

Background: We earlier reported novel mutations in PKM2 that reduce its activity.
Results: These mutations promoted cancer features and tumor growth in a dominant negative manner.
Conclusion: Impaired PKM2 activity due to mutations benefits cancer.
Significance: This study provides the first evidence linking natural mutations in PKM2 with cancer.
Keywords: Cancer Tumor Promoter, Enzyme Mutation, Glycolysis, Pyruvate Kinase, Tumor Metabolism, Bloom Syndrome, Dominant Negative Mutation, PKM2
Abstract
The present study was designed to examine the functional relevance of two heterozygous mutations (H391Y and K422R), observed earlier by us in the Bloom syndrome condition. Cells stably expressing exogenous wild-type or mutant PKM2 (K422R or H391Y) or co-expressing both wild type and mutant (PKM2-K422R or PKM2-H391Y) were assessed for cancer metabolism and tumorigenic potential. Interestingly, cells co-expressing PKM2 and mutant (K422R or H391Y) showed significantly aggressive cancer metabolism as compared with cells expressing either wild-type or mutant PKM2 independently. A similar trend was observed for oxidative endurance, tumorigenic potential, cellular proliferation, and tumor growth. These observations signify the dominant negative nature of mutations. Remarkably, PKM2-H391Y co-expressed cells showed a maximal effect on all the studied parameters. Such a dominant negative impaired function of PKM2 in tumor development is not known; this study demonstrates for the first time the possible predisposition of Bloom syndrome patients with impaired PKM2 activity to cancer and the importance of studying genetic variations in PKM2 in the future to understand their relevance in cancer in general.
Introduction
Dominant negative mutation refers to a state where a mutant recessive allele adversely affects the normal function of a wild-type allele by interacting with it, eventually contributing to pathological phenotype. This situation is often present in oligomeric proteins, such as collagen, where it results in skin and bone fragility (1, 2). Dominant negative mutations in ataxia telangiectasia-mutated (ATM) and p53 are associated with cancer progression (3, 4).
The M2 isoform of pyruvate kinase (PKM2),3 which catalyzes the last but pace-making step of glycolysis, i.e. irreversible dephosphorylation of phosphoenolpyruvate (PEP) to pyruvate, has recently been identified as a crucial player in cancer progression (5–9). The switch to PKM2 isoform is considered essential for cancer metabolism and tumor growth. In fact, switching from PKM2 to PKM1 is reported to reverse the aerobic glycolysis or Warburg effect, which is characterized by high glucose uptake and lactate production even in the presence of oxygen (6, 10). There are four PK isoforms in mammals: L and R are expressed in liver and RBCs, respectively; M1 is present in skeletal muscle, heart, and brain; and M2 isoform predominates in proliferating embryonic and tumor cells (10). Out of the four PK isoforms, PKM2 has the potential of reprogramming the glycolytic flux, thus controlling the shift between catabolism and anabolism. This ability of PKM2 emanates from its existence in high and low activity oligomeric forms, which are subjected to diverse but tight regulation (11–13). In cancer cells, a low activity PKM2 form predominates, as a requirement, to cause accumulation of glycolytic intermediates, which are then channeled to biosynthetic pathways such as the pentose phosphate pathway (PPP), generating building blocks for daughter cells (12–15). Moreover, expression of the low activity PKM2 form is associated with prevention of entry of pyruvate into mitochondria by its conversion into lactate, through a largely unknown mechanism (12). Besides its conventional metabolic attributes, recent studies show the involvement of PKM2 in a variety of non-metabolic functions such as transcriptional regulation of genes that are needed for cancer cell growth and survival (16, 17).
Previously, our laboratory has reported two rare missense mutations in intersubunit contact domain (ISCD) of PKM2, coded by exon 10, in a Bloom syndrome cell line (H391Y) and an Indian Bloom patient (K422R) (18). Subsequent work from our laboratory showed that these mutations resulted in down-regulated PKM2 activity (19). Later, our laboratory revealed that wild-type and mutant PKM2 monomer cross-interact to form stable hetero-oligomers and promote cellular growth and polyploidy, in a dominant negative manner (20). However, studies of the effect of these mutations on cancer metabolism, tumor growth, and other important pro-cancerous features have not been attempted. Thus, the biological relevance of these mutations was unclear.
Studying the biological implications of rare genetic aberrations in PKM2 in a naturally occurring pathological condition is crucial to understand its role as a “key enzyme” in cancer. Because Bloom syndrome patients are prone to a variety of cancers spontaneously (21) and because of the recent role attributed to PKM2 in cancer progression, it was pertinent to study the functional relevance of these mutations.
