

Background: Steroid sulfatase (STS)-mediated desulfation regulates the chemical and functional homeostasis of estrogens.
Results: Overexpression of STS in the liver improved metabolic functions in mouse models of obesity and type 2 diabetes through sex-specific mechanisms.
Conclusion: STS-mediated estrogen reactivation is beneficial in energy and glucose metabolism.
Significance: Liver-specific activation of estrogen signaling may represent a novel approach to manage metabolic syndrome.
Keywords: Diabetes, Energy Metabolism, Estrogen, Obesity, Steroid Hormone, Transgenic Mice
Abstract
The steroid sulfatase (STS)-mediated desulfation is a critical metabolic mechanism that regulates the chemical and functional homeostasis of endogenous and exogenous molecules. In this report, we first showed that the liver expression of Sts was induced in both the high fat diet (HFD) and ob/ob models of obesity and type 2 diabetes and during the fed to fasting transition. In defining the functional relevance of STS induction in metabolic disease, we showed that overexpression of STS in the liver of transgenic mice alleviated HFD and ob/ob models of obesity and type 2 diabetes, including reduced body weight, improved insulin sensitivity, and decreased hepatic steatosis and inflammation. Interestingly, STS exerted its metabolic benefit through sex-specific mechanisms. In female mice, STS may have increased hepatic estrogen activity by converting biologically inactive estrogen sulfates to active estrogens and consequently improved the metabolic functions, whereas ovariectomy abolished this protective effect. In contrast, the metabolic benefit of STS in males may have been accounted for by the male-specific decrease of inflammation in white adipose tissue and skeletal muscle as well as a pattern of skeletal muscle gene expression that favors energy expenditure. The metabolic benefit in male STS transgenic mice was retained after castration. Treatment with the STS substrate estrone sulfate also improved metabolic functions in both the HFD and ob/ob models. Our results have uncovered a novel function of STS in energy metabolism and type 2 diabetes. Liver-specific STS induction or estrogen/estrogen sulfate delivery may represent a novel approach to manage metabolic syndrome.
Introduction
Steroid sulfatase (STS)2-mediated desulfation and sulfotransferase (SULT)-mediated sulfation represent critical metabolic mechanisms to regulate the chemical and functional homeostasis of endogenous chemicals, including the estrogens. Estrogen is not only a key hormone in reproduction, but it also has great implications in glucose and energy metabolism. Estrogens exhibit an overall protective role in preventing obesity and type 2 diabetes (1). Postmenopausal women face increased risk of developing obesity and insulin resistance. On the other hand, estrogen replacement therapies ameliorate metabolic disorders in both women and animal models (2–4). Estrogen receptor α (ERα) knock-out mice developed impaired insulin sensitivity and obesity (5). Mice deficient in aromatase, the enzyme that converts androgens to estrogens, exhibited increased adiposity and fatty liver, which were normalized upon estrogen treatment (6, 7). Human subjects lacking ERα or aromatase also developed insulin resistance (8, 9). Estrone sulfate is the major active component in conjugated equine estrogens that are commonly used in hormone replacement therapy. Treatment with conjugated equine estrogens improved glycemic control and blood lipid profiles in postmenopausal, type 2 diabetic women (10). Despite its known metabolic benefit, a systemic treatment with estrogens is limited by its potential side effects, such as risk for cardiovascular disease and tumor promotion (11).
The estrogen homeostasis is tightly regulated by sulfation and desulfation. Estrogens can be sulfonated and deactivated by the estrogen sulfotransferase (EST; also called SULT1E1) (12). Unlike the parent estrogens, estrogen sulfates cannot bind to ER and thus are biologically inactive, but they have higher concentrations and prolonged half-life in the circulation, acting as a reservoir for regenerating active estrogens by the STS-mediated desulfation (13). It is believed that STS is the only enzyme responsible for desulfating estrogen sulfates. STS gene deletion or mutation caused X-linked ichthyosis, which is often associated with reproductive abnormalities due to disrupted steroid hormone homeostasis (14).
We have previously reported that EST affected mouse models of type 2 diabetes in a sex-specific manner (15). Loss of Est in female mice improved metabolic function in ob/ob and HFD-fed mice as a result of decreased estrogen deprivation and increased estrogenic activity in the liver. In contrast, Est ablation in ob/ob males exacerbated the diabetic phenotype due to the loss of pancreatic β-cell mass as a result of increased white adipose tissue inflammation. Although STS is known to counteract with EST to enhance estrogen activity, whether STS can affect the pathogenesis of obesity and type 2 diabetes through its regulation of estrogen homeostasis and whether the effect of STS has sex specificity have not been reported.
In this report, we showed that hepatic overexpression of STS elicited metabolic benefits in both sexes but via distinct mechanisms. The metabolic benefit in female STS mice was probably mediated by increased hepatic estrogen activity, whereas the protective effect of STS in males may have been accounted for by decreased inflammation in white adipose tissue and skeletal muscle. Our results have uncovered a novel function of STS in regulating energy metabolism and improving insulin sensitivity.
EXPERIMENTAL PROCEDURES
Generation of STS Transgenic Mice, Diet and Drug Treatment, Body Composition Analysis, and Indirect Calorimetry
The human STS cDNA was cloned into the TRE-SV40 transgene cassette (16) to construct the TRE-STS transgene. Transgenic production was performed at the University of Pittsburgh Transgenic Core Facility. The TRE-STS/FABP-tTA double transgenic or “STS” mice were generated by cross-breeding the TRE-STS mice with the FABP-tTA mice (17) and maintained in the C57BL/6J background. The majority of the subsequent mating was set up between the TRE-STS/FABP-tTA double transgenic mice and FABP-tTA single transgenic mice of the opposite sex, in which we observed 55.3% of female pups and 52.7% of male pups were double transgenic and expressed the STS transgene. These mating results suggested a Mendelian segregation of the transgene. The obS mice (ob/ob mice expressing the STS transgene) were generated by breeding the transgene into the ob/ob background. When necessary, doxycycline (DOX; 2 mg/ml) was given in drinking water 1 week before the HFD treatment and until the completion of the experiment. For estrone sulfate treatment, mice were treated with 75 mg/kg/day estrone 3-sulfate from Sigma for 5 days by oral gavage (18). HFD (catalog no. S3282) with 60% of total calories coming from animal fat was purchased from Bio-serv (Frenchtown, NJ). Body composition by EchoMRI and indirect calorimetry by the Oxymax indirect calorimetry system were performed as we have described previously (19). The use of mice in this study complied with relevant federal guidelines and institutional policies.
