

Background: It is not clear how BMP-induced chondrocyte maturation is cell-autonomously terminated.
Results: BMP-2 induced the ceramide-generating enzyme neutral sphingomyelinase 2 (nSMase2) in chondrocytes, whereas silencing of nSMase2 enhanced maturation in an Akt signaling-dependent manner.
Conclusion: nSMase2 signaling regulates BMP-induced chondrocyte maturation as a negative feedback mechanism.
Significance: This study elucidated the novel link between BMP and lipid signaling in chondrogenesis.
Keywords: Akt, Bone Morphogenetic Protein (BMP), Ceramide, Chondrogenesis, Sphingomyelinase, Hyaluronan Synthase 2, Sphingomyelin Phosphodiesterase 3
Abstract
Although bone morphogenic protein (BMP) signaling promotes chondrogenesis, it is not clear whether BMP-induced chondrocyte maturation is cell-autonomously terminated. Loss of function of Smpd3 in mice results in an increase in mature hypertrophic chondrocytes. Here, we report that in chondrocytes the Runx2-dependent expression of Smpd3 was increased by BMP-2 stimulation. Neutral sphingomyelinase 2 (nSMase2), encoded by the Smpd3 gene, was detected both in prehypertrophic and hypertrophic chondrocytes of mouse embryo bone cartilage. An siRNA for Smpd3, as well as the nSMase inhibitor GW4869, significantly enhanced BMP-2-induced differentiation and maturation of chondrocytes. Conversely, overexpression of Smpd3 or C2-ceramide, which mimics the function of nSMase2, inhibited chondrogenesis. Upon induction of Smpd3 siRNA or GW4869, phosphorylation of both Akt and S6 proteins was increased. The accelerated chondrogenesis induced by Smpd3 silencing was negated by application of the Akt inhibitor MK2206 or the mammalian target of rapamycin inhibitor rapamycin. Importantly, in mouse bone culture, GW4869 treatment significantly promoted BMP-2-induced hypertrophic maturation and calcification of chondrocytes, which subsequently was eliminated by C2-ceramide. Smpd3 knockdown decreased the apoptosis of terminally matured ATDC5 chondrocytes, probably as a result of decreased ceramide production. In addition, we found that expression of hyaluronan synthase 2 (Has2) was elevated by a loss of Smpd3, which was restored by MK2206. Indeed, expression of Has2 protein decreased in nSMase2-positive hypertrophic chondrocytes in the bones of mouse embryos. Our data suggest that the Smpd3/nSMase2-ceramide-Akt signaling axis negatively regulates BMP-induced chondrocyte maturation and Has2 expression to control the rate of endochondral ossification as a negative feedback mechanism.
Introduction
Over 95% of bone formation during the embryonic and developmental stages is achieved through endochondral ossification. This process is primed by the condensation of mesenchymal progenitor cells expressing the chondrogenic master regulator Sox9 (1), after which cells further differentiate into proliferating chondrocytes that are able to express a specific marker, Col2a1, encoding type II collagen (2). These chondrocytes then mature to hypertrophic chondrocytes, which eventually mineralize the surrounding cartilage matrix to be replaced by bone-forming osteoblasts (3). The maturation of chondrocytes into hypertrophic chondrocytes, which are able to express the type X collagen-encoding gene Col10a1, is mainly governed by the Runx2 transcription factor (4, 5). The rate of proliferation and differentiation of chondrocytes in vivo is tightly regulated by a signaling network between Indian hedgehog, parathyroid hormone-related protein, fibroblast growth factor (FGF), and bone morphogenetic protein (BMP)3 signaling (6).
BMPs belong to the transforming growth factor-β (TGF-β) family, which transduces signals through type II and type I receptors to activate receptor-regulated Smads. Upon ligand binding, BMP type I receptors phosphorylate Smad1/5/8 in the cytoplasm. Phosphorylated Smads form a trimeric complex with Smad4 (co-Smad) to translocate into the nucleus to directly or indirectly regulate the transcription of target genes (7, 8). BMPs and their receptors are expressed throughout the growth plate and perichondrium of developing bone (9). BMPs maintain expression of Sox9 and subsequent cartilage matrix production (10, 11). BMPs are also required in vitro for the induction of Col2a1 and Col10a1 to promote chondrocyte maturation at later stages (12–14). Chondrocyte-specific overexpression of the extracellular BMP antagonist, Noggin, in transgenic mice resulted in no cartilage formation (15). Similarly, mice with cartilage-specific combined deletions of two BMP type I receptor genes (Bmpr1a and Bmpr1b), or with a double knock-out of Smad1 and Smad5, showed severely impaired chondrogenesis (16, 17). At a later maturation stage, BMP signaling directly accelerates the expression of Col10a1 in concert with Runx2 (13, 18, 19). Forced expression of constitutively active Bmpr1a in cartilage promoted the maturation and hypertrophy of chondrocytes in mice (20). The evidence clearly demonstrates the accelerating roles of BMP signaling in chondrocyte commitment, proliferation, and hypertrophic maturation both in vivo and in vitro.
Chondrogenesis in the endochondral ossification process is a promising cellular event for application in cartilage and bone regeneration, and it is a process that can be artificially engineered from human mesenchymal stem/stroma cells (MSCs) (21). Regarding the treatment of articular cartilage defects, the engineered chondrocytes must arrest maturation processes, because abnormally matured hypertrophic chondrocytes, expressing COL10A1, mineralize cartilage matrix to cause pathological conditions such as osteoarthritis (OA) (22–24). This is a major problem of cartilage tissue engineering in an in vitro culture system, where MSCs rapidly express COL10A1 in a monolayer or pellet culture before the cells express hypertrophic phenotypes, suggesting that the mechanism by which MSCs induce type X collagen expression in vitro is different from that in chondrogenesis in vivo (25, 26). In bone engineering events, MSCs have formed bone trabeculae in vivo only when they had developed hypertrophic chondrocyte structure in vitro prior to implantation (21). Therefore, controlling the maturation and hypertrophy of chondrocytes is crucial for regenerative medicine of both cartilage and bone.
Although the effects of BMP signaling in promoting chondrocyte maturation should be eliminated for cartilage regeneration, they may be artificially enhanced to promote the efficiency of bone engineering. Although the BMP-Smad pathway directly induces the inhibitory Smad, Smad6, to inhibit the phosphorylation of Smad1/5/8 as a negative feedback mechanism (27), and Smurf1 targets Smad6 and the BMP type I receptors to block BMP signaling (28, 29), these inhibitory molecules are not specifically expressed in maturing chondrocytes. Recently, we reported that expression of the transcriptional repressor SnoN gradually increases in BMP-induced differentiating chondrocytes to suppress BMP signaling and the subsequent chondrocyte hypertrophic maturation (30). However, because SnoN partially blocked chondrocyte maturation, the mechanism regulating the rate of BMP signaling-driven chondrocyte differentiation in a cell-autonomous manner remains unclear.
The membrane-bound enzyme neutral sphingomyelinase 2 (nSMase2), encoded by the sphingomyelin phosphodiesterase 3 (Smpd3) gene, cleaves sphingomyelin to generate the lipid second messenger ceramide, which affects a variety of cellular process, including proliferation, apoptosis, and differentiation (31–34). A deletion mutation in the Smpd3 gene was identified in fragilitas ossium (fro), a mouse model of osteogenesis imperfecta (35). fro/fro mice are characterized by retarded skeletal growth and a severely hypomineralized skeleton with normal osteoblast differentiation; bone mineralization can be rescued by osteoblast-specific overexpression of Smpd3, suggesting an important role of nSMase2 in the mineralization of the extracellular matrix (36). Interestingly, the number and area of hypertrophic chondrocytes were significantly increased, and conversion of cartilage into bone was retarded in fro/fro bone. Smpd3 knock-out mice also showed fragile bones and dwarfism, with retarded transition of proliferative chondrocytes into hypertrophic chondrocytes (37, 38). Importantly, the knee joint cartilage of adult Smpd3-null mice showed severe deformity with exostosis, the phenotype of OA (38). These findings, from two lines of nSMase2 loss-of-function mice, suggest that Smpd3/nSMase2 plays an important role in suppressing hypertrophic maturation of chondrocytes and OA initiation. However, there is currently no information regarding the molecular mechanism for the function of nSMase2 in chondrocyte differentiation. In C2C12 myoblasts, BMP-2-induced Runx2 directly binds to the Smpd3 promoter to up-regulate expression, suggesting that BMP signaling could elevate Smpd3 level in a certain context (39).
In this study, we report that Smpd3 is continuously up-regulated by BMP-2 stimulation of chondrocytes during the maturation stage to suppress the late differentiation step via the Akt pathway. We found that this induction of Smpd3 by BMP-2 was Runx2-dependent and that the transcription factor is crucial for the hypertrophic maturation of chondrocytes. Loss- and gain-of-function experiments revealed that Smpd3/nSMase2 or ceramide signaling cell-autonomously suppressed expression of Col2a1, Col10a1, and hyaluronate synthase 2 (Has2), as well as accumulation of glycosaminoglycan. The Akt-S6 pathway was found to be responsible for the action of Smpd3/nSMase2. Importantly, application of an inhibitor compound for nSMase into the mouse bone organ culture system significantly increased the hypertrophic conversion and extracellular matrix calcification of cartilage.