EXPERIMENTAL PROCEDURES
Cell Culture, Transfections, PKM2 Knockdown, Kill Curve, Soft Agar, and Cellular Proliferation Assay
H1299 (non-small cell lung cancer) cell line, a kind gift from Dr. Uttam Pati, School of Biotechnology, Jawaharlal Nehru University, New Delhi, was used for the experiments. It was maintained in DMEM (Sigma) with 10% heat-inactivated fetal bovine serum (FBS) from Biowest (France), 1% penicillin/streptomycin (Sigma), at 37 °C and 5% CO2 in a humidified atmosphere (Thermo Scientific). The plasmid DNA (transfection grade, isolated using the Qiagen plasmid midi kit) was transfected using Lipofectamine LTX (Invitrogen). PKM2 silencing was done using short hairpin RNA (shRNA) as described (22). Kill curves analysis was performed for selecting antibiotic concentrations. For the cellular proliferation assay, transfected cells were counted using a hemocytometer, seeded, and then trypsinized followed by counting. Cells transfected with vector only (pcDNA) or with FLAG-tagged wild-type PKM2 (pcDNA-FLAG-PKM2) or with HA-tagged mutant K422R (pcDNA-HA-K422R) or with H391Y (pcDNA-HA-H391Y) or co-transfected wild-type PKM2 and mutant K422R (pcDNA-FLAG-PKM2+ pcDNA-HA-K422R) or wild-type PKM2 and mutant H391Y (pcDNA-FLAG-PKM2+ pcDNA-HA-H391Y) were first selected in Geneticin for 2–3 weeks. The selected cells were transfected with control and pGIPZ-shRNAPKM2 vector to knock down endogenous PKM2 followed by selection in puromycin for 1 week. shRNA was designed against UTR to prevent silencing of transfected PKM2 and its mutants. Confirmation of transfection was done by Western blotting. For the soft agar assay, equal numbers of stably transfected H1299 cells (∼5000) were seeded in each well of a six-well plate in 0.35% soft agar in DMEM plus 10% FBS at 37 °C for 12 days. 2 μg of DNA was used for transfections of a single plasmid, whereas in the case of co-transfections, an equal amount (1 μg) of each plasmid was used so that total amount remained constant (2 μg).
Cell Lysate Preparation and PKM2 Activity Assay
Whole cell protein, for PKM2 activity analysis, was extracted from transfected cells using 50 mm Tris, pH 7.5, as described (22). Protein quantitation was done using the bicinchoninic acid assay (BCA assay) kit from Thermo Scientific. PKM2 activity was measured spectrophotometrically (UV-1800, Shimadzu, Kyoto, Japan) using the NADH/lactate dehydrogenase-coupled assay as described (22).
Cancer Cell Metabolism Analysis
Used medium was collected from wells harboring transfected cells and then subjected to clearing and deproteinization as described (22). Glucose and lactate were measured spectrophotometrically using kits from Sigma and BioVision, respectively. Metabolite extract was prepared as described (22). Alternatively, metabolites were extracted with 5% trichloroacetic acid (TCA). PEP was assessed using an NADH/lactate dehydrogenase-coupled assay with 30 ng of recombinant PKM2. Fructose 1,6-bisphosphate (FBP) was measured spectrophotometrically using the enzyme assay as described earlier (23). For both FBP and PEP, concentration was determined against the standard curve prepared. NADPH was analyzed spectrophotometrically using the protocol prescribed by the kit manufacturer (BioVision). All measurements were normalized to cell numbers.
Fluorescence Spectrophotometry and ROS Analysis
Reactive oxygen species (ROS) production was analyzed by fluorescence spectrophotometry. ROS generation was determined in cells using 2′, 7′-dichlorofluorescein-diacetate as described (22).
Xenografting and Tumor Growth Analysis
3 million cells of stably transfected H1299 cells were washed with Hepes-buffered saline, resuspended in 100 μl of Hepes-buffered saline, and then injected subcutaneously in 6-week-old female nude mice. A total of 20 mice were used in the study. Mice were sacrificed after 4 weeks, and perpendicular diameters of tumors were measured. Tumor area was calculated using the formula D1/2 × D2/2 × π, where D1 and D2 are two diameters (24).
Data Analysis
All experiments were repeated at least three times, and data are expressed as mean ± S.E. p values were calculated using ordinary one-way analysis of variance. p < 0.05 was considered statistically significant.
RESULTS
Generation of Cells Stably Expressing Independent and Co-expressed Exogenous Wild-type and Mutant PKM2
H1299 cells were transfected independently with wild-type or mutant PKM2 and co-transfected with wild-type and mutant PKM2 to mimic the heterozygous state of mutations as observed in the Bloom syndrome condition (18). The immunoblot shows the confirmation of transfection of PKM2 and its mutants, K422R and H391Y, independently and together, and the knockdown of endogenous PKM2, to enrich the effect of ectopic wild-type and mutant PKM2 expression on studied parameters (Fig. 1).
FIGURE 1.
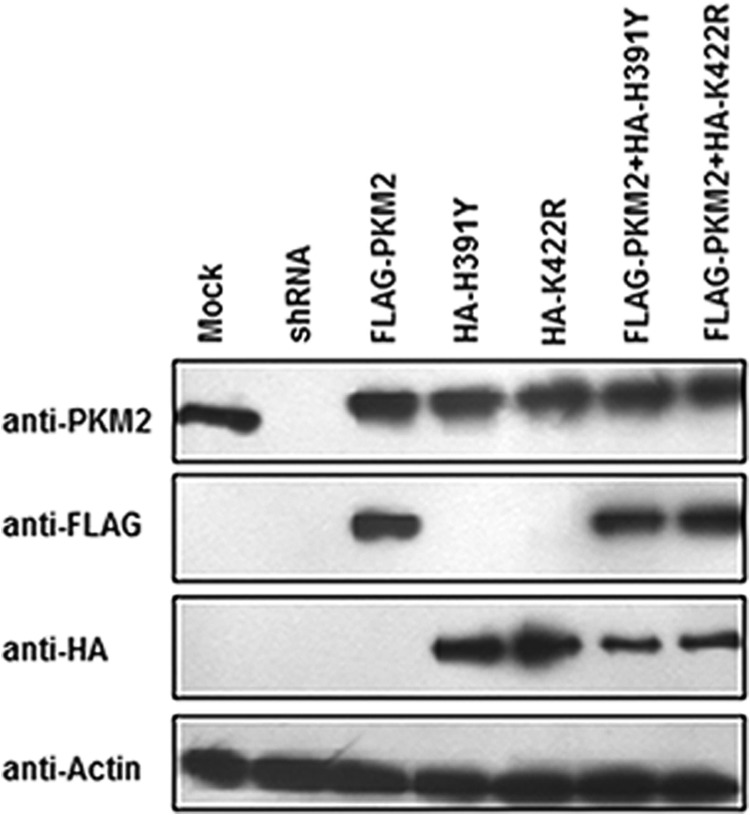
Transfection confirmation. Immunoblot of H1299 cells stably expressing FLAG-PKM2 or HA-H391Y or HA-K422R or FLAG-PKM2/HA-H391Y or FLAG-PKM2/HA-K422R or PKM2-shRNA was performed. Cells for stable transfection were selected for 2 weeks in 400 μg/ml Geneticin (G418). After selection, cells were transfected with control or shRNAPKM2 followed by further selection in 2.5 μg/ml puromycin for a week.
Enhanced Warburg Effect in Cells Co-expressing PKM2 and Mutants
Cancer cells need a large and continuous supply of glucose for biomass production and to produce lactate to subvert the entry of pyruvate into mitochondria (25). Aerobic glycolysis, which refers to high glucose uptake and lactate production even in the presence of oxygen, is therefore a hallmark of cancer cells (26). To measure glucose uptake and lactate production, medium was collected from wells harboring stably transfected H1299 cells and subjected to the protocol described under “Experimental Procedures.” Cells with overexpressed PKM2 showed ∼2.3-fold higher glucose uptake as compared with mock transfected control (Fig. 2). Overexpression of mutants K422R and H391Y further increased the glucose uptake, although slightly, as compared with wild-type PKM2 transfected cells. Interestingly, consumption of glucose increased maximally when PKM2 and mutants were co-expressed, showing highest increase (∼2.22-fold, as compared with PKM2) by cells co-expressing PKM2 and H391Y (Fig. 2A). A similar trend was observed in lactate production, showing maximum production of lactate (∼2.25-fold higher, as compared with wild-type PKM2 expressing cells) by cells co-expressing PKM2 and H391Y (Fig. 2B). Measurements were normalized to cell numbers and expressed as nanomoles per million cells per minute (6).
FIGURE 2.
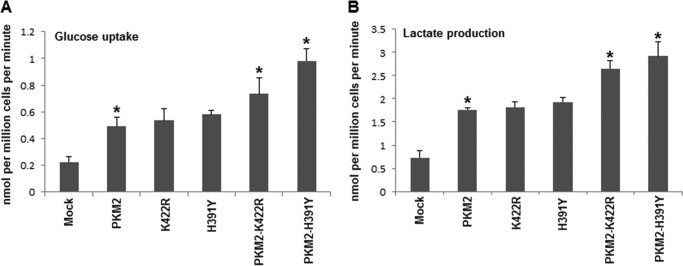
Effect of PKM2 and its mutants on aerobic glycolysis. A, glucose uptake by H1299 cells stably expressing independent and co-expressed wild-type and mutant PKM2. The highest uptake of glucose was demonstrated by PKM2-H391Y co-transfected cells. B, lactate production followed same pattern with maximal production by PKM2-H391Y co-transfected H1299 cells. *, p < 0.05.
Co-expressed Wild-type and Mutant PKM2 Increased Accumulation of Glycolytic Intermediates and NADPH
Decreased PKM2 activity results in characteristic buildup of glycolytic intermediates, which in turn increases the availability of substrates for PPP (13–15). Because independent and co-transfected mutants have lower activity than wild-type PKM2 (20), an enhanced accumulation of glycolytic intermediates was expected. Metabolite extract was prepared from stably transfected cells followed by measurement of intracellular concentrations of FBP and PEP (27). Expression of mutants alone did not result in noticeable accumulation of FBP and PEP; however, substantial accumulation occurred in the case of wild-type and mutant co-transfection. Co-expression of PKM2 and H391Y showed highest (∼2-fold) increase in intracellular concentration of both FBP and PEP (Fig. 3). Furthermore, intracellular NADPH, a product of PPP that provides reducing power for anabolic synthesis (28), accumulated markedly in wild-type and mutant co-transfected cells with peak reaching in PKM2-H391Y co-transfected cells (Fig. 3). The prominent effect of wild-type and mutant co-transfections on metabolic behavior of cells correlated with the stability of hetero-oligomers, preferentially hetero-dimers, supporting the dominant negative status of these mutations, as reported by our laboratory previously (18, 20).
FIGURE 3.
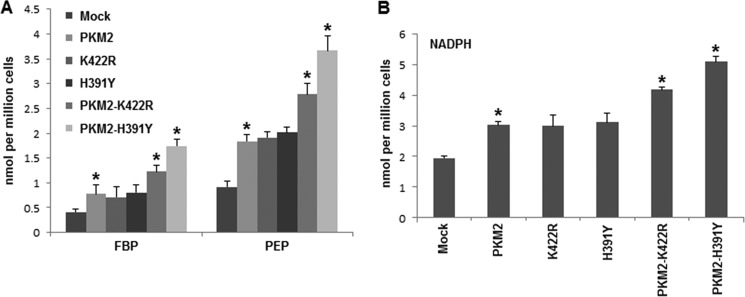
Changes in accumulation of glycolytic intermediates and NADPH. A, PKM2 and mutants, when co-expressed, increased FBP and PEP accumulation in H1299 cells, which indicates increased glycolytic accumulation. B, higher NADPH accumulated in co-expressed situation. Glycolytic and NADPH accumulation suggested enhanced macromolecular synthesis. *, p < 0.05.
Decreased ROS Production in Cells Co-expressing Wild-type and Mutant PKM2
Oxidative stress essentially represents a disproportion between the production of intracellular ROS and the ability of the cell to detoxify them. PKM2 has been reported recently to counter the oxidative stress by virtue of its decreased activity due to oxidation, which generates enough reducing potential (NADPH) through macromolecular synthesis to cause the detoxification of ROS (11). To measure intracellular ROS, stably transfected H1299 cells were incubated in 2′, 7′-dichlorofluorescein-diacetate (dye that binds H2O2) for 30 min followed by fluorescence measurements as detailed under “Experimental Procedures.” Substantial decrease in ROS, as evident from decreased fluorescence, was observed upon PKM2 overexpression (Fig. 4; compare Mock and PKM2). This result supported the notion that PKM2 is important in controlling ROS production in cancer cells. Strikingly, further decrease in fluorescence due to ROS (∼4–5-fold; p < 0.05) was observed in cells co-expressing PKM2 and mutants, especially H391Y (Fig. 4). These results demonstrated a protective effect imparted by mutations against oxidative damage, thus enhancing the survival of cancer cells.
FIGURE 4.
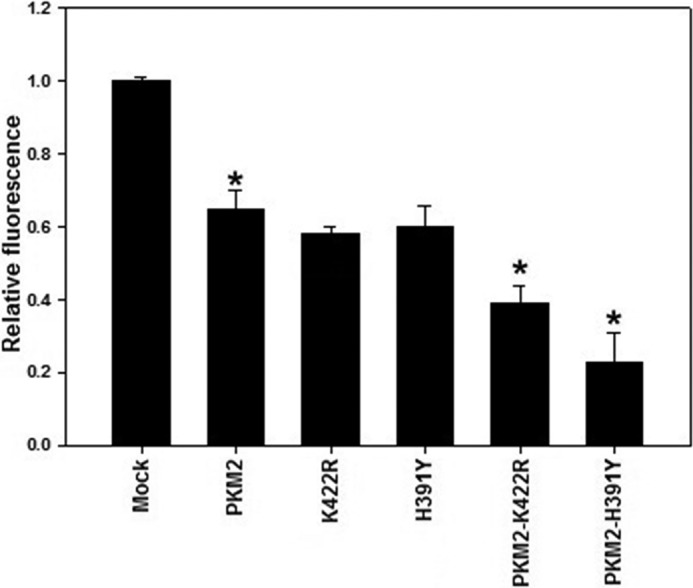
Mutants reduce oxidative stress. PKM2 overexpression decreased intracellular ROS production; however, when PKM2 was co-expressed along with mutants, a further decrease in ROS production was observed. *, p < 0.05.
Wild-type and Mutant PKM2 Co-expression Promoted Anchorage-independent Growth
Anchorage-independent growth is one of the hallmarks of cell transformation. The soft agar colony formation assay is a common method to monitor anchorage-independent growth, which measures proliferation of cancer cells in a semisolid culture media by counting colonies formed.
The soft agar assay was performed, as described under “Experimental Procedures,” to assess the effect of mutants on malignant transformation. After 12 days of seeding an equal number of differentially transfected H1299 cells, colonies obtained were counted under a microscope in a plate dissection manner (29, 30). An increasing gradient of colony number (from mock to co-transfected cells through independent wild-type and mutant PKM2) was obtained with the highest number, i.e. 1232 ± 42, in co-transfected PKM2-H391Y cells (Fig. 5). The number of colonies developed in co-transfected PKM2-K422R was 943 ± 32, ∼31% lesser than PKM2-H391Y, but higher than that observed in independently transfected PKM2 or mutants. Not much difference in the number of colonies was observed between independently transfected PKM2 and its mutants (Fig. 5). These results suggested the importance of PKM2-centered metabolic transformation in promoting malignancy.
FIGURE 5.

Soft agar assay. A, microscopic field images at 10× representing the actual density of colonies obtained after 12 days of seeding independent and co-transfected H1299 cells. Colony numbers in the entire well are given below. B, bar graph depicting the differences between the number of colonies obtained in each case.
Co-expression of PKM2 and Its Mutants Promoted Cell Proliferation and Tumor Growth
As co-expressed wild type and mutants showed stimulating effects on cancer metabolism and inhibitory effects on oxidative stress, subsequent implications on cellular proliferation and tumor growth were analyzed. Results showed that cellular proliferation was enhanced significantly in PKM2-K422R and PKM2-H391Y transfected H1299 cells with maximum increase in PKM2-H391Y (Fig. 6A) (20), which correlated with the observed effects on cancer metabolism and oxidative stress. However, no significant increase in proliferation, as compared with wild-type PKM2 transfected cells, was observed in cells expressing mutants alone (Fig. 6A). Encouraged by this result, the effect of wild-type and mutant co-transfections was analyzed for xenograft tumor growth in mice. Stably transfected H1299 cells were injected subcutaneously near the right posterior thigh of mice. Tumor growth in wild-type and mutant co-transfection (PKM2-K422R and PKM2-H391Y) was found to be higher as compared with only wild-type PKM2 transfects (Fig. 6B). Further, in correlation with other results, PKM2-H391Y transfected H1299 cells showed the highest mean tumor area (Fig. 6B). These outcomes further substantiated the tumor-promoting ability of the mutants. Importantly, these results emphasize the role of oligomer status of PKM2 in tumor progression.
FIGURE 6.

Effect of PKM2 mutations on cellular proliferation and tumor growth. A, enhanced proliferation was observed in co-transfected cells; however, proliferation observed was highest in cells co-expressing PKM2-H391Y. B, tumor growth increased in wild-type and mutant co-transfected cells with maximum increase by PKM2-H391Y co-transfected cells. p value for the differences among the means of four groups was statistically significant (*, p < 0.05).
DISCUSSION
The functional implications of the studied mutations revealed their importance in altering the role of PKM2 in cancer, especially in the wild-type and mutant co-transfected situation. The same role essentially could be extrapolated to the Bloom syndrome condition, where these mutations were found in a heterozygous condition behaving in a dominant negative fashion.
Higher glucose uptake and lactate production by PKM2 transfected cells (Fig. 2), a feature associated with pro-cancerous state, was consistent with previous observations (6, 9). Explanations for this phenomenon could be understood from the fact that PKM2 is known to transactivate expression of genes such as GLUT1 (glucose transporter) and LDHA (lactate dehydrogenase A), which are responsible for glucose uptake and lactate production in proliferating cells (16). Down-regulated PKM2 activity as a result of dimer formation has also been reported to enhance lactate production through poorly understood mechanism (12). One possible explanation for high lactate production by virtue of decreased activity could be preferential interaction of dimeric PKM2 with lactate dehydrogenase, thus facilitating the conversion of pyruvate to lactate (6). Although mutants have lower activity as compared with wild-type PKM2 (20), H1299 cells expressing mutants alone did not show much change in glucose uptake and lactate production as compared with that of wild-type PKM2 stably transfected cells (Fig. 2). This is possibly due to the inability of mutants to form stable homo-dimers (19, 20), required to promote aerobic glycolysis (10, 12). Interestingly, the co-expression of wild type and mutants showed maximum increase in glucose uptake and lactate production. This is possibly due to preferential hetero-dimerization when wild-type and mutant PKM2 were co-expressed (20). In fact, hetero-dimers of wild-type and mutant monomer have higher stability as well as probability of formation than homo-dimers of wild-type or mutant PKM2, and the activity of hetero-dimers is 60–85% less than that of wild-type PKM2 (20). Aerobic glycolysis supports diversion of the glycolytic flux into biosynthetic pathways such as PPP and also helps in survival of tumor cells that are exposed to lower oxygen concentrations (26, 31). The prominent effect of co-expressed wild type and mutants on glycolytic and NADPH accumulation (Fig. 3) correlated with the inherent tendency of wild-type and mutant monomers to cross-interact to produce stable hetero-dimers (20). These results provide functional explanation to our previous work where mutations in PKM2 were shown to exist in a heterozygous condition and behave in a dominant negative fashion (18).
As is known, ROS damages cellular components such as DNA and so forth, and therefore cancer cells have to decrease ROS to favor their survival. The effect of independent and co-expressed wild-type and mutant PKM2 on ROS production followed the same trend as observed in the metabolic behavior of cells (Figs. 2–4). Apparently, a decrease in ROS in cells harboring co-transfections was due to enhanced accumulation of NADPH (Fig. 3). NADPH, the major byproduct of PPP, is the principal intracellular reductant, which helps in detoxification of ROS by maintaining ROS scavenger glutathione in its reduced form (32). Because NADPH was markedly accumulated in wild-type and mutant co-transfections, the consequent reduction of ROS was therefore highest in these co-transfections. It appears that stabilized hetero-dimers with low activity are mimicking the effect of ROS-induced oxidation of cysteine residue in PKM2 to reduce its activity to eventually generate NADPH for ROS detoxification (11).
Anchorage-independent growth is one of the hallmarks of cellular transformation (33). Therefore, a soft agar assay was performed to assess whether the pro-cancerous changes in metabolism and oxidative stress by wild-type and mutant PKM2, in different combinations, could enhance the transformation potential of cells. Approximately 2.5 times higher number of colonies in PKM2-only transfected cells as compared with that of mock transfected control suggested the importance of PKM2 in malignant transformation (Fig. 5). Also, this observation is in agreement with increased cancer metabolism and reduced oxidative stress in PKM2 transfects. Independently expressed mutants showed slightly higher number of colonies than that of wild-type PKM2 alone, with a remarkable increase in colony formation observed in wild-type and mutant co-transfections (5.5 times by PKM2-K422R and 7 times by PKM2-H391Y, as compared with wild-type PKM2-only) (Fig. 5). These data suggested that effect of co-transfections on cancer metabolism and oxidative stress contributed proportionally and positively to cell transformation. Furthermore, it could also be extrapolated that changes in cancer metabolism and oxidative stress affect the status of malignant transformation of cells accordingly.
Because co-expressed cells with wild-type and mutant PKM2 showed maximal effects on different parameters studied, it was pertinent to understand the potential of such cells in proliferation and tumor growth. Consistent with our previous results (20), cellular proliferation significantly increased in co-transfections as compared with wild-type and mutant PKM2-only transfected cells (Fig. 6A). Moreover, xenograft tumor growth in nude mice also showed a similar pattern (Fig. 6B). Collectively, these results substantiated the ability of heterozygous mutations to promote cancer and emphasized the need for studying the PKM2 biology to further unravel its role in cancer.
Out of the two co-transfections, the consistent maximal effect of PKM2-H391Y on all the parameters studied was an interesting observation. The underlying reason could be understood from the earlier observation of our laboratory where energy minimization studies showed the higher stability of hetero-dimers of PKM2 and H391Y with substantially high probability (∼65%) of formation of these hetero-dimers (20). Importantly, the PKM2-H391Y hetero-dimers are 2.5 times more stable than homo-dimers of PKM2 (20). Furthermore, the higher number of unique hydrogen bonds within the intersubunit contact of PKM2 and H391Y as compared with that of PKM2 and K422R also explains higher stability of PKM2-H391Y hetero-dimer (20). It seems that PKM2-H391Y hetero-dimer, which is even more stable than PKM2 homo-dimer, affected these functions far more greatly than PKM2 homo-dimer itself. Furthermore, ∼85% lesser activity of PKM2-H391Y hetero-dimer possibly explains high lactate production and macromolecular synthesis, reduced oxidative stress, and tumor growth, the attributes that are linked to down-regulated activity of PKM2 (12, 13, 15). Apparently, what is novel in the observations is how the heterozygous dominant negative state imparted higher morbidity, facilitating tumor development as compared with the homozygous-recessive mutant state.
Notably, our results correlate with recent findings that activity-impairing heterozygous PKM2 mutations are present in human cancers (34), where they may provide metabolic plasticity to tumors through differential PKM2 activity (35). The implications of studied heterozygous mutations in PKM2 have not only added to its significance as an important enzyme in cancer metabolism but also corroborated the importance of exon 10-coded intersubunit contact domain in affecting PKM2 activity. The variety of pro-cancerous implications of studied missense mutations highlights the importance of lower PKM2 activity in benefiting cancer and also validates PKM2 as an attractive therapeutic target.
Acknowledgments
We thank Dr. Rita Mulherkar and Ganesh Joshi, Advanced Centre for Treatment, Research and Education in Cancer (ACTREC), Mumbai, India for help in nude mice experiments.
Footnotes
- PKM2
- pyruvate kinase M2
- PEP
- phosphoenolpyruvate
- FBP
- fructose 1,6-bisphosphate
- PPP
- pentose phosphate pathway
- ROS
- reactive oxygen species.
REFERENCES
- 1. Fritsch A., Spassov S., Elfert S., Schlosser A., Gache Y., Meneguzzi G., Bruckner-Tuderman L. (2009) Dominant-negative effects of COL7A1 mutations can be rescued by controlled overexpression of normal collagen VII. J. Biol. Chem. 284, 30248–30256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gajko-Galicka A. (2002) Mutations in type I collagen genes resulting in osteogenesis imperfecta in humans. Acta Biochim. Pol. 49, 433–441 [PubMed] [Google Scholar]
- 3. Chenevix-Trench G., Spurdle A. B., Gatei M., Kelly H., Marsh A., Chen X., Donn K., Cummings M., Nyholt D., Jenkins M. A., Scott C., Pupo G. M., Dörk T., Bendix R., Kirk J., Tucker K., McCredie M. R., Hopper J. L., Sambrook J., Mann G. J., Khanna K. K. (2002) Dominant negative ATM mutations in breast cancer families. J. Natl. Cancer Inst. 94, 205–215 [DOI] [PubMed] [Google Scholar]
- 4. Brachmann R. K., Vidal M., Boeke J. D. (1996) Dominant-negative p53 mutations selected in yeast hit cancer hot spots. Proc. Natl. Acad. Sci. U.S.A. 93, 4091–4095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chaneton B., Gottlieb E. (2012) Rocking cell metabolism: revised functions of the key glycolytic regulator PKM2 in cancer. Trends Biochem. Sci. 37, 309–316 [DOI] [PubMed] [Google Scholar]
- 6. Christofk H. R., Vander Heiden M. G., Harris M. H., Ramanathan A., Gerszten R. E., Wei R., Fleming M. D., Schreiber S. L., Cantley L. C. (2008) The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 452, 230–233 [DOI] [PubMed] [Google Scholar]
- 7. Luo W., Semenza G. L. (2012) Emerging roles of PKM2 in cell metabolism and cancer progression. Trends Endocrinol. Metab. 23, 560–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tamada M., Suematsu M., Saya H. (2012) Pyruvate kinase M2: multiple faces for conferring benefits on cancer cells. Clin. Cancer Res. 18, 5554–5561 [DOI] [PubMed] [Google Scholar]
- 9. Sun Q., Chen X., Ma J., Peng H., Wang F., Zha X., Wang Y., Jing Y., Yang H., Chen R., Chang L., Zhang Y., Goto J., Onda H., Chen T., Wang M. R., Lu Y., You H., Kwiatkowski D., Zhang H. (2011) Mammalian target of rapamycin up-regulation of pyruvate kinase isoenzyme type M2 is critical for aerobic glycolysis and tumor growth. Proc. Natl. Acad. Sci. U.S.A. 108, 4129–4134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mazurek S., Boschek C. B., Hugo F., Eigenbrodt E. (2005) Pyruvate kinase type M2 and its role in tumor growth and spreading. Semin. Cancer Biol. 15, 300–308 [DOI] [PubMed] [Google Scholar]
- 11. Anastasiou D., Poulogiannis G., Asara J. M., Boxer M. B., Jiang J. K., Shen M., Bellinger G., Sasaki A. T., Locasale J. W., Auld D. S., Thomas C. J., Vander Heiden M. G., Cantley L. C. (2011) Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science 334, 1278–1283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hitosugi T., Kang S., Vander Heiden M. G., Chung T. W., Elf S., Lythgoe K., Dong S., Lonial S., Wang X., Chen G. Z., Xie J., Gu T. L., Polakiewicz R. D., Roesel J. L., Boggon T. J., Khuri F. R., Gilliland D. G., Cantley L. C., Kaufman J., Chen J. (2009) Tyrosine phosphorylation inhibits PKM2 to promote the Warburg effect and tumor growth. Sci. Signal. 2, ra73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lv L., Li D., Zhao D., Lin R., Chu Y., Zhang H., Zha Z., Liu Y., Li Z., Xu Y., Wang G., Huang Y., Xiong Y., Guan K. L., Lei Q. Y. (2011) Acetylation targets the M2 isoform of pyruvate kinase for degradation through chaperone-mediated autophagy and promotes tumor growth. Mol. Cell 42, 719–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Deberardinis R. J., Sayed N., Ditsworth D., Thompson C. B. (2008) Brick by brick: metabolism and tumor cell growth. Curr. Opin. Genet. Dev. 18, 54–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Christofk H. R., Vander Heiden M. G., Wu N., Asara J. M., Cantley L. C. (2008) Pyruvate kinase M2 is a phosphotyrosine-binding protein. Nature 452, 181–186 [DOI] [PubMed] [Google Scholar]
- 16. Luo W., Hu H., Chang R., Zhong J., Knabel M., O'Meally R., Cole R. N., Pandey A., Semenza G. L. (2011) Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell 145, 732–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gao X., Wang H., Yang J. J., Liu X., Liu Z. R. (2012) Pyruvate kinase M2 regulates gene transcription by acting as a protein kinase. Mol. Cell 45, 598–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Anitha M., Kaur G., Baquer N. Z., Bamezai R. (2004) Dominant negative effect of novel mutations in pyruvate kinase-M2. DNA Cell Biol. 23, 442–449 [DOI] [PubMed] [Google Scholar]
- 19. Akhtar K., Gupta V., Koul A., Alam N., Bhat R., Bamezai R. N. (2009) Differential behavior of missense mutations in the intersubunit contact domain of the human pyruvate kinase M2 isozyme. J. Biol. Chem. 284, 11971–11981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gupta V., Kalaiarasan P., Faheem M., Singh N., Iqbal M. A., Bamezai R. N. (2010) Dominant negative mutations affect oligomerization of human pyruvate kinase M2 isozyme and promote cellular growth and polyploidy. J. Biol. Chem. 285, 16864–16873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. German J. (1997) Bloom's syndrome. XX. The first 100 cancers. Cancer Genet. Cytogenet. 93, 100–106 [DOI] [PubMed] [Google Scholar]
- 22. Iqbal M. A., Siddiqui F. A., Gupta V., Chattopadhyay S., Gopinath P., Kumar B., Manvati S., Chaman N., Bamezai R. N. (2013) Insulin enhances metabolic capacities of cancer cells by dual regulation of glycolytic enzyme pyruvate kinase M2. Mol. Cancer 12, 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ryu H., Walker J. K., Kim S., Koo N., Barak L. S., Noguchi T., Kang B. Y., Kim K. M. (2008) Regulation of M2-type pyruvate kinase mediated by the high-affinity IgE receptors is required for mast cell degranulation. Br. J. Pharmacol. 154, 1035–1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gutman M., Couillard S., Labrie F., Candas B., Labrie C. (2003) Effect of treatment sequence with radiotherapy and the antiestrogen EM 800 on the growth of ZR 75 1 human mammary carcinoma in nude mice. Int. J. Cancer 103, 268–276 [DOI] [PubMed] [Google Scholar]
- 25. Vander Heiden M. G., Cantley L. C., Thompson C. B. (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gatenby R. A., Gillies R. J. (2004) Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 4, 891–899 [DOI] [PubMed] [Google Scholar]
- 27. Zwerschke W., Mazurek S., Massimi P., Banks L., Eigenbrodt E., Jansen-Dürr P. (1999) Modulation of type M2 pyruvate kinase activity by the human papillomavirus type 16 E7 oncoprotein. Proc. Natl. Acad. Sci. U.S.A. 96, 1291–1296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cairns R. A., Harris I. S., Mak T. W. (2011) Regulation of cancer cell metabolism. Nat. Rev. Cancer 11, 85–95 [DOI] [PubMed] [Google Scholar]
- 29. Yoshioka N., Wang L., Kishimoto K., Tsuji T., Hu G. F. (2006) A therapeutic target for prostate cancer based on angiogenin-stimulated angiogenesis and cancer cell proliferation. Proc. Natl. Acad. Sci. U.S.A. 103, 14519–14524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lee C. C., Jia Y., Li N., Sun X., Ng K., Ambing E., Gao M. Y., Hua S., Chen C., Kim S., Michellys P. Y., Lesley S. A., Harris J. L., Spraggon G. (2010) Crystal structure of the ALK (anaplastic lymphoma kinase) catalytic domain. Biochem. J. 430, 425–437 [DOI] [PubMed] [Google Scholar]
- 31. Lunt S. Y., Vander Heiden M. G. (2011) Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 27, 441–464 [DOI] [PubMed] [Google Scholar]
- 32. Tian W. N., Braunstein L. D., Pang J., Stuhlmeier K. M., Xi Q. C., Tian X., Stanton R. C. (1998) Importance of glucose-6-phosphate dehydrogenase activity for cell growth. J. Biol. Chem. 273, 10609–10617 [DOI] [PubMed] [Google Scholar]
- 33. Mori S., Chang J. T., Andrechek E. R., Matsumura N., Baba T., Yao G., Kim J. W., Gatza M., Murphy S., Nevins J. R. (2009) Anchorage-independent cell growth signature identifies tumors with metastatic potential. Oncogene 28, 2796–2805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Israelsen W. J., Dayton T. L., Davidson S. M., Fiske B. P., Hosios A. M., Bellinger G., Li J., Yu Y., Sasaki M., Horner J. W., Burga L. N., Xie J., Jurczak M. J., DePinho R. A., Clish C. B., Jacks T., Kibbey R. G., Wulf G. M., Di Vizio D., Mills G. B., Cantley L. C., Vander Heiden M. G. (2013) PKM2 isoform-specific deletion reveals a differential requirement for pyruvate kinase in tumor cells. Cell 155, 397–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. (2013) Regulation of PKM2 activity enables metabolic adaptation in tumors. Cancer Discov. 3, OF18 [Google Scholar]