

Background: p75 neurotrophin receptor (p75NTR) is an important mediator of invasion of malignant gliomas, but its role in glioma proliferation is unknown.
Results: p75NTR mediates proliferation of brain tumor-initiating cells (BTICs) via its cleavage and release of an intracellular domain.
Conclusion: p75NTR also regulates proliferation of BTICs.
Significance: p75NTR is a potential target for the treatment of malignant gliomas.
Keywords: ADAM, ADAMTS, Brain Tumors, Cancer Stem Cells, Cell Surface Receptor, Glioblastoma, Neurotrophins, Proliferation, Nerve Growth Factor, p75 Neurotrophin Receptor
Abstract
Malignant gliomas are highly invasive, proliferative, and resistant to treatment. Previously, we have shown that p75 neurotrophin receptor (p75NTR) is a novel mediator of invasion of human glioma cells. However, the role of p75NTR in glioma proliferation is unknown. Here we used brain tumor-initiating cells (BTICs) and show that BTICs express neurotrophin receptors (p75NTR, TrkA, TrkB, and TrkC) and their ligands (NGF, brain-derived neurotrophic factor, and neurotrophin 3) and secrete NGF. Down-regulation of p75NTR significantly decreased proliferation of BTICs. Conversely, exogenouous NGF stimulated BTIC proliferation through α- and γ-secretase-mediated p75NTR cleavage and release of its intracellular domain (ICD). In contrast, overexpression of the p75NTR ICD induced proliferation. Interestingly, inhibition of Trk signaling blocked NGF-stimulated BTIC proliferation and p75NTR cleavage, indicating a role of Trk in p75NTR signaling. Further, blocking p75NTR cleavage attenuated Akt activation in BTICs, suggesting role of Akt in p75NTR-mediated proliferation. We also found that p75NTR, α-secretases, and the four subunits of the γ-secretase enzyme were elevated in glioblastoma multiformes patients. Importantly, the ICD of p75NTR was commonly found in malignant glioma patient specimens, suggesting that the receptor is activated and cleaved in patient tumors. These results suggest that p75NTR proteolysis is required for BTIC proliferation and is a novel potential clinical target.
Introduction
Malignant gliomas remain largely incurable with a poor prognosis (1). Glioblastoma multiformes (GBMs)5 are highly proliferative, invasive, and resistant to treatment, and patients have an average survival of ∼1 year (2–4). How malignant gliomas arise is not clear; genetic characterization of GBMs has identified four important signaling pathways, including the p53, retinoblastoma protein, receptor tyrosine kinase, and NF1 pathways (5). Mutations in IDH1 are uncommon in primary GBM (<5%) but more common in grade II/III gliomas and in secondary GBMs (6, 7). Isolation of glioma stem cells, which we designate as brain tumor-initiating cells (BTICs), from patient tumors suggest that gliomas may arise from these cells (8, 9). Glioma stem cells share several features with neuronal stem cells, including expression of neuronal stem cell markers (10, 11) and the capability to self-renew and to undergo multilineage differentiation (12–14). Glioma stem cells develop tumors when transplanted into immunodeficient mice that phenotypically and molecularly resemble MGs in patients (8, 14). These are relatively resistant to radiation and chemotherapies (15–17), suggesting their role in tumor progression and treatment resistance.
We recently used an unbiased in vivo selection strategy to identify genes required for glioma invasion (18) and found that p75 neurotrophin receptor (p75NTR) was up-regulated in the highly invasive glioma cells. p75NTR-overexpressing cells were more migratory and invasive in vitro and in vivo, and receptor proteolysis of p75NTR was required for glioma invasion (19).
The p75NTR is a multifunctional signaling protein that regulates a variety of biological effects, which are highly cell type- and context-specific. Its effect ranges from neurite outgrowth to myelin formation to cell survival and death (20, 21). p75NTR is also implicated in regulating proliferation and differentiation of neuronal and non-neuronal cells (22, 23). Importantly, p75NTR mediates neuronal proliferation and differentiation when bound by brain-derived neurotrophic factor (BDNF) during neurogenesis (24). However, the role of p75NTR and its signaling pathways in glioma stem cell proliferation is unknown. Here we show that p75NTR is required for glioma stem cell proliferation, and this function is associated with Trk (tropomyosin receptor kinase)-dependent p75NTR proteolysis by α-secretase and γ-secretases, which releases its intracellular domain and ultimately triggers proliferation. Further, p75NTR, α-secretase, and γ-secretase cleavage products are elevated in malignant glioma tumor specimens, suggesting that this may be relevant clinically and target two hallmarks of cancer (invasion and proliferation).
EXPERIMENTAL PROCEDURES
Brain Tumor-initiating Cell Culture
In this study, we used four different glioma stem cell lines/BTICs isolated from human GBMs (lines 54, 31, and G144) and a giant cell GBM (G179). Lines 54 and 31 were isolated by Dr. Michael Cooper (Vanderbilt University). Lines G144 and G179 were generated by Dr. Peter Dirks (The Hospital for Sick Children) (14), deposited in the Biorep cell bank (Milan, Italy). We then purchased these lines from the Biorep cell bank (catalogue no. NS00013*A (G144) and catalogue no. NS00011*B (G179)). BTICs were cultured under adherent conditions on Laminin (Sigma, catalogue no. L2020)-coated plates in serum-free neuronal stem cell medium (Stem Cell Technology) supplemented with 1% N2 (containing insulin, apotransferrin, BSA, progesterone, putrescine, and sodium selenite) and 1% B27 (Invitrogen), 2 mm l-glutamine, 1 mm sodium pyruvate, 100 units/ml penicillin, 100 μg/ml streptomycin, and 20 ng/ml FGF and EGF (Stem Cell Technology), as described previously (14). BTICs were supplied with new medium every 4–5 days and were dissociated using Accutase (Sigma, catalogue no. A6964).
U251 glioma cell lines stably expressing p75NTR were cultured in Dulbecco's modified Eagle's medium (Invitrogen) containing 10% FBS, 2 mm l-glutamine, 1 mm sodium pyruvate, 100 μg/ml streptomycin, 100 units/ml penicillin, and 400 μg/ml G418.
Total RNA Isolation, cDNA Synthesis, RT-PCR Analysis, and Quantitative PCR
Total RNA was isolated from BTICs using an RNeasy minikit (Qiagen, catalogue no. 74104) as per the manufacturer's protocol. Total RNA (1 μg) was reverse transcribed into cDNA using a SuperScript VILO cDNA synthesis kit (Invitrogen, catalogue no. 11754) for 60 min at 42 °C, and then the resulting cDNA (1 μl) was used as templates for subsequent PCR amplification using Platinum PCR super mix high fidelity (Invitrogen, catalogue no. 12532) and specific primer sets against human NGFR/p75NTR (Qiagen, catalog no. QT00056756, NM_002507, 118 bp), NTRK1/TrkA (Qiagen, catalogue no. QT00054110, NM_002529, 112 bp), NTRK2/TrkB (Qiagen, catalogue no. QT00082033, NM_006180, 103 bp), NTRK3/TrkC (Qiagen, catalogue no. QT00052906, NM_002530, 143 bp), NGF (Qiagen, catalogue no. QT00001589, NM_002506, 73 bp), BDNF (Qiagen, catalogue no. QT00235368, NM_001143805, 120 bp), neurotrophin 3 (NT3) (Qiagen, catalogue no. QT00204218, NM_001102654, 104 bp) and human Actin B (Qiagen, catalogue no. QT01680476, NM_001101, 104 bp). PCR conditions were as follows: 5 min 95 °C for initial PCR activation; 35 cycles of 94 °C for 30 s, 58 °C for 30 s, and 72 °C for 1 min; and 72 °C for 10 min for final extension. The same amount of human brain total RNA (Clontech, catalogue no. 636530) for cDNA synthesis and PCRs was used as a positive control. After amplification, PCR products were run on a 2% agarose gel, and gel images were captured for further analysis. Quantitative PCR was conducted with a real-time PCR detection system (Bio-Rad) using cDNA, SYBR Green PCR master mix (Applied Biosystems, catalogue no. 4309155), and primers for TrkA, (Qiagen, catalogue no. QT00054110, NM_002529, 112 bp), ADAM17 (Qiagen, catalogue no. QT00055580, NM_003183, 109 bp), and actin (Qiagen, catalogue no. QT01680476, NM_001101, 104 bp). All samples were run in triplicates, data were analyzed at the appropriate cycle number, and expression was calculated as ΔΔCt and normalized to actin.
Transfection of Brain Tumor-initiating Cells
BTICs were dissociated using Accutase as described previously (14). The cell suspensions were then transfected with Stealth control siRNA (Invitrogen, catalogue no. 452001) or Stealth siRNAs to p75NTR with p75NTR duplex siRNA with the following sequence: p75NTR-siRNA 1, CACUUCUGACCACACUUCCUGUCCA (sense) and AAAUAAAUACACCCAGACUCUGUCC (antisense); p75NTR siRNA 2, GGACAGAGUCUGGGUGUAUUUAUUU (sense) and AAAUAAAUACACCCAGACUCUGUCC (antisense). Cells were transfected with 40 nmol of p75NTR-siRNA 1 or p75NTR-siRNA 2 or control siRNA by using the Amaxa electroporation kit (Lonza, catalogue no. VPG-1004) and the T-030 program on an Amaxa electroporation device. For Western blotting analysis, 3 days after electroporation, cells were lysed and used for p75NTR Western blotting. For proliferation assays, cells were used 4 days after electroporation; for MTT assays, cells were washed and added with MTT reagent and lysed; for the trypan blue assay, cells were collected and added with trypan blue; and for immunostaining, cells were fixed and immunostained with Ki67 antibody.
In some of the experiments, the BTICs were transfected with 2 μg of wild type p75NTR, or γ-secretase-resistant mutant p75NTR (p75FasTM) (25) (kindly provided by Dr. Moses V. Chao, Skirball Institute, New York University) using the Amaxa electroporation method as described above. Three days after the electroporation, cells were treated with the proteosome inhibitor epoxomycin (1 μm; Cabiochem, catalogue no. 324800) alone or along with 100 ng/ml NGF (Harlan, catalogue no. BT3061) for 6 h, and then cells were lysed and subjected to p75NTR Western blotting. For assessing proliferation, 48 h after transfection, cells were treated with 100 ng/ml NGF or left untreated for 3 days and then fixed using 4% paraformaldehyde and stained for Ki67, and Ki67-positive cells were scored for proliferation.
In some of the other experiments, BTICs were electroporated with control siRNA, p75NTR siRNAs 1 and 2, or p75FasTM as described above. Cells were maintained in neurobasal medium without EGF and FGF for 48 h, and then cells were switched to medium containing EGF and FGF for 6 h, lysed, and subjected to p75NTR, phospho-Akt (1:1000; Cell Signaling, catalogue no. 4056), and actin (1:1000; Cell signaling, catalogue no. 4967) Western blotting analysis. BTICs were also electroporated with GFP alone or with GFP and p75NTR intracellular domain (ICD) together (kindly provided by Dr. Philip Barker, McGill University, Montreal, Canada), and 2 days later, cells were lysed and subjected to p75NTR ICD and tubulin Western blotting. For examining the proliferation, 3 days following transfection, cells were fixed and stained with Ki67 antibody.
Western Blotting Analysis
BTICs were cultured under neuronal stem cell medium as described above, and then cells were harvested, lysed in radioimmune precipitation assay buffer (10 mm Tris-HCl, 1 mm EDTA, 0.4 mm EGTA, 0.1% SDS, 140 mm sodium chloride, 0.1% sodium deoxycholate, 1% Triton X-100, supplemented with 1 mm Na3VO4, 1 mm phenylmethylsulfonyl fluoride, aprotinin, and leupeptin), and lysates were subjected to Western blotting analysis using antibodies to p75NTR (1:3000; provided from Dr. Bruce Carter, Vanderbilt University), TrkB (1:1000; Cell signaling, catalogue no. 4603), TrkC (1:1000; Cell Signaling, catalogue no. 3376), and tubulin (1:1000; Calbiochem, catalogue no. CP06). In some experiments, to detect the ICD of the receptor, cells were washed and treated with the proteosome inhibitor epoxomycin. Epoxomycin (1 μm; Calbiochem, catalogue no. 324800) was added to cells with or without γ-secretase inhibitor DAPT (200 nm; Calbiochem, catalogue no. 565770) or metalloprotease inhibitor TAPI-2 (500 nm; Calbiochem, catalogue no. 579052) or Trk inhibitor K252a (200 nm; Sigma, catalogue no. K2015) in the presence or absence of 50–100 ng/ml NGF (Harlan, catalogue no. BT3060) to activate p75NTR. Then cells were lysed in radioimmune precipitation assay buffer, and lysates were subjected to p75NTR Western blot analysis (1:3000 dilution of p75NTR antibody raised against intracellular domain) as described previously (26, 27). The lysates were collected following transfection of various p75NTR constructs were also subjected to p75NTR ICD Western blotting. These blots were reprobed for the loading control tubulin and actin (1:1000; Cell Signaling, catalogue no. 4967).
Enzyme-linked Immunosorbent Assay for Neurotrophins
BTICs 54, 31, G144, and G179 were maintained in neuronal stem cell medium for 5 days, and then medium was collected and filtered through a syringe filter and used for measuring neurotrophins by the ELISA method. ELISA was performed for NGF (human NGF ELISA kit, Boster Immunoleader, catalogue no. EK0469), BDNF (human BDNF ELISA kit, Boster Immunoleader, catalogue no. EK0307), and NT3 (human neurotrophin-3 ELISA kit, Boster Immunoleader, catalogue no. EK0472) according to the manufacturer's instructions. Data are expressed as ng of neurotrophins secreted/ml of medium.
MTT and Trypan Blue Assay for Cell Proliferation/Cell Viability
Cell proliferation or cell viability of BTICs was measured using an MTT assay. BTICs were dissociated and transfected with control siRNA or p75NTR siRNAs as described above and then plated onto 24-well plates coated with laminin. Four days after transfection, cells were washed with 1× PBS and then incubated with 240 μl of MTT solution (1 mg/ml; Sigma) at 37 °C. 2 h later, MTT solution was removed, and cells were lysed, adding 240 μl of isopropyl alcohol containing 0.04 m HCl and 160 mm NaoH, and incubated for 10 min at room temperature. Then absorbance was measured at 570 and 630 nm. Each sample was run in triplicate, and the data were presented as a percentage of control.
For the trypan blue assay, cells were collected in an Eppendorf tubes and added with an equal volume of 0.4% trypan blue solution (Sigma, catalogue no. T8154), and the total number of live cells was measured using a TC10TM automated cell counter (Bio-Rad). Data were expressed as total number of cells/ml.
Ki67 Immunostaining for Cell Proliferation
BTICs were transfected with control siRNA, p75NTR siRNA, wild type p75NTR, or cleavage-resistant mutant p75NTR (p75FasTM); and cells were also treated with DAPT (200 nm), TAPI-2 (500 nm), or K252a (200 nm) and with NGF (25, 50, and 100 ng/ml), BDNF (50 ng/ml; Peprotech, catalogue no. 450-02), NT3 (50 ng/ml; Millipore, catalogue no. GF031), anti-NGF (0.1 μg/ml; Millipore, catalogue no. MAB5260Z), or control IgG (0.1 μg/ml; Calbiochem, catalogue no. N101), fixed in 4% paraformaldehyde, and processed for the proliferative marker Ki67 by immunostaining. Cells were permeabilized with 0.1% Triton X-100 and 0.1% sodium citrate for 10 min at room temperature, washed with 1× PBS, and blocked with 10% normal goat serum for 1 h at room temperature. Then cells were incubated with the Ki67 antibody (1:100 dilution; Biocare Medical, catalogue no. CRM 325C) in PBS containing 0.1% Triton X-100 overnight at 4 °C. This was followed by incubation with anti-rabbit Alexa 488 (1:500 dilution; Molecular Probes, catalogue no. A11008) or anti-rabbit Alexa 546 (1:500 dilution; Molecular Probes, catalogue no. A11010) in 1× PBS for 1 h at room temperature and staining with DAPI (Vector Labs) to visualize nuclei. Cells were viewed and captured using an LSM 710 Meta confocal microscope (Zeiss) at ×40. The number of Ki67-positive (mainly nuclear) cells was quantified by examining the total number of cells counted (by DAPI staining), and proliferation was expressed as a percentage.
Bioinformatics Analysis
Gene expression analysis from two different data sets was used in this study. One contains microarray expression from the Cancer Genome Atlas data portal, and level 3 expression data on the Affymetrix U133A chip was used in this analysis. The other data set contains microarray expression data for 108 GBM patients from Moffitt's Total Cancer Care program. A customized Affymetrix chip was used to generate the expression profile, and MAS5 was used to normalize the expression data. The R package was used to analyze gene expression data, in which the gene expression level was categorized into three groups, up-regulated, intermediate, and down-regulated, according to the quartile of normalized expression data.
Tumor Samples
Malignant glioma tumor specimens were obtained from the Total Cancer Care program of the Moffitt Cancer Center after obtaining informed consent, following guidelines and permission from the Institutional Review Board. Tumors were obtained from patients who were either previously irradiated or newly diagnosed. Samples were immediately snap frozen and graded according to the World Health Organization. Tumor samples were used to isolate total RNA for PCR analysis of p75NTR and NGF; for Western blotting, samples were lysed in radioimmune precipitation assay buffer, debris was removed by centrifugation, and lysates were subjected to Western blotting using p75NTR ICD and tubulin antibodies as described above.
RESULTS
Neurotrophin Receptors and Ligands Are Expressed in Brain Tumor-initiating Cells
Neurotrophins and their receptors (p75NTR, TrkA, TrkB, and TrkC) play important roles in regulating cell survival and proliferation in multiple cell types (20, 21, 28). p75NTR signaling is required for survival and proliferation of breast cancer cells (29–31). We used four different patient-derived BTICs, designated as 54, 31, G144, and G179, and we investigated whether p75NTR regulates their proliferation. We cultured BTICs on laminin-coated plates with EGF and FGF (14) and confirmed that they express the neuronal stem cells markers Sox2 and Nestin (14) (data not shown). RT-PCR analysis demonstrated that all four BTICs express the neurotrophin receptors p75NTR, TrkB, and TrkC, but TrkA was expressed only in G179 (Fig. 1, A and B). However, quantitative PCR analysis data demonstrated the expression of TrkA in all BTICs examined (Fig. 1C). Western blotting of p75NTR, TrkB, and TrkC shows that BTICs express neurotrophin receptor proteins (Fig. 1, D–F). For TrkB and TrkC, with the antibodies we used (Cell Signaling, 80E3, catalogue no. 4603 for TrkB and C44H5, catalogue no. 3376 for TrkC), we detected bands at 140 kDa for full-length receptors and additional bands at 90 kDa for truncated forms of the receptors (Fig. 1, E and F). However, expression patterns of TrkB and TrkC full-length receptors and their truncated forms are different in each BTIC. These data indicate that neurotrophin receptors are widely expressed in BTICs.
FIGURE 1.

Brain tumor-initiating cells express neurotrophin receptors p75NTR, TrkA, TrkB, and TrkC and neurotrophins NGF, BDNF, and NT3. BTICs (lines 54, 31, G144, and G179) isolated from glioblastoma patient tumors were maintained under proliferative conditions with EGF and FGF, and expression of neurotrophin receptors and expression and secretion of neurotrophic factors were examined. RT-PCR analysis of p75NTR (A) and of TrkA, TrkB, and TrkC (B) was performed using human-specific primers. Actin acts as an internal control. Human brain total RNA obtained from Clontech was used as positive control (+ve). C, quantitative PCR was performed for TrkA and internal control actin, and expression of TrkA was calculated as ΔΔCt against the actin. Data are the mean of three independent experiments. Lysates isolated from BTICs were subjected to Western blot analysis (using 60 μg of lysate from each cell line) using antibodies against the p75NTR (D), TrkB (E), TrkC (F), and tubulin. FL, full-length receptor; T, truncated form of receptor. RT-PCR analysis (G) using human specific primers and ELISA (H) using human specific ELISA kits were performed for neurotrophic factors NGF, BDNF, and NT3 to determine the expression and secretion, and levels of neurotrophins in the media were expressed as ng/ml of media. Data are means ± S.D. (error bars), n = 3 independent experiments (*, p < 0.05).
Mammalian neurotrophins, such as NGF, BDNF, and NT3, are secretory proteins that regulate neuronal survival and growth by binding to Trks and the p75NTR. NGF exhibits specific affinity for TrkA receptor, BDNF for TrkB receptor, and NT3 for TrkC receptor. However, every neurotrophin binds to p75NTR with equal affinity (21). Therefore, we hypothesized that BTICs express and secrete neurotrophins. RT-PCR analysis showed that neurotrophic factors NGF, BDNF, and NT3 are expressed in all BTICs we examined (Fig. 1G). Estimation of secreted neurotrophins in culture media by ELISA demonstrated that all neurotrophins were secreted into the media; however, NGF (22.40 ng/ml) secretion was ∼10-fold higher than BDNF (2.33 ng/ml) and NT3 (2.50 ng/ml) secretion (Fig. 1H). These results show that neurotrophins are secreted by BTICs and may activate receptors in an autocrine and/or paracrine fashion to mediate BTIC invasion, survival, and proliferation.
Knockdown of p75NTR Decreases Brain Tumor-initiating Cell Proliferation
Because we have previously shown that p75NTR mediates glioma invasion (18, 19), we wanted to determine whether it mediated other aspects of the malignant phenotype, such as proliferation. To address this, we used siRNAs to down-regulate p75NTR expression in BTICs. We transfected BTICs with control siRNA or two different p75NTR-siRNAs and then 3 days later confirmed the knockdown of p75NTR protein by Western blotting (Fig. 2, A and B). We then assessed the effects of p75NTR knockdown on proliferation. First, we used the MTT and trypan blue assays to measure proliferation (32, 33). Knockdown of p75NTR significantly decreased proliferation of all four BTICs as measured by the MTT assay (Fig. 2, C–F) and trypan blue assay (Fig. 2, G–I), suggesting that p75NTR normally facilitates BTIC proliferation.
FIGURE 2.

Knockdown of p75NTR decreases proliferation of brain tumor-initiating cells in vitro. A, BTICs (line 54) were transfected with control siRNA (Con-siRNA) or two different p75NTR-siRNAs (p75NTR-siRNA(1) and p75NTR-siRNA(2)) using the Amaxa nucleofector device and Amaxa electroporation kit. After 3 days in culture, the cells were lysed and subjected to immunoblot analysis (WB) with anti-p75NTR antibody. B, quantitation of p75NTR Western blots expressed as relative expression level with actin. Results are means ± S.D. of three independent experiments (*, p < 0.05). BTICs, such as line 54 (C), line 31 (D), line G144 (E), and line G179 (F), were transfected with p75NTR siRNAs or control siRNA (Con-siRNA) using the Amaxa electroporation method. 4 days later, cells were subjected to an MTT assay to assess proliferation. Proliferation is expressed as a percentage of control. n = 4 independent experiments (*, p < 0.05). BTIC lines 54 (G), 31 (H), and G179 (I) were transfected with p75NTR siRNAs or control siRNA (Con-siRNA), and 4 days later, cells were subjected to a trypan blue assay to assess cell proliferation. Proliferation is expressed as the total number of cells/ml. n = 3 independent experiments (*, p < 0.05). Error bars, S.D.
To confirm the role of p75NTR in BTIC proliferation, we also used the Ki67 immunostaining method. Ki67 is a nuclear protein highly linked with cell proliferation and extensively used as a proliferation marker in human tumors (34, 35). Following p75NTR knockdown by two p75NTR-siRNAs, there was a decrease in the proliferation rate of BTICs in vitro (Fig. 3). The decrease in proliferation rate after p75NTR knockdown for BTIC-54 was 37.68 and 36.28% (Fig. 3, A and B); for BTIC-31, it was 32.31 and 35.36% (Fig. 3C); for BTIC-G144, it was 25.22 and 28.83% (Fig. 3D); and for G179, it was 31.21 and 32.69% (Fig. 3E) for p75NTR-siRNA-1 and p75NTR-siRNA-2, respectively. Taken together, these results demonstrate that p75NTR is expressed in patient-derived BTICs and is required for BTIC proliferation.
FIGURE 3.

Down-regulation of p75NTR decreases brain tumor-initiating cell proliferation in vitro. BTIC lines 54 (A and B), 31 (C), G144 (D), and G179 (E) were transfected with p75NTR-siRNAs or control siRNA (con-siRNA), and after 4 days in culture, the cells were fixed and processed for immunostaining with the proliferative marker Ki67 and mounted with DAPI-containing media. A, Ki67 (green) and DAPI (blue) staining were imaged by confocal microscopy. Scale bar, 100 μm. B–E, proliferation was assessed by calculating the number of Ki67-positive cells remaining after 4 days in culture as a percentage of the total number of DAPI-stained cells, and data are expressed as a percentage of Ki67-positive cells. n = 3 independent experiments (*, p < 0.05). Error bars, S.D.
NGF Stimulates Brain Tumor-initiating Cell Proliferation and This Is Blocked by NGF-blocking Antibody
p75NTR and TrkA form a high affinity binding complex when they are co-expressed, and this increases the binding affinity of NGF to the TrkA receptor and enhances tyrosine kinase activity and survival (36–39). However, p75NTR can also promote survival independently of TrkA, and binding of NGF to p75NTR alone supports cell survival through activation of NF-κB (40–42). All four BTICs that we examined secrete 10-fold higher levels of NGF (the ligand for both p75NTR and TrkA) compared with levels of BDNF (the ligand for both p75NTR and TrkB) and NT3 (the ligand for both p75NTR and TrkC) (Fig. 1H). Therefore, we hypothesized that NGF-mediated activation of p75NTR and TrkA independently, or as a p75NTR-TrkA complex, stimulates BTIC proliferation. NGF has been implicated in tumor cell proliferation and survival (31); because our BTICs secrete ∼25 ng/ml NGF, we next examined the effects of multiple concentrations (such as 25, 50, and 100 ng/ml) of NGF and anti-NGF on BTIC proliferation. We treated the BTICs with different concentrations of NGF for 3 days and assessed the proliferation by Ki67 immunostaining. Interestingly, NGF at all concentrations significantly enhanced proliferation of BTICs (Fig. 4, A–E). The enhanced proliferation following treatment with NGF in BTIC-54 was 24.79% at 25 ng/ml, 38.26% at 50 ng/ml, and 34.22% at 100 ng/ml (Fig. 4B); in BTIC-31, it was 17.65% at 25 ng/ml, 35.09% at 50 ng/ml, and 16.62% at 100 ng/ml (Fig. 4C); in BTIC-G144, it was 10.26% at 25 ng/ml, 33.49% at 50 ng/ml, and 27.97% at 100 ng/ml (Fig. 4D); and in G179, it was 28.23% at 25 ng/ml, 39.11% at 50 ng/ml, and 31.25% at 100 ng/ml (Fig. 4E). In order to examine the effects of endogenously secreted NGF on proliferation, we treated the cells with control-IgG or NGF antibody to sequester secreted NGF (antibodies were added to every 24 h). NGF antibody treatment significantly decreased the proliferation of all BTICs, with a 41.76% decrease in BTIC 54 (Fig. 4F), 21.58% in BTIC 31 (Fig. 4G), 39.41% in BTIC G144 (Fig. 4H), and 43.29% in BTIC G179 (Fig. 4I); control IgG had no effect on proliferation (Fig. 4F), suggesting the autocrine and/or paracrine action of NGF in BTIC proliferation. Our results indicate that NGF acts through p75NTR/TrkA to stimulate BTIC proliferation.
FIGURE 4.

Neurotrophin NGF stimulates brain tumor-initiating cell proliferation, and this is blocked by anti-NGF. BTICs (lines 54, 31, G144, and G179) were maintained in neuronal stem cell expansion medium and treated with 25, 50, and 100 ng/ml NGF for 3 days (NGF was added to cells at 24-h intervals). The cells were then fixed and immunostained for Ki67 (green) and DAPI (blue) and imaged using confocal microscopy (A). Scale bar, 100 μm. Then proliferation was assessed by calculating the number of Ki67-positive cells remaining after 3 days in culture as a percentage of the total number of DAPI-stained cells. n = 3 independent experiments (B–E) (*, p < 0.05). In some experiments, BTICs were treated with NGF antibody or control IgG (Con-IgG) (F–I) for 4 days without the addition of NGF, and then the proliferation was assessed as described above (*, p < 0.05). Error bars, S.D.
BTICs also express TrkB and TrkC receptors (Fig. 1, B, E, and F) and their ligands BDNF and NT3 (Fig. 1G), but the secretion levels of BDNF and NT3 were dramatically lower compared with NGF (Fig. 1H). We next examined the effect of exogenous BDNF and NT3 on BTIC proliferation. We treated the BTICs with 50 ng/ml BDNF and NT3 (BDNF and NT3 were added every 24 h), and 3 days later these were examined using Ki67 immunostaining. Interestingly, both BDNF (Fig. 5, A and C) and NT3 (Fig. 5, B and D) increased proliferation in all BTICs. These results indicate that all neurotrophins are capable of mediating BTIC proliferation in vitro.
FIGURE 5.

Neurotrophins BDNF and NT3 stimulate brain tumor-initiating cell proliferation. BTICs 54, 31, G144, and G179 were maintained in neuronal stem cell expansion medium with growth factors and treated with neurotrophin BDNF (50 ng/ml) for 3 days (BDNF was added to cells at 24-h intervals). The cells were then fixed and immunostained for proliferative marker Ki67 (green) and DAPI (blue) and imaged using confocal microscopy (A). Scale bar, 100 μm. The proliferation rates for all lines (C) were assessed by calculating the number of Ki67-positive cells remaining after 3 days in culture as a percentage of the total number of DAPI-stained cells. Data are expressed as a percentage of Ki67-positive cells. n = 3 independent experiments (*, p < 0.05). BTICs 54, 31, G144, and G179 were maintained in neuronal stem cell expansion medium with growth factors EGF and FGF and treated with 50 ng/ml NT3 for 3 days (NT3 was added to cells at 24-h intervals). Three days later, cells were fixed and immunostained for Ki67, a proliferative marker (green) and DAPI (blue) and imaged using microscopy (B) (scale bar, 100 μm). The proliferation was assessed by calculating the number of Ki67-positive cells as a percentage of the total number of DAPI-stained cells and expressed as a percentage of Ki67-positive cells (D). n = 3 independent experiments (*, p < 0.05). Error bars, S.D.
NGF Triggers p75NTR Cleavage, Which Is Necessary for Proliferation of Brain Tumor-initiating Cells
p75NTR is cleaved by the metalloproteinase, TACE/ADAM17, and produces a soluble extracellular domain and the carboxyl-terminal fragment (CTF) (25, 27, 43–46). The released CTF can be further processed by γ-secretase within the transmembrane domain, to release the intracellular domain (ICD) into the cytosol (26, 27, 47–50). The released p75NTR ICD generates signals to regulate several physiological functions that are cell type-specific. In cerebellar granular neurons, the ICD inhibits MAG (myelin-associated glycoprotein)-induced neurite outgrowth (49), and in sympathetic neurons, it mediates proapoptotic ligand-induced cell death (26, 27) and also mediates Trk receptor-mediated cell neuronal survival (46). The p75NTR ICD has also been implicated in cell division and proliferation of spiral ganglion Schwann cells (51). Further, the ICD translocates to the nucleus and modulates gene expression (52). We have previously shown that p75NTR cleavage and release of the ICD is necessary for receptor-mediated glioma invasion in vitro and in vivo (19). Therefore, we next wanted to determine if NGF induces α- and γ-secretase-dependent p75NTR proteolysis and if this is required for BTIC proliferation.
We first used a pharmacological approach to test whether p75NTR is cleaved by α-secretase and γ-secretase in response to NGF treatment. We washed the BTICs (line 54) with fresh medium and pretreated them with 500 nm α-secretase inhibitor TAPI-2 or with 200 nm γ-secretase inhibitor DAPT or left them untreated and then added 100 ng/ml NGF for 6 h. The cells were also treated with the proteosome inhibitor epoxomycin (1 μm) to enhance detection of the cleaved fragments of the receptor, such as the ICD, which is quickly degraded by proteosome following its release (48, 47, 26, 27, 19). We found that NGF treatment of the BTICs causes accumulation of the ICD and that DAPT, which blocks the release of ICD, leads to CTF accumulation. TAPI-2 blocks both CTF generation and ICD release (Fig. 6A). These data show that NGF treatment leads to α- and γ-secretase-mediated cleavage of the p75NTR in BTICs.
FIGURE 6.

NGF stimulates p75NTR cleavage, and inhibition of cleavage by α- and γ-secretase inhibitors blocks NGF-mediated receptor proteolysis and proliferation of brain tumor-initiating cells. A, BTICs (line 54) were maintained in neurobasal medium with EGF and FGF for 2 days, and then cells were washed twice with fresh medium and treated with 1 μm epoxomycin (proteosome inhibitor) alone or together with 100 ng/ml NGF in the presence or absence of γ-secretase inhibitor DAPT (200 nm) or metalloprotease inhibitor TAPI-2 (500 nm) for 6 h. Then cells were lysed and performed Western blot analysis using antibodies against the p75NTR ICD and tubulin. U251 expressing p75NTR treated with epoxomycin was used as positive control. FL, full-length p75NTR. Data are representative of three independent experiments. Cell lines 54 (B and C), 31 (D and E), G144 (F and G), and G179 (H and I) were cultured in neurobasal medium for 12 h, and then cells were treated with NGF alone or along with γ-secretase inhibitor DAPT (200 nm) (B, D, F, and H) or metalloprotease inhibitor TAPI-2 (500 nm) (C, E, G, and I) or left untreated for 3 days. Cells were added with NGF, DAPT and TAPI-2 for every 24 h. Three days later, cells were fixed and stained for Ki67 and DAPI, and Ki67-positive cells were quantified. Data shown are means ± S.D., n = 3 independent experiments (*, p < 0.05); **, significant difference from control with p < 0.05. Error bars, S.D.
In order to determine whether p75NTR cleavage was required for NGF-induced BTIC proliferation, we left BTIC lines untreated or exposed them to 500 nm α-secretase inhibitor TAPI-2 or 200 nm γ-secretase inhibitor DAPT; cells were then treated with 100 ng/ml NGF for 3 days (DAPT, TAPI-2, and NGF were added to cells every 24 h), and the effect of NGF on BTIC proliferation was measured by Ki67 staining. We found that both TAPI-2 and DAPT completely blocked the NGF-mediated stimulation of proliferation (Fig. 6, B–I). Somewhat surprisingly, inhibition of α-secretase or γ-secretase reduced proliferation even when unstimulated by exogenous NGF (Fig. 6, B–I). These results demonstrate that p75NTR cleavage and release of ICD is necessary to stimulate NGF-mediated proliferation of BTICs.
Expression of Cleavage-resistant Mutant p75NTRs Prevents NGF-stimulated Brain Tumor-initiating Cell Proliferation
Because α-secretase and γ-secretase are known to cleave many other transmembrane proteins to generate signaling intracellular domains (55), it is also likely that inhibitors block the cleavage of other non-p75NTR substrates. To confirm that BTIC proliferation by NGF is due to cleavage of p75NTR and release of ICD by γ-secretase, we used cleavage-resistant mutant p75NTR, p75FasTM, which is resistant to γ-secretase-mediated cleavage and can block p75NTR-dependent apoptosis (26, 27) and invasion of glioma cells (19). BTICs were transfected with wild-type p75NTR or p75FasTM by electroporation or left untransfected; 3 days later, the transfected cells were washed twice with media and treated with NGF and epoxomycin for 6 h, and receptor cleavage was measured. NGF-induced p75NTR cleavage was found in BTICs left untransfected or transfected with wild type p75NTR, whereas in BTICs transfected with p75FasTM, the receptor cleavage and ICD release were completely blocked (Fig. 7A). These results demonstrate that mutant p75NTR blocks the processing of wild type p75NTR in BTICs.
FIGURE 7.
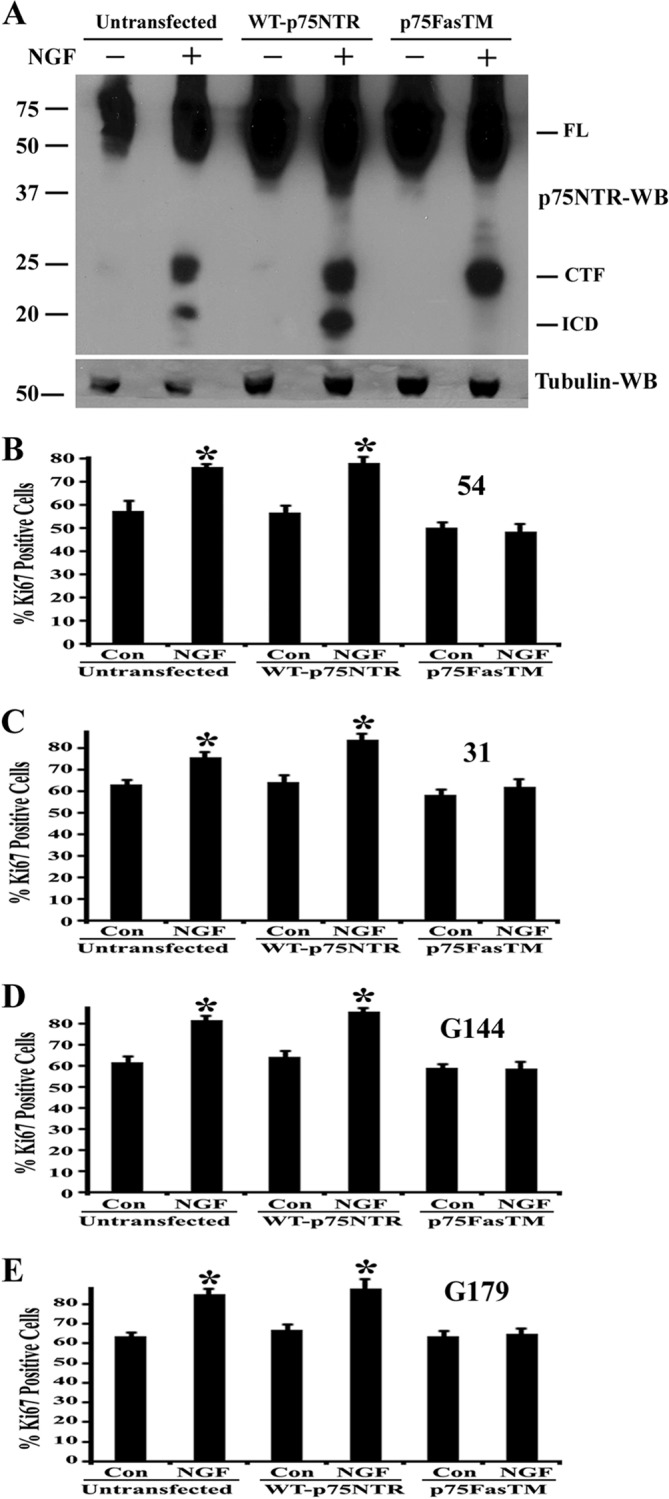
Expression of γ-secretase-resistant mutant p75NTR blocks proliferation of brain tumor-initiating cells. A, BTICs (line 54) were transfected with wild type p75NTR or γ-secretase-resistant mutant p75NTR (p75FasTM) using the Amaxa nucleofector device and Amaxa electroporation kit or left untransfected. After 3 days, the cells were washed twice with fresh media and treated with 100 ng/ml NGF and 1 μm epoxomycin for 6 h, and then cells were lysed and subjected to Western blot analysis with p75NTR ICD antibody and tubulin. FL, full-length p75NTR. Data are representative of three independent experiments. BTICs, such as lines 54 (B), 31 (C), G144 (D), and G179 (E), were transfected as above with wild type p75NTR or p75FasTM or left untransfected. Two days after transfection, the cells were treated with 100 ng/ml NGF for 3 days (NGF was added to medium every 24 h). Three days after NGF treatment, cells were fixed and stained for Ki67 and DAPI, and Ki67-positive cells were quantified, and data were expressed as a percentage of Ki67-positive cells. Data shown are means ± S.D., n = 3 independent experiments (*, p < 0.05). Error bars, S.D.
To assess whether p75FasTM could block NGF-p75NTR-mediated cell proliferation, BTICs were transfected with these constructs by electroporation, and 2 days after transfection, cells were treated with NGF (NGF was added every 24 h) for 3 days. Then we measured the proliferation by Ki67 immunostaining. In cells left untransfected or transfected with wild type-p75NTR, NGF stimulated proliferation; however, the γ-secretase-resistant mutant completely blocked the NGF-induced proliferation (Fig. 7, B–E). These results show that γ-secretase-mediated cleavage of p75NTR and release of its ICD is essential for the NGF-mediated proliferation of BTICs.
Inhibition of Trk Signaling Blocks NGF-mediated Brain Tumor-initiating Cell Proliferation and p75NTR Cleavage
NGF is known to signal through both p75NTR and TrkA to mediate physiological functions, such as neuronal survival, proliferation, and differentiation (21). The BTICs we used in this study express both p75NTR and TrkA and also secrete NGF (Fig. 1). Inhibition of secreted NGF decreases BTIC proliferation, and treating these cells with exogenous NGF stimulate proliferation (Fig. 4). Further, NGF stimulated the α- and γ-secretase-mediated p75NTR cleavage, which is necessary for BTIC proliferation (Figs. 6 and 7). However, the role of TrkA signaling in this context was not known. Recent studies have demonstrated that activation of TrkA receptor with NGF regulates α-secretase- and γ-secretase-mediated p75NTR cleavage and releases the intracellular domain (56, 50), which is required for NGF-mediated cell survival (50, 46). Based on these observations, we hypothesized that NGF binding to the TrkA receptor in BTICs activates signals to mediate p75NTR cleavage, which stimulate BTIC proliferation. Therefore, we examined the role of Trk signaling in BTIC proliferation and p75NTR cleavage. We treated BTICs with 200 nm Trk inhibitor K252a or left them untreated with or without 50 ng/ml NGF for 3 days (K252a and NGF were added to cells for every 24 h), and proliferation was measured by Ki67 immunostaining. Fig. 8, A–D, shows that K252a completely blocked NGF-mediated BTIC proliferation.
FIGURE 8.

Inhibition of Trk signaling attenuates NGF-stimulated brain tumor-initiating cell proliferation and p75NTR cleavage. BTIC lines 54 (A), 31 (B), G144 (C), and G179 (D) were split and maintained in neurobasal medium for 12 h and then treated with 50 ng/ml NGF alone or along with Trk inhibitor K252a (200 nm) or left untreated for 3 days. Cells were added with NGF and K252a every 24 h. Three days later, cells were fixed and stained for Ki67 and DAPI, and Ki67-positive cells were quantified. Data shown are means ± S.D. (error bars), n = 3 independent experiments (*, p < 0.05). E, BTIC line 54 was maintained in neurobasal medium for 2 days, then washed with fresh medium and treated with 50 ng/ml of NGF in the presence or absence of Trk inhibitor K252a (200 nm) and with 1 μm epoxomycin for 6 h. Then cells were lysed and subjected to Western blotting (WB) using p75NTR ICD and tubulin antibodies. FL, full-length p75NTR. Data are representative of three independent experiments. F, BTIC line 54 was maintained in neuronal stem cell expansion medium containing EGF and FGF and treated with 50 ng/ml NGF for 2 and 6 h. Then total RNA was isolated and reverse-transcribed, and quantitative PCR was performed for ADAM17/TACE and actin. Expression of ADAM17 was calculated as ΔΔCt against the actin. Data are the mean of two independent experiments.
To test whether Trk signaling regulates p75NTR cleavage, BTIC line 54 was left untreated or treated with 200 nm Trk inhibitor K252a and then exposed to 50 ng/ml NGF or vehicle for 6 h. In the presence of epoxomycin, K252a blocked accumulation of the CTF and ICD (Fig. 8E), indicating that Trk signaling is necessary for p75NTR cleavage in brain tumor-initiating cells.
Studies have shown that TACE/ADAM17 is required for release of the p75NTR extracellular domain and the CTF via its up-regulation or phosphorylation at threonine 735 in response to neurotrophin treatment (45, 27, 46). Further, knockdown of ADAM17 blocked p75NTR cleavage-mediated activation of Jun kinase and Akt and also mediates cell death and survival (27, 46). Therefore, we examined the expression levels of ADAM17 in response to NGF in BTICs. Quantitative PCR analysis demonstrates that ADAM17 mRNA was increased by 2.5-fold following NGF treatment (Fig. 8F). It is possible that ADAM17 might also be phosphorylated following NGF treatment as reported recently (46). Considering the role of ADAM17 in tumor formation (53, 54), these results suggest that ADAM17 might be involved in neurotrophin-mediated BTIC proliferation and tumor development.
Expression of the p75NTR ICD Alone Is Sufficient to Stimulate Brain Tumor-initiating Cell Proliferation
In order to determine whether γ-secretase-released p75NTR ICD is sufficient to induce BTIC proliferation and to rule out the possible involvement of other substrates of γ-secretase in this process, we expressed the ICD in BTICs by Amaxa electroporation and examined the effect on proliferation. The same ICD construct has been shown to induce apoptotic cell death in PC12 cells (57) and also in sympathetic neurons (26) and activate Jun kinase (27) and Akt (46), suggesting signaling capabilities of ICD. Expression of the ICD in BTIC line 54 was confirmed 2 days after transfection (Fig. 9A), and proliferation was assessed 1 day later. We found that expression of ICD stimulated the proliferation of brain tumor-initiating cells (Fig. 9, B and C). These data clearly show that ICD alone is sufficient to mediate proliferation without involvement from other substrates of the γ-secretase.
FIGURE 9.
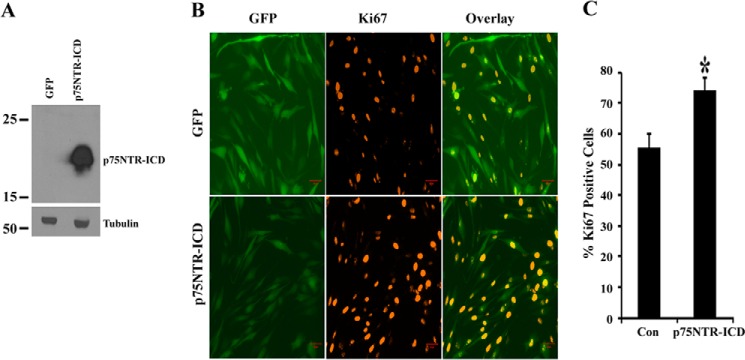
p75NTR ICD expression induces proliferation of brain tumor-initiating cells. BTIC line 54 was transfected with GFP alone or co-transfected with GFP and p75NTR intracellular domain construct (p75ICD) using the Amaxa electroporation method, and cells were maintained in medium with growth factors. Two days later, cells were lysed, and p75ICD and tubulin Western blotting analysis was performed (A). Three days after transfection, cells were fixed, stained for Ki67 (red), and imaged using microscopy (B) (scale bar, 50 μm). The proliferation was scored by calculating the number of Ki67-positive cells in GFP-positive cells (C). Data shown are means ± S.D. (error bars), n = 3 independent experiments (*, p < 0.05).
p75NTR Cleavage Is Required for Akt Activation in Brain Tumor-initiating Cells
The Akt pathway is necessary for glioma cell proliferation and invasion and for glioma development and progression (58, 59). p75NTR cleavage by α- and γ-secretases has also been implicated in regulating neurotrophin-dependent survival through activation of Akt signaling (50, 46). Therefore, we examined the Akt activation when p75NTR was down-regulated and receptor cleavage was blocked. We electroporated control siRNA and p75NTR siRNAs (siRNA 1 and 2 combined) to knock down p75NTR or γ-secretase-resistant mutant p75NTR (p75FasTM) shown to block endogenous p75NTR cleavage and proliferation (Fig. 7). Cells were maintained in medium without growth factors for 2 days and then replaced with the medium with growth factors EGF and FGF for 6 h. We found that knockdown of p75NTR or blocking of its γ-secretase-dependent cleavage significantly decreased Akt activation (Fig. 10, A and B), suggesting that p75NTR cleavage mediates BTIC proliferation via Akt activity.
FIGURE 10.
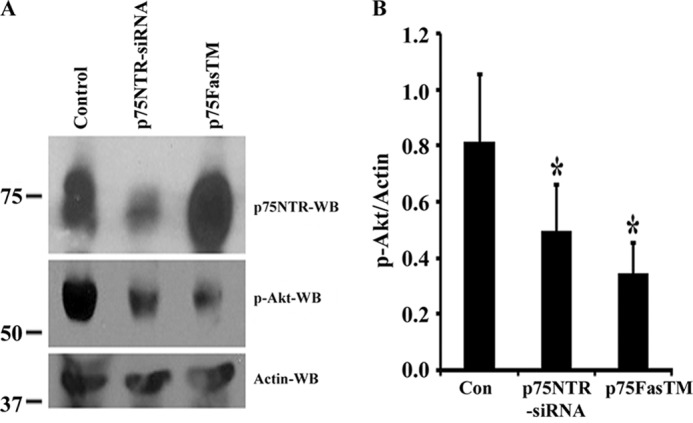
p75NTR cleavage is required for Akt activation in brain tumor-initiating cells. A, BTIC line 54 was electroporated using the Amaxa electroporation method with control siRNA (Control), p75NTR-siRNAs, or γ-secretase-mediated resistant mutant p75NTR (p75FasTM), and cells were maintained in neurobasal medium without growth factors for 2 days. Then cells were added with growth factors EGF and FGF for 6 h and lysed, and Western blotting (WB) was performed for p75NTR, phospho-Akt, and actin. B, phospho-Akt (p-Akt) and actin Western blots were quantified and expressed as relative phospho-Akt/actin ratio. n = 3 independent experiments (*, p < 0.05).
p75NTR, α-Secretase, and γ-Secretase Components Are Expressed, and p75NTR Cleavage Occurs in Vivo in Malignant Glioma Patient Tumors
p75NTR is expressed in high grade malignant gliomas (18, 60), indicating its potential role in glioma development and progression in humans; however, the mechanisms that underlie this are not known. Several recent studies have shown that p75NTR undergoes regulated intramembrane proteolytic cleavage via an α-secretase and then by a γ-secretase in a manner analgous to Notch, amyloid precursor protein, and Erb4 (48, 47, 55). Further, the p75NTR ICD has signaling potential and inhibits neurite outgrowth in cerebellar granular neurons through the Rho pathway (49), induces ligand-dependent cell death in sympathetic neurons via activation of JNK (26, 27), mediates cell survival and growth arrest through Akt activation (50, 46), regulates angiogenesis via stabilization of HIF-1α (61), and also has been implicated in cell division and proliferation of spiral ganglion Schwann cells (51). We have also found that p75NTR cleavage was required for glioma invasion in vivo and in vitro (19) and proliferation of glioma-initiating cells in vitro (Figs. 6 and 7). We therefore hypothesized that α-secretase and γ-secretase enzymes are expressed in human brain tumors and p75NTR cleavage occurs in vivo in human malignant glioma specimens.
We used microarray expression data from the Cancer Genome Atlas (Fig. 11A), which has gene expression data from 528 GBM patients and 10 normal brain tissues, and validated our findings in an independent data set derived from the Moffitt Cancer Center Total Cancer Care data set (Fig. 11B), which has no expression data from normal brain tissue. We examined the expression levels of p75NTR (NGFR), TrkA (NTRK1), NGF, and genes related to p75NTR processing, such as α-secretases (ADAM10 and ADAM17/TACE) and the γ-secretase enzyme subunits (PSEN1, PSEN2, NCSTN, and APH1). In the Cancer Genome Atlas data, p75NTR, α-secretase, and γ-secretase enzyme levels were elevated in GBM tissue compared with normal brain tissue (Fig. 11A), and the Moffitt Cancer Center Total Cancer Care data set showed a similar pattern of expression (Fig. 11B). However, TrkA levels were not expressed at higher levels than controls (Fig. 11A). These results indicate that p75NTR and its cleavage-related signaling components are present and elevated in the tumors derived from patients with GBMs and that our observations above are not an artifact of tissue culture conditions.
FIGURE 11.

p75NTR and signaling components associated with receptor proteolysis and p75NTR cleaved products, CTF, and ICD are elevated in malignant glioma patient tumor specimens. Shown is the microarray data set of 528 GBM patients and 10 normal tissues from the Cancer Genome Atlas (A) and Moffitt Cancer Center data set of 108 GBM patients (B) for expression analysis of p75NTR, its co-receptor TrkA/NTRK1, ligand NGF, α-secretases (ADAM10 and ADAM17), and γ-secretase components, such as presenelin 1 and 2 (PSEN1 and PSEN2), nicastrin (NCSTN), and APH1. We considered values above 2 of log2 expression as significantly higher. C, frozen tumor specimens from grade I to grade IV of malignant gliomas obtained from the Moffitt Cancer Center. We isolated total RNA, from frozen tissue and performed RT-PCR analysis for p75NTR, NGF, and actin using human-specific primers. Human brain total RNA obtained from Clontech served as a positive control (+ve). D, frozen tumor specimens were lysed and subjected to Western blotting analysis (WB) of p75NTR using p75NTR ICD antibody, which recognizes full-length (FL) receptor and the CTF and ICD of the receptor. Tubulin was used as a loading control, and U251 cells expressing p75NTR were used as positive control for p75NTR. JPA1, juvenile pilocytic astrocytoma; GA, gemistocytic astrocytoma; LGA, low grade astrocytoma; LGO, low grade oligodendroglioma; AOA, anaplastic oligoastrocytoma; AA, anaplastic astrocytoma. RT and Chemo, patient tumors treated with radiation and temozolomide (chemotherapy) before surgery.
In order to both validate the expression data at the level of RNA and protein expression and determine directly whether p75NTR is cleaved in malignant glioma tumor specimens, we examined snap frozen tumor specimens from a subset of patients with malignant gliomas. We used tumors from glioma patients (ranging from WHO grade I to grade IV tumors) and performed RT-PCR analysis for p75NTR and NGF using human-specific primers and p75NTR Western blotting using an antibody raised against the ICD of the receptor. NGF and p75NTR were expressed in glioma tumors (Fig. 11C), and CTF and ICD fragments were found in every tumor we examined; however, the cleaved products were more highly expressed in GBM patients as compared with low grade gliomas (Fig. 11D).
DISCUSSION
Malignant gliomas are a very aggressive type of brain tumors that have a very poor prognosis. Their etiology is unknown. The identification and characterization of a subset of poorly differentiated stemlike cells, called glioma stem cells or BTICs (62, 13, 63), offer an opportunity to understand the genesis of gliomas. Identification of the factors that regulate BTIC proliferation, invasion, and tumor development is essential to identify novel therapeutic targets for these aggressive tumors.
By using unbiased serial in vivo selection, we identified p75NTR as a novel mediator of glioma (18) and BTIC invasion (19). Expression of p75NTR in human gliomas can make a subset of non-invasive gliomas highly invasive; this effect is enhanced with NGF stimulation. Conversely, the siRNA knockdown of p75NTR reduced the migration of highly invasive glioma cells and BTICs. p75NTR is also implicated in regulating cell survival and proliferation of other types of cancers. In malignant melanoma, p75NTR is highly expressed, and neurotrophin binding increases melanoma cell invasiveness and promotes survival (64–66). p75NTR activation by NGF promotes survival and proliferation of breast cancer cells (29–31). However, in prostate cancer, bladder cancer, and gastric cancer cells, p75NTR acts as a tumor suppressor by inducing apoptosis and inhibiting cell proliferation and invasion (67–70). Taken together, these findings suggest that p75NTR expression on some tumor cells enhances their survival and invasion, and in other tumor cells it inhibits survival and invasion. The role of p75NTR in cellular proliferation of human BTICs has not been described previously. In this study, we found that knockdown of p75NTR by siRNA decreased BTIC proliferation, and binding of NGF to TrkA receptor led to Trk signaling-dependent p75NTR receptor activation by regulated intramembrane proteolysis, and this was necessary for BTIC proliferation. NGF and TrkA signaling is known to promote breast cancer cell proliferation, invasion, and metastasis (31, 71, 72). We discovered that inhibition of Trk signaling blocked p75NTR cleavage and BTIC proliferation, suggesting the role of Trks in glioma cell proliferation. It is not clear how Trk signaling regulates p75NTR processing.
According to the cancer stem cell hypothesis, glioma stem cells/BTICs are responsible for malignant brain tumor development (62, 73). They express neuronal stem cell markers, such as Nestin and Sox2, self-renew, potentially differentiate into many cell types that make up the tumor (10–14, 62, 74, 75), and develop tumors when transplanted into immunodeficient mice, which resemble MGs found in human patients (8, 13, 14). The inhibition of proliferation pathways, such as the Notch (76) and hedgehog pathways (77–79), in glioma stem cells abrogate glioma formation. Similarly, we demonstrate here that the inhibition of p75NTR signaling in BTICs inhibits proliferation and, from our previous work, invasion and migration of BTICs (19). These findings indicate that targeting p75NTR in BTICs may be a unique therapeutic strategy for MGs.
Others have reported on the roles of the enzymes ADAM-10 and -17 (which cleave the extracellular domain of p75NTR and liberate its CTF) in gliomas. The ADAM metalloproteases are required for numerous cellular functions and may be involved in glioma pathogenesis (80, 81). ADAM10, -12, and -17 are implicated in invasion and proliferation of GBM (53, 54, 82–84). TGFβ1 mediates glioma invasion through up-regulation of ADAM17/TACE (53). These findings suggest that increased expression and activity of ADAM family members is associated with gliomagenesis. p75NTR is known to be processed by ADAM17/TACE (27, 85) and that p75NTR signaling increases ADAM17 expression, which is necessary for apoptotic cell death in sympathetic neurons (27). We found that NGF stimulation of BTICs increases ADAM17 expression (Fig. 8F), and ADAM10 and ADAM17 levels are increased in malignant glioma (Fig. 11, A and B). Here we show that the p75NTR-CTF (the cleaved product of ADAM cleavage) is increased in MG patient tumors (Fig. 11D). It is also known that ADAMs are required for glioma stem cell sphere formation (86). We found that inhibition of the ADAM metalloproteases blocked p75NTR proteolysis, prevented glioma invasion in vitro and in vivo (19), and reduced proliferation of BTICs (Fig. 6). ADAM family proteins are required for glioma proliferation and invasion (53, 54, 82–84). Although p75NTR up-regulates ADAM17 levels through Jun kinase activity (27), and ADAM17 enzymatic activity is increased through phosphorylation by the MAPK p38 (87), ERK (88), and MEK (46) at Thr735, it is not yet clear which ADAMs are involved in p75NTR processing in malignant glioma and whether they are transcriptionally up-regulated or post-translationally modified.
γ-Secretase is known to process many membrane receptors (e.g. Notch, amyloid precursor protein, Erb-4, and E-cadherin) and to release ICDs that have signaling capabilities; this has been identified as a novel mechanism in receptor signaling biology (55, 89). It is known that inhibition of γ-secretase blocks glioma stem cell sphere formation, proliferation, and survival and increases the differentiation and cell death in vitro and tumor formation in vivo (90–93). It has been speculated that these effects are due to γ-secretase-mediated Notch signaling (92, 93). However, several studies have now found that p75NTR is also cleaved by the γ-secretase and is required for receptor signaling in neurons, where it mediates processes that include inhibition of neurite outgrowth, cell death, and cell survival (25, 26, 46–49, 61). Here we expand the contextual repertoire of p75NTR cleavage by the γ-secretase and show that it mediated cellular proliferation in BTICs (Fig. 6 and 7).
Here we show that p75NTR activation is also required for proliferation of brain tumor-initiating cells in addition to its role in invasion. In our efforts to understand the mechanisms underlying p75NTR-regulated glioma invasion and proliferation, we recently (19) and here demonstrated that the γ-secretase-mediated cleavage of the receptor and release of intracellular domain are required for receptor-mediated invasion and proliferation. How receptor cleavage mediates these effects was not known. p75NTR cleavage is necessary for Akt activation and neurotrophin survival in PC12 cells and neurons (46, 50). Further, Akt is implicated in glioma cell proliferation, migration, and invasion and in glioma development and progression (58, 59). Here we have shown that Akt activation was attenuated in BTICs when receptor proteolysis was blocked (Fig. 10).
We previously studied the effect of p75NTR in gliomas (19), largely focusing on U87 and U251 human glioma cell lines, which do not express endogenous p75NTR (19) and have been in cell culture for decades. Here, we regarded the short term culture of patient-derived BTICs as more likely to maintain the critical factors and pathways involved in tumor formation. In addition, we did not perform p75NTR knockdown in the single BTIC we studied, so the effects on proliferation were not apparent. In our current study, we have used four different glioma stem cell lines to understand the role of p75NTR in glioma stem cell proliferation, which we examined thoroughly here.
p75NTR signaling is very complex. The receptor interacts with several co-receptors and multiple ligands and activates multiple signaling pathways, such as NF-κB and JNK with important physiological consequences (21). The p75NTRs exist as dimers that are linked through conserved cysteine 257 in the transmembrane domain. Upon binding of neurotrophins to p75NTR, dimers activate the receptor function via conformational rearrangement, which leads to recruitment of various interactors to the p75NTR ICD and initiation of downstream signaling (94). Several interactors have been identified that can bind to its ICD, including NRIF (95), TRAF6 (42), Rho-GDI (96), NRAGE (97, 98), SC1 (99), RIP2 (41), Bex1 (100), and Fascin (101). There are several examples of the effects of p75NTR cleavage and its interactions with various interactors that are cell type-specific. For example, p75NTR undergoes proteolytic cleavage and releases its ICD, which is required for 1) receptor-dependent inhibition of neurite outgrowth through Rho activation (49), 2) induction of apoptosis via TRAF6-dependent ubiquitination, nuclear translocation of NRIF, and activation of JNK (26, 27, 102), 3) cell survival through Akt activation (46), and 4) neoangiogenesis via HIF-1α stabilization (61). Interestingly, Akt (59), JNK (103, 104), and HIF-1α (105, 106) are also required for glioma cell proliferation, migration, and invasion. The identity of the proteins that interact with the ICD of p75NTR in MGs to mediate its effects on invasion and proliferation will be an important subject for further study.
Acknowledgments
We thank Dr. Moses Chao (New York University) for plasmids expressing wild type and p75FasTM. We also thank Dr. D. L. Alkon, S. Phares, and Dr. C. Lim (Blanchette Rockefeller Neuroscience Institute) and Dr. Fortenbery (Moffitt Cancer Center) for assistance.
This work was supported, in whole or in part, by National Institutes of Health Grants R01NS038220 (to B. D. C.) and 1 I01 BX000744-1 (to M. K. C.). This work was also supported by grants from the Moffitt Foundation (to P. A. F. and R. S. K.).
- GBM
- glioblastoma multiforme
- BTIC
- brain tumor-initiating cell
- p75NTR
- p75 neurotrophin receptor
- ICD
- p75 neurotrophin receptor intracellular domain
- MG
- malignant glioma
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- ICD
- intracellular domain
- BDNF
- brain-derived neurotrophic factor
- NT3
- neurotrophin 3
- CTF
- carboxyl-terminal fragment
- DAPT
- N-[N-3,5-difluorophenacetyl-l-analyl]-S-phenyl glycine t-butyl ester.
REFERENCES
- 1. Louis D. N., Ohgaki H., Wiestler O. D., Cavenee W. K., Burger P. C., Jouvet A., Scheithauer B. W., Kleihues P. (2007) The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 114, 97–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kleihues C., Cavenee W. K. (2007) Tumors of the Nervous System, 4th Ed., IARC Press, Lyon, France [Google Scholar]
- 3. Holland E. C. (2001) Gliomagenesis: genetic alterations and mouse models. Nat. Rev. Genet. 2, 120–129 [DOI] [PubMed] [Google Scholar]
- 4. Stupp R., Hegi M. E., Mason W. P., van den Bent M. J., Taphoorn M. J., Janzer R. C. (2009) Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet. Oncol. 10, 459–466 [DOI] [PubMed] [Google Scholar]
- 5. Cancer Genome Atlas Research Network (2008) Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455, 1061–1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Parsons D. W., Jones S., Zhang X., Lin J. C., Leary R. J., Angenendt P. (2008) An integrated genomic analysis of human glioblastoma multiforme. Science 321, 1807–1812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yan H., Parsons D. W., Jin G., McLendon R., Rasheed B. A., Yuan W. (2009) IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 360, 765–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Singh S. K., Hawkins C., Clarke I. D., Squire J. A., Bayani J., Hide T., Henkelman R. M., Cusimano M. D., Dirks P. B. (2004) Identification of human brain tumour initiating cells. Nature 432, 396–401 [DOI] [PubMed] [Google Scholar]
- 9. Clarke M. F., Dick J. E., Dirks P. B., Eaves C. J., Jamieson C. H., Jones D. L., Visvader J., Weissman I. L., Wahl G. M. (2006) Cancer stem cells perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 66, 9339–9344 [DOI] [PubMed] [Google Scholar]
- 10. Gangemi R. M., Griffero F., Marubbi D., Perera M., Capra M. C., Malatesta P., Ravetti G. L., Zona G. L., Daga A., Corte G. (2009) SOX2 silencing in glioblastoma tumor initiating cells causes stop of proliferation and loss of tumorigenicity. Stem Cells 27, 40–48 [DOI] [PubMed] [Google Scholar]
- 11. Mangiola A., Lama G., Giannitelli C., De Bonis P., Anile C., Lauriola L., La Torre G., Sabatino G., Maira G., Jhanwar-Uniyal M., Sica G. (2007) Stem cell marker nestin and c-Jun NH2-terminal kinases in tumor and peritumor areas of glioblastoma multiforme. Possible prognostic implications. Clin. Cancer Res. 13, 6970–6977 [DOI] [PubMed] [Google Scholar]
- 12. Galli R., Binda E., Orfanelli U., Cipelletti B., Gritti A., De Vitis S., Fiocco R., Foroni C., Dimeco F., Vescovi A. (2004) Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 64, 7011–7021 [DOI] [PubMed] [Google Scholar]
- 13. Lee J., Kotliarova S., Kotliarov Y., Li A., Su Q., Donin N. M., Pastorino S., Purow B. W., Christopher N., Zhang W., Park J. K., Fine H. A. (2006) Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell 9, 391–403 [DOI] [PubMed] [Google Scholar]
- 14. Pollard S. M., Yoshikawa K., Clarke I. D., Danovi D., Stricker S., Russell R., Bayani J., Head R., Lee M., Bernstein M., Squire J. A., Smith A., Dirks P. (2009) Glioma stem cell lines expanded in adherent culture have tumor-specific phenotypes and are suitable for chemical and genetic screens. Cell Stem Cell 4, 568–580 [DOI] [PubMed] [Google Scholar]
- 15. Bao S., Wu Q., McLendon R. E., Hao Y., Shi Q., Hjelmeland A. B., Dewhirst M. W., Bigner D. D., Rich J. N. (2006) Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 444, 756–760 [DOI] [PubMed] [Google Scholar]
- 16. Bleau A. M., Hambardzumyan D., Ozawa T., Fomchenko E. I., Huse J. T., Brennan C. W., Holland E. C. (2009) PTEN/PI3K/Akt pathway regulates the side population phenotype and ABCG2 activity in glioma tumor stem-like cells. Cell Stem Cell 4, 226–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen J., Li Y., Yu T. S., McKay R. M., Burns D. K., Kernie S. G., Parada L. F. (2012) A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 488, 522–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Johnston A. L., Lun X., Rahn J. J., Liacini A., Wang L., Hamilton M. G., Parney I. F., Hempstead B. L., Robbins S. M., Forsyth P. A., Senger D. L. (2007) The p75 neurotrophin receptor is a central regulator of glioma invasion. PLoS Biol. 5, e212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang L., Rahn J. J., Lun X., Sun B., Kelly J. J., Weiss S., Robbins S. M., Forsyth P. A., Senger D. L. (2008) γ-Secretase represents a therapeutic target for the treatment of invasive glioma mediated by the p75 neurotrophin receptor. PLoS Biol. 6, e289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chao M. V. (2003) Neurotrophins and their receptors. A convergence point for many signalling pathways. Nat. Rev. Neurosci. 4, 299–309 [DOI] [PubMed] [Google Scholar]
- 21. Reichardt L. F. (2006) Neurotrophin-regulated signalling pathways. Philos. Trans. R. Soc. Lond. B Biol. Sci. 361, 1545–1564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cattaneo E., McKay R. (1990) Proliferation and differentiation of neuronal stem cells regulated by nerve growth factor. Nature 347, 762–765 [DOI] [PubMed] [Google Scholar]
- 23. Seidl K., Erck C., Buchberger A. (1998) Evidence of the participation of nerve growth factor and its low affinity receptor (p75NTR) in regulation of the myogenic program. J. Cell Physiol. 176, 10–21 [DOI] [PubMed] [Google Scholar]
- 24. Young K. M., Merson T. D., Sotthibundhu A., Coulson E. J., Bartlett P. F. (2007) p75 neurotrophin receptor expression defines a population of BDNF responsive neurogenic precursor cells. J. Neurosci. 27, 5146–5155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zampieri N., Xu C. F., Neubert T. A., Chao M. V. (2005) Cleavage of p75 neurotrophin receptor by α-secretase and γ-secretase requires specific receptor domains. J. Biol. Chem. 280, 14563–14571 [DOI] [PubMed] [Google Scholar]
- 26. Kenchappa R. S., Zampieri N., Chao M. V., Barker P. A., Teng H. K., Hempstead B. L., Carter B. D. (2006) Ligand-dependent cleavage of the P75 neurotrophin receptor is necessary for NRIF nuclear translocation and apoptosis in sympathetic neurons. Neuron 50, 219–232 [DOI] [PubMed] [Google Scholar]
- 27. Kenchappa R. S., Tep C., Korade Z., Urra S., Bronfman F. C., Yoon S. O., Carter B. D. (2010) P75NTR mediated apoptosis in sympathetic neurons involves a biphasic activation of JNK and up regulation of TACE/ADAM17. J. Biol. Chem. 285, 20358–20368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nykjaer A., Willnow T. E., Petersen C. M. (2005) p75NTR: live or let die. Curr. Opin. Neurobiol. 15, 49–57 [DOI] [PubMed] [Google Scholar]
- 29. Descamps S., Pawlowski V., Révillion F., Hornez L., Hebbar M., Boilly B., Hondermarck H., Peyrat J. P. (2001) Expression of nerve growth factor receptors and their prognostic value in human breast cancer. Cancer Res. 61, 4337–4340 [PubMed] [Google Scholar]
- 30. Verbeke S., Meignan S., Lagadec C., Germain E., Hondermarck H., Adriaenssens E., Le Bourhis X. (2010) Overexpression of p75(NTR) increases survival of breast cancer cells through p21(waf1). Cell. Signal. 22, 1864–18673 [DOI] [PubMed] [Google Scholar]
- 31. Hondermarck H. (2012) Neurotrophins and their receptors in breast cancer. Cytokine Growth Factor Rev. 23, 357–365 [DOI] [PubMed] [Google Scholar]
- 32. Weichert H., Blechschmidt I., Schroder S., Ambrosius H. (1991) The MTT-assay as a rapid test for cell proliferation and cell killing: application to human peripheral blood lymphocytes (PBL). Allerg. Immunol. (Leipz.) 37, 139–144 [PubMed] [Google Scholar]
- 33. Wang Y. Q., Zhang F., Tian R., Ji W., Zhou Y., Sun X. M., Liu Y., Wang Z. Y., Niu R. F. (2012) Tyrosine 23 phosphorylation of annexin A2 promotes proliferation, invasion, and Stat3 phosphorylation in the nucleus of human breast cancer SK-BR-3 cells. Cancer Biol. Med. 9, 248–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schlüter C., Duchrow M., Wohlenberg C., Becker M. H., Key G., Flad H. D., Gerdes J. (1993) The cell proliferation-associated antigen Ki-67: a very large, ubiquitous nuclear protein with numerous repeated elements, representing a new kind of cell cycle-maintaining proteins. J. Cell Biol. 123, 513–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schnell O., Romagna A., Jaehnert I., Albrecht V., Eigenbrod S., Juerchott K., Kretzschmar H., Tonn J. C., Schichor C. (2012) Krüppel-like factor 8 (KLF8) is expressed in gliomas of different WHO grades and is essential for tumor cell proliferation. PLoS One 7, e30429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hempstead B. L., Martin-Zanca D., Kaplan D. R., Parada L. F., Chao M. V. (1991) High affinity NGF binding requires co-expression of the trk proto-oncogene and the low-affinity NGF receptor. Nature 350, 678–683 [DOI] [PubMed] [Google Scholar]
- 37. Barker P. A., Shooter E. M. (1994) Disruption of NGF binding to the low affinity neurotrophin receptor p75LNTR reduces NGF binding to TrkA in PC12 cells. Neuron 13, 203–215 [DOI] [PubMed] [Google Scholar]
- 38. Epa W. R., Markovska K., Barrett G. L. (2004) The p75 neurotrophin receptor enhances TrkA signaling by binding to Shc and augmenting its phosphorylation. J. Neurochem. 89, 344–353 [DOI] [PubMed] [Google Scholar]
- 39. Kuruvilla R., Zweifel L. S., Glebova N. O., Lonze B. E., Valdez G., Ye H., Ginty D. D. (2004) A neurotrophin signaling cascade coordinates sympathetic neuron development through differential control of TrkA trafficking and retrograde signaling. Cell 39, 57–68 [DOI] [PubMed] [Google Scholar]
- 40. Carter B. D., Kaltschmidt C., Kaltschmidt B., Offenhäuser N., Böhm-Matthaei R., Baeuerle P. A., Barde Y. A. (1996) Selective activation of NF-κB by nerve growth factor through the neurotrophin receptor p75. Science 272, 542–555 [DOI] [PubMed] [Google Scholar]
- 41. Khursigara G., Bertin J., Yano H., Moffett H., DiStefano P. S., Chao M. V. (2001) A prosurvival function for the p75 receptor death domain mediated via the caspase recruitment domain receptor-interacting protein 2. J. Neurosci. 21, 5854–5863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Khursigara G., Orlinick J. R., Chao M. V. (1999) Association of the p75 neurotrophin receptor with TRAF6. J. Biol. Chem. 274, 2597–2600 [DOI] [PubMed] [Google Scholar]
- 43. DiStefano P. S., Chelsea D. M. (1990) Regulation of Schwann cell surface and truncated nerve growth factor receptors in vitro by axonal components. Brain Res. 534, 340–344 [DOI] [PubMed] [Google Scholar]
- 44. DiStefano P. S., Chelsea D. M., Schick C. M., McKelvy J. F. (1993) Involvement of a metalloprotease in low-affinity nerve growth factor receptor truncation: inhibition of truncation in vitro and in vivo. J. Neurosci. 13, 2405–2414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Weskamp G., Schlöndorff J., Lum L., Becherer J. D., Kim T. W., Saftig P., Hartmann D., Murphy G., Blobel C. P. (2004) Evidence for a critical role of the tumor necrosis factor α convertase (TACE) in ectodomain shedding of the p75 neurotrophin receptor (p75NTR). J. Biol. Chem. 279, 4241–4249 [DOI] [PubMed] [Google Scholar]
- 46. Kommaddi R. P., Thomas R., Ceni C., Daigneault K., Barker P. A. (2011) Trk-dependent ADAM17 activation facilitates neurotrophin survival signaling. FASEB J. 25, 2061–2070 [DOI] [PubMed] [Google Scholar]
- 47. Kanning K. C., Hudson M., Amieux P. S., Wiley J. C., Bothwell M., Schecterson L. C. (2003) Proteolytic processing of the p75 neurotrophin receptor and two homologs generates C-terminal fragments with signaling capability. J. Neurosci. 23, 5425–5436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jung K. M., Tan S., Landman N., Petrova K., Murray S., Lewis R., Kim P. K., Kim D. S., Ryu S. H., Chao M. V., Kim T. W. (2003) Regulated intramembrane proteolysis of the p75 neurotrophin receptor modulates its association with the TrkA receptor. J. Biol. Chem. 278, 42161–42169 [DOI] [PubMed] [Google Scholar]
- 49. Domeniconi M., Zampieri N., Spencer T., Hilaire M., Mellado W., Chao M. V., Filbin M. T. (2005) MAG induces regulated intramembrane proteolysis of the p75 neurotrophin receptor to inhibit neurite outgrowth. Neuron 46, 849–855 [DOI] [PubMed] [Google Scholar]
- 50. Ceni C., Kommaddi R. P., Thomas R., Vereker E., Liu X., McPherson P. S., Ritter B., Barker P. A. (2010) The p75NTR intracellular domain generated by neurotrophin-induced receptor cleavage potentiates Trk signaling. J. Cell Sci. 123, 2299–2307 [DOI] [PubMed] [Google Scholar]
- 51. Provenzano M. J., Minner S. A., Zander K., Clark J. J., Kane C. J., Green S. H., Hansen M. R. (2011) p75(NTR) expression and nuclear localization of p75(NTR) intracellular domain in spiral ganglion Schwann cells following deafness correlate with cell proliferation. Mol. Cell Neurosci. 47, 306–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Parkhurst C. N., Zampieri N., Chao M. V. (2010) Nuclear localization of the p75 neurotrophin receptor intracellular domain. J. Biol. Chem. 285, 5361–5368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lu Y., Jiang F., Zheng X., Katakowski M., Buller B., To S. S., Chopp M. (2011) TGF-β1 promotes motility and invasiveness of glioma cells through activation of ADAM17. Oncol. Rep. 25, 1329–1335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zheng X., Jiang F., Katakowski M., Zhang Z. G., Lu Q. E., Chopp M. (2009) ADAM17 promotes breast cancer cell malignant phenotype through EGFR-PI3K-AKT activation. Cancer Biol. Ther. 8, 1045–1054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Fortini M. E. (2002) γ-Secretase mediated proteolysis in cell-surface-receptor signaling. Nat. Rev. Mol. Cell Biol. 3, 673–684 [DOI] [PubMed] [Google Scholar]
- 56. Urra S., Escudero C. A., Ramos P., Lisbona F., Allende E., Covarrubias P., Parraguez J. I., Zampieri N., Chao M. V., Annaert W., Bronfman F. C. (2007) TrkA receptor activation by nerve growth factor induces shedding of the p75 neurotrophin receptor followed by endosomal γ-secretase-mediated release of the p75 intracellular domain. J. Biol. Chem. 282, 7606–7615 [DOI] [PubMed] [Google Scholar]
- 57. Roux P. P., Bhakar A. L., Kennedy T. E., Barker P. A. (2001) The p75 neurotrophin receptor activates Akt (protein kinase B) through a phosphatidylinositol 3-kinase-dependent pathway. J. Biol. Chem. 276, 23097–24104 [DOI] [PubMed] [Google Scholar]
- 58. Gallia G. L., Tyler B. M., Hann C. L., Siu I. M., Giranda V. L., Vescovi A. L., Brem H., Riggins G. J. (2009) Inhibition of Akt inhibits growth of glioblastoma and glioblastoma stem-like cells. Mol. Cancer Ther. 8, 386–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Eyler C. E., Foo W. C., LaFiura K. M., McLendon R. E., Hjelmeland A. B., Rich J. N. (2008) Brain cancer stem cells display preferential sensitivity to Akt inhibition. Stem Cells 26, 3027–3036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Xiong J., Zhou L., Yang M., Lim Y., Zhu Y. H., Fu D. L., Li Z. W., Zhong J. H., Xiao Z. C., Zhou X. F. (2013) Pro-BDNF and its receptors are upregulated in glioma and inhibit the growth of glioma cells in vitro. Neuro. Oncol. 15, 990–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Le Moan N., Houslay D. M., Christian F., Houslay M. D., Akassoglou K. (2011) Oxygen-dependent cleavage of the p75 neurotrophin receptor triggers stabilization of HIF-1α. Mol. Cell 44, 476–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Singh S. K., Clarke I. D., Terasaki M., Bonn V. E., Hawkins C., Squire J., Dirks P. B. (2003) Identification of a cancer stem cell in human brain tumors. Cancer Res. 63, 5821–5828 [PubMed] [Google Scholar]
- 63. Nakano I., Kornblum H. I. (2006) Brain tumor stem cells. Pediatr. Res. 59, 54R–8R [DOI] [PubMed] [Google Scholar]
- 64. Herrmann J. L., Menter D. G., Hamada J., Marchetti D., Nakajima M., Nicolson G. L. (1993) Mediation of NGF-stimulated extracellular matrix invasion by the human melanoma low-affinity p75 neurotrophin receptor: melanoma p75 functions independently of trkA. Mol. Biol. Cell 4, 1205–1216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Mattei S., Colombo M. P., Melani C., Silvani A., Parmiani G., Herlyn M. (1994) Expression of cytokine/growth factors and their receptors in human melanoma and melanocytes. Int. J. Cancer. 56, 853–857 [DOI] [PubMed] [Google Scholar]
- 66. Marchetti D., Aucoin R., Blust J., Murry B., Greiter-Wilke A. (2004) p75 neurotrophin receptor functions as a survival receptor in brain-metastatic melanoma cells. J. Cell Biochem. 91, 206–215 [DOI] [PubMed] [Google Scholar]
- 67. Khwaja F., Tabassum A., Allen J., Djakiew D. (2006) The p75(NTR) tumor suppressor induces cell cycle arrest facilitating caspase mediated apoptosis in prostate tumor cells. Biochem. Biophys. Res. Commun. 341, 1184–1192 [DOI] [PubMed] [Google Scholar]
- 68. Tabassum A., Khwaja F., Djakiew D. (2003) The p75(NTR) tumor suppressor induces caspase-mediated apoptosis in bladder tumor cells. Int. J. Cancer 105, 47–52 [DOI] [PubMed] [Google Scholar]
- 69. Jin H., Pan Y., Zhao L., Zhai H., Li X., Sun L., He L., Chen Y., Hong L., Du Y., Fan D. (2007) p75 neurotrophin receptor suppresses the proliferation of human gastric cancer cells. Neoplasia 9, 471–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Jin H., Pan Y., He L., Zhai H., Li X., Zhao L., Sun L., Liu J., Hong L., Song J., Xie H., Gao J., Han S., Li Y., Fan D. (2007) p75 neurotrophin receptor inhibits invasion and metastasis of gastric cancer. Mol. Cancer Res. 5, 423–433 [DOI] [PubMed] [Google Scholar]
- 71. Lagadec C., Meignan S., Adriaenssens E., Foveau B., Vanhecke E., Romon R., Toillon R. A., Oxombre B., Hondermarck H., Le Bourhis X. (2009) TrkA overexpression enhances growth and metastasis of breast cancer cells. Oncogene 28, 1960–1970 [DOI] [PubMed] [Google Scholar]
- 72. Descamps S., Toillon R. A., Adriaenssens E., Pawlowski V., Cool S. M., Nurcombe V., Le Bourhis X., Boilly B., Peyrat J. P., Hondermarck H. (2001) Nerve growth factor stimulates proliferation and survival of human breast cancer cells through two distinct signaling pathways. J. Biol. Chem. 276, 17864–17870 [DOI] [PubMed] [Google Scholar]
- 73. Hemmati H. D., Nakano I., Lazareff J. A., Masterman-Smith M., Geschwind D. H., Bronner-Fraser M., Kornblum H. I. (2003) Cancerous stem cells can arise from pediatric brain tumors. Proc. Natl. Acad. Sci. U.S.A. 100, 15178–15183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Fael Al-Mayhani T. M., Ball S. L., Zhao J. W., Fawcett J., Ichimura K., Collins P. V., Watts C. (2009) An efficient method for derivation and propagation of glioblastoma cell lines that conserves the molecular profile of their original tumours. J. Neurosci. Methods 176, 192–199 [DOI] [PubMed] [Google Scholar]
- 75. Piccirillo S. G., Reynolds B. A., Zanetti N., Lamorte G., Binda E., Broggi G., Brem H., Olivi A., Dimeco F., Vescovi A. L. (2006) Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature 444, 761–765 [DOI] [PubMed] [Google Scholar]
- 76. Fan X., Matsui W., Khaki L., Stearns D., Chun J., Li Y. M., Eberhart C. G. (2006) Notch pathway inhibition depletes stem-like cells and blocks engraftment in embryonal brain tumors. Cancer Res. 66, 7445–7452 [DOI] [PubMed] [Google Scholar]
- 77. Bar E. E., Chaudhry A., Lin A., Fan X., Schreck K., Matsui W., Piccirillo S., Vescovi A. L., DiMeco F., Olivi A., Eberhart C. G. (2007) Cyclopamine-mediated hedgehog pathway inhibition depletes stem-like cancer cells in glioblastoma. Stem Cells 25, 2524–2533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Clement V., Sanchez P., de Tribolet N., Radovanovic I., Ruiz i Altaba A. (2007) HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr. Biol. 17, 165–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Sarangi A., Valadez J. G., Rush S., Abel T. W., Thompson R. C., Cooper M. K. (2009) Targeted inhibition of the Hedgehog pathway in established malignant glioma xenografts enhances survival. Oncogene 28, 3468–3476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Wildeboer D., Naus S., Amy Sang Q. X., Bartsch J. W., Pagenstecher A. (2006) Metalloproteinase disintegrins ADAM8 and ADAM19 are highly regulated in human primary brain tumors and their expression levels and activities are associated with invasiveness. J. Neuropathol. Exp. Neurol. 65, 516–527 [DOI] [PubMed] [Google Scholar]
- 81. Mochizuki S., Okada Y. (2007) ADAMs in cancer cell proliferation and progression. Cancer. Sci. 98, 621–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Kohutek Z. A., diPierro C. G., Redpath G. T., Hussaini I. M. (2009) ADAM-10-mediated N-cadherin cleavage is protein kinase C-α dependent and promotes glioblastoma cell migration. J. Neurosci. 29, 4605–4615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Kodama T., Ikeda E., Okada A., Ohtsuka T., Shimoda M., Shiomi T., Yoshida K., Nakada M., Ohuchi E., Okada Y. (2004) ADAM12 is selectively overexpressed in human glioblastomas and is associated with glioblastoma cell proliferation and shedding of heparin-binding epidermal growth factor. Am. J. Pathol. 165, 1743–1753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Hart S., Fischer O. M., Ullrich A. (2004) Cannabinoids induce cancer cell proliferation via tumor necrosis factor α-converting enzyme (TACE/ADAM17)-mediated transactivation of the epidermal growth factor receptor. Cancer Res. 64, 1943–1950 [DOI] [PubMed] [Google Scholar]
- 85. Srinivasan B., Wang Z., Brun-Zinkernagel A. M., Collier R. J., Black R. A., Frank S. J., Barker P. A., Roque R. S. (2007) Photic injury promotes cleavage of p75NTR by TACE and nuclear trafficking of the p75 intracellular domain. Mol. Cell Neurosci. 36, 449–461 [DOI] [PubMed] [Google Scholar]
- 86. Bulstrode H., Jones L. M., Siney E. J., Sampson J. M., Ludwig A., Gray W. P., Willaime-Morawek S. (2012) A-disintegrin and metalloprotease (ADAM) 10 and 17 promote self-renewal of brain tumor sphere forming cells. Cancer Lett. 326, 79–87 [DOI] [PubMed] [Google Scholar]
- 87. Xu P., Derynck R. (2010) Direct activation of TACE-mediated ectodomain shedding by p38 MAP kinase regulates EGF receptor-dependent cell proliferation. Mol. Cell 37, 551–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Díaz-Rodriguez E., Montero J. C., Esparís-Ogando A., Yuste L., Pandiella A. (2002) Extracellular signal regulated kinase phosphorylates tumor necrosis factor α-converting enzyme at threonine 735: a potential role in regulated shedding. Mol. Biol. Cell 13, 2031–2044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Ebinu J. O., Yankner B. A. (2002) A RIP tide in neuronal signal transduction. Neuron 34, 499–502 [DOI] [PubMed] [Google Scholar]
- 90. Dai L., Liu Y., He J., Flack C. G., Talsma C. E., Crowley J. G., Muraszko K. M., Fan X., Lubman D. M. (2011) Differential profiling studies of N-linked glycoproteins in glioblastoma cancer stem cells upon treatment with γ-secretase inhibitor. Proteomics 11, 4021–4028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Hu Y. Y., Zheng M. H., Cheng G., Li L., Liang L., Gao F., Wei Y. N., Fu L. A., Han H. (2011) Notch signaling contributes to the maintenance of both nomal neural stem cells and patient-derived glioma stem cells. BMC Cancer 11, 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Wang J., Wakeman T. P., Lathia J. D., Hjelmeland A. B., Wang X. F., White R. R., Rich J. N., Sullenger B. A. (2010) Notch promotes radio-resistance of glioma stem cells. Stem Cells 28, 17–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Fan X., Khaki L., Zhu T. S., Soules M. E., Talsma C. E., Gul N., Koh C., Zhang J., Li Y. M., Maciaczyk J., Nikkhah G., Dimeco F., Piccirillo S., Vescovi A. L., Eberhart C. G. (2010) NOTCH pathway blockade depletes CD133-positive glioblastoma cells and inhibits growth of tumor neurospheres and xenografts. Stem Cells 28, 5–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Vilar M., Charalampopoulos I., Kenchappa R. S., Simi A., Karaca E., Reversi A., Choi S., Bothwell M., Mingarro I., Friedman W. J., Schiavo G., Bastiaens P. I., Verveer P. J., Carter B. D., Ibáñez C. F. (2009) Activation of the p75 neurotrophin receptor through conformational rearrangement of disulfide-linked receptor dimers. Neuron 62, 72–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Casademunt E., Carter B. D., Benzel I., Frade J. M., Dechant G., Barde Y. A. (1999) The zinc finger protein NRIF interacts with the neurotrophin receptor (p75NTR) and participates in programmed cell death. EMBO J. 18, 6050–6061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Yamashita T., Tohyama M. (2003) The p75 receptor acts as a displacement factor that releases Rho from Rho-GDI. Nat. Neurosci. 6, 461–467 [DOI] [PubMed] [Google Scholar]
- 97. Salehi A. H., Roux P. P., Kubu C. J., Zeindler C., Bhakar A., Tannis L. L., Verdi J. M., Barker P. A. (2000) NRAGE, a novel MAGE protein, interacts with the p75 neurotrophin receptor and facilitates nerve growth factor-dependent apoptosis. Neuron 27, 279–288 [DOI] [PubMed] [Google Scholar]
- 98. Bertrand M. J., Kenchappa R. S., Andrieu D., Leclercq-Smekens M., Nguyen H. N., Carter B. D., Muscatelli F., Barker P. A., De Backer O. (2008) NRAGE, a p75NTR adaptor protein, is required for developmental apoptosis in vivo. Cell Death Differ. 15, 1921–1929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Chittka A., Arevalo J. C., Rodriguez-Guzman M., Pérez P., Chao M. V., Sendtner M. (2004) The p75NTR-interacting protein SC1 inhibits cell cycle progression by transcriptional repression of cyclin E. J. Cell Biol. 164, 985–996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Vilar M., Murillo-Carretero M., Mira H., Magnusson K., Besset V., Ibáñez C. F. (2006) Bex1, a novel interactor of the p75 neurotrophin receptor, links neurotrophin signaling to the cell cycle. EMBO J. 25, 1219–1230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Shonukan O., Bagayogo I., McCrea P., Chao M., Hempstead B. (2003) Neurotrophin-induced melanoma cell migration is mediated through the actin-bundling protein fascin. Oncogene 22, 3616–3623 [DOI] [PubMed] [Google Scholar]
- 102. Geetha T., Kenchappa R. S., Wooten M. W., Carter B. D. (2005) TRAF6-mediated ubiquitination regulates nuclear translocation of NRIF, the p75 receptor interactor. EMBO J. 24, 3859–3868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Antonyak M. A., Kenyon L. C., Godwin A. K., James D. C., Emlet D. R., Okamoto I., Tnani M., Holgado-Madruga M., Moscatello D. K., Wong A. J. (2002) Elevated JNK activation contributes to the pathogenesis of human brain tumors. Oncogene 21, 5038–5046 [DOI] [PubMed] [Google Scholar]
- 104. Cui J., Han S. Y., Wang C., Su W., Harshyne L., Holgado-Madruga M., Wong A. J. (2006) c-Jun NH2-terminal kinase 2α2 promotes the tumorigenicity of human glioblastoma cells. Cancer Res. 66, 10024–10031 [DOI] [PubMed] [Google Scholar]
- 105. Majmundar A. J., Wong W. J., Simon M. C. (2010) Hypoxia-inducible factors and the response to hypoxic stress. Mol Cell. 40, 294–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Amberger-Murphy V. (2009) Hypoxia helps glioma to fight therapy. Curr. Cancer Drug Targets 9, 381–390 [DOI] [PubMed] [Google Scholar]