EXPERIMENTAL PROCEDURES
Cell Culture and Differentiation
The mouse chondrogenic cell line ATDC5, established from a differentiated culture of the teratocarcinoma stem cell line AT805 on the basis of chondrogenic potential (40), was obtained from the RIKEN BioResource Center. Human chondrocyte cell line C28/I2 was a kind gift from Dr. Mary Goldring (41). The cells were cultured in Dulbecco's modified Eagle's medium (DMEM)/Ham's F-12 (1:1) (Invitrogen), containing 5% fetal bovine serum (FBS) with 100 units/ml penicillin G and 100 μg/ml streptomycin. The mouse C3H10T1/2 cell line was obtained from the ATCC. The cells were cultured in Eagle's basal medium (Sigma) with 2 mm l-glutamine, 10% FBS, 100 units/ml penicillin G, and 100 μg/ml streptomycin. Primary chondrocytes were harvested from 2-day-old mice with a C57BL/6J background. Briefly, articular cartilage of the femoral heads, femoral condyles, and tibial plateaus was isolated from mice and digested by 3 mg/ml collagenase D (Roche Applied Science) for 45 min, followed by 0.5 mg/ml collagenase D overnight. The chondrocytes were filtered through a sterile 40-μm cell strainer and cultured in DMEM/Ham's F-12 (1:1) containing 10% FBS, 100 units/ml penicillin G, and 100 μg/ml streptomycin. Differentiation of the cells, cultured in monolayer, was induced by the addition of recombinant human BMP-2 (PeproTech) at a concentration of 300 ng/ml, with or without insulin/transferrin/selenium (ITS) supplement (Sigma) on collagen type I-coated culture plates (Iwaki).
Embryonic Bone Organ Culture
Metatarsal bone rudiments were harvested from C57BL/6J mouse embryos at 16.5 days post-coitum (E16.5) and cultured in DMEM/Ham's F-12 (1:1) supplemented with 10% FBS, and 100 units/ml penicillin G, and 100 μg/ml streptomycin, as described (42). Cultured bones were stained with Alcian blue and alizarin red dyes according to a standard protocol for skeletal preparation. Briefly, bones fixed in 96% ethanol were stained with 0.015% Alcian blue 8GX (Sigma) in a mixture solution of 96% ethanol/acetic acid (4:1) for 1 day, followed by a dehydration step in 100% ethanol. Dehydrated bones were immersed briefly in 1% potassium hydroxide (KOH), followed by staining in 0.001% alizarin red S (Sigma) in 1% KOH for 1 day. Images were captured with stereomicroscope M165FC (Leica). Four bones per group were analyzed. Animal experiments were approved by the Institutional Animal Care and Use Committee of Kagoshima University (number MD12043).
RNA Interference
Dharmacon siRNA ON-TARGETplus SMARTpool, a mixture of four independent siRNAs against mouse Runx1, Runx2, Runx3, Smpd3, or Has2, and the control reagent were purchased from Thermo Scientific. siRNAs were transfected into cells using Lipofectamine RNAiMax (Invitrogen). BMP-2 and compounds were added to the culture simultaneously after an overnight transfection of siRNA.
Plasmids and Adenovirus
Mouse Smpd3 cDNA was cloned from ATDC5 by employing an RT-PCR-based technique, subcloned into the entry vector, pENTR, and further transferred into the C-terminally V5-tagged expression vector, pEF-DEST51, by attL-attR recombination (Invitrogen). To generate adenovirus-carrying Smpd3 cDNA, the Smpd3 gene in the pENTR-Smpd3 vector was transferred into the C-terminally V5-tagged adenovirus expression vector pAd/CMV/V5-DEST by attL-attR recombination (Invitrogen) and further transfected into the adenovirus-producing cell line 293A according to the manufacturer's protocol. pAd/CMV/V5-GW/lacZ adenovirus expression vector was used as a control to express β-galactosidase. Adenovirus infection into ATDC5 cells was performed at a multiplicity of infection of 10. These experiments were approved by the Kagoshima University safety control committee for gene recombination techniques (number 22053).
Chemical Inhibitor Compounds and C2-ceramide
All of the following agents were resolved in dimethyl sulfoxide (DMSO), and DMSO was used as the mock control. Cycloheximide was purchased from Sigma and applied at a concentration of 10 mm for 2 h, prior to BMP-2 stimulation. The nSMase inhibitor, GW4869 (Sigma), or C2-ceramide (Enzo Life Sciences) was applied at the same time of BMP-2 induction, at 1 or 10 μm, respectively. The PI3K inhibitor, LY294002 (Sigma), Akt inhibitor, MK2206 (Chemie Tek), and the mammalian target of rapamycin inhibitor (Sigma) were applied at the same time of BMP-2 induction, at the indicated concentrations.
Immunoblotting and RTK Signaling Antibody Array Analysis
Cells were lysed in M-PER lysis buffer (Thermo Scientific) supplemented with aprotinin, sodium orthovanadate, and phenylmethylsulfonyl fluoride (PMSF) and subjected to SDS-PAGE, membrane transfer, and chemiluminescence, using standard protocol. Blots were incubated with the following: anti-Runx2 (1:1,000, 8G5, MBL); anti-nSMase2 (1:500, H-195, Santa Cruz Biotechnology); anti-aggrecan (1:500, H-300, Santa Cruz Biotechnology); anti-collagen type II α1 (1:1,000, LSBio); anti-phospho-Akt (Ser-473) (1:1,000, 587F11, Cell Signaling); anti-Akt (1:1,000, 5G3, Cell Signaling); anti-phospho-S6 ribosomal protein (Ser-235/236) (1:1,000, D57.2.2E, Cell Signaling); anti-S6 ribosomal protein (1:1,000, 54D2, Cell Signaling); anti-phospho-PI3K (1:1,000, number 4228, Cell Signaling); anti-PI3K (p85) (1:1,000, number 610045, BD Transduction Laboratories); anti-phospho-Smad1/5/8 (1:1,000, number 9511, Cell Signaling); anti-Smad1 (1:1,000, number 9743, Cell Signaling); and horseradish peroxidase (HRP)-conjugated anti-rabbit secondary antibody, anti-mouse secondary antibody (1:10,000) (Cell Signaling), or anti-tubulin antibody (1:1,000, DM1A, T9026, Sigma). Signals were detected using the LAS 4000 Mini Image Analyzer (Fujifilm). We employed PathScan® RTK signaling antibody array kit (Cell Signaling) to analyze the signaling pathway influenced by loss of Smpd3/nSMase2 function. This system is a slide-based antibody array, founded upon the sandwich immunoassay principle and allowing for the simultaneous detection of 28 receptor tyrosine kinases and 11 important signaling nodes when phosphorylated at tyrosine or other residues, was employed to analyze the signaling pathway influenced by loss of Smpd3/nSMase2 function. The experiment was performed according to the manufacturer's manual, and the chemiluminescent readout was performed with LAS 4000 Mini Image Analyzer.
Immunocytochemistry and Immunohistochemistry
For immunocytochemistry, cells were fixed with 4% paraformaldehyde in PBS for 30 min and treated with 0.2% Triton X-100. CAS block (Zymed Laboratories Inc.) was used for blocking. For immunohistochemistry, formalin-fixed mouse E17.5 embryo humeri were embedded in paraffin blocks, which were sliced at a 4-μm thickness. The antigen was retrieved by the Liberate Antibody Binding solution (Polysciences). A CAS block was used for blocking. Cells or bone sections were incubated with anti-aggrecan (1:100, H-300, Santa Cruz Biotechnology), anti-collagen type II α1 (1:100, LSBio), anti-nSMase2 rabbit polyclonal antibody (1:100, H-195, Santa Cruz Biotechnology), and anti-Has2 mouse monoclonal antibody (1:100, D-8, Santa Cruz Biotechnology). Anti-mouse Alexa Fluor 488 (1:200, A11001) or anti-rabbit Alexa Fluor 568 (1:200, A11011) (Invitrogen) was used to detect signals. Normal rabbit or mouse IgG was used as negative control. Hyaluronan (HA) detection involved the following protocol. Samples were blocked with a streptavidin/biotin blocking kit (Vector Laboratories), according to the manufacturer's protocol, and digested sequentially with 0.05% of trypsin and 1 unit/ml of chondroitinase ABC (Sigma). Samples were further blocked with CAS block and sequentially incubated with biotin-conjugated HA-binding protein (1:1,000, Hokudo) and 1 μg/ml Alexa Fluor 488-conjugated streptavidin (Invitrogen). Images of immunocytochemistry and immunohistochemistry were captured with microscope AX80 and digital camera DP70 (Olympus). Animal experiments were approved by the Institutional Animal Care and Use Committee of Kagoshima University (number MD12043).
Real Time Quantitative PCR Assay
Cells were lysed with the TRIzol reagent (Invitrogen) to purify RNA, and 1 μg of RNA was reverse-transcribed into cDNA using the Verso cDNA kit (Thermo Scientific). The relative amount of gene transcripts was determined by real time PCR using the SYBR premix Ex TaqII (Takara) and the Thermal Cycler Dice TP850 (Takara). PCRs were performed in duplicate per sample, and the measured expression level of each gene was normalized to that of Hprt1. ΔCt values were calculated by subtracting Ct values of Hprt1 from Ct values of target genes. Sequence information of primers used is listed in supplemental Table 1.
TUNEL Assay
For detecting ATDC5 cells that underwent apoptosis, we used the ApopTag® peroxidase in situ apoptosis detection kit (Merck), which detects apoptotic cells in situ by labeling and detecting DNA strand breaks by the TUNEL method. The apoptotic cells were visualized by immunoperoxidase staining. Four independent experiments were performed, and four fields per well were evaluated for the number of apoptotic cells.