Intraperitoneal Glucose Tolerance Test (IPGTT), Insulin Tolerance Test (ITT), and Euglycemic-Hyperinsulinemic Clamp
IPGTT was performed in 16-h fasted mice with an intraperitoneal injection of d-glucose at 1 g/kg body weight for ob/ob mice or at 2 g/kg body weight for other genotypes. ITT was performed in 6-h fasted mice with an intraperitoneal injection of insulin at 1.5 units/kg body weight for ob/ob mice, or at 0.5 unit/kg body weight for other genotypes. For euglycemic-hyperinsulinemic clamp study, 16-h fasted mice were constantly infused with [3-3H]glucose at 0.05 μCi/min through a right jugular vein catheter at the basal state. During the clamp state, the mice were infused with a primed dose of human insulin from Novo Nordisk (Princeton, NJ) at 300 milliunits/kg body weight, followed by a constant insulin infusion at 2.5 milliunits/kg/min. [3-3H]glucose was infused at 0.1 μCi/min. A variable rate of 20% glucose was infused at the same time to maintain a blood glucose range between 120 and 140 mg/dl. Blood glucose levels were monitored every 10 min. Twenty μl of blood were sampled at the end of the basal state and clamp state for plasma [3-3H]glucose measurement. Liver, skeletal muscle, and adipose tissue were harvested at the end of the clamp experiment to assess the insulin signaling (20).
Measurement of Hepatic Estrogen Activities by Liver Luciferase Reporter Gene Transfection
Mouse livers were transfected with the estrogen receptor-responsive tk-ERE-Luc reporter gene by hydrodynamic injection. Hepatic luciferase activity was assessed 16 h after transfection and normalized to the protein concentrations (21). When necessary, ovariectomized and tk-ERE-Luc-transfected mice were subcutaneously injected with 2.5 mg/kg estrone sulfate 6 h after transfection.
UPLC-MS/MS Analysis of Hepatic Estrogens and Estrogen Sulfates
UPLC-MS/MS was carried out with a Waters Acquity UPLC system connected with the Xevo TQ triple quadrupole mass spectrometer, as we have described previously (22).
Measurement of Serum Biochemistry
Serum levels of estradiol (Diagnostic Systems Laboratories, Webster, TX), total triglycerides and cholesterol (Stanbio Laboratory, Boerne, TX), and insulin and leptin (Crystal Chem, Downers Grove, IL) were measured by using commercial assay kits.
Hepatocyte Glucose Production Assay
Forskolin-stimulated glucose production was measured from mouse primary hepatocytes as described, and the data were normalized to protein concentrations (23).
Northern Blotting, Real-time PCR Analysis, Western Blotting, and Immunohistochemistry
Northern blot analysis and real-time PCR were performed as described previously (24). Primary antibodies used for Western blot analysis include phospho-Akt (serine 473) (catalog no. 9271) and total Akt (catalog no. 9272) from Cell Signaling (Danvers, MA) and STS (catalog no. ab62219) from Abcam (Cambridge, MA). The insulin antibody (catalog no. 3014) used for immunohistochemistry was purchased from Cell Signaling (Danvers, MA). The Islet area was quantified using the ImageJ software from the National Institutes of Health (Bethesda, MD).
Statistical Analysis
Results are expressed as means ± S.D. Statistical significance between groups was determined by unpaired two-tailed Student's t test or by two-way analysis of variance. A p value less than 0.05 is considered as statistically significant.
RESULTS
The Hepatic Expression of STS Was Induced in Obese Mice and by Fasting and the Creation of Transgenic Mice Expressing STS in the Liver
HFD-fed mice and chow diet-fed ob/ob mice are commonly used rodent models of obesity and type 2 diabetes. In profiling the gene expression in HFD-fed WT mice and ob/ob mice, we found that the hepatic expression of endogenous Sts was increased in both models (Fig. 1A). Sts was also induced in chow-fed mice by fasting (Fig. 1B), during which the mouse liver accumulates large amounts of triglycerides (25).
FIGURE 1.

The hepatic expression of STS was induced in obese mice and by fasting and creation of transgenic mice expressing STS in the liver. A, hepatic mRNA expression of mouse Sts in chow-diet fed WT mice, HFD-fed WT mice, and ob/ob mice. B, effect of 16-h fasting on hepatic Sts mRNA expression in chow diet-fed WT mice. C, schematic representation of the Tet-off STS transgenic system. FABP, fatty acid binding protein; tTA, tetracycline-controlled transactivator; SV40 poly(A), simian virus 40 polyadenylation signal; TRE, tetracycline-response element; Pmin CMV, minimal human cytomegalovirus promoter. D, mRNA expression of the transgene was determined by Northern blot analysis using the STS cDNA as the probe. The 18 S rRNA was used as a loading control. WT, WT mice; TG, STS transgenic mice; LIV, liver; INT, small intestine; BAT, brown adipose tissue. E, the expression of STS protein was measured by Western blotting. F, the hepatic STS enzymatic activity was determined by an estrone sulfate conversion assay and was normalized against protein concentrations. G, the hepatic expression of STS in female and male transgenic mice was measured by real-time PCR. n ≥ 4 for each group. *, p < 0.05. Error bars, S.D.
To understand the functional relevance of STS induction and to investigate the metabolic role of STS in vivo, we created the tetracycline-responsive STS transgenic mice by crossing two transgenic mouse lines. The FABP-tTA line expresses the tetracycline transactivator (tTA) in the liver and small intestine under the fatty acid-binding protein (FABP) gene promoter (17), and the TRE-STS line expresses STS under the control of the tetracycline-response element (TRE). In the double transgenic mice, the tTA protein binds to tetracycline-response element and initiates the expression of the transgenic STS, whereas administration of DOX can silence the transgene expression (Fig. 1C). The expression of the transgenic STS and its silencing by DOX in the liver and small intestine were confirmed by Northern blotting (Fig. 1D). The transgene was absent in a panel of none-targeting tissues, including brain, kidney, skeletal muscle, brown adipose tissue, and white adipose tissue. The protein expression of the transgene in the liver was confirmed by Western blotting (Fig. 1E). The transgenic expression in the liver was also confirmed at the enzymatic level by using the tritium-labeled estrone sulfate as substrate (Fig. 1F). It is noted that the hepatic expression of the transgene was comparable between the male and female TG mice (Fig. 1G).
Overexpression of STS Ameliorated HFD-induced Obesity, Insulin Resistance, and Hepatic Steatosis in Female Mice
When analyzing the metabolic phenotype, we found no significant changes in body weight, body composition (Fig. 2A), IPGTT (Fig. 2B, left), and ITT (Fig. 2B, right). However, when challenged with HFD, the female STS mice gained less body weight compared with their WT counterparts (Fig. 2C). Body composition analysis showed that the reduced body weight gain in STS mice was mainly due to decreased fat mass, because the lean mass was comparable between the two genotypes (Fig. 2C). The serum level of leptin was decreased in STS mice (Table 1), consistent with their decreased adiposity. The body weight is maintained by balanced energy intake and energy expenditure. Although the food intake was comparable between the two genotypes, the oxygen consumption and energy expenditure were significantly higher in STS mice (Fig. 2D).
FIGURE 2.

Overexpression of STS alleviated HFD-induced obesity and improved insulin sensitivity. All mice are female. A and B, mice maintained on a chow diet were analyzed for body weight and body composition (A) and IPGTT and ITT (B). C–H, mice were fed with HFD for 20 weeks before analysis. C–E, body weight and body composition (C); food intake, oxygen consumption, and energy expenditure (D); and IPGTT and ITT (E) of HFD-fed mice. The quantifications of the IPGTT and ITT results are shown as the area under the curve. F, primary hepatocytes were measured for forskolin (FSK)-stimulated glucose production. G, glucose infusion rate (left) and hepatic glucose production (right). H, Western blot analysis of insulin-stimulated Akt phosphorylation in the liver, WAT, and skeletal muscle at the completion of euglycemic-hyperinsulinemic clamp. Arbitrary units in the bottom panels represent the ratio of phosphorylated Akt (p-AKT) to total Akt. I, hepatic expression of gluconeogenic enzyme genes was measured by real-time PCR. n ≥ 4 for each group. *, p < 0.05; **, p < 0.01, TG versus WT, or the comparisons are labeled. Error bars, S.D.
TABLE 1.
Metabolic profile of HFD-fed WT and STS transgenic female mice
| WT | TG | |
|---|---|---|
| Serum leptin (ng/ml) | 29.56 ± 0.36 | 22.76 ± 2.96a |
| Fasting glucose (mg/dl) | 153.3 ± 14.67 | 112.4 ± 7.05a |
| Fasting insulin (ng/ml) | 2.1 ± 0.52 | 1.69 ± 0.36 |
| Serum triglyceride (mg/dl) | 104.5 ± 7.65 | 99.99 ± 4.88 |
| Serum total cholesterol (mg/dl) | 125.1 ± 8.21 | 116.8 ± 5.06 |
a p < 0.05.
Obesity is closely associated with insulin resistance. We went on to measure the insulin sensitivity in HFD-fed mice by IPGTT. The STS mice had a lower glucose level after a 16-h fast, but their serum insulin level was comparable with that of the WT counterparts (Table 1), and they also showed improved IPGTT performance (Fig. 2E, left two panels). Interestingly, the STS mice and WT mice performed similarly in ITT (Fig. 2E, right two panels), which may have been accounted for by the unchanged insulin sensitivity in the skeletal muscle and WAT (see below). The inhibition of hepatic glucose production by STS was recapitulated in isolated mouse primary hepatocytes because the hepatocytes isolated from the STS mice showed blunted forskolin-induced glucose production (Fig. 2F). We then performed hyperinsulinemic euglycemic clamp to further assess the effect of STS on hepatic and peripheral insulin sensitivity. The STS mice showed a 50% higher glucose infusion rate compared with WT mice during the clamp period (Fig. 2G, left), indicating better insulin sensitivity. The STS mice also displayed lower glucose production at the basal state, which may have accounted for the reduced fasting hyperglycemia. More dramatic suppression of hepatic glucose production in STS mice was observed during the insulin-stimulated clamp state (Fig. 2G, right). To gain insight into the improved insulin sensitivity, we measured the insulin-stimulated Akt phosphorylation in three major metabolic tissues: liver, skeletal muscle, and WAT. Increased Akt phosphorylation was observed in the liver but not in the skeletal muscle and WAT of STS mice (Fig. 2H), suggesting a liver-specific improvement of insulin sensitivity. The hepatic expression of the gluconeogenic enzymes glucose-6-phosphatase (G6pase) and phosphoenolpyruvate carboxykinase (Pepck) was suppressed in STS mice (Fig. 2I), which may explain the reduced hepatic glucose production and improved glucose homeostasis in this genotype.
The HFD-fed STS mice also showed relief of hepatic steatosis, as evidenced by a reduced number and size of lipid droplets within the hepatocytes (Fig. 3A) as well as a decreased liver triglyceride level (Fig. 3B). Interestingly, the expression of major genes responsible for lipogenesis (Fig. 3C) and fatty acid oxidation (Fig. 3D) was not different between WT and STS mice. The circulating levels of triglycerides and cholesterol were also not affected (Table 1). The hepatic fatty acid translocase (FAT/CD36), a fatty acid transporter responsible for the uptake of fatty acids from the circulation (17), was down-regulated in STS mice (Fig. 3E), which may have contributed to the relief of steatosis. Sustained liver steatosis is known to cause inflammation. Consistent with their relief of steatosis, the STS mice showed decreased hepatic expression of the inflammatory marker genes, such as tumor necrosis factor α (Tnfα) and monocyte chemoattractant protein-1 (Mcp-1) (Fig. 3F). The suppression of inflammatory gene expression was liver-specific, because the expression of Tnfα and Mcp-1 was not affected in the WAT and skeletal muscle of STS mice (Fig. 3F). The metabolic benefit in HDF-fed female STS mice was achieved without affecting the pancreatic islet morphology and volume (Fig. 3G).
FIGURE 3.

Overexpression of STS alleviated HFD-induced hepatic steatosis and inflammation. Mice are the same as those shown in Fig. 2, C–H. A, histological analysis of liver sections by H&E staining. B, hepatic lipid levels. C–E, the hepatic expression of genes responsible for lipogenesis (C), fatty acid oxidation (D), and fatty acid uptake (E) in the liver was measured by real-time PCR. F, the expression of proinflammatory genes and/or macrophage marker genes in the liver (left), WAT (middle), and skeletal muscle (right) was measured by real-time PCR. G, immunostaining of insulin and quantification of total islet area. Arrowheads indicate islets. n ≥ 4 for each group. *, p < 0.05, TG versus WT. Error bars, S.D.
The metabolic benefit was transgene-dependent, because silencing the transgene by treating the STS mice with DOX normalized the body weight and body composition (Fig. 4A), energy expenditure (Fig. 4B), IPGTT performance (Fig. 4C), gluconeogenic gene expression (Fig. 4D), and inflammation marker gene expression (Fig. 4E).
FIGURE 4.
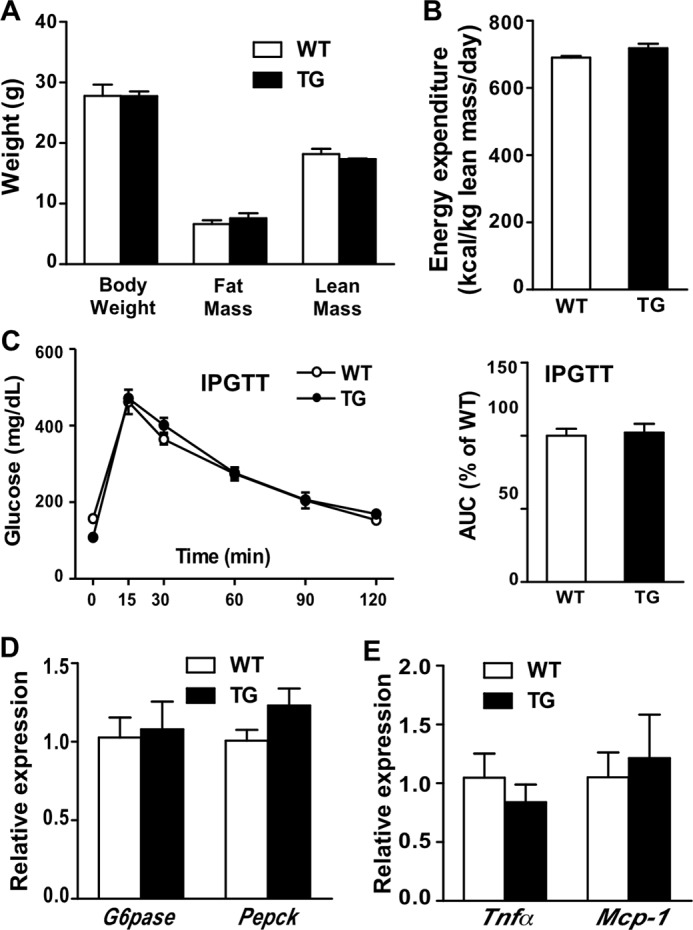
Silencing the transgene expression abolished the metabolic benefit. A–E, body weight and body composition (A), energy expenditure (B), IPGTT (C), and the expression of hepatic gluconeogenic genes (D) and inflammation marker genes (E) in doxycycline-treated and HFD-fed female WT and STS mice. n = 4 for each group. Error bars, S.D.
The Metabolic Benefit in Female STS Mice Was Estrogen-dependent
Estrogen sulfates are the preferred STS substrates existing in high concentrations in vivo (13, 26). Because estrogens are known for their activity in improving metabolic functions (1), we hypothesized that the STS transgene may have exerted its metabolic benefit by converting estrogen sulfates to active estrogens and thus enhancing estrogen signaling in the liver. Indeed, the STS mice showed increased and decreased liver concentrations of estrone (E1) and estrone sulfate (E1S), respectively (Fig. 5A). The enhanced hepatic estrogen activity in STS mice was also supported by the observation that in the presence or absence of estrone sulfate treatment, the activity of an estrogen-responsive luciferase reporter gene, tk-ERE-Luc, was increased when transfected into the mouse liver (Fig. 5B). Despite the increased hepatic estrogen activity, neither the serum estradiol concentration nor the length of estrous cycle was affected in STS mice (Fig. 5C). These results suggested the liver-specific increase of estrogen activity in STS mice.
FIGURE 5.

The metabolic benefit in female STS mice was estrogen-dependent. All mice are female. A, increased hepatic estrogen levels in HFD-fed female STS transgenic mice. The estrogen and estrogen sulfate were extracted from mouse liver and analyzed by the LC-MS method. Shown are the chromatographs of the estrone and estrone sulfate standards and the mouse liver extracts (left) and quantification of the results (right). The data were normalized against liver weight. E1, estrone; E1S, estrone sulfate. B, enhanced hepatic estrogen activity in female STS mice treated with vehicle (VEH) or estrogen sulfate (ES), as shown by a liver-specific transfection of the estrogen-responsive luciferase reporter gene tk-ERE-Luc. The data were expressed as the percentage of VEH-treated WT mice. C, serum estradiol levels (left) and estrus cycle length (right) were measured in intact mice. The length of vaginal estrus cycle was determined by vaginal smears. D–F, body weight and body composition (D), IPGTT (E), and the expression of hepatic gluconeogenic genes (F, left), inflammation markers (F, middle) and fatty acid transporter (F, right) in ovariectomized mice fed with HFD for 20 weeks. n ≥ 4 for each group. *, p < 0.05, TG versus WT. Error bars, S.D.
To determine whether the metabolic benefit was mediated by estrogens, we eliminated the primary source of endogenous estrogens from 5-week-old prepubertal female mice by ovariectomy before challenging them with HFD. Ovariectomy completely abolished the metabolic benefit of the STS transgene in body weight and body composition (Fig. 5D), IPGTT performance (Fig. 5E), and hepatic expression of gluconeogenic genes, inflammatory marker genes, and Cd36 (Fig. 5F). These results suggested that the metabolic benefit in female STS mice was estrogen-dependent.
Overexpression of STS Improved Metabolic Functions in ob/ob Mice
The leptin-deficient ob/ob mice represent a genetic model of obesity and type 2 diabetes. To determine the metabolic effect of STS on this model, we bred the STS transgene into the ob/ob background. The resulting ob/ob-STS transgenic mice were termed “obS” mice. Both ob/ob and obS mice were maintained on a chow diet. Although they were not less obese, the female obS mice showed an improved body composition by having increased lean mass (Fig. 6A), improved IPGTT performance (Fig. 6B), and suppression of gluconeogenic (Fig. 6C) and proinflammatory (Fig. 6D) gene expression in the liver. The obS mice also exhibited ameliorated liver steatosis (Fig. 6E) without affecting the expression of genes involved in lipogenesis and fatty acid oxidation (Fig. 6F). The serum levels of triglyceride and cholesterol were not affected either (data not shown). The morphology and area of pancreatic islets were indistinguishable between the ob/ob and obS mice (Fig. 6G). These phenotypes were consistent with those observed in the HFD model.
FIGURE 6.

Overexpression of STS improved metabolic function in ob/ob mice. The female ob/ob and obS mice were maintained on a chow diet. A–D, body weight and body composition (A), IPGTT and ITT (B), and the expression of hepatic gluconeogenic genes (C) and proinflammatory marker genes (D). The quantifications of the IPGTT and ITT results are shown as the area under the curve. E, histological analysis of liver sections by H&E staining (left) and measurement of hepatic triglyceride and cholesterol levels (right). F, expression of genes responsible for hepatic lipogenesis (left) and fatty acid oxidation (right), as determined by real-time PCR. G, immunostaining of insulin and quantification of total islet area. Arrowheads, islets. n = 4 for each group. *, p < 0.05, obS versus ob/ob. Error bars, S.D.
Treatment with Estrogen Sulfate Improved Insulin Sensitivity in ob/ob and HFD Models
Estrogen administration has been reported to improve insulin sensitivity in both ob/ob mice and HFD-fed mice (27, 28). The induction of STS in the ob/ob and HFD models (Fig. 1A) led to our hypothesis that administration of estrone sulfate in these models may result in the same estrogenic benefit due to the increased STS-mediated conversion of estrone sulfates to estrogens in the liver. As shown in Fig. 7A, administration of estrone sulfate gradually reduced the fed glucose levels in ob/ob mice and maximized its glucose-lowering effect by day 5. The estrone sulfate-treated mice also showed improved IPGTT performance in both the ob/ob (Fig. 7B, top two panels) and HFD (Fig. 7C, left two panels) models. Interestingly, the estrone sulfate treatment did not improve ITT performance in ob/ob mice (Fig. 7B, bottom two panels) but did so in the HFD model (Fig. 7C, right two panels).
FIGURE 7.

Estrogen sulfate improved insulin sensitivity in ob/ob mice and HFD-fed mice. A and B, female ob/ob mice were maintained on a chow diet. Shown are fed glucose levels (A) and IPGTT and ITT (B) of vehicle (VEH) or estrone sulfate (ES)-treated ob/ob mice. C, WT female mice were fed with HFD for 16 weeks. Shown are IPGTT and ITT of mice treated with VEH or ES. The quantifications of the IPGTT and ITT results are shown as the area under the curve. n ≥ 4 for each group. *, p < 0.05, ES versus VEH. Error bars, S.D.
Overexpression of STS Improved Metabolic Function in HFD-fed Male Mice with Distinct Mechanisms
Unlike their female counterparts, the male STS mice showed no increased activation of the tk-ERE-Luc reporter gene when transfected into their livers (Fig. 8A). Having demonstrated the estrogen-dependent metabolic benefit in STS females, we were surprised to find that the STS males also showed protection from the HFD challenge. Although their body weight was not affected, the HFD-fed STS males had reduced fat mass and increased lean mass (Fig. 8B). In addition, the HFD-fed STS males showed increased oxygen consumption and energy expenditure (Fig. 8C), improved IPGTT performance but unchanged ITT performance (Fig. 8D), and suppression of the gluconeogenic genes and proinflammatory genes (Fig. 8E) in their livers. The HFD-fed STS males also showed inhibition of hepatic steatosis, as revealed by liver histology and hepatic triglyceride measurement (data not shown). These results were in general agreement with those observed in the HFD-fed STS females. The improvement of IPGTT performance and inhibition of gluconeogenic gene expression were retained in HFD-fed STS males following castration (data not shown), suggesting that the sex hormones are dispensable for the metabolic benefit of STS in males.
FIGURE 8.

Overexpression of STS improved metabolic functions in male mice via distinct mechanisms. All mice are male. A, lack of change in hepatic estrogen activity in male STS mice as shown by a liver-specific transfection of the estrogen-responsive luciferase reporter gene tk-ERE-Luc. B–E, mice were fed with HFD for 20 weeks before analysis. Body weight and body composition (B), oxygen consumption and energy expenditure (C), IPGTT and ITT (D), and expression of hepatic gluconeogenic genes (E, left) and inflammatory marker genes (E, right). F, H&E staining (left) and expression of macrophage and inflammatory marker genes (right) in abdomen adipose tissue. Arrowheads (left), crown-like structures. G, gene expression in the skeletal muscle as measured by real-time PCR. n = 4 for each group. H, quantification of total islet area based on insulin immunostaining. *, p < 0.05, TG versus WT. ns, statistically not significant. Error bars, S.D.
In understanding the mechanism for the metabolic benefit of STS in male mice, we found that the WAT of STS males showed fewer signs of inflammation, including reduced crown-like structures (Fig. 8F, left), and decreased expression of macrophage and inflammation marker genes (Fig. 8F, right). The inhibition of WAT inflammation was not seen in STS females (Fig. 3F). In addition, we observed a decreased expression of macrophage and inflammation marker genes, increased expression of the glucose transporter Glut4, and increased expression of genes responsible for mitochondrial biogenesis and fatty acid oxidation in the skeletal muscle of STS males (Fig. 8G), which was also absent in STS females (Fig. 3F) (data not shown). The transgene had little effect on the area of pancreatic islets either in the HFD model (Fig. 8H, left) or the obS model (Fig. 8F, right).
DISCUSSION
In this study, we reported the induction of hepatic STS in mouse models of obesity and insulin resistance as well as during the transition from fed to fasting. The induction of STS may represent a protective response against metabolic stress, because overexpression of STS in the liver improved metabolic function in both the HFD and ob/ob models.
The metabolic benefit of the STS transgene in female mice may have resulted from enhanced hepatic estrogen production, as evidenced by the increased estrogen level and activity in the livers of STS mice, whereas ovariectomy abolished the protective effect of STS. The sulfation-desulfation pathways are important to maintain local estrogen homeostasis. Under normal conditions, the mouse liver has a low basal expression of EST but a substantial constitutive expression of STS, suggesting that a dominant estrogenic activity is required to maintain hepatic estrogen levels and normal physiology. However, in type 2 diabetes, EST is dramatically and specifically induced in the liver (29), which may have increased estrogen deprivation and contributed to the development of insulin resistance. In our transgenic mice, overexpression of STS may have regenerated active estrogens, compensated for reduced estrogen signaling, and thus protected mice from insulin resistance.
Although estrogens are known for their activities in preventing metabolic syndrome, their effector tissues or cell types remain to be clearly defined. It has been suggested that estrogen signaling in the central nervous system controls food intake, energy metabolism, and body weight homeostasis (30). In the pancreas, estrogen actions protect β-cells from death and promote insulin biosynthesis and secretion (31). Estrogen decreases lipogenesis and adipogenesis in adipose tissue and enhances fatty acid oxidation in skeletal muscle. Estrogen also reduces inflammation in the muscle and adipose tissue, which altogether improves insulin sensitivity in these peripheral tissues (1). Our results provided a direct evidence that a liver-specific enhancement of estrogen activity was sufficient to confer the metabolic benefit. The enhanced estrogen activity in our STS mice was probably localized to the liver, because the circulating levels of estradiol were similar between WT and STS mice, and the increased insulin-stimulated Akt phosphorylation in STS mice was also liver-specific. The confinement of insulin sensitization to the liver may also explain why improved IPGTT performance, but not ITT performance, was observed in HFD-fed STS mice and chow-fed obS mice.
A systemic treatment with estrogens has been limited by their potential side effects (11). Our results raised the promise of using liver-specific activation of estrogen signaling to harness the metabolic benefit of estrogens and avoid unwanted side effects. It is encouraging that targeted estrogen deliveries have been explored. In one such example, targeted estrogen delivery was achieved by fusing the glucagon-like peptide-1 with estrogen, which improved glucose and lipid metabolism without inducing endocrine toxicity and oncogenicity (32). In addition to a direct use of estrogens, our results suggested that delivery of the hormonally inactive estrogen sulfate may also be a practical approach to enhance estrogen activity in tissues bearing a high basal or inducible expression of STS, such as the liver.
Having demonstrated the sex-specific effect of Est ablation on glucose homeostasis (15), it was surprising to note that overexpression of STS improved glucose homeostasis in both female and male mice. EST is expressed in both the liver and adipose tissue. The anti-diabetic and pro-diabetic effect of Est ablation in females and males may be due to the loss of Est in the liver and adipose tissue, respectively (15). In the current study, the STS transgene was expressed in the liver but not in the adipose tissue, which may explain the consistent benefit in both sexes. Interestingly, the mechanism for the protective effect of STS appeared to be sex-specific. The protection in female mice was estrogen-dependent, but the protection in males may be independent of sex hormones, because the hepatic estrogen activity did not increase, and castration failed to abolish the protective effect of STS. Instead, the metabolic benefit in males might have been accounted for by the male-specific inhibition of inflammation in the adipose tissue and skeletal muscle as well as a pattern of skeletal muscle gene expression that favors energy expenditure. Another notable difference between the STS transgenic and Est null model is their impact on the pancreatic islets and β-cell mass. Est ablation caused a male-specific loss of β-cell mass in the ob/ob background, which was reasoned to be due to the WAT inflammation (15). In contrast, the STS transgene had little effect on the pancreatic islets either in the HFD model or ob/ob model regardless of the sex of the mice.
The STS transgene was not targeted to the adipose tissue and skeletal muscle, so we cannot exclude the possibility that the metabolic benefit of STS in these two tissues was secondary. Indeed, we found that the serum concentration of dehydroepiandrosterone, which can be converted from dehydroepiandrosterone sulfate by STS, was increased in male STS mice (data not shown). Dehydroepiandrosterone is known to have multiple beneficial effects on liver and extrahepatic tissues in preventing the progression of metabolic syndrome (33).
X-linked ichthyosis results from inactivation of STS by complete deletion or point mutations (34). STS polymorphisms have been associated with vulnerability to attention deficit/hyperactivity disorder (35, 36). It would be interesting to know whether the polymorphisms of STS are associated with altered estrogen signaling and susceptibility to obesity and insulin resistance in human populations. In addition, several STS inhibitors are being developed for the treatment of breast cancer in postmenopausal women (37, 38). It is imperative to evaluate the effect of STS inhibitors on glucose and energy metabolism in order to avoid their potential side effects.
The 60 kcal% HFD we used, with 60% of total calories from animal fat, is a widely used research diet to induce obesity and type 2 diabetes in rodents (39). However, we understand that the 60 kcal% HFD has been criticized as non-physiological and can be toxic to non-adipose tissues, including the pancreatic β-cells (40). A future use of HFD with lower fat content may help to exclude the possibility that the toxicity of the 60 kcal% HFD may have impacted the phenotypic exhibition or interpretation.
In summary, the current study has uncovered a novel function of STS in regulating energy metabolism and improving insulin sensitivity through sex-specific mechanisms. Liver-specific activation of the estrogen signaling pathway was sufficient to confer metabolic benefit. We propose that liver-specific STS induction or estrogen/estrogen sulfate delivery may represent novel approaches to manage metabolic syndrome.
This work was supported, in whole or in part, by National Institutes of Health Grants DK083952 and HD073070 (to W. X.) and DK058855 and DK072162 (to R. O. D.). This work was also supported in part by the University of Pittsburgh Metabolic Diseases Research Center.
- STS
- steroid sulfatase
- DOX
- doxycycline
- ER
- estrogen receptor
- EST
- estrogen sulfotransferase
- HFD
- high fat diet
- IPGTT
- intraperitoneal glucose tolerance test
- ITT
- insulin tolerance test
- SULT
- sulfotransferase
- WAT
- white adipose tissue
- TG
- transgenic.
REFERENCES
- 1. Mauvais-Jarvis F., Clegg D. J., Hevener A. L. (2013) The role of estrogens in control of energy balance and glucose homeostasis. Endocr. Rev. 34, 309–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Carr M. C. (2003) The emergence of the metabolic syndrome with menopause. J. Clin. Endocrinol. Metab. 88, 2404–2411 [DOI] [PubMed] [Google Scholar]
- 3. Margolis K. L., Bonds D. E., Rodabough R. J., Tinker L., Phillips L. S., Allen C., Bassford T., Burke G., Torrens J., Howard B. V. (2004) Effect of oestrogen plus progestin on the incidence of diabetes in postmenopausal women. Results from the Women's Health Initiative Hormone Trial. Diabetologia 47, 1175–1187 [DOI] [PubMed] [Google Scholar]
- 4. Zhu L., Brown W. C., Cai Q., Krust A., Chambon P., McGuinness O. P., Stafford J. M. (2013) Estrogen treatment after ovariectomy protects against fatty liver and may improve pathway-selective insulin resistance. Diabetes 62, 424–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Heine P. A., Taylor J. A., Iwamoto G. A., Lubahn D. B., Cooke P. S. (2000) Increased adipose tissue in male and female estrogen receptor-α knockout mice. Proc. Natl. Acad. Sci. U.S.A. 97, 12729–12734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jones M. E., Thorburn A. W., Britt K. L., Hewitt K. N., Wreford N. G., Proietto J., Oz O. K., Leury B. J., Robertson K. M., Yao S., Simpson E. R. (2000) Aromatase-deficient (ArKO) mice have a phenotype of increased adiposity. Proc. Natl. Acad. Sci. U.S.A. 97, 12735–12740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hewitt K. N., Pratis K., Jones M. E., Simpson E. R. (2004) Estrogen replacement reverses the hepatic steatosis phenotype in the male aromatase knockout mouse. Endocrinology 145, 1842–1848 [DOI] [PubMed] [Google Scholar]
- 8. Smith E. P., Boyd J., Frank G. R., Takahashi H., Cohen R. M., Specker B., Williams T. C., Lubahn D. B., Korach K. S. (1994) Estrogen resistance caused by a mutation in the estrogen-receptor gene in a man. N. Engl. J. Med. 331, 1056–1061 [DOI] [PubMed] [Google Scholar]
- 9. Morishima A., Grumbach M. M., Simpson E. R., Fisher C., Qin K. (1995) Aromatase deficiency in male and female siblings caused by a novel mutation and the physiological role of estrogens. J. Clin. Endocrinol. Metab. 80, 3689–3698 [DOI] [PubMed] [Google Scholar]
- 10. Friday K. E., Dong C., Fontenot R. U. (2001) Conjugated equine estrogen improves glycemic control and blood lipoproteins in postmenopausal women with type 2 diabetes. J. Clin. Endocrinol. Metab. 86, 48–52 [DOI] [PubMed] [Google Scholar]
- 11. Billeci A. M., Paciaroni M., Caso V., Agnelli G. (2008) Hormone replacement therapy and stroke. Curr. Vasc. Pharmacol. 6, 112–123 [DOI] [PubMed] [Google Scholar]
- 12. Falany C. N. (1997) Enzymology of human cytosolic sulfotransferases. FASEB J. 11, 206–216 [DOI] [PubMed] [Google Scholar]
- 13. Reed M. J., Purohit A., Woo L. W., Newman S. P., Potter B. V. (2005) Steroid sulfatase. Molecular biology, regulation, and inhibition. Endocr. Rev. 26, 171–202 [DOI] [PubMed] [Google Scholar]
- 14. Hazan C., Orlow S. J., Schaffer J. V. (2005) X-linked recessive ichthyosis. Dermatol. Online J. 11, 12. [PubMed] [Google Scholar]
- 15. Gao J., He J., Shi X., Stefanovic-Racic M., Xu M., O'Doherty R. M., Garcia-Ocana A., Xie W. (2012) Sex-specific effect of estrogen sulfotransferase on mouse models of type 2 diabetes. Diabetes 61, 1543–1551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Saini S. P., Sonoda J., Xu L., Toma D., Uppal H., Mu Y., Ren S., Moore D. D., Evans R. M., Xie W. (2004) A novel constitutive androstane receptor-mediated and CYP3A-independent pathway of bile acid detoxification. Mol. Pharmacol. 65, 292–300 [DOI] [PubMed] [Google Scholar]
- 17. Zhou J., Zhai Y., Mu Y., Gong H., Uppal H., Toma D., Ren S., Evans R. M., Xie W. (2006) A novel pregnane X receptor-mediated and sterol regulatory element-binding protein-independent lipogenic pathway. J. Biol. Chem. 281, 15013–15020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Borthwick E. B., Houston M. P., Coughtrie M. W., Burchell A. (2001) The antihyperglycemic effect of estrone sulfate in genetically obese-diabetic (ob/ob) mice is associated with reduced hepatic glucose-6-phosphatase. Horm. Metab. Res. 33, 721–726 [DOI] [PubMed] [Google Scholar]
- 19. Gao J., He J., Zhai Y., Wada T., Xie W. (2009) The constitutive androstane receptor is an anti-obesity nuclear receptor that improves insulin sensitivity. J. Biol. Chem. 284, 25984–25992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cani P. D., Knauf C., Iglesias M. A., Drucker D. J., Delzenne N. M., Burcelin R. (2006) Improvement of glucose tolerance and hepatic insulin sensitivity by oligofructose requires a functional glucagon-like peptide 1 receptor. Diabetes 55, 1484–1490 [DOI] [PubMed] [Google Scholar]
- 21. Al-Dosari M. S., Knapp J. E., Liu D. (2006) Activation of human CYP2C9 promoter and regulation by CAR and PXR in mouse liver. Mol. Pharm. 3, 322–328 [DOI] [PubMed] [Google Scholar]
- 22. Gaikwad N. W. (2013) Ultra performance liquid chromatography-tandem mass spectrometry method for profiling of steroid metabolome in human tissue. Anal. Chem. 85, 4951–4960 [DOI] [PubMed] [Google Scholar]
- 23. He J., Gao J., Xu M., Ren S., Stefanovic-Racic M., O'Doherty R. M., Xie W. (2013) PXR ablation alleviates diet-induced and genetic obesity and insulin resistance in mice. Diabetes 62, 1876–1887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xie W., Barwick J. L., Downes M., Blumberg B., Simon C. M., Nelson M. C., Neuschwander-Tetri B. A., Brunt E. M., Guzelian P. S., Evans R. M. (2000) Humanized xenobiotic response in mice expressing nuclear receptor SXR. Nature 406, 435–439 [DOI] [PubMed] [Google Scholar]
- 25. Lin X., Yue P., Chen Z., Schonfeld G. (2005) Hepatic triglyceride contents are genetically determined in mice. Results of a strain survey. Am. J. Physiol. Gastrointest. Liver Physiol. 288, G1179–G1189 [DOI] [PubMed] [Google Scholar]
- 26. Hawkins R. A., Oakey R. E. (1974) Estimation of oestrone sulphate, oestradiol-17β and oestrone in peripheral plasma. Concentrations during the menstrual cycle and in men. J. Endocrinol. 60, 3–17 [DOI] [PubMed] [Google Scholar]
- 27. Gao H., Bryzgalova G., Hedman E., Khan A., Efendic S., Gustafsson J. A., Dahlman-Wright K. (2006) Long-term administration of estradiol decreases expression of hepatic lipogenic genes and improves insulin sensitivity in ob/ob mice. A possible mechanism is through direct regulation of signal transducer and activator of transcription 3. Mol. Endocrinol. 20, 1287–1299 [DOI] [PubMed] [Google Scholar]
- 28. Bryzgalova G., Lundholm L., Portwood N., Gustafsson J. A., Khan A., Efendic S., Dahlman-Wright K. (2008) Mechanisms of antidiabetogenic and body weight-lowering effects of estrogen in high-fat diet-fed mice. Am. J. Physiol. Endocrinol. Metab. 295, E904–E912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Leiter E. H., Chapman H. D. (1994) Obesity-induced diabetes (diabesity) in C57BL/KsJ mice produces aberrant trans-regulation of sex steroid sulfotransferase genes. J. Clin. Invest. 93, 2007–2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xu Y., Nedungadi T. P., Zhu L., Sobhani N., Irani B. G., Davis K. E., Zhang X., Zou F., Gent L. M., Hahner L. D., Khan S. A., Elias C. F., Elmquist J. K., Clegg D. J. (2011) Distinct hypothalamic neurons mediate estrogenic effects on energy homeostasis and reproduction. Cell Metab. 14, 453–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tiano J. P., Mauvais-Jarvis F. (2012) Importance of oestrogen receptors to preserve functional beta-cell mass in diabetes. Nat. Rev. Endocrinol. 8, 342–351 [DOI] [PubMed] [Google Scholar]
- 32. Finan B., Yang B., Ottaway N., Stemmer K., Müller T. D., Yi C. X., Habegger K., Schriever S. C., García-Caceres C., Kabra D. G., Hembree J., Holland J., Raver C., Seeley R. J., Hans W., Irmler M., Beckers J., de Angelis M. H., Tiano J. P., Mauvais-Jarvis F., Perez-Tilve D., Pfluger P., Zhang L., Gelfanov V., DiMarchi R. D., Tschöp M. H. (2012) Targeted estrogen delivery reverses the metabolic syndrome. Nat. Med. 18, 1847–1856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Aoki K., Nakajima A., Mukasa K., Osawa E., Mori Y., Sekihara H. (2003) Prevention of diabetes, hepatic injury, and colon cancer with dehydroepiandrosterone. J. Steroid Biochem. Mol. Biol. 85, 469–472 [DOI] [PubMed] [Google Scholar]
- 34. Webster D., France J. T., Shapiro L. J., Weiss R. (1978) X-linked ichthyosis due to steroid-sulphatase deficiency. Lancet 1, 70–72 [DOI] [PubMed] [Google Scholar]
- 35. Brookes K. J., Hawi Z., Kirley A., Barry E., Gill M., Kent L. (2008) Association of the steroid sulfatase (STS) gene with attention deficit hyperactivity disorder. Am. J. Med. Genet. B Neuropsychiatr. Genet. 147B, 1531–1535 [DOI] [PubMed] [Google Scholar]
- 36. Kent L., Emerton J., Bhadravathi V., Weisblatt E., Pasco G., Willatt L. R., McMahon R., Yates J. R. (2008) X-linked ichthyosis (steroid sulfatase deficiency) is associated with increased risk of attention deficit hyperactivity disorder, autism and social communication deficits. J. Med. Genet. 45, 519–524 [DOI] [PubMed] [Google Scholar]
- 37. Maltais R., Poirier D. (2011) Steroid sulfatase inhibitors. A review covering the promising 2000–2010 decade. Steroids 76, 929–948 [DOI] [PubMed] [Google Scholar]
- 38. Stanway S. J., Purohit A., Woo L. W., Sufi S., Vigushin D., Ward R., Wilson R. H., Stanczyk F. Z., Dobbs N., Kulinskaya E., Elliott M., Potter B. V., Reed M. J., Coombes R. C. (2006) Phase I study of STX 64 (667 Coumate) in breast cancer patients. The first study of a steroid sulfatase inhibitor. Clin. Cancer Res. 12, 1585–1592 [DOI] [PubMed] [Google Scholar]
- 39. Petro A. E., Cotter J., Cooper D. A., Peters J. C., Surwit S. J., Surwit R. S. (2004) Fat, carbohydrate, and calories in the development of diabetes and obesity in the C57BL/6J mouse. Metabolism 53, 454–457 [DOI] [PubMed] [Google Scholar]
- 40. Schaffer J. E. (2003) Lipotoxicity. When tissues overeat. Curr. Opin. Lipidol. 14, 281–287 [DOI] [PubMed] [Google Scholar]