols (8), and Npc1nmf164 mice were genotyped based on PCR followed by digestion with BstEII (29). In this study, unless otherwise indicated, Npc1nih mice were used.
RNA Extraction
In mice, formalin-fixed paraffin-embedded tissue was sectioned (4–5 μm), and total RNA was isolated using an RNeasy FFPE kit (Qiagen, Germantown, MD), which included treatment with DNase. Frozen human livers and cerebella from four NPC patients and four age-, gender-, and ethnicity-matched controls were obtained from the NICHD Brain and Tissue Bank for Developmental Disorders (University of Maryland, Baltimore, MD) as approved by the Institutional Review Board of the University of Notre Dame (FWA 00002462). Total RNA was isolated using an RNeasy kit (Qiagen). Eluted RNA was further digested with RNase-free DNase I and repurified using an RNeasy column. The quality of RNA was checked using a Bioanalyzer chip (Agilent Technologies, Santa Clara, CA), and the quantity was determined using a Nanodrop 2000 (Thermo Fisher Scientific, Waltham, MA).
Quantitative PCR
qPCR was performed using a Power SYBR Green RNA-to-CT 1-Step kit and an ABI Prism 7500 Fast Real-time PCR System (Applied Biosystems). The reaction was carried out in 20 μl using 100 nm primers and 5–100 ng of total RNA as template. The thermal cycling parameters were as follows: step 1, 48 °C for 30 min; step 2, 95 °C for 10 min; step 3, 95 °C for 15 s; step 4, 60 °C for 15 s. Steps 3 and 4 were repeated for 40 cycles followed by melt curve analysis. The nucleotide sequences of gene-specific primers and their sources are listed in Table 1. Specific amplification was validated by analysis of template titration, melt curves, and agarose gel electrophoresis. In both mouse and human tissues, the mRNA levels were normalized to the housekeeping gene glyceraldehyde 3-phosphate dehydrogenase (Gapdh). -Fold change was calculated using a relative standard curve method after correcting for PCR efficiency. In mice, -fold change in expression levels of different genes in Npc1−/− was calculated relative to average levels of expression in Npc1+/− mice. In human tissues, -fold change in transcript expression in NPC liver and cerebellum was expressed relative to average expression in age-matched controls.
TABLE 1.
Primers used in qPCR studies
F, forward; R, reverse.
| Genes | Species | Sequence | Source/Ref. |
|---|---|---|---|
| Ctss | Mouse | F, 5′-ACCTACCAAGTGGGCATGAACGAT-3′ | 55 |
| R, 5′-TCGGGGAATTCTCAGAGCACCCAT-3′ | |||
| Ctsd | Mouse | F, 5′-CTGAGTGGCTTCATGGGAAT-3′ | 55 |
| R, 5′-CCTGACAGTGGAGAAGGAGC-3′ | |||
| Ctsb | Mouse | F, 5′-AAATCAGGAGTATACAAGCATGA-3′ | 55 |
| R, 5′-GCCCAGGATGCGGATGG-3′ | |||
| Gapdh | Mouse | F, 5′-TCCATGACAACTTTGGCATTG-3′' | 56 |
| R, 5′-CAGTCTTCTGGGTGGCAGTGA-3′' | |||
| Itgax (Cd11c) | Mouse | F, 5′-CTTCATTCTGAAGGGCAACCT-3′ | 57 |
| R, 5′-CACTCAGGAGCAACACCTTTTT-3′ | |||
| Cola1 | Mouse | F, 5′-CTCCAAGGAAATGGCAACTCAG-3′ | 10 |
| R, 5′-TCCTCATCCAGGTACGCAATG-3′ | |||
| Cd68 | Mouse | F, 5′-CCTCCACCCTCGCCTAGTC-3′ | 22 |
| R, 5′-TTGGGTATAGGATTCGGATTTGA-3′ | |||
| Mip-1α(Ccl3) | Mouse | F, 5′-TTCATCGTTGACTATTTTGAAACCA-5′ | 22 |
| R, 5′-GCCGGTTTCTCTTAGTCAGGA-5′ | |||
| Gfap | Mouse | F, 5′-TGCTGGAGGGCGAAGAAA-3′ | 57 |
| R, 5′-CGGATCTGGAGGTTGGAGAA-3′ | |||
| Lyz1 | Mouse | F, 5′-AAGAATGCCTGTGGGATCAA-3′ | mouseprimerdepot.nci.nih.gov/ |
| R, 5′-CGGTTTTGACATTGTGTTCG-3′ | |||
| Cyp46a1 | Mouse | F, 5′-GCTATGAGCACATCCCCG-3′ | mouseprimerdepot.nci.nih.gov/ |
| R, 5′-AACACATCTTGGAGCACACG-3′ | |||
| CTSB | Human | F, 5′-CCAAGTGTAGCAAGATCTGTGAG-3′ | This study |
| R, 5′-GTAGGAATTGTATCCGTAGTGCTT-3′ | |||
| CTSD | Human | F, 5′-AGAGGACTACACGCTCAAGGT-3′ | This study |
| R, 5′-CGGTCAAACACAGTGTAGTAGC-3′ | |||
| CTSS | Human | F, 5′-CCAGTGTCTGTTGGTGTAGATG-3′ | This study |
| R, 5′-TTCCCATTAAGATCACCATAGC-3′ | |||
| LYZ | Human | F, 5′-TGTAATGATGGCAAAACCCC-3′ | primerdepot.nci.nih.gov/ |
| R, 5′-ATCACGGACAACCCTCTTTG-3′ | |||
| CYP46A1 | Human | F, 5′-CTGTCCCAGGCAGTGAAACT-3′ | primerdepot.nci.nih.gov/ |
| R, 5′-AATGCTCTCCCGGACCTC-3′ | |||
| GAPDH | Human | F, 5′-CTCTGACTTCAACAGCGACAC-3′ | This study |
| R, 5′-GTTGTCATACCAGGAAATGAGC-3′ |
Lysozyme Activity Assay
Lysozyme activity in plasma was measured using a fluorescence-based lysozyme assay kit (EnzCheck, Invitrogen) as describer earlier (19). Plasma corresponding to 25 μg of protein from female and male Npc1nih mice was used in a 100-μl reaction volume. The reaction was carried out at 37 °C for 24 h. Fluorescence was read using excitation/emission of 494/518 nm in a multiwell plate reader (Spectramax M2, Molecular Devices). The values obtained were normalized by dividing them by the mean value of lysozyme obtained among untreated Npc1+/− mice. Purified chicken egg white lysozyme was used as a positive control.
Cathepsin S ELISA
Total plasma cathepsin S was determined using an ELISA Duo Set kit (DY1183) from R&D Systems (Minneapolis, MN) according to the manufacturer's instructions. Plasma of Npc1+/+ and Npc1+/− mice of both Npc1nih and Npc1nmf164 strains was diluted to 1:10, whereas that of Npc1−/− mice of both strains was diluted to 1:20. All measurements were done in triplicate wells. For normalization, the raw absorbance values were divided by the average absorbance of Npc1+/− mice of each strain of a given age group.
24(S)-Hydroxycholesterol (24(S)-HC) ELISA
Plasma 24(S)-HC concentration was determined using an ELISA kit from Enzo Life Sciences (Farmingdale, NY) according to the manufacturer's instructions. Plasma was diluted to 1:1000 in the supplied buffer, and measurements were done in triplicate wells. Pure 24(S)-HC (supplied with the kit) was used to prepare the standard curve. 24(S)-HC concentration was normalized to plasma protein content.
Organ Harvest and Immunohistochemistry
Mice were sacrificed by asphyxiation using CO2. The circulatory bed was washed with PBS (pH 7.4) and subsequently perfused with 10% neutral buffered formalin (∼4% formaldehyde). The organs (brain and liver) were surgically harvested and stored in 4% formaldehyde at room temperature until transfer to paraffin. Paraffin-embedded tissue sections (3–4 μm) were dewaxed in xylene and alcohol. For Ly-6G and calbindin staining, antigen retrieval was done by preincubating deparaffinized samples with 0.05% proteinase K (Dako, Germany) in 50 mm Tris-HCl (pH 7.5) for 8 min at room temperature. CTSS and lysozyme were retrieved by boiling the sections in acidic conditions for 30 min. Sections were incubated with anti-Ly-6G (20 μg/ml), anti-calbindin (1:1000), anti-CTSS (20 μg/ml), or anti-lysozyme (1:20) overnight at 4 °C. Reagents were prepared according to the manufacturer's instructions (Vector Laboratories). The staining protocol used was described previously (19). The secondary antibody for neutrophil staining was biotinylated rabbit anti-rat IgG (mouse absorbed, Vector Laboratories) and for Purkinje neurons was biotinylated horse anti-mouse IgG (Vector Laboratories).
For fluorescence microscopy, FITC-conjugated IgG (MP Biomedicals, Solon, OH) was the secondary antibody. Sections stained only with secondary antibodies served as controls. Bright field images were acquired on a Nikon Olympus microscope using a Nikon digital DS-Fi1-U2 camera controlled by NIS-Elements F3.0 Nikon software (all from Nikon Instruments Inc., Tokyo, Japan). Images were visualized with an A10 PL 10×/0.25, DPlan Apo 40×/1.00 oil immersion, or DPlan Apo 100×/1.30 oil immersion objective lens (Nikon). Fluorescence microscopy and digital image collection were performed using an Olympus IX inverted fluorescence microscope and a Photometrix cooled charge-coupled device camera (CH350/LCCD) driven by DeltaVision software from Applied Precision (Seattle, WA). DeltaVision software (softWoRx) was used to deconvolve these images. Images were visualized with a 40× oil immersion objective lens and are single optical sections. NIH ImageJ software was used to process and quantify the fluorescence intensity of CTSS and lysozyme.
Drug Injections and Blood Withdrawal
Starting at P21and once a week thereafter, Npc1nih and Npc1nmf164 mice were injected intraperitoneally with 20% HPβCD (4000 mg/kg) prepared in 0.2% DMSO and 0.9% saline. Control mice received 0.2% DMSO in 0.9% saline. Blood was collected in EDTA tubes (BD Biosciences) either via cheek bleed or terminal heart bleed from mice. Plasma was separated by centrifugation at 2500 rpm for 15 min and stored at −70 °C until used.
Statistical Tests
Student's t test was carried out to determine the statistical significance of the data. p < 0.05 was considered significant.
RESULTS
Validation of Cathepsins B (Ctsb), D (Ctsd), and S (Ctss) in Liver and Brain of NPC Mice and NPC Patients
Of 12 potential biomarker genes identified in our previous study (19), three belonged to the cathepsin family. These were Ctsb, Ctsd, and Ctss. Although there is no information about cathepsin S in NPC disease, cathepsins B and D have been reported to be overexpressed in the cerebellar neurons in Npc1−/− mouse brain and have been linked to increased neurodegeneration (30–32), suggesting that the family may be suitable for further investigation.
Disease progression as a function of age in Npc1−/− mice is shown schematically in Fig. 1A. Our microarray data suggested that the -fold up-regulation of Ctsb, Ctsd, and Ctss was 1.5, 3.2, and 6.2, respectively, in the liver of late stage Npc1−/− mice compared with age-matched control mice (Table 2). Similarly, transcript levels of Ctsb, Ctsd, and Ctss were 2.8-, 1.9-, and 2.7-fold higher in the brain of Npc1−/− mice compared with controls (Table 2). To validate the microarray data, we performed qPCR to determine transcript increases for Ctsb, Ctsd, and Ctss in liver and brain of Npc1−/− mice at a late symptomatic stage (Fig. 1). As shown in Fig. 1B (panels i–iii), in the liver, the -fold change was 4.4 for Ctsb, 11.7 for Ctsd, and 60.6 for Ctss in Npc1−/− compared with age-matched control mice. In the brain, the -fold increase of Ctsb was 1.5, that of Ctsd was 3.7, and that of Ctss was 3.4 (Fig. 1C, panels i–iii).
FIGURE 1.

Elevated expression of cathepsin S, D, and B genes in NPC mice and patients. A, diagrammatic representation of the onset of phenotypic symptoms and life span of Npc1nih mice. qPCR reveals that Ctss, Ctsd, and Ctsb transcripts are elevated in liver (B, panels i–iii) and brain (C, panels i–iii) of Npc1−/− (−/−) mice compared with Npc1+/− (+/−) counterparts at 70–83 days. Each group consisted of four mice. The data represent mean triplicate values ±S.D. (error bars). D, panels i–iii, expression analysis of CTSS, CTSD, and CTSB in liver and cerebellum of human NPC patients. Total RNA was isolated from frozen livers and cerebella from four NPC and four control subjects. Expression levels of cathepsins were determined by qPCR. -Fold change is relative to the average value of control subjects. Change above 1 (shown by dotted line) represents the extent of overexpression. For both mouse and human qPCR studies, Gapdh was used as an internal control. *, p < 0.005.
TABLE 2.
-Fold up-regulation in the transcript level of different cathepsins
Data were taken from Ref. 19. Genome-wide gene expression analysis was done using an Affymetrix chip, and -fold differences in the Npc1−/− were calculated relative to Npc1+/− mice.
| Marker (transcript) | -Fold increase in Npc1−/− liver (67–71 days) | -Fold increase in Npc1−/− brain (81–84 days) |
|---|---|---|
| Cathepsin B | 1.53 | 2.82 |
| Cathepsin D | 3.22 | 1.95 |
| Cathepsin S | 6.26 | 2.77 |
The -fold change detected by qPCR was not the same as seen in the microarrays. Many factors such as mRNA extraction and stability, hybridization efficiency, and differences in the efficiency of cDNA synthesis may contribute to this discrepancy. Although microarrays are useful in obtaining trends of change, qPCR provides the quantitative confirmatory data.
Cluzeau et al. (20) have reported that plasma cathepsin D is elevated in NPC patients. However, information on levels of cathepsins D, S, and B in human organs is not available. Therefore, we obtained frozen livers and cerebella from four NPC and four control subjects matched for age, gender, and ethnicity. As shown in Fig. 1D, panel i, we detected increased transcripts of CTSS in liver (1.4-, 1.8-, and 2.6-fold) as well as in cerebellum (1.4-, 1.7-, and 2.8-fold) of three NPC patients. In the fourth NPC patient, CTSS transcript was unchanged in liver but decreased in cerebellum compared with controls (Fig. 1D, panel i). In contrast, CTSD expression was not increased in liver, but increases (1.3- and 7.6-fold) were seen in two NPC cerebella (Fig. 1D, panel ii). CTSB showed elevation (5.2-fold) in one of four liver samples and in the cerebellum of all four NPC patients (1.3-, 1.3-, 1.5-, and 1.7-fold) (Fig. 1D, panel iii). Because the sample size is small, the data do not rule out CTSD or CTSB as potential markers. Nevertheless, because there was an increase in CTSS in three of four patient samples for both organs and CTSS showed the greatest change in the mouse liver (60.6-fold), we investigated it as the lead marker of interest in subsequent work.
Characterization of Plasma Cathepsin S Levels in Npc1nih and Npc1nmf Mice and the Response to HPβCD
In NPC mice, weight is a central parameter to follow disease progression. The data in Fig. 2A show the weight curves of male and female Npc1nih mice as a function of their age in days. As shown in Fig. 2B, plasma CTSS in Npc1−/− mice was significantly elevated at all ages compared with age-matched Npc1+/+ and Npc1+/− mice. At the first three time points (21–28, 35–42, and 49–56 days), the levels were ∼2-fold higher (p < 0.00001) and at later times (63–70 days) became further elevated to an ∼2.5-fold increase (p < 0.00001) (Fig. 2B). The data shown in Fig. 2B are derived from both male and female animals, suggesting that elevation of CTSS was independent of gender.
FIGURE 2.

Plasma cathepsin S levels are elevated in Npc1nih and Npc1nmf164 mice and reduced after cyclodextrin treatment. A, weight as a function of age for Npc1nih male and female mice. Homozygous mutant Npc1nih (Npc1−/−) mice begin weight loss at 49–56 days followed by further decreases and death at 77–84 days. B, elevated cathepsin S detected in plasma of Npc1nih Npc1−/− mice as determined by ELISA (“Experimental Procedures”). C, weight as a function of age of Npc1nmf male and female mice. Homozygous mutant Npc1nmf (Npc1−/−) mice lose weight from 84–91 days onward followed by further decreases and death at 119–126 days. D, elevated cathepsin S level in plasma of Npc1nmf Npc1−/− mice. E, cathepsin S levels in Npc1nih Npc1−/− (−/−) mice treated with saline or HPβCD compared with Npc1+/− (+/−). F, cathepsin S levels in Npc1nmf Npc1−/− (−/−) mice treated with saline or HPβCD compared with Npc1+/− (+/−). In B, D, E, and F, blood plasma was sampled at the indicated time points. -Fold change in cathepsin S is expressed relative to average levels of activity in Npc1+/− mouse plasma. The data represent mean triplicate values ±S.D. (error bars). Median values are shown by horizontal lines. Statistical significance was determined using Student's t test. *, p < 0.0001 in B, p < 0.05 in D, and p < 0.00001 in E. NS indicates not significant. gm, grams; AU, arbitrary units.
We further examined plasma from Npc1nmf mouse. Previous studies suggested that Npc1nmf mice in the C57BL/6J background have a life span of ∼112 days and develop progressive disease (29). BALB/c Npc1nmf have a comparable life span (∼120–125 days) and exhibited weight loss from 85 to 90 days (19). As shown in Fig. 2C, plasma CTSS levels were indeed elevated ∼1.4–1.6-fold (p < 0.05) in both early (∼75 days) and later (100 days) symptomatic stages. Remarkably, HPβCD reduced levels of CTSS at late stages (80–114 days) in Npc1nih (Fig. 2E) and mild to moderately symptomatic Npc1nmf mice to those seen in healthy controls (Fig. 2, E and F). These findings were surprising because HPβCD-treated Npc1nih and Npc1nmf mice manifest disease at 100 days of age.
Effect of HPβCD Treatment on Ctss, Ctsd, Ctsb, and Other Inflammatory Marker Expression in Liver and the Pathologies of the Organ in Npc1nih Mice
To investigate whether cathepsin levels in the plasma of Npc1−/− mice reflect disease status of the liver and its response to HPβCD, we studied the effect of treatment on (i) the expression levels of Ctss, Ctsd, and Ctsb and (ii) liver pathology. After HPβCD treatment, the expression of Ctss in the liver of late stage Npc1−/− mice was markedly reduced and equivalent to that in control mice (Fig. 3A). Similar trends were also observed in the expression of Ctsd and Ctsb (Fig. 3, B and C).
FIGURE 3.

Effect of cyclodextrin treatment on the expression of cathepsins and additional markers of inflammation and fibrosis in mouse liver. Ncp1nih Npc1−/− mice (−/−) were given weekly HPβCD injections. Npc1+/− (+/−) animals remained untreated. Animals were sacrificed at the indicated ages, organs were removed and processed, and total RNA was extracted. qPCR was undertaken to determine the expression level of Ctss (A), Ctsd (B), Ctsb (C), Cd68 (D), Itgax (E), and Cola1 (F). Each group consisted of four mice except for the Cola1 assay. Cola1 included six Npc1+/−, six untreated Npc1−/−, and seven HPβCD-treated Npc1−/− mice. -Fold changes shown indicate transcript levels in Npc1−/− relative to Npc1+/− mice. Gapdh was used as an internal control. The data represent the mean ± S.D. (error bars). The data shown for untreated Npc1−/− mice in A–C are identical to those shown in Fig. 1B to enable comparisons across the study. *, Npc1+/− versus untreated Npc1−/−, p < 0.005; **, untreated Npc1−/− versus treated Npc1−/−, p < 0.005.
We undertook analysis of additional inflammatory markers and histology. We studied the expression of two inflammatory genes, Cd68 (macrophage marker) and Itgax (a marker of activated macrophage, granulocytes, dendritic cells, etc.; also known as Cd11c). qPCR analysis showed that Cd68 was up-regulated by ∼88-fold (Fig. 3D) and Itgax was up-regulated by ∼400-fold (Fig. 3E) in Npc1−/− mice at late stages (70–83 days) in the liver. These were reduced to normal levels after HPβCD treatment (Fig. 3, D and E), suggesting amelioration of liver inflammation. A third marker, Cola1 (procollagen type a1), shown to be up-regulated during liver fibrosis (10) was increased (∼1.3-fold) in Npc1−/−. HPβCD induced anomalous reduction in its expression (Fig. 3F), suggesting that although HPβCD treatment reduced inflammation it may also adversely change levels of important molecular determinants of the liver.
To study the expression of CTSS protein and its localization in the liver, sections were subjected to immunohistochemistry using anti-CTSS antibodies. The liver of Npc1+/− mice (age, 80 days) showed healthy hepatocyte architecture (Fig. 4A). In contrast, numerous large foamy macrophages containing high levels of CTSS were seen in Npc1−/− mice of the same age (Fig. 4B, blue arrows). Saline had no effect (Fig. 4C), but HPβCD treatment eliminated accumulation of foamy macrophages and dramatically reduced CTSS accumulation in Npc1−/− (Fig. 4D).
FIGURE 4.

Treatment with cyclodextrin reduces the CTSS level, accumulation of neutrophils, and lysozyme in Npc1nih mouse liver. A–D, formalin-fixed paraffin-embedded liver sections (4–5 μm) of Npc1+/− and Npc1−/− mice at the indicated ages were stained with anti-CTSS antibodies. The micrographs show the labeling of CTSS in untreated Npc1+/− mice (A), untreated Npc1−/− mice (B), Npc1−/− mice treated with saline (C), and Npc1−/− mice treated with HPβCD (D). CTSS staining (brown) was seen in the foamy macrophages (blue arrows) of untreated and saline-treated Npc1−/− mice. HPβCD injection eliminated the foamy macrophages and CTSS staining. To visualize neutrophils, sections were stained with anti-Ly-6G antibodies. In E, liver sections are shown from Npc1+/− mice at 54 days (E1) and 80 days (E2). One to two neutrophils (cells stained in brown) are infrequently seen in these sections. F, detection of giant foci of neutrophils (cluster of brown cells; blue arrows) in the liver of an Npc1−/− mouse at age 54 days (F1). Increased sizes of neutrophil clusters were seen as mice aged to 80 days (F2). G, large foci of neutrophils were also seen in liver sections of Npc1−/− mice treated with saline at 50 (G1) and 80 days (G2). H, neutrophils were barely detected in liver sections from Npc1−/− mice treated with HPβCD at either 50 or 80 days (H1 and H2). Original magnifications, ×40. Representative images are shown. I, mRNA levels of Lyz1 in liver. Npc1+/− (+/−) and Npc1−/− (−/−) mice treated with saline or HPβCD were sacrificed between 70 and 95 days. Total RNA was extracted from liver, and the expression of Lyz1 was quantified by qPCR (as described under “Experimental Procedures”). Gapdh was used as an internal control. -Fold changes shown are relative to average levels of Lyz1 transcripts detected in Npc1+/− mice. The data represent mean triplicate values ±S.D. (error bars). Data were subjected to the Student's t test for statistical significance. J–M, immunofluorescence analyses of lysozyme in liver: effects of HPβCD. Liver sections of Npc1+/− and Npc1−/− mice were stained with anti-mouse lysozyme antibodies. Immunostaining shows the expression of lysozyme in the liver sections of untreated Npc1+/− mice (J), untreated Npc1−/− mice (K), Npc1−/− mice treated with saline (L), and Npc1−/− mice treated with HPβCD (M). Enhanced lysozyme staining (green) was seen in the foamy macrophages (white arrows) of untreated and saline-treated Npc1−/− mice. d, days.
We reported previously that giant foci of neutrophils accumulate in liver of Npc1−/− mice, suggesting that they contribute to the inflammatory response (19). Therefore, we also examined the effects of HPβCD on neutrophil accumulation in the liver. Immunohistochemical analyses did not show neutrophil infiltration in healthy mice at 54 and 80 days (Fig. 4, E1 and E2). In contrast, in diseased mice, clusters of neutrophils were clearly seen (Fig. 4, F1, Ly-6G-positive cells in brown, shown by blue arrows). The number of neutrophils increased in the liver of diseased mice at 80 days (Fig. 4, F2). Administration of saline had no effect (Fig. 4, G1 and G2), but HPβCD treatment reduced neutrophil clusters at both 50 (Fig. 4, H1) and 80 days (Fig. 4, H2). However, there were differences in hepatocyte architecture (decreased cytoplasmic staining with slightly irregular plasma membrane) in animals treated with HPβCD, suggesting that treatment does not completely restore all aspects of liver health (at least as judged by histochemistry).
Our previous microarray studies also reported the up-regulation of lysozyme transcripts in the liver of diseased animals. As shown in Fig. 4I, we detected ∼85-fold up-regulation of Lysozyme1 (Lyz1) gene in the Npc1−/− liver at late stages of disease compared with age-matched control mice. HPβCD treatment abrogated Lyz1 overexpression in the liver of Npc1−/− mice, bringing transcript levels back to those seen in healthy mice (Fig. 4I). To confirm that lysozyme protein levels were also elevated and determine the site(s) of concentration, we undertook immunohistochemical analyses of liver sections using antibodies to mouse lysozyme (see “Experimental Procedures”). Large foamy macrophages contained high levels of lysozyme in the liver of Npc1−/− mice (age 80 days). They were absent in healthy animals (Fig. 4, J and K). HPβCD treatment (Fig. 4M) but not saline (Fig. 4L) largely eliminated the macrophages containing lysozyme from the liver. A low, basal level of lysozyme expression was seen in hepatocytes but at levels comparable with that in healthy mice (Fig. 4M).
Together, these data provide new markers (such as CTSS and lysozyme) to confirm prior findings that HPβCD treatment improves inflammation in the liver. Furthermore, they show that HPβCD treatment returns CTSS levels in the liver of diseased animals to that seen in healthy counterparts (Figs. 3 and 4), which is analogous to effects seen with this marker in plasma (Fig. 2).
Effect of HPβCD Treatment on Ctss, Ctsd, and Ctsb Expression in Brain and the Pathologies of the Organ in Npc1nih Mice
In the brain, HPβCD treatment resulted in partial reduction of Ctss and Ctsd (Fig. 5, A and B), whereas Ctsb expression returned to normal levels (Fig. 5C). Activation of microglia and astrocytes in the brain has been associated with neuroinflammation and neurodegeneration in NPC disease (14, 33, 34). qPCR analyses of associated markers revealed ∼24-fold up-regulation of Cd68 in Npc1−/− mice (70–83 days) compared with age-matched Npc1+/− mice (Fig. 5D). The expression of Cd68 in Npc1−/− mice treated with HPβCD was reduced significantly, but there was still ∼6-fold higher transcript relative to Npc1+/− mice (Fig. 5D). Furthermore, mutant animals showed an ∼44-fold increase in Mip-1α transcript, which was reduced to ∼27-fold upon treatment with HPβCD (Fig. 5E). Untreated Npc1−/− mice at late stages showed ∼9-fold higher Gfap transcript compared with age-matched controls (Fig. 5F) that was slightly reduced in HPβCD-treated animals (Fig. 5F). These results are consistent with the data with Ctsb, Ctsd, and Ctss as markers and prior reports that HPβCD partially alleviates neuroinflammation in Npc1−/− mice (23, 35–37).
FIGURE 5.

Effect of cyclodextrin treatment on the expression of cathepsins and inflammatory markers in Npc1nih mouse brain. Total RNA from the brain was prepared, and qPCR analysis was carried out for Ctss (A), Ctsd (B), Ctsb (C), Cd68 (D), Mip-1α (E), and Gfap (F). For Ctss, Ctsd, and Ctsb (A–C), there were four mice per group. For Cd68, Mip-1α, and Gfap (E–F), there were six mice per group. -Fold change in Npc1−/− is expressed relative to transcript levels in Npc1+/− mice. The data represent mean triplicate values ±S.D. (error bars). Gapdh was used as an internal control. The data shown for untreated Npc1−/− mice in A–C are identical to those shown in Fig. 1C to enable comparisons across the study. *, Npc1+/− versus untreated Npc1−/−, p < 0.005; **, untreated Npc1−/− versus treated Npc1−/−, p < 0.05. G–J, immunofluorescence micrographs showing the expression of CTSS in the hippocampal neurons of untreated Npc1+/− mice (G), untreated Npc1−/− mice (H), Npc1−/− mice treated with saline (I), and Npc1−/− mice treated with HPβCD (J). Enhanced CTSS staining was seen in untreated and saline-treated Npc1−/− mice. Original magnifications, ×40. Representative images are shown. K, quantification of CTSS fluorescence using NIH ImageJ software. Eight sections (two mice/group, four sections/mouse) were analyzed. The data represent the mean ± S.D. (error bars). *, p < 0.05. d, days.
Immunohistochemical analyses of brain sections showed enhanced labeling of CTSS in the pyramidal neurons of hippocampus of Npc1−/− mice (Fig. 5, G–J), recognized by their characteristic morphology. Quantitative analysis using ImageJ revealed an 1.7-fold (p < 0.05) increase in CTSS in Npc1−/− mice. HPβCD treatment slightly reduced CTSS levels (∼20%), whereas saline had no effect (Fig. 5K). CTSS was expressed at low levels in the rest of the brain (not shown), but there were no differences between Npc1+/− and Npc1−/− mice elsewhere including the cerebellum (data not shown).
Loss of Purkinje neurons in the cerebellum is a characteristic feature of NPC disease and has been used as a benchmark to study brain pathology (24, 34, 38, 39). To study the effect of HPβCD on Purkinje neuron death, immunohistochemical staining of sagittal sections of brain of Npc1−/− mice was carried out using anti-calbindin (a marker of Purkinje neurons) antibody. Mouse cerebellum is composed of 10 (I–X) different lobules. All cerebellar sections were examined; however, in Fig. 6, micrographs corresponding to IX lobule are shown as representative images. Numerous Purkinje neurons (Fig. 6A, stained in brown, shown by arrows) were clearly seen in the cerebellar section of Npc1+/− mouse at 80 days, whereas in Npc1−/− mice of the same age, the numbers were markedly reduced (five to eight/lobule) (Fig. 6B). This was unchanged upon treatment with saline (Fig. 6C). However, treatment with HPβCD preserved additional Purkinje neurons in the IX lobule of the cerebellum (Fig. 6D) although at lower levels than in Npc1+/− animals. The intensity of calbindin-positive neurons in HPβCD-treated Npc1−/− mice were reduced compared with Npc1+/− animals (Fig. 6, compare intensity of brown staining seen in A1 and A2 with that in D1 and D2). Purkinje neurons were also seen in the X lobule (data not shown). Even after HPβCD treatment, Purkinje neurons were barely seen in the rest of the cerebellar regions of Npc1−/− mice (data not shown). A semiquantitative analysis of Purkinje neurons in a cerebellar section showed they were significantly reduced in number in Npc1−/− (39 ± 11) mice as compared with Npc1+/− mice (473 ± 75). Saline treatment showed no effect (40 ± 8), whereas HPβCD treatment showed an increased number of Purkinje neurons (100 ± 16) (Fig. 6E). The data are consistent with prior results (22, 24, 40) that HPβCD treatment may to a small degree resolve neuroinflammation and inhibit loss of Purkinje neurons. Furthermore, the data are consistent with our marker analysis for inflammatory proteins including the cathepsins.
FIGURE 6.

Effects of cyclodextrin on the Npc1nih mouse brain as determined by immunohistochemistry. Formalin-fixed paraffin-embedded brains were sectioned (sagittally, 4–5 μm) and stained using anti-mouse calbindin antibodies to visualize Purkinje neurons in the entire cerebellum. Micrographs shown are representative images of the IX lobule of the cerebellum. A, Purkinje neurons (stained in brown) indicated by black arrows are evident in Npc1+/− mice (age, 80 days). A1 and A2 are the magnified areas boxed in A. B, loss of Purkinje cells in the cerebellum of an Npc1−/− mouse (age, 80 days). Calbindin immunoreactivity was barely detected across the different lobules of cerebellum. B1 and B2 are magnified areas boxed in B. C, cerebellar section of an Npc1−/− mouse (age, 80 days) injected with saline was devoid of Purkinje neurons. C1 and C2 are magnified areas boxed in C. D, chronic HPβCD treatment (that partially rescued inflammation) also partially recovered Purkinje neurons in Npc1−/− mice. A few lightly brown stained Purkinje neurons (indicated by arrows) are seen. D1 and D2 are magnified areas boxed in D. Original magnifications, ×40. E, semiquantitative analysis of Purkinje neurons in an Npc1nih mouse brain. Numbers of Purkinje neurons in the calbindin-labeled cerebellar sections from four mice (age, 80 days) in each group were counted. HPβCD treatment resulted in a small but significant increase in the number of Purkinje neurons. The data represent the mean ± S.D. (error bars). NS, not significant.
Characterization of Lysozyme Levels in Plasma and Brain in HPβCD-treated Npc1nih Mice at Terminal Stages of Disease: Localization of Lysozyme Elevation in the Cerebellum and Development of a Composite Scale to Distinguish among Four Distinct States of Cerebral and Liver Disease
Our prior study (19) identified lysozyme transcripts as the most highly elevated in the brain of Npc1nih mice. We further validated the elevation of lysozyme in the plasma of Npc1nih as well as a second mouse model, Npc1nmf. Additionally, we showed that plasma lysozyme levels elevated in asymptomatic Npc1nmf mice (age, ∼50 days) were reduced by HPβCD treatment and rendered comparable with untreated wild type animals (19).
To examine time points of advanced disease, we returned to the shorter Npc1nih model. This model typically manifests phenotypic symptoms (weight loss, gait, tremor, etc.) from ∼50 to 55 days and survives up to ∼80–84 days (Fig. 7A). Previous studies have shown that weekly injections of HPβCD to Npc1nih reduce disease and extend survival (23, 24). Therefore, we treated Npc1−/− mice with HPβCD or vehicle control (0.2% DMSO in 0.9% saline) by once a week drug injections (4000 mg/kg) starting at age P21. Npc1nih mice treated with this regime showed delayed onset of symptoms (from ∼49–56 to ∼70–80 days) and survived ∼105–112 days (diagrammatically represented in Fig. 7A). As reported earlier, the plasma lysozyme activity of vehicle-treated Npc1−/− mice was elevated on average 2-fold in early symptomatic animals (50–60 days) (Fig. 7B) and remained elevated at the late symptomatic stage (70 days) compared with age-matched controls (Fig. 7C). Furthermore, HPβCD treatment at 50–60 days reduced plasma lysozyme activity levels seen in wild type mice (Fig. 7B). At 80+ days, HPβCD-treated animals showed a reduction of plasma lysozyme compared with mock-treated animals but nonetheless displayed a 1.5-fold increase compared with normal animals (Fig. 7C).
FIGURE 7.

Cyclodextrin partially reduces lysozyme levels in plasma and brain of Npc1nih mice. A, diagrammatic representation of the onset of phenotypic symptoms and life span of Npc1nih mice (upper panel) and their improvement upon treatment with HPβCD (lower panel). Plasma lysozyme activity was determined for Npc1−/− mice (−/−) treated with saline or HPβCD compared with untreated Npc1+/− mice (+/−) at 49–56 (B) and 80–114 days (C). -Fold changes shown are relative to average levels of lysozyme activity detected in Npc1+/− mice. Horizontal lines indicate median values. D, mRNA levels of Lyz1 in brain. Npc1+/− (+/−) and Npc1−/− (−/−) mice treated with saline or HPβCD were sacrificed between 70 and 95 days. Total RNA was extracted from liver and brain, and the expression of Lyz1 was quantified by qPCR (as described under “Experimental Procedures”). Gapdh was used as an internal control. -Fold changes shown are relative to average levels of Lyz1 transcripts detected in Npc1+/− mice. Data represent the mean of three experiments ±S.E. (error bars). Data were subjected to the Student's t test for statistical significance. NS, not significant. Un, untreated; AU, arbitrary units. *, p < 0.05 in B and p < 0.0005 in C.
This persistent elevation of plasma lysozyme could not be derived from the liver because as shown previously HPβCD treatment restored lysozyme transcript and protein to normal levels in the liver (Figs. 3 and 4). Therefore, we examined the brain where qPCR revealed an ∼81-fold up-regulation of Lyz1 in Npc1−/− mice at late stages of disease (Fig. 7D). Moreover, after HPβCD treatment, Lyz1 expression remained elevated by ∼28-fold (Fig. 7D).
In immunolocalization studies by fluorescence microscopy, low levels of lysozyme were detected throughout the brains of normal and diseased mice except in the cerebellum where there was a marked increase in the mutant animals (Fig. 8, B–K). Numerous cells highly positive for lysozyme were seen in the cerebellar white matter of Npc1−/− mice (80 days) but not in Npc1+/− mice (Fig. 8, B and C). HPβCD treatment had no significant effect on the lysozyme levels in the cerebellum (Fig. 8, E and F). Lysozyme was also elevated in the molecular layer of the cerebellum (Fig. 8, G–K). In this region, healthy mice showed minimal levels of lysozyme staining (Fig. 8G), whereas elevated fibrillar staining was seen in the Npc1−/− mice (Fig. 8H). Saline treatment had no effect, but HPβCD treatment resulted in a minor reduction (∼25%; Fig. 8, I–K). Although CTSS was increased in the hippocampus of mutant mice (Fig. 5, G–J) lysozyme was unchanged here (Fig. 8, L–P) and as indicated earlier in the rest of the brain (data not shown).
FIGURE 8.

Immunohistochemical analyses of lysozyme in brain of Npc1nih mice. A, schematic illustration of the structure of mouse cerebellum. gcl, granule cell layer; wm, white matter; ml, molecular layer. Brain sections of Npc1+/− and Npc1−/− mice were stained with anti-mouse lysozyme antibodies. B–E, immunohistochemical micrographs corresponding to the granule cell layer and white matter of the cerebellum of untreated Npc1+/− mice (B), untreated Npc1−/− mice (C), Npc1−/− mice treated with saline (D), and Npc1−/− mice treated with HPβCD (E). Numerous lysozyme-positive cells (green; indicated by white arrows) were primarily seen in the white matter of the cerebellum of Npc1−/− mice. F, bar diagram showing the total lysozyme intensity in the cerebellum corresponding to the region shown in B–E. G–J, region corresponding to the granule cell layer and molecular layer of the cerebellum of untreated Npc1+/− mice (G), untreated Npc1−/− mice (H), Npc1−/− mice treated with saline (I), and Npc1−/− mice treated with HPβCD (J). Enhanced lysozyme staining in the molecular layer of the cerebellum of untreated and saline-treated Npc1−/− mice was seen. K, bar diagram showing the total lysozyme intensity in the cerebellum corresponding to the region shown in G–J. L–O, immunohistochemical micrographs showing the staining of lysozyme in the hippocampus of untreated Npc1+/− mice (L), untreated Npc1−/− mice (M), Npc1−/− mice treated with saline (N), and Npc1−/− mice treated with HPβCD (O). P, quantification of lysozyme fluorescence in the hippocampus corresponding to the region shown in L–O. Nuclei (blue) are stained with DAPI. Treatment and age of the mouse are shown. Original magnifications, ×40. Representative images are shown. NIH ImageJ was used for the quantification of lysozyme fluorescence. In cerebellum, 20 different fields (two mice/group, 10 fields from each mouse) were analyzed. In hippocampus, eight sections (two mice/group, four sections/mouse) were analyzed. The data represent the mean ± S.D. (error bars). *, p < 0.05. Q, schematic summarizing the elevation of lysozyme in the cerebellum of the Npc1−/− mouse brain. NS, not significant. d, days.
In several instances, -fold changes in plasma levels of both cathepsin and lysozyme were considerably lower than their transcript levels in brain and liver but more in keeping with changes seen by immunohistochemistry, which is to be expected because the latter is a readout of protein levels in tissue. Taken together, these data suggest that HPβCD given postweaning into the body cavity can deplete lysozyme in the liver. It may also reduce to a small extent lysozyme in the brain, but significant levels persist. Remarkably, lysozyme elevation in the brain of diseased animals appears concentrated in the cerebellum whose function is prominently compromised in NPC (summarized in Fig. 8Q).
Nonetheless, prior to HPβCD treatment, plasma levels of lysozyme likely reflect inflammation in the brain as well as the liver in mice and humans (Figs. 4, 7, 8, and 9A). To estimate the contribution from the inflamed liver, we needed a second marker whose levels in plasma solely reflect that of the liver (such as CTSS). Thus, we considered that lysozyme along with CTSS may contribute to a composite, quartile scale for both cerebral disease and inflamed liver (Fig. 9B, panels i–iii). The first two quartiles reflect elevated lysozyme and thus potential contribution from cerebral disease (Fig. 9B). However, simultaneous elevation of CTSS in the second quartile suggests liver inflammation that can also contribute lysozyme to the plasma. Accordingly, HPβCD, which abrogates CTSS, decreases lysozyme by ∼50%, reflecting that one-half of lysozyme activity is contributed by the liver and the other half is contributed by the brain. Thus, for the same lysozyme levels, quartile 2 is expected to reflect more moderate levels of cerebral disease compared with quartile 1 (Fig. 9B). Quartile 4 reflects low disease, whereas quartile 3 is indicative of just liver disease (Fig. 9B).
FIGURE 9.
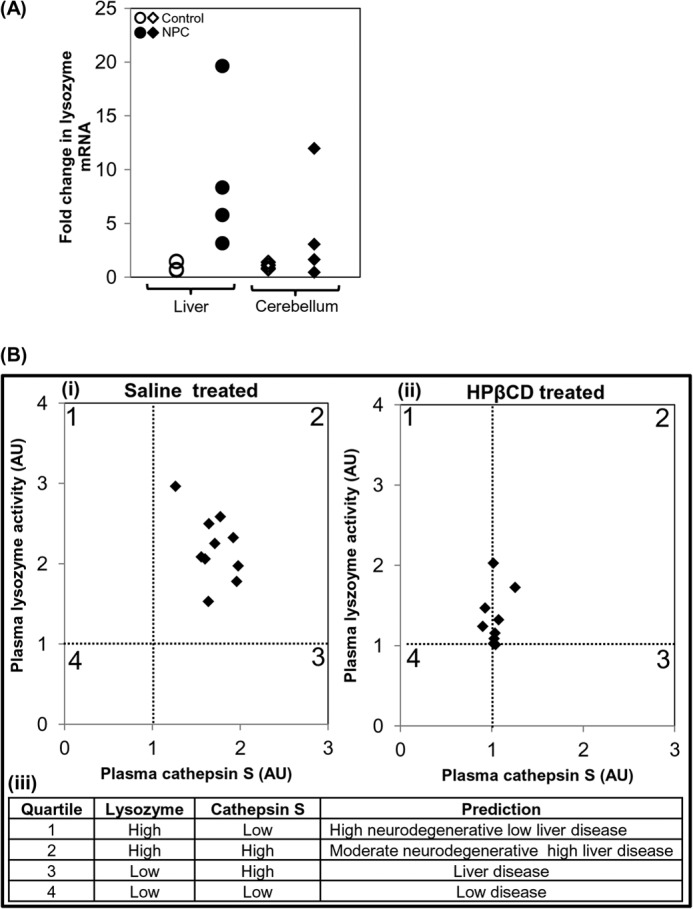
Evidence of lysozyme elevation in patients and development of a composite scale to distinguish among four distinct states of cerebral and liver disease. A, expression analysis of LYZ in livers and cerebella of four human NPC patients. Expression levels of LYZ were determined by qPCR. -Fold change is relative to the average value of control subjects. GAPDH was used as an internal control. B, correlation between plasma lysozyme activity and cathepsin S level in NPC mice treated with saline (panel i), HPβCD (panel ii), and a derived quartile score predictive of four distinct states of neurodegeneration and liver disease (panel iii), suggesting composite plasma diagnostic of neuroinflammation. AU, arbitrary units.
Comparison with Oxysterol Markers
Oxysterol species are emerging as markers of NPC disease (41, 42). Plasma oxysterols (7-ketocholetserol and 3β,5α,6β-triol) generated by non-enzymatic pathways are largely produced by the liver (43, 44) and thus are likely to be more useful to understand liver pathology rather than brain pathology. However, 24(S)-HC is derived from cholesterol by an enzyme, 24-hydroxylase, which is primarily expressed in the neurons of the central nervous system (45, 46). As shown in Fig. 10A, we detected an ∼25% reduction in the expression of Cyp46a1 (24-hydroxylase) gene in the brain of Npc1−/− mice at late stage disease. This is consistent with a slight reduction of 24(S)-HC reported in NPC patients (42), but our analysis of human cerebellum in four patients suggested variability in transcript levels (Fig. 10B). Unexpectedly, the levels of 24(S)-HC in the plasma of Npc1−/− mice were elevated and remained largely unaffected after HPβCD treatment (Fig. 10C). Thus, plasma 24(S)-HC may not assess neuropathology in mouse models.
FIGURE 10.
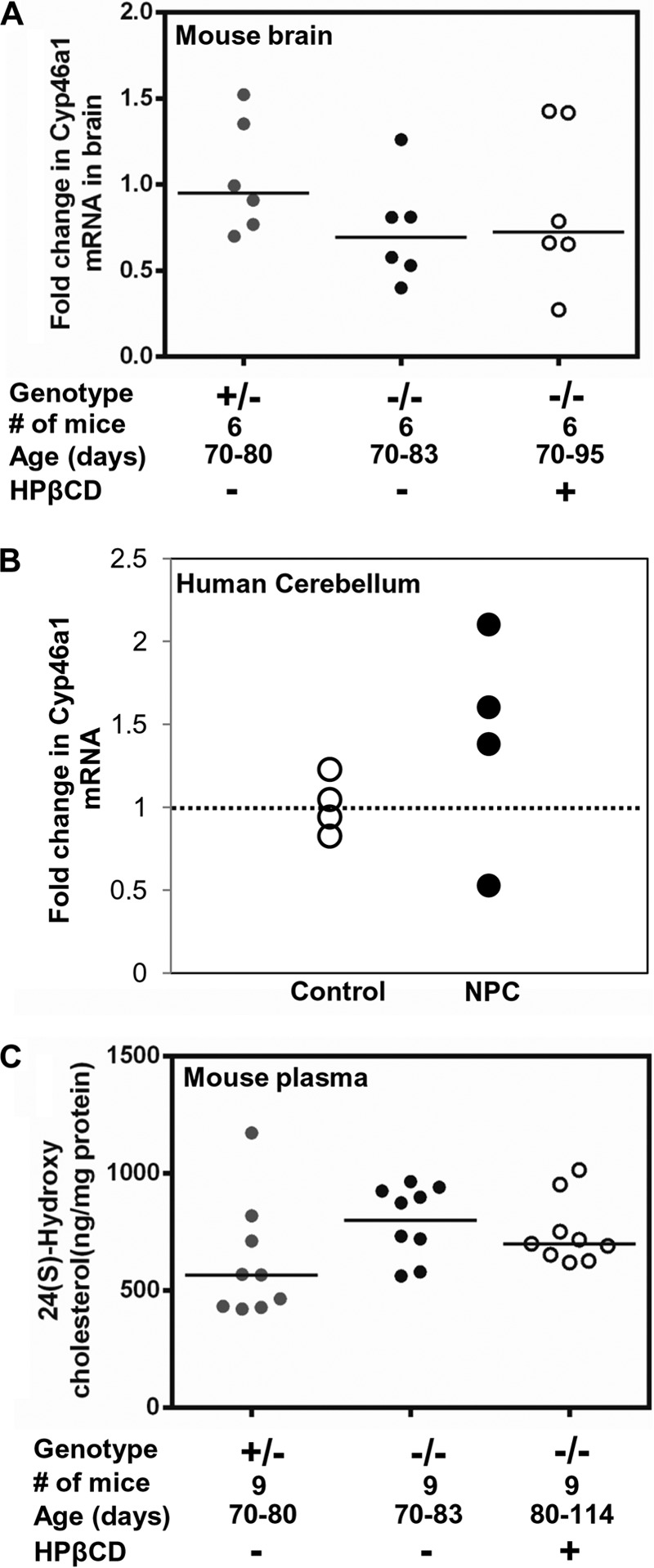
Expression of 24-hydroxylase gene and plasma 24(S)-hydroxycholesterol level in NPC. A, expression of 24-hydroxylase (Cyp46a1) gene in NPC mouse brain. The Cyp46a1 RNA level was determined by qPCR with total RNA extracted from the brain of Npc1+/− (+/−; n = 6), Npc1−/− (−/−; n = 6), and HPβCD-treated Npc1−/− (−/−; n = 6) mice. -Fold increase is expressed relative to transcript levels in Npc1+/− mice. B, expression of CYP46A1 gene in the cerebella of four NPC and four control subjects. Expression levels were determined by qPCR. -Fold change is relative to the average value of control subjects. Change above and below 1 (shown by the dotted line) represents the extent of up- and down-regulation, respectively. For both mouse and human qPCR studies, Gapdh was used as an internal control. Horizontal bars show the median values. C, 24(S)-HC levels in Npc1nih Npc1−/− (−/−) mice treated with saline or HPβCD compared with Npc1+/− (+/−). The plasma concentration of 24(S)-HC was determined by ELISA (see “Experimental Procedures”).
DISCUSSION
Oxysterols are emerging as sensitive blood-based biomarkers for NPC (42). However, they are largely products of the liver, not the brain. In addition, the disease is heterogeneous with respect to both neurological and metabolic symptoms as well as age of onset, which strongly argues for the need for multiple markers.
Elevation of several cathepsins including CTSB, CTSD, and CTSS has been implicated in neurodegenerative diseases (47). The level and activity of CTSB and CTSD are elevated in the hippocampal, cerebellar, and cortical neurons (30, 31, 34) of Npc1−/− mice. By immunohistochemistry, CTSS can be detected in almost all regions of the brain. However, CTSS was elevated only in hippocampal neurons of Npc1−/− mice compared with healthy counterparts. It is possible that the hippocampal neurons can tolerate a minor elevation of these proteases and remain resistant to degeneration. Increased cytosolic levels of CTSB and CTSD have been shown to activate the autophagic pathways, thereby leading to neuronal death in Npc1−/− cells or mice (31, 32). CTSS may do the same. Additionally, activated microglia can release CTSB and CTSD that along with CTSS can induce neuronal death through digestion of extracellular matrix (48).
But importantly, CTSS detected in plasma of NPC mouse models does not reflect cerebral disease but is derived largely from the liver. Our studies suggest that among the cathepsins, cathepsin S appears to be the best candidate biomarker for liver disease. Although transcript analysis in mouse organs suggests that Ctss increases gradually, direct measurements in plasma revealed high levels from the outset. The marked elevation of Ctss in the liver and its concomitant responsiveness to HPβCD treatment in plasma and liver suggest that it may be a preferred marker of early liver disease. This is of value because although neurodegeneration is a prominent feature and linked to fatal disease, NPC is recognized as a significant cause of liver disease in early life (5–7). A history of neonatal jaundice or persisting hepatosplenomegaly is common among patients with early and late infantile onset disease. NPC is the second most common cause of neonatal cholestasis resulting in liver failure and death of ∼10% patients (49, 50). Thus, along with oxysterols, plasma CTSS may also help in the diagnosis of NPC, particularly in a newborn child or infants manifesting cholestatic jaundice along with hepatomegaly or splenomegaly.
The Purkinje cell layer in the cerebellum contains two types of cells, Purkinje neurons and Bergmann glial cells. At advanced disease states, the Purkinje neurons are largely lost in Npc1−/− mice. This suggests that increased lysozyme in the Purkinje cell layer and molecular layer is due to its expression and secretion by Bergmann glial cells. Activated microglia and Bergmann glial cells may secrete a higher level of lysozyme that may also play a role in the loss of Purkinje neurons in Npc1−/− mice (through mechanisms that remain undefined). Lysozyme at higher concentration has been shown to be amyloidogenic (51), and exposure of cultured rat neurons to oligomers of hen egg white lysozyme has been found to induce hyperphosphorylation of tau (52). In fact, neurons expressing lysozyme have been shown to have increased hyperphosphorylated tau in the mucopolysaccharidosis type IIIB mouse brain (28). Therefore, it is plausible that overexpression of lysozyme may allow it to reach a critical concentration at which it either oligomerizes or aggregates and serves as a template for the aggregation of tau and its phosphorylation in the cerebellum. Importantly, cerebellar ataxia is a major clinical symptom of NPC.
Prior studies have suggested that macrophage activation and accumulation in the liver are responsive to HPβCD treatment (22, 23). We confirm that with two new markers, CTSS and lysozyme, and also show that neutrophil accumulation is reduced, suggesting that both types of inflammatory cells respond to lipid accumulation. One possibility is that anomalous neutrophil migration occurs in response to changes in lipid gradients to inflict inflammatory damage, which is then removed by macrophage action. At late stages of disease, reduction of inflammatory proteins lysozyme and CTSS in plasma closely corresponds to reduction of inflammation in the liver. However, the liver is not completely “normal.” The observed reduction of collagen in liver can be correlated with compromised cellular organization, suggesting that high levels of HPβCD in circulation may also have adverse effects on the liver. Nonetheless, the dramatic reduction in inflammation may outweigh adverse effects, resulting in a net benefit. Further studies are required to establish improved liver function.
HPβCD injections have been shown previously to slightly but detectably improve brain pathology and levels of inflammatory markers (21–24, 35, 37). Our data are consistent with these findings both with respect to organ pathologies and marker analysis. Nonetheless, the improvement in liver pathology after HPβCD treatment far exceeds that in the brain. In the initial microarray analysis of the age-dependent increase in transcripts, lysozyme was the topmost transcript hit. Because both brain pathology and plasma lysozyme levels are relatively refractory to intraperitoneal HPβCD injections, it is likely they are linked. Indeed, HPβCD-treated animals, although rescued in liver pathology, nonetheless die of cerebral disease.
How loss of NPC protein function leads to neuroinflammation is poorly understood. One possibility is that lysosomal functions are compromised due to harmful accumulation of cholesterol and other lipids. In response, cellular systems may compensate the functional loss by overexpressing lysosomal proteins such as cathepsins and lysozyme. This may be a general phenomenon as neuroinflammation is the hallmark of almost all lysosomal storage diseases (2, 53). Malfunctioning of the lysosomal system may hamper phagocytosis, rapid membrane synthesis, and recycling in macrophages and microglial cells, which in turn may lead to their activation and subsequent overexpression of markers of neuroinflammation.
Inflammatory proteins corresponding to members of chemokines and cytokines family have been explored in cerebrospinal fluid of NPC patients; however, further investigation is required to establish their usefulness as biomarkers (54). Oxysterols largely reflect liver function, although 24(S)-HC has been proposed as a marker for neuronal disease in humans because it is produced in the brain. However, the Npc1−/− mouse model failed to provide insights into the utility of this marker for human disease.
Rather, our data show that plasma lysozyme is derived from the brain and overexpressed in the cerebellum. This is important because cerebellar ataxia is a major symptom of NPC. Lysozyme in conjunction with CTSS may be used to distinguish distinct states of brain and liver disease that have hitherto not been possible but would be very helpful to monitor the progression and management of human disease. In this regard, mouse models may be particularly helpful in dissecting the differential response of major disease organs to emerging therapeutics in both preclinical and clinical studies.
Acknowledgments
We thank Professor Tomas Ganz (University of California at Los Angeles) for antibodies to mouse lysozyme. Studies with human tissue and mice were performed with respective approval and authorization from the Institutional Review Board (IRB) and the Animal Care and Use Committee of University of Notre Dame, Indiana. Human tissues were obtained from the NICHD Brain and Tissue Bank for Developmental Disorders at the University of Maryland, Baltimore, MD. The IRB approval number for experimentation with human tissues was FWA 00002462. We thank Professor Robert P. Erickson, University of Arizona Health Sciences Center, Tucson, AZ, for providing a breeding pair of Npc1nmf164.
This work was supported, in whole or in part, by National Institutes of Health Grant P01HL078826 (to K. H.). This work was also supported by the Center for Rare and Neglected Diseases at the University of Notre Dame, Indiana.
- NPC
- Niemann-Pick Type C
- CTSS
- cathepsin S
- Lyz
- lysozyme
- HPβCD
- 2-hydroxy-propyl-β-cyclodextrin
- 24(S)-HC
- 24(S)-hydroxycholesterol
- qPCR
- quantitative PCR
- Ctsb
- cathepsin B
- Ctsd
- cathepsin D.
REFERENCES
- 1. Cappellano G., Carecchio M., Fleetwood T., Magistrelli L., Cantello R., Dianzani U., Comi C. (2013) Immunity and inflammation in neurodegenerative diseases. Am. J. Neurodegener. Dis. 2, 89–107 [PMC free article] [PubMed] [Google Scholar]
- 2. Parkinson-Lawrence E. J., Shandala T., Prodoehl M., Plew R., Borlace G. N., Brooks D. A. (2010) Lysosomal storage disease: revealing lysosomal function and physiology. Physiology 25, 102–115 [DOI] [PubMed] [Google Scholar]
- 3. Wilms H., Zecca L., Rosenstiel P., Sievers J., Deuschl G., Lucius R. (2007) Inflammation in Parkinson's diseases and other neurodegenerative diseases: cause and therapeutic implications. Curr. Pharm. Des. 13, 1925–1928 [DOI] [PubMed] [Google Scholar]
- 4. Vanier M. T. (2010) Niemann-Pick disease type C. Orphanet J. Rare Dis. 5, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Patterson M. C., Hendriksz C. J., Walterfang M., Sedel F., Vanier M. T., Wijburg F., and NP-C Guidelines Working Group (2012) Recommendations for the diagnosis and management of Niemann-Pick disease type C: an update. Mol. Genet. Metab. 106, 330–344 [DOI] [PubMed] [Google Scholar]
- 6. Garver W. S., Francis G. A., Jelinek D., Shepherd G., Flynn J., Castro G., Walsh Vockley C., Coppock D. L., Pettit K. M., Heidenreich R. A., Meaney F. J. (2007) The National Niemann-Pick C1 disease database: report of clinical features and health problems. Am. J. Med. Genet. A 143A, 1204–1211 [DOI] [PubMed] [Google Scholar]
- 7. Imrie J., Dasgupta S., Besley G. T., Harris C., Heptinstall L., Knight S., Vanier M. T., Fensom A. H., Ward C., Jacklin E., Whitehouse C., Wraith J. E. (2007) The natural history of Niemann-Pick disease type C in the UK. J. Inherit. Metab. Dis. 30, 51–59 [DOI] [PubMed] [Google Scholar]
- 8. Loftus S. K., Morris J. A., Carstea E. D., Gu J. Z., Cummings C., Brown A., Ellison J., Ohno K., Rosenfeld M. A., Tagle D. A., Pentchev P. G., Pavan W. J. (1997) Murine model of Niemann-Pick C disease: mutation in a cholesterol homeostasis gene. Science 277, 232–235 [DOI] [PubMed] [Google Scholar]
- 9. Rimkunas V. M., Graham M. J., Crooke R. M., Liscum L. (2008) In vivo antisense oligonucleotide reduction of NPC1 expression as a novel mouse model for Niemann Pick type C-associated liver disease. Hepatology 47, 1504–1512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sayre N. L., Rimkunas V. M., Graham M. J., Crooke R. M., Liscum L. (2010) Recovery from liver disease in a Niemann-Pick type C mouse model. J. Lipid Res. 51, 2372–2383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Smith D., Wallom K. L., Williams I. M., Jeyakumar M., Platt F. M. (2009) Beneficial effects of anti-inflammatory therapy in a mouse model of Niemann-Pick disease type C1. Neurobiol. Dis. 36, 242–251 [DOI] [PubMed] [Google Scholar]
- 12. Vázquez M. C., del Pozo T., Robledo F. A., Carrasco G., Pavez L., Olivares F., González M., Zanlungo S. (2011) Alteration of gene expression profile in Niemann-Pick type C mice correlates with tissue damage and oxidative stress. PLoS One 6, e28777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Beltroy E. P., Richardson J. A., Horton J. D., Turley S. D., Dietschy J. M. (2005) Cholesterol accumulation and liver cell death in mice with Niemann-Pick type C disease. Hepatology 42, 886–893 [DOI] [PubMed] [Google Scholar]
- 14. Pressey S. N., Smith D. A., Wong A. M., Platt F. M., Cooper J. D. (2012) Early glial activation, synaptic changes and axonal pathology in the thalamocortical system of Niemann-Pick type C1 mice. Neurobiol. Dis. 45, 1086–1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schrantz N., Sagiv Y., Liu Y., Savage P. B., Bendelac A., Teyton L. (2007) The Niemann-Pick type C2 protein loads isoglobotrihexosylceramide onto CD1d molecules and contributes to the thymic selection of NKT cells. J. Exp. Med. 204, 841–852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sagiv Y., Hudspeth K., Mattner J., Schrantz N., Stern R. K., Zhou D., Savage P. B., Teyton L., Bendelac A. (2006) Cutting edge: impaired glycosphingolipid trafficking and NKT cell development in mice lacking Niemann-Pick type C1 protein. J. Immunol. 177, 26–30 [DOI] [PubMed] [Google Scholar]
- 17. Reddy J. V., Ganley I. G., Pfeffer S. R. (2006) Clues to neuro-degeneration in Niemann-Pick type C disease from global gene expression profiling. PLoS One 1, e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. De Windt A., Rai M., Kytömäki L., Thelen K. M., Lütjohann D., Bernier L., Davignon J., Soini J., Pandolfo M., Laaksonen R. (2007) Gene set enrichment analyses revealed several affected pathways in Niemann-pick disease type C fibroblasts. DNA Cell Biol. 26, 665–671 [DOI] [PubMed] [Google Scholar]
- 19. Alam M. S., Getz M., Safeukui I., Yi S., Tamez P., Shin J., Velázquez P., Haldar K. (2012) Genomic expression analyses reveal lysosomal, innate immunity proteins, as disease correlates in murine models of a lysosomal storage disorder. PLoS One 7, e48273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cluzeau C. V., Watkins-Chow D. E., Fu R., Borate B., Yanjanin N., Dail M. K., Davidson C. D., Walkley S. U., Ory D. S., Wassif C. A., Pavan W. J., Porter F. D. (2012) Microarray expression analysis and identification of serum biomarkers for Niemann-Pick disease, type C1. Hum. Mol. Genet. 21, 3632–3646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liu B., Li H., Repa J. J., Turley S. D., Dietschy J. M. (2008) Genetic variations and treatments that affect the lifespan of the NPC1 mouse. J. Lipid Res. 49, 663–669 [DOI] [PubMed] [Google Scholar]
- 22. Liu B., Turley S. D., Burns D. K., Miller A. M., Repa J. J., Dietschy J. M. (2009) Reversal of defective lysosomal transport in NPC disease ameliorates liver dysfunction and neurodegeneration in the npc1−/− mouse. Proc. Natl. Acad. Sci. U.S.A. 106, 2377–2382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ramirez C. M., Liu B., Taylor A. M., Repa J. J., Burns D. K., Weinberg A. G., Turley S. D., Dietschy J. M. (2010) Weekly cyclodextrin administration normalizes cholesterol metabolism in nearly every organ of the Niemann-Pick type C1 mouse and markedly prolongs life. Pediatr. Res. 68, 309–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Davidson C. D., Ali N. F., Micsenyi M. C., Stephney G., Renault S., Dobrenis K., Ory D. S., Vanier M. T., Walkley S. U. (2009) Chronic cyclodextrin treatment of murine Niemann-Pick C disease ameliorates neuronal cholesterol and glycosphingolipid storage and disease progression. PLoS One 4, e6951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Leto G., Tumminello F. M., Pizzolanti G., Montalto G., Soresi M., Ruggeri I., Gebbia N. (1996) Cathepsin D serum mass concentrations in patients with hepatocellular carcinoma and/or liver cirrhosis. Eur. J. Clin. Chem. Clin. Biochem. 34, 555–560 [DOI] [PubMed] [Google Scholar]
- 26. Herszényi L., Farinati F., Cardin R., István G., Molnár L. D., Hritz I., De Paoli M., Plebani M., Tulassay Z. (2008) Tumor marker utility and prognostic relevance of cathepsin B, cathepsin L, urokinase-type plasminogen activator, plasminogen activator inhibitor type-1, CEA and CA 19-9 in colorectal cancer. BMC Cancer 8, 194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lv B. J., Lindholt J. S., Cheng X., Wang J., Shi G. P. (2012) Plasma cathepsin S and cystatin C levels and risk of abdominal aortic aneurysm: a randomized population-based study. PLoS One 7, e41813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ohmi K., Kudo L. C., Ryazantsev S., Zhao H. Z., Karsten S. L., Neufeld E. F. (2009) Sanfilippo syndrome type B, a lysosomal storage disease, is also a tauopathy. Proc. Natl. Acad. Sci. U.S.A. 106, 8332–8337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Maue R. A., Burgess R. W., Wang B., Wooley C. M., Seburn K. L., Vanier M. T., Rogers M. A., Chang C. C., Chang T. Y., Harris B. T., Graber D. J., Penatti C. A., Porter D. M., Szwergold B. S., Henderson L. P., Totenhagen J. W., Trouard T. P., Borbon I. A., Erickson R. P. (2012) A novel mouse model of Niemann-Pick type C disease carrying a D1005G-Npc1 mutation comparable to commonly observed human mutations. Hum. Mol. Genet. 21, 730–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liao G., Yao Y., Liu J., Yu Z., Cheung S., Xie A., Liang X., Bi X. (2007) Cholesterol accumulation is associated with lysosomal dysfunction and autophagic stress in Npc1 −/− mouse brain. Am. J. Pathol. 171, 962–975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Amritraj A., Peake K., Kodam A., Salio C., Merighi A., Vance J. E., Kar S. (2009) Increased activity and altered subcellular distribution of lysosomal enzymes determine neuronal vulnerability in Niemann-Pick type C1-deficient mice. Am. J. Pathol. 175, 2540–2556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Amritraj A., Wang Y., Revett T. J., Vergote D., Westaway D., Kar S. (2013) Role of cathepsin D in U18666A-induced neuronal cell death: potential implication in Niemann-Pick type C disease pathogenesis. J. Biol. Chem. 288, 3136–3152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lopez M. E., Klein A. D., Dimbil U. J., Scott M. P. (2011) Anatomically defined neuron-based rescue of neurodegenerative Niemann-Pick type C disorder. J. Neurosci. 31, 4367–4378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. German D. C., Liang C. L., Song T., Yazdani U., Xie C., Dietschy J. M. (2002) Neurodegeneration in the Niemann-Pick C mouse: glial involvement. Neuroscience 109, 437–450 [DOI] [PubMed] [Google Scholar]
- 35. Liu B., Ramirez C. M., Miller A. M., Repa J. J., Turley S. D., Dietschy J. M. (2010) Cyclodextrin overcomes the transport defect in nearly every organ of NPC1 mice leading to excretion of sequestered cholesterol as bile acid. J. Lipid Res. 51, 933–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Grammatikakis I., Evangelinakis N., Salamalekis G., Tziortzioti V., Samaras C., Chrelias C., Kassanos D. (2009) Prevalence of severe pelvic inflammatory disease and endometriotic ovarian cysts: a 7-year retrospective study. Clin. Exp. Obstet. Gynecol. 36, 235–236 [PubMed] [Google Scholar]
- 37. Ramirez C. M., Liu B., Aqul A., Taylor A. M., Repa J. J., Turley S. D., Dietschy J. M. (2011) Quantitative role of LAL, NPC2, and NPC1 in lysosomal cholesterol processing defined by genetic and pharmacological manipulations. J. Lipid Res. 52, 688–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ko D. C., Milenkovic L., Beier S. M., Manuel H., Buchanan J., Scott M. P. (2005) Cell-autonomous death of cerebellar Purkinje neurons with autophagy in Niemann-Pick type C disease. PLoS Genet. 1, 81–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yu T., Lieberman A. P. (2013) Npc1 acting in neurons and glia is essential for the formation and maintenance of CNS myelin. PLoS Genet. 9, e1003462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Taylor A. M., Liu B., Mari Y., Liu B., Repa J. J. (2012) Cyclodextrin mediates rapid changes in lipid balance in Npc1−/− mice without carrying cholesterol through the bloodstream. J. Lipid Res. 53, 2331–2342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jiang X., Sidhu R., Porter F. D., Yanjanin N. M., Speak A. O., te Vruchte D. T., Platt F. M., Fujiwara H., Scherrer D. E., Zhang J., Dietzen D. J., Schaffer J. E., Ory D. S. (2011) A sensitive and specific LC-MS/MS method for rapid diagnosis of Niemann-Pick C1 disease from human plasma. J. Lipid Res. 52, 1435–1445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Porter F. D., Scherrer D. E., Lanier M. H., Langmade S. J., Molugu V., Gale S. E., Olzeski D., Sidhu R., Dietzen D. J., Fu R., Wassif C. A., Yanjanin N. M., Marso S. P., House J., Vite C., Schaffer J. E., Ory D. S. (2010) Cholesterol oxidation products are sensitive and specific blood-based biomarkers for Niemann-Pick C1 disease. Sci. Transl. Med. 2, 56ra81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Olsen B. N., Schlesinger P. H., Ory D. S., Baker N. A. (2012) Side-chain oxysterols: from cells to membranes to molecules. Biochim. Biophys. Acta 1818, 330–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Brown A. J., Jessup W. (2009) Oxysterols: sources, cellular storage and metabolism, and new insights into their roles in cholesterol homeostasis. Mol. Aspects Med. 30, 111–122 [DOI] [PubMed] [Google Scholar]
- 45. Hughes T. M., Rosano C., Evans R. W., Kuller L. H. (2013) Brain cholesterol metabolism, oxysterols, and dementia. J. Alzheimers Dis. 33, 891–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Leoni V., Caccia C. (2011) Oxysterols as biomarkers in neurodegenerative diseases. Chem. Phys. Lipids 164, 515–524 [DOI] [PubMed] [Google Scholar]
- 47. Pišlar A., Kos J. (2013) Cysteine cathepsins in neurological disorders. Mol. Neurobiol. 10.1007/s12035-013-8576-6 [DOI] [PubMed] [Google Scholar]
- 48. Nakanishi H. (2003) Microglial functions and proteases. Mol. Neurobiol. 27, 163–176 [DOI] [PubMed] [Google Scholar]
- 49. Kelly D. A., Portmann B., Mowat A. P., Sherlock S., Lake B. D. (1993) Niemann-Pick disease type C: diagnosis and outcome in children, with particular reference to liver disease. J. Pediatr. 123, 242–247 [DOI] [PubMed] [Google Scholar]
- 50. Yerushalmi B., Sokol R. J., Narkewicz M. R., Smith D., Ashmead J. W., Wenger D. A. (2002) Niemann-pick disease type C in neonatal cholestasis at a North American Center. J. Pediatr. Gastroenterol. Nutr. 35, 44–50 [DOI] [PubMed] [Google Scholar]
- 51. Trexler A. J., Nilsson M. R. (2007) The formation of amyloid fibrils from proteins in the lysozyme family. Curr. Protein Pept. Sci. 8, 537–557 [DOI] [PubMed] [Google Scholar]
- 52. Vieira M. N., Forny-Germano L., Saraiva L. M., Sebollela A., Martinez A. M., Houzel J. C., De Felice F. G., Ferreira S. T. (2007) Soluble oligomers from a non-disease related protein mimic Aβ-induced tau hyperphosphorylation and neurodegeneration. J. Neurochem. 103, 736–748 [DOI] [PubMed] [Google Scholar]
- 53. Vitner E. B., Platt F. M., Futerman A. H. (2010) Common and uncommon pathogenic cascades in lysosomal storage diseases. J. Biol. Chem. 285, 20423–20427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cologna S. M., Cluzeau C. V., Yanjanin N. M., Blank P. S., Dail M. K., Siebel S., Toth C. L., Wassif C. A., Lieberman A. P., Porter F. D. (2014) Human and mouse neuroinflammation markers in Niemann-Pick disease, type C1. J. Inherit. Metab. Dis. 37, 83–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Vitner E. B., Dekel H., Zigdon H., Shachar T., Farfel-Becker T., Eilam R., Karlsson S., Futerman A. H. (2010) Altered expression and distribution of cathepsins in neuronopathic forms of Gaucher disease and in other sphingolipidoses. Hum. Mol. Genet. 19, 3583–3590 [DOI] [PubMed] [Google Scholar]
- 56. Hruz T., Wyss M., Docquier M., Pfaffl M. W., Masanetz S., Borghi L., Verbrugghe P., Kalaydjieva L., Bleuler S., Laule O., Descombes P., Gruissem W., Zimmermann P. (2011) RefGenes: identification of reliable and condition specific reference genes for RT-qPCR data normalization. BMC Genomics 12, 156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Repa J. J., Li H., Frank-Cannon T. C., Valasek M. A., Turley S. D., Tansey M. G., Dietschy J. M. (2007) Liver X receptor activation enhances cholesterol loss from the brain, decreases neuroinflammation, and increases survival of the NPC1 mouse. J. Neurosci. 27, 14470–14480 [DOI] [PMC free article] [PubMed] [Google Scholar]