Statistical Analysis
Data in this study are expressed as mean ± S.D. of three independent experiments, unless otherwise noted. The statistical comparisons between the different treatments were performed using an unpaired Student's t test in which p < 0.05 was considered significant and p < 0.01 was highly significant.
RESULTS
Smpd3 Is Induced by BMP-2 in Chondrocytes and Is Detected in Mature Chondrocytes in Vivo
We hypothesized that BMP signaling cell-autonomously activated unknown mechanisms to terminate BMP-induced chondrocyte differentiation through a negative feedback pathway. We focused on the Smpd3 gene, because it could be up-regulated by BMP-2 in C2C12 myoblasts of mesenchymal origin (39); loss-of-function models for Smpd3 in mice showed an increased number of hypertrophic chondrocytes or retarded transition of proliferative chondrocytes into hypertrophic chondrocytes. Molecular mechanisms for both cartilage phenotypes are unclear (37, 38), suggesting a relationship between BMP signaling and Smpd3/nSMase2 in chondrogenesis. To investigate the possible cell-autonomous roles of Smpd3 in chondrocyte maturation, we mainly employed the clonal chondrogenic mouse cell line ATDC5 because it is an excellent in vitro model for skeletal development, which can be stimulated by BMP-2 (43, 44). We also used the normal human chondrocyte cell line C28/I2, mouse chondrogenic cell line C3H10T1/2, and mouse primary articular chondrocytes. We used BMP-2 to stimulate chondrogenic differentiation because, in addition to existing evidence, we and others have observed strong expression of the BMP-2 protein in proliferating and mature chondrocytes in the developing bones of rodents (45, 46). We first checked if these cell lines could differentiate into chondrocytes and mature into hypertrophic chondrocytes and not fibrochondrocytes. Upon BMP-2 induction, ATDC5 cells could produce chondrocyte-specific proteins, aggrecan and type II collagen, in the extracellular matrix (Fig. 1A). The early chondrocyte differentiation marker aggrecan (Acan) was elevated after day 1 and was maintained at elevated levels for 2 weeks, whereas Col10a1 increased mildly from day 3 and was strongly up-regulated after day 7. Col1a1 and Col3a1 gene expression was not elevated, but rather decreased, suggesting that these cells did not differentiated into fibrochondrocytes (Fig. 1B). Similar results were obtained in C28/I2 cells (Fig. 1, C and D) and C3H10T1/2 cells (Fig. 1E). We also confirmed chondrogenic differentiation of primary chondrocytes by BMP-2 (Fig. 1F). ΔCt values of quantitative PCR data indicated that primary chondrocytes express higher levels of chondrocyte marker genes than the three chondrogenic cell lines, suggesting that primary chondrocytes are already committed and differentiated into chondrocytes without BMP-2 induction, whereas the cell lines are relatively immature to differentiate into chondrocytes upon BMP stimulation. From these expression profiles, we considered these cell lines to be suitable models for chondrocyte differentiation. In ATDC5 cells, Smpd3 showed a varied expression pattern from that of Acan and Col10a1, in that it was strongly elevated from day 1 and showed a second peak of increase after day 7 (Fig. 1G). We also observed the BMP-2-induced increment of the Smpd3 gene in C28/I2, C3H10T1/2, and primary chondrocytes (Fig. 1H). Importantly, the primary chondrocytes also expressed higher levels of Smpd3, suggesting its functional importance in chondrocytes. If Smpd3/nSMase2 has an in vivo role in chondrogenesis or cartilage maintenance, it would be expressed in cartilage, although its expression in cartilage has not been characterized in detail. Quantitative PCR analysis on a panel of cDNAs from multiple tissues generated from 3-month-old mice revealed that Smpd3 was expressed at the highest levels in the thymus, intestine, skin, fat, and bone, although it was almost absent in the heart, liver, and kidney (Fig. 1I). Smpd3 was moderately expressed in cartilage tissue. This expression profile of Smpd3 is essentially similar to the results reported for 2-week-old mice (36). In embryonic bone, we detected prominent expression of Smpd3-coding nSMase2 protein in the bone collar and trabecular bone by immunofluorescence (Fig. 1J). In the cartilage, little nSMase2 expression was noted in resting and columnar proliferating chondrocytes, whereas relatively high expression was observed in the prehypertrophic layer and hypertrophic chondrocytes (Fig. 1J), with an expression pattern resembling that of Runx2 (4).
FIGURE 1.

Expression of Smpd3/nSMase2 is promoted by BMP-2 treatment in maturing chondrocytes in vitro and is increased in prehypertrophic and hypertrophic chondrocytes in a growth plate in vivo. A, chondrogenic differentiation of mouse ATDC5 chondrocytes was induced by application of BMP-2 (300 ng/ml) for 7 days (d). Immunofluorescence for aggrecan or type II collagen was performed, with normal IgG as negative control. Scale bar, 100 μm. B, ATDC5 cells were cultured in the presence of BMP-2 (300 ng/ml) for the indicated periods. Expression levels of Acan, Col10a1, Col1a1, and Col3a1 were examined by quantitative RT-PCR. C, chondrogenic differentiation of human C28/I2 chondrocytes was induced by the application of BMP-2 (300 ng/ml) for 7 days. Immunocytochemistry for aggrecan or type II collagen was performed, with normal IgG as negative control. Scale bar, 100 μm. D, C28/I2 chondrocytes were cultured in presence of BMP-2 (300 ng/ml) and ITS supplement for 14 days. Expression of COL1A1 or COL3A1 was evaluated by quantitative RT-PCR. E, mouse C3H10T1/2 cells were cultured with BMP-2 (300 ng/ml) for 48 h. Expression levels of Col2a1, Col10a1, Col1a1, and Col3a1 were evaluated by quantitative RT-PCR. F, mouse primary chondrocytes (Pr.Ch.) cells were cultured in the presence of BMP-2 (300 ng/ml) for 6 days. Expression levels of Acan, Col2a1, and Col10a1 were evaluated by quantitative RT-PCR. G, ATDC5 cells were cultured in presence of BMP-2 (300 ng/ml) for the indicated periods. Expression of Smpd3 was examined by quantitative RT-PCR. H, quantitative RT-PCR for Smpd3 was performed on samples in D, E, and F. I, real time PCR for Smpd3 was performed on a tissue cDNA panel of a 3-month-old mouse. J, expression of nSMase2 in mouse E17.5 humerus cartilage was evaluated by immunofluorescence. Normal IgG was used as negative control. r, resting chondrocytes; p, proliferating chondrocytes; ph, prehypertrophic chondrocytes; h, hypertrophic chondrocytes; b, bone. Scale bar, 200 μm. *, p < 0.05; **, p < 0.01. ΔCt values (Ct target − Ct Hprt1) of quantitative RT-PCR are indicated in the graphs.
BMP-2-induced Increase of Smpd3 Expression Is Runx2-dependent
We wondered if this BMP-2-induced expression of Smpd3 in ATDC5 was directed by the BMP-Smad signaling axis, although the expression pattern was not the typical rapid pattern of a gene that is directly targeted (47). To test this, we treated ATDC5 cells with cycloheximide (CHX), an inhibitor of de novo protein synthesis, before adding BMP-2. As expected, 24 h after BMP-2 stimulation, the induction of Id1, a representative direct-target gene of the BMP-Smad pathway, was maintained, even in the presence of CHX (Fig. 2A). Induction of Id1 was higher in CHX-treated cells, likely because of the suppression of inhibitory Smad6 synthesis (27). However, expression of Col2a1 was eliminated by CHX treatment (Fig. 1A). This basal suppression by CHX probably results from an inhibition of the constitutive expression of Sox9 and downstream Sox5 and Sox6, transcription factors that cooperatively activate the promoter of Col2a1 (2, 48). However, existing Sox proteins should be responsible for the partially increased expression of Col2a1 by BMP-2. The basal expression level of Smpd3 was also suppressed by CHX treatment. Unlike Col2a1, however, the CHX-eliminated basal expression of Smpd3 was not up-regulated by BMP-2 (Fig. 2A) suggesting that a de novo protein other than the Sox trio was necessary for BMP-induced expression of Smpd3 in ATDC5 cells. Because the Runx2 protein is a master regulator of chondrocyte maturation (4, 5), and its direct interaction with the Smpd3 promoter is important for its expression in myoblasts (39), we investigated the relationship between Runx2 and expression of Smpd3 in chondrocytes. We confirmed the increment of the Runx2 gene during BMP-induced maturation of ATDC5 chondrocytes (Fig. 2B). To address this question, we transfected ATDC5 cells with Runx2 siRNA. At day 2 of BMP-2 application, Runx2 was weakly induced, and its expression was knocked down by the siRNA to about 40% that of the control (Fig. 2C). Expression of Id1 was not decreased by siRunx2, but rather it was increased, suggesting that Runx2 is inhibitory of the BMP pathway during this early differentiation stage. Interestingly, loss of Runx2 not only suppressed basal expression of Smpd3 but also completely blocked induction by BMP-2 treatment (Fig. 2C). This effect of siRunx2 was confirmed by analyzing protein expression of nSMase2 using immunoblotting (Fig. 2D). To assess the functional specificity of Runx2 among the three Runx isoforms, siRNAs for Runx1 and Runx3 were tested in ATDC5 cells. Although we could obtain efficient knockdown of Runx1 and Runx3, the BMP-2-induced increase of Smpd3 was not blocked by the corresponding siRNA (Fig. 2E). In primary chondrocytes, knockdown of Runx2 completely abrogated the BMP-stimulated up-regulation of Smpd3 (Fig. 2F). In contrast to ATDC5 cells, silencing of Runx1 or Runx3 could mildly suppress expression of Smpd3 in primary chondrocytes (Fig. 2F), suggesting that Runx1 and Runx3 were partially responsible for Smpd3 expression. These data demonstrate that BMP signaling increases the expression of Smpd3/nSMase2 in chondrocytes in cooperation with Runx2, especially in the maturation stages when the level of Runx2 is elevated.
FIGURE 2.

BMP-2-induced increase of Smpd3 expression in chondrocytes is Runx2-dependent. A, CHX was applied to ATDC5 cells at a concentration of 10 mm for 2 h prior to BMP-2 (300 ng/ml) stimulation. Cells were harvested 24 h after BMP-2 induction to perform quantitative RT-PCR analysis for Id1, Col2a1, and Smpd3. B, ATDC5 cells were cultured with BMP-2 (300 ng/ml) for the indicated times. Expression of Runx2 was examined by quantitative RT-PCR. C, ATDC5 chondrocytes were transfected with control siRNA (siCont) or Runx2 siRNA (siRunx2) for 16 h and then treated with or without BMP-2 (300 ng/ml) for 48 h. Quantitative RT-PCR analysis was performed for Runx2, Id1, and Smpd3. D, ATDC5 cells were transfected with control siRNA (siCont) or Runx2 siRNA (siRunx2) for 16 h and stimulated with BMP-2 (300 ng/ml) for 48 h. Cells were subjected to immunoblot analysis for the indicated antibodies. Tubulin served as a loading control. E, ATDC5 cells were transfected with control siRNA (siCont), Runx1 siRNA (siRunx1), or Runx3 siRNA (siRunx3) for 16 h and then treated with or without BMP-2 (300 ng/ml) for 48 h. Quantitative RT-PCR analysis was performed for Runx1, Runx3, and Smpd3. F, mouse primary chondrocytes were transfected with control siRNA (siCont), Runx1 siRNA (siRunx1), Runx2 siRNA (siRunx2), or Runx3 siRNA (siRunx3) for 16 h and then treated with or without BMP-2 (300 ng/ml) for 48 h. Quantitative RT-PCR analysis was performed for Runx1, Runx2, Runx3, and Smpd3. *, p < 0.05; **, p < 0.01; n.s., not significant.
Smpd3/nSMase2 and C2-ceramide Inhibit Chondrogenic Differentiation and Maturation of Chondrocytes
We used a knockdown assay to investigate whether Smpd3/nSMase2 has a role in the differentiation of ATDC5 chondrocytes. The potent induction of Smpd3 by BMP treatment was silenced by siSmpd3 at a level of ∼25% of that in cells treated with control siRNA at day 6 (Fig. 3A). Loss of Smpd3 mildly increased the basal level of Col2a1 expression and significantly enhanced the BMP-2-induced increment (Fig. 3A). Although expression of Col10a1 was not significantly elevated by BMP-2 at day 6, it was dramatically increased by siSmpd3 in the presence of BMP-2. Similar effects of siSmpd3 on the expression of Col2a1 and Col10a1 were observed in primary chondrocytes (Fig. 5F). This effect of Smpd3 knockdown against expression of Col2a1 was mimicked by the addition of 1 μm GW4869, a specific inhibitor compound for nSMase (Fig. 3B) (49), suggesting that the data from the siSmpd3 experiment resulted from down-regulation of nSMase2. GW4869 also enhanced the production of the cartilage-specific extracellular component, glycosaminoglycan, by BMP-2 stimulation for 17 days, as assessed by Alcian blue staining, both in ATDC5 cells and in primary chondrocytes (Fig. 3C). Because nSMase2 generates ceramide as a lipid second messenger from the cell membrane, and the total level of ceramide was decreased in fro/fro bone (36), we challenged the cell membrane-permeable C2-ceramide to mimic nSMase-ceramide signaling. Combined application of GW4869 and C2-ceramide completely eliminated the GW4869-mediated enhancement observed by Alcian blue staining (Fig. 3C). The effect of GW4869 or C2-ceramide on Alcian blue staining was confirmed by immunoblotting for aggrecan (Fig. 3C). Although C2-ceramide showed no effect on BMP-2-induced expression of Smpd3 at day 14, it significantly suppressed the expression of both Col2a1 and Col10a1 (Fig. 3D). As another gain-of-function approach, adenovirus-mediated overexpression of Smpd3 in ATDC5 cells was performed to yield transgene expression levels that were ∼100 times those of endogenous levels even after 8 days of induction (Fig. 3E). Infection of Smpd3-expressing adenovirus presented similar results as those of the C2-ceramide experiment, in which overexpression inhibited the BMP-2-mediated elevation of both Col2a1 and Col10a1 expression (Fig. 3E). Similarly, Smpd3-expressing adenovirus suppressed maturation of primary chondrocytes (Fig. 3F). These loss- or gain-of-function experiments suggest a cell-autonomous inhibitory action of the Smpd3/nSMase2-ceramide axis on maturation of chondrocytes.
FIGURE 3.
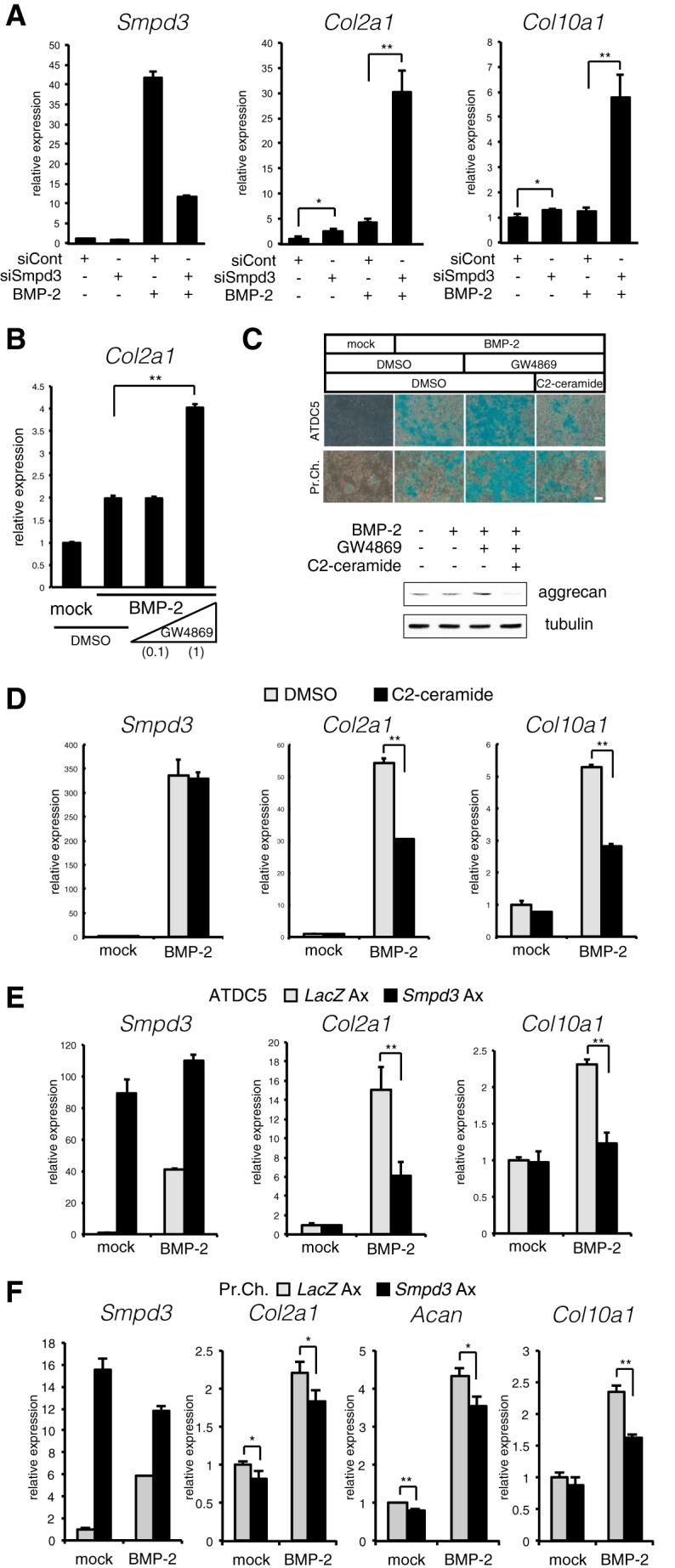
Loss or gain of Smpd3/nSMase2 function promotes or suppresses BMP-2-induced chondrogenic maturation, respectively. A, ATDC5 chondrocytes were transfected with control siRNA (siCont) or Smpd3 siRNA (siSmpd3) for 16 h and then treated with or without BMP-2 (300 ng/ml) for 6 days. Quantitative RT-PCR analysis was performed for Smpd3, Col2a1, and Col10a1. B, ATDC5 cells were treated with BMP-2 (300 ng/ml) in combination with GW4869 at a concentration of 0.1 or 1 μm for 4 days to analyze expression of Col2a1 by quantitative RT-PCR. C, ATDC5 cells or primary chondrocytes were stimulated with BMP-2 (300 ng/ml) in combination with GW4869 (1 μm) and C2-ceramide (10 μm) for 17 days. Cells were subjected to Alcian blue staining. Scale bar, 200 μm. A parallel experiment was done with ATDC5 with a stimulation time of 7 days, and immunoblotting was performed for aggrecan and tubulin. D, ATDC5 cells were stimulated with BMP-2 (300 ng/ml) in combination with C2-ceramide at 10 μm for 14 days. E, ATDC5 chondrocytes were infected with adenovirus (Ax) carrying LacZ or Smpd3 for 2 h, and further cultured with or without BMP-2 (300 ng/ml) for 7 days. Expression of Smpd3, Col2a1, and Col10a1 was evaluated by quantitative RT-PCR. F, mouse primary chondrocytes were infected with adenovirus carrying lacZ or Smpd3 for 2 h and further cultured with or without BMP-2 (300 ng/ml) for 6 days. Expression of Smpd3, Col2a1, and Col10a1 was evaluated by quantitative RT-PCR. *, p < 0.05; **, p < 0.01.
FIGURE 5.

Blocking the Akt or PI3K pathway negates the Smpd3 siRNA-mediated acceleration of chondrogenesis initiated by BMP-2 in ATDC5 cells. A, ATDC5 cells were transfected with control siRNA (siCont) or Smpd3 siRNA (siSmpd3) for 16 h and stimulated by BMP-2 (300 ng/ml) with or without MK2206 at the indicated concentrations (micromolar) for 6 days. Expression of Smpd3, Acan, Col2a1, and Col10a1 was evaluated by quantitative RT-PCR. B, ATDC5 cells were transfected with control siRNA (siCont) or Smpd3 siRNA (siSmpd3) for 16 h, and then cultured in the presence of BMP-2 (300 ng/ml) with or without MK2206 (10 μm) for 9 days. Alcian blue staining was performed. Scale bar, 300 μm. C, ATDC5 cells were transfected with control siRNA (siCont) or Smpd3 siRNA (siSmpd3) for 16 h and stimulated by BMP-2 (300 ng/ml) with or without rapamycin at the indicated concentrations (micromolar) for 3 days. Expression of Smpd3 and Col10a1 was evaluated by quantitative RT-PCR. D, ATDC5 cells were transfected with control siRNA (siCont) or Smpd3 siRNA (siSmpd3) for 16 h and stimulated by BMP-2 (300 ng/ml) for 24 h, and then immunoblotted for the indicated antibodies. Tubulin served as a loading control. E, ATDC5 cells were transfected with control siRNA (siCont) or Smpd3 siRNA (siSmpd3) for 16 h and further stimulated by BMP-2 (300 ng/ml) with or without LY294002 at the indicated concentrations (μm) for 6 days. Expression of Smpd3, Acan, Col2a1, and Col10a1 was evaluated by quantitative RT-PCR analysis. F, mouse primary chondrocytes were transfected with control siRNA (siCont) or Smpd3 siRNA (siSmpd3) for 16 h, and were further stimulated by BMP-2 (300 ng/ml) with or without LY294002 (LY, 25 μm), MK2206 (MK, 5 μm), or rapamycin (Rp, 0.5 μm) for 7 days. Expression of Smpd3, Acan, Col2a1, and Col10a1 was evaluated by quantitative RT-PCR analysis. *, p < 0.05; **, p < 0.01; n.s., not significant.
Smpd3 Suppresses the Activity of the Akt-S6 Pathway during Chondrogenesis in Vitro
We sought the molecular mechanism by which Smpd3/nSMase2 suppresses chondrogenesis and focused on the Akt signaling pathway for the following reasons. First, phosphorylation of Akt and the downstream ribosomal protein S6 (rpS6) was increased in fro/fro fibroblasts (50). Second, genetic approaches revealed that the IGF-IGF receptor-PI3K-Akt pathway plays key roles in skeletal growth and endochondral ossification and that overexpression of the activated form of Akt in the cartilage of transgenic mice promoted chondrocyte differentiation and maturation, whereas forced expression of its dominant-negative form delayed these cellular events (51). To evaluate the specificity of Akt among the various RTK signaling pathways, we examined the possible correlation of the Akt pathway in Smpd3/nSMase2 signaling during chondrocyte differentiation by performing an RTK signaling antibody array assay. Because receptors for insulin or insulin-like growth factor (IGF), which promote chondrogenic differentiation of ATDC5 cells (40), are RTKs, and a mixture of ITS is preferentially used to prepare the chondrogenic condition of ATDC5 cells (52), we first checked the effect of application of the ITS supplement alone and found no effect on the RTK array (Fig. 4A). Interestingly, 8 h after the addition of BMP-2, phosphorylation of rpS6 was significantly strengthened. More importantly, BMP-2-enhanced rpS6 phosphorylation was further increased by GW4869 or Smpd3 knockdown, and these loss-of-function conditions for Smpd3/nSMase2 induced mild phosphorylation of Akt (Fig. 4A), suggesting that Akt and rpS6 are the specific targets of inhibition. This notion was further supported by an immunoblot assay using ATDC5 cells treated with siSmpd3 or Smpd3-expressing adenovirus. Application of BMP-2, in combination with the ITS supplement, dramatically induced expression of the nSMase2 protein, which was clearly diminished by transfecting with siRNA for Smpd3 (Fig. 4B), even after 20 h of stimulation. Phosphorylation of Akt, as well as of rpS6, was significantly increased by BMP-2 stimulation, although these proteins became more phosphorylated by the loss of Smpd3 (Fig. 4B). Importantly, siSmpd3 increased the weak basal phosphorylation of both Akt and rpS6 in mock control cells, suggesting that neither ITS nor BMP-2 is crucial for the function of nSMase2. Overexpression of Smpd3 weakened the induced phosphorylation of both Akt and rpS6 (Fig. 4C), whereas similar effects of Smpd3 adenovirus were observed in primary chondrocytes (Fig. 4D). Increased phosphorylation of Akt and rpS6 by the loss of Smpd3 was also observed in primary chondrocytes (Fig. 4D) and C3H10T1/2 (Fig. 4E). The phosphorylation level of Smad1/5/8 was not altered by Smpd3 knockdown (Fig. 4E), suggesting that the accelerated chondrogenic differentiation by siSmpd3 (Fig. 3A) was not due to an enhancement of BMP signaling. These results demonstrate an inhibitory function of Smpd3/nSMase2 against activation of Akt and rpS6 and a positive effect of BMP-2 in chondrocytes.
FIGURE 4.
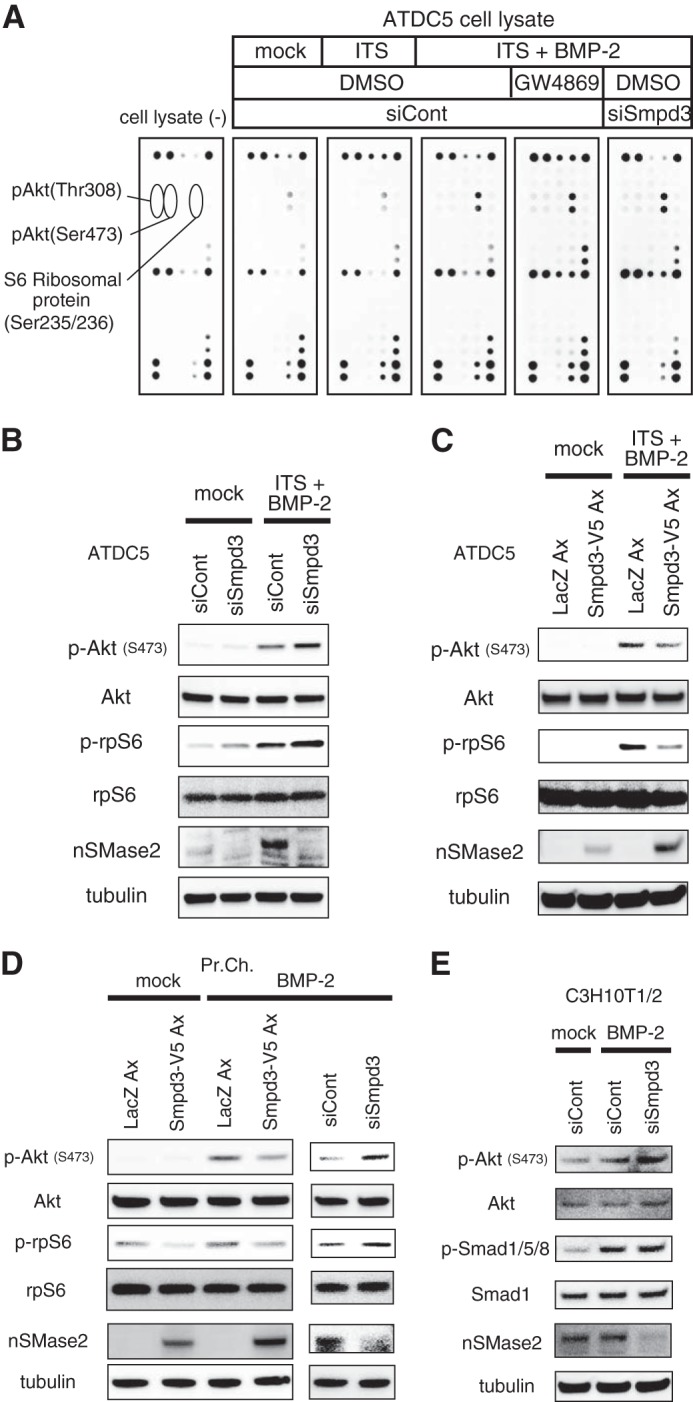
Akt pathway is activated or repressed by loss or gain of Smpd3 function, respectively. A, ATDC5 cells were transfected with control siRNA (siCont) or Smpd3 siRNA (siSmpd3) for 16 h, and then stimulated by a combination of ITS supplement and BMP-2 (300 ng/ml), with or without GW4869 (1 μm), for 8 h. Cells were analyzed by a PathScan® RTK signaling antibody array. B and C, ATDC5 cells were transfected with control siRNA (siCont) or Smpd3 siRNA (siSmpd3) for 16 h (B) or infected with adenovirus (Ax) carrying lacZ or Smpd3 for 2 h (C), and stimulated with the combination of ITS supplement and BMP-2 (300 ng/ml) for 20 h. Cells were subjected to immunoblot analysis for the indicated antibodies. Tubulin served as a loading control. D, mouse primary chondrocytes were infected with adenovirus carrying lacZ or Smpd3 for 2 h or transfected with control siRNA (siCont) or Smpd3 siRNA (siSmpd3) for 16 h and stimulated with BMP-2 (300 ng/ml) for 8 h. Cells were subjected to immunoblot analysis for the indicated antibodies. Tubulin served as a loading control. E, C3H10T1/2 cells were transfected with control siRNA (siCont) or Smpd3 siRNA (siSmpd3) for 8 h and stimulated with BMP-2 (300 ng/ml) for 16 h. Cells were subjected to immunoblot analysis for the indicated antibodies. Tubulin served as a loading control.
Smpd3 Suppresses Maturation of ATDC5 Chondrocytes via the PI3K-Akt Pathway
We next investigated the role of the Akt pathway in nSMase2-mediated inhibition of chondrogenesis in ATDC5 cells by employing specific inhibitor compounds. MK2206, an inhibitor for Akt, was tested for its ability to negate the enhanced chondrogenesis caused by loss of Smpd3. Expression of Smpd3 in siSmpd3-treated cells was not further altered by MK2206 at concentrations between 1 and 3 μm, although 10 μm of the MK compound suppressed it (Fig. 5A). At day 6 of BMP-2 induction, MK2206 successfully suppressed the Smpd3 siRNA-mediated increase of Acan, Col2a1, and Col10a1, in a dose-dependent fashion, at concentrations between 1 and 10 μm (Fig. 5A). Alcian blue staining revealed that the BMP-2-induced production of glycosaminoglycan, which was further stimulated by siSmpd3, was eliminated by the addition of MK2206 at 10 μm (Fig. 5B). We also investigated the participation of mammalian target of rapamycin, a downstream effector of Akt, by using its specific inhibitor, rapamycin. Although rapamycin suppressed the expression of Smpd3, it could block the siSmpd3-mediated up-regulation of Col10a1 at 1 μm (Fig. 5C). Because total protein expression of PI3K, the upstream mediator of Akt, was significantly increased in fro/fro fibroblasts, which resulted in an up-regulated phosphorylation level of PI3K, we evaluated these in ATDC5 chondrocytes by an immunoblot assay. Indeed, phosphorylated, as well as total, PI3K protein was increased upon transfection with Smpd3 siRNA (Fig. 5D). Therefore, a specific inhibitor for PI3K, LY294002, was tested with siSmpd3. LY294002 did not change the expression of Smpd3 at concentrations between 1 and 5 μm, but a 25 μm concentration led to suppression. However, LY294002 did suppress the elevated expression of Acan, Col2a1, and Col10a1 caused by the loss of Smpd3, in a dose-dependent manner at concentrations between 1 and 25 μm (Fig. 5E). The role of the Akt pathway was confirmed in primary chondrocytes by applying LY294002 (25 μm), MK2206 (5 μm), and rapamycin (0.5 μm); only MK2206 suppressed Smpd3 expression (Fig. 5F). Hence, none of these inhibitor compounds increased expression of Smpd3, indicating that the inhibitory action on chondrocyte maturation was independent of Smpd3 expression level. These data suggest that Smpd3/nSMase2 suppresses chondrocyte maturation, at least in part, via the PI3K-Akt pathway.
GW4869 or C2-ceramide Promotes or Eliminates, Respectively, Terminal Hypertrophic Maturation of Chondrocytes in Mouse Bone Organ Culture
To further examine the role of the nSMase-ceramide signaling axis in relatively physiological conditions, we employed an ex vivo organ culture system of mouse embryonic metatarsal bone, a widely used method that permits the study of a complex chondrogenic process in a three-dimensional structure, in the context of native cell-cell and cell-extracellular matrix interactions and cellular signaling (42). The cartilage matrix was stained by Alcian blue, and the extracellular matrix calcified by mature hypertrophic chondrocytes was stained by alizarin red. The clear zone represents layers of uncalcified hypertrophic chondrocytes (Fig. 6A). All zone lengths were measured after image capturing (Fig. 6B). Blocking the function of nSMase by GW4869 solely enlarged both the clear zone and the calcified zone in a mild but statistically significant manner, a result similar to that seen by treatment with BMP-2 alone (Fig. 6, A and B, 2nd and 3rd lanes). Combined treatment with GW4869 and BMP-2 showed an additive effect (Fig. 6, A and B, 4th lane), whereas C2-ceramide eliminated the BMP-2-induced increase of the hypertrophic zone and, especially, the terminally differentiated calcified zone (Fig. 6, A and B, 5th lane). Hence, GW4869 and C2-ceramide exhibited opposite actions against BMP-2-driven acceleration in the hypertrophic conversion and terminal maturation of chondrocytes. In addition, C2-ceramide clearly cancelled the additive promotion induced by GW4869 and BMP-2 (Fig. 6, A and B, 6th lane). These results indirectly demonstrate the physiologically suppressive role of the nSMase2-ceramide pathway on chondrocyte maturation in cartilage/bone rudiments. Moreover, these data suggest a new strategy to control the rate of hypertrophic maturation in cartilage and bone regenerative medicine.
FIGURE 6.
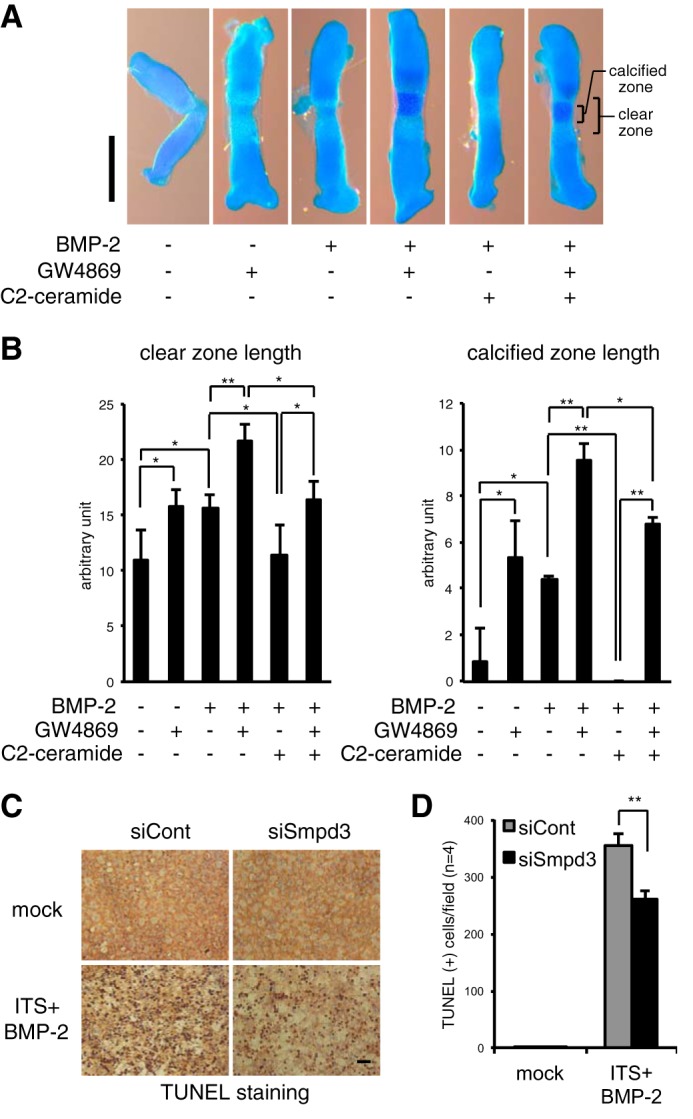
Blocking of nSMase2 function by GW4869 promotes, whereas mimicking the function by C2-ceramide suppresses, hypertrophic maturation of chondrocytes in ex vivo mouse cartilage rudiment culture and loss of Smpd3 decreased apoptosis of ATDC5 chondrocytes. A and B, metatarsal bones from E16.5 mouse embryo were cultured with BMP-2 (300 ng/ml) in combination with GW4869 (1 μm) and/or C2-ceramide (10 μm) for 3 days. The cartilage matrix was stained with Alcian blue, and the chondrocyte matrix calcified by mature hypertrophic chondrocytes was stained by alizarin red (A). The clear zone represents hypertrophic chondrocytes. Scale bar, 500 μm. The length of the hypertrophic clear zone and the calcified zone were measured (n = 4) (B). C and D, ATDC5 cells were transfected with control siRNA (siCont) or Smpd3 siRNA (siSmpd3) for 16 h and further stimulated by ITS supplement and BMP-2 (300 ng/ml) for 6 days. Apoptotic cells were visualized by TUNEL immunoperoxidase staining (C). Scale bar, 300 μm. The number of apoptotic cells was counted (n = 4) (D). *, p < 0.05; **, p < 0.01.
Apoptosis of terminally matured hypertrophic chondrocytes was reduced in the bone of fro/fro mice, a phenotype that accounted for the delayed onset of bone formation (36), suggesting an accelerating role for Smpd3/nSMase2 in the apoptosis of chondrocytes. To investigate whether this is a cell-autonomous event, we knocked down Smpd3 in ATDC5 chondrocytes and performed a TUNEL assay to evaluate the effect on apoptosis (Fig. 6C). TUNEL-positive cells were counted after image capturing (Fig. 6D). Matured ATDC5 chondrocytes, stimulated by BMP-2 with ITS supplement for 6 days, showed hypertrophic morphology, and a substantial number of cells underwent apoptosis (Fig. 6, C and D). Indeed, transfection of Smpd3 siRNA into maturing ATDC5 cells resulted in a statistically significant reduction in apoptosis (Fig. 6, C and D), suggesting that nSMase2 cell-autonomously accelerates apoptosis of hypertrophic chondrocytes.
Smpd3/nSMase2 Suppresses Expression of Has2 during Chondrogenesis via the PI3K-Akt Axis
Chondrocyte maturation is supported by hyaluronan, and embryonic limb mesoderm-specific ablation of hyaluronan synthase 2 (Has2) in mice resulted in reduced formation of zones for prehypertrophic and hypertrophic chondrocytes (53), suggesting a major role of Has2 in the three Has isoforms involved in the production of hyaluronan in cartilage. Recent studies have reported a significant level of Has2 expression and hyaluronan synthesis in fro/fro fibroblasts and that nSMase2 suppressed production of Has2 via inactivation of Akt (50). Taken together, if Smpd3/nSMase2 also regulates expression of Has2 in chondrocytes, Has2 might be another target for the inhibitory action of Smpd3/nSMase2 on chondrocyte hypertrophic maturation. We confirmed the crucial role of Has2 in chondrocyte differentiation and maturation; siRNA-mediated knockdown of Has2 resulted in a decline in expression of Col2a1 and Col10a1, both in ATDC5 cells (Fig. 7A) and primary chondrocytes (Fig. 7B). In ATDC5 chondrocytes, upon BMP-2 stimulation the expression of Has2 was down-regulated by half at day 6, whereas silencing of Smpd3 recovered the decline (Fig. 7C). Although expression of Has1 and Has3 was also suppressed by BMP-2 induction, Smpd3 siRNA did not rescue the decrease (Fig. 7C), suggesting that only Has2, among the three Has isoforms, is a specific target of Smpd3 signaling in chondrocytes. This finding was confirmed using immunofluorescence for protein expression levels in ATDC5 cells (Fig. 7D) and primary chondrocytes (Fig. 7E), which indicated that although nSMase2 accumulated due to BMP-2 induction, the signals of Has2 protein were diminished. The merged images show the mutually exclusive expression of nSMase2 and Has2. Importantly, Smpd3 knockdown rescued the weakened expression of Has2 protein (Fig. 7, D and E). The expression level of Has2 protein was reflected to the production of hyaluronan in primary chondrocytes (Fig. 7E). In vivo, both nSMase2 and Has2 were strongly expressed and co-localized in bone (Fig. 7F). In cartilage, however, Has2 was widely expressed in proliferating and resting chondrocytes with moderate strength, although it was diminished in the hypertrophic zone, where the expression pattern contrasted with that of nSMase2 being dominant in hypertrophic chondrocytes (Fig. 7F). Hyaluronan not only localized to the extracellular matrix of Has2-expressing chondrocytes in immature cartilage but also existed in the matrix of hypertrophic chondrocytes (Fig. 7F), suggesting that the low turnover rate may have caused its retention in the cartilage matrix, even after a decrease in Has2. Finally, we checked if the PI3K or Akt pathway was involved in the suppressive action of Smpd3/nSMase2 on Has2. The accelerated expression of Has2 by silencing of Smpd3 in the presence of BMP-2 treatment was negated by the addition of LY294002 or MK2206, suggesting that the Has2 gene is under the control of the PI3K or Akt pathway, respectively (Fig. 7G). These data suggest that Has2 plays a role in the Smpd3/nSMase2-mediated inhibition of chondrocyte maturation via PI3K-Akt signaling.
FIGURE 7.

Expression of Has2 is suppressed by nSMase2 via the PI3K or Akt pathway in ATDC5 cells, whereas localization of nSMase2 and Has2 is mutually exclusive in the growth plate cartilage of mouse embryo. A and B, ATDC5 cells (A) or mouse primary chondrocytes (B) were transfected with control siRNA (siCont) or Has2 siRNA (siHas2) for 16 h and then treated with BMP-2 (300 ng/ml) for 6 days. Quantitative RT-PCR analysis was performed for Col2a1 and Col10a1. C, ATDC5 chondrocytes were transfected with control siRNA (siCont) or Smpd3 siRNA (siSmpd3) for 16 h and then treated with BMP-2 (300 ng/ml) for 6 days. Quantitative RT-PCR analysis was performed for Has1, Has2, and Has3. D, immunofluorescence for nSMase2 or Has2 was performed in ATDC5 chondrocytes. IgG was used as negative control. Scale bar, 50 μm. E, immunofluorescence for nSMase2 or Has2 was performed on mouse primary chondrocytes. Biotin-conjugated hyaluronan-binding protein (HABP) and Alexa Fluor 488-conjugated streptavidin were applied to detect hyaluronan. IgG was the negative control. Scale bar, 50 μm. F, expression of nSMase2 or Has2 in mouse E17.5 humerus cartilage was evaluated by immunofluorescence. Biotin-conjugated HA-binding protein and Alexa Fluor 488-conjugated streptavidin were used to detect hyaluronan. IgG was the negative control. r, resting chondrocytes; p, proliferating chondrocytes; ph, prehypertrophic chondrocytes; h, hypertrophic chondrocytes. Scale bar, 250 μm. G, ATDC5 cells were transfected with control siRNA (siCont) or Smpd3 siRNA (siSmpd3) for 16 h and further stimulated by BMP-2 (300 ng/ml) with or without LY294002 (LY, 1 μm) or MK2206 (MK, 1 μm) for 6 days. Expression of Has2 was evaluated by quantitative RT-PCR analysis. *, p < 0.05; **, p < 0.01; n.s., not significant.
DISCUSSION
Previous reports had suggested that Smpd3/nSMase2 may have a crucial role in in vivo chondrogenesis (36–38). We observed a moderate level of Smpd3 expression in the brains of adult mice (Fig. 1I), consistent with the finding that Smpd3−/− mice showed a defect in the hypothalamus-pituitary growth axis, which likely accounted for the dwarfism (37). However, the enlarged hypertrophic zone and retarded apoptosis in the chondrocytes of mutant mice cannot be explained by the reduced production of growth hormone and IGF (37). In this study, we present evidence for a cell-autonomous role of the nSMase-ceramide axis in regulating Akt signaling and the subsequent chondrogenic marker expression and differentiation. The induction of Smpd3 by BMP-2 was a common feature among the tested chondrogenic cells, including primary articular chondrocytes, but Smpd3 did not seem to be a direct target of the BMP-Smad pathway. Its coding protein, nSMase2, was dominant in mature hypertrophic chondrocytes in vivo (Fig. 1J), with an expression pattern resembling that of Runx2, whereas the loss of Runx2 suppressed expression of Smpd3 (Fig. 2, C, D and F). Taken together with the evidence that Runx2 directly interacts with and activates the promoter of Smpd3 in C2C12 myoblasts (39), Runx2 seems to be mainly responsible for the spatiotemporal expression of Smpd3 in chondrocytes, in concert with BMP signaling. In addition, it should be noted that the maximum expression of Smpd3/nSMase2 in vivo was observed in bone tissue, where Runx2 is highly expressed. So far, the molecular mechanism by which BMP-2 increases Runx2-dependent expression of Smpd3 remains unclear. It is likely that a mechanism similar to that of Col10a1 gene induction, in which BMP-activated Smads interact with Runx2 to enhance the Col10a1 promoter-activating ability of Runx2 to drive chondrocyte maturation (13), may take place on the Smpd3 promoter.
PI3K and its downstream Akt are activated by a large number of receptors, but most notably by tyrosine kinases, such as the IGF-1 receptor. The majority of published studies suggest that PI3K or Akt signaling is required for normal hypertrophic cell maturation and endochondral bone growth during cartilage development (51, 54, 55), although the precise molecular mechanisms for this remain unclear. We demonstrated that the loss or gain of Smpd3/nSMase2 function in chondrocytes increased or decreased the phosphorylation of both PI3K and Akt, respectively. In an RTK signaling antibody array, only phosphorylation of Akt and rpS6 was strengthened by the loss of Smpd3 (Fig. 4A), demonstrating their specificity as downstream targets of nSMase2. Importantly, the increase in Akt phosphorylation was induced by the addition of BMP-2 and not by ITS alone (Fig. 4, A, D and E). A similar enhancement in the phosphorylation of Akt was observed within 1 h of BMP-2 application in gastric cancer cells, although the precise mechanism by which the BMP-2 signaling pathway induced Akt activity was unclear (56). We expect the Akt pathway to take part in BMP-2-induced chondrogenesis because this pathway promotes chondrocyte differentiation.
The GW4869-mediated blockade of nSMase2 function accelerated differentiation of ATDC5 chondrocytes, as well as hypertrophic conversion and calcification of chondrocytes, in bone ex vivo culture; both phenotypes were cancelled by application of C2-ceramide (Figs. 3C and 6, A and B). nSMase2 hydrolyzes the phosphodiester bond of the membrane sphingolipid sphingomyelin to yield ceramide and phosphocholine (57). Ceramides have been shown to reduce the level of Akt phosphorylation by activating protein phosphatase 2A (PP2A) (58). The phosphorylation level of PP2A in fro/fro fibroblasts is reduced (50). Taken together, in the maturing phase of chondrogenesis, BMP-2-induced nSMase2 is thought to release ceramide, which in turn activates PP2A to inactivate Akt and the subsequent chondrogenic molecular cascades (Fig. 8). Thus, the Smpd3/nSMase2-ceramide axis negatively regulates BMP-2-induced activation of the Akt pathway through a negative feedback mechanism.
FIGURE 8.
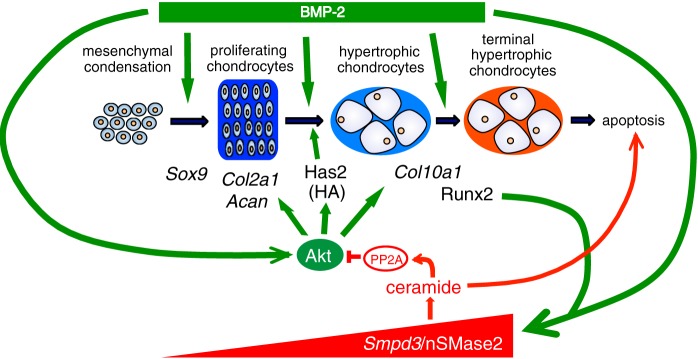
Proposed model for the negative or positive regulation of chondrocyte maturation or apoptosis by Smpd3/nSMase2, respectively. BMP-2 promotes chondrogenesis by multiple pathways, including activation of Akt signaling and the subsequent induction of Has2. During chondrocyte maturation, up-regulated Runx2 induces Smpd3 in concert with BMP signaling. nSMase2 releases ceramide, which activates PP2A to dephosphorylate Akt. This blockade of the Akt pathway interferes not only with chondrocyte maturation but also with Has2-mediated production of HA.
nSMase2 is one of the major intracellular regulators of sphingolipids, and many reports have implicated nSMase2 activation in ceramide-mediated apoptosis (49, 59–61). Sphingomyelinase-released ceramide is essential for the clustering of the death receptors CD95 or DR5 in membrane rafts to trigger apoptosis (62, 63). Indeed, silencing of Smpd3 in mature ATDC5 chondrocytes reduced the number of apoptotic cells (Fig. 6, C and D), suggesting that delayed apoptosis in fro/fro cartilage was a cell-autonomous effect of the loss of function of nSMase2 (36). Because apoptosis of terminally matured hypertrophic chondrocytes is a crucial step in the transition of chondrogenic stage to the bone formation stage in the endochondral ossification system, Smpd3/nSMase2 probably plays a key role in regulating the timing of osteogenesis onset.
HA is a linear high molecular weight glycosaminoglycan and is composed of disaccharide repeats of glucuronic acid and N-acetylglucosamine. It is produced in the plasma membrane by three hyaluronan synthases (Has1–3); Has2 is the crucial hyaluronan synthase involved in the endochondral ossification process (53). The Akt-rpS6 pathway is important in the expression of Has2 in MCF-7 breast cancer cells (64), although nSMase2 suppresses production of Has2 via inactivation of Akt in mouse dermal fibroblasts (50). In chondrocytes, Has2 expression was decreased by BMP-2 stimulation and was then recovered by silencing of Smpd3, demonstrating the importance of BMP-induced Smpd3/nSMase2 in the suppression of Has2 (Fig. 7, C–E). Because an inhibitor compound for PI3K or Akt cancelled this effect (Fig. 7G), Has2 expression is also considered to be under the control of PI3K-Akt signaling. In vivo, expression of Has2 was diminished in hypertrophic chondrocytes, whereas nSMase2 was strongly expressed in the same cells (Fig. 7F). Taken together, these results indicate that Has2 is another mediator of Smpd3/nSMase2-induced inhibition of the hypertrophic maturation of chondrocytes, downstream of Akt signaling (Fig. 8).
Studies of articular cartilage suggest that ceramide plays a role in cartilage degeneration and the disruption of cartilage matrix homeostasis to decrease the levels of type II collagen (65, 66). Farber disease, in which a lack of ceramidase causes excess ceramide accumulation within the cartilage and bone, is associated with arthritis-like joint degeneration (67). Moreover, tumor necrosis factor α (TNFα), a proinflammatory cytokine that is widely implicated in the pathogenesis of arthritic diseases (68), can increase the level of ceramide through hydrolysis of the cell membrane lipid sphingomyelin by endosomal acidic and membrane-bound neutral sphingomyelinases (69). In chondrocytes, we observed a decrease of Col2a1 expression by induction of C2-ceramide or Smpd3-expressing adenovirus. Conversely, Smpd3 knock-out mice showed an enlarged hypertrophic zone in the growth plate of the joints and, in adulthood, a severe OA-phenotype with osteophytes in the knee joint (38). Similarly, in chondrocytes, we observed increase of hypertrophic phenotype (Col10a1) by induction of Smpd3 siRNA. Accordingly, an excess level of nSMase2 leads to the degradation of cartilage matrix proteins, whereas loss of nSMase2 introduces a hypertrophic change in chondrocytes, and both circumstances may result in the progression of OA. Therefore, the expression of Smpd3/nSMase2 must be fine-tuned to maintain cartilage homeostasis that is, at least in part, controlled by Runx2 and BMP signaling.
In the case of cartilage regenerative medicine, pharmacological manipulation of steps of the nSMase2-ceramide-PP2A-Akt pathway may improve the efficiency and quality of generated tissues. As an indication, it is noteworthy that we could manipulate hypertrophic conversion and calcification in ex vivo cartilage rudiment culture using combinations of BMP-2, GW4869, and C2-ceramide (Fig. 6, A and B).
In summary, our study has provided a cell-autonomous pivotal role for Smpd3/nSMase2 in determining the rate of chondrocyte maturation in chondrocytes. As illustrated in Fig. 8, BMP-2 accelerates general chondrogenesis through multiple approaches, including activation of the Akt pathway, which involves induction of Has2 and a subsequent production of HA. Meanwhile, increased Runx2 in maturating chondrocytes induces Smpd3 in concert with BMP-2. nSMase2, coded by Smpd3, releases ceramide from the cell membrane to activate PP2A, which in turn dephosphorylates Akt. This inactivation of the Akt pathway suppresses not only chondrocyte differentiation and subsequent maturation but also production of HA via Has2. We propose that Smpd3/nSMase2 is a molecular target in cartilage and bone medicine that constitutes a negative feedback loop in BMP-induced chondrogenesis.
Supplementary Material
Acknowledgments
Human chondrocyte C28/I2 was kindly provided by Dr. Mary Goldring. We gratefully acknowledge the technical assistance of Hui Gao.
This work was supported by Japan Society for the Promotion of Science KAKENHI Grants 25462376 (to H. K.), 23592221 (to S. M.), and 23592222 (to Y. I.), a grant from the Hip Joint Foundation of Japan (to S. M.), and a grant from the Cell Science Research Foundation (to S. M.).

This article contains supplemental Table S1.
- BMP
- bone morphogenic protein
- nSMase2
- neutral sphingomyelinase 2
- MSC
- mesenchymal stem/stroma cell
- CHX
- cycloheximide
- RTK
- receptor tyrosine kinase
- OA
- osteoarthritis
- ITS
- insulin/transferrin/selenium
- PP2A
- protein phosphatase 2A
- rpS6
- ribosomal protein S6.
REFERENCES
- 1. Bi W., Deng J. M., Zhang Z., Behringer R. R., de Crombrugghe B. (1999) Sox9 is required for cartilage formation. Nat. Genet. 22, 85–89 [DOI] [PubMed] [Google Scholar]
- 2. Akiyama H., Chaboissier M. C., Martin J. F., Schedl A., de Crombrugghe B. (2002) The transcription factor Sox9 has essential roles in successive steps of the chondrocyte differentiation pathway and is required for expression of Sox5 and Sox6. Genes Dev. 16, 2813–2828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Provot S., Schipani E. (2005) Molecular mechanisms of endochondral bone development. Biochem. Biophys. Res. Commun. 328, 658–665 [DOI] [PubMed] [Google Scholar]
- 4. Enomoto H., Enomoto-Iwamoto M., Iwamoto M., Nomura S., Himeno M., Kitamura Y., Kishimoto T., Komori T. (2000) Cbfa1 is a positive regulatory factor in chondrocyte maturation. J. Biol. Chem. 275, 8695–8702 [DOI] [PubMed] [Google Scholar]
- 5. Zheng Q., Zhou G., Morello R., Chen Y., Garcia-Rojas X., Lee B. (2003) Type X collagen gene regulation by Runx2 contributes directly to its hypertrophic chondrocyte-specific expression in vivo. J. Cell Biol. 162, 833–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kronenberg H. M. (2003) Developmental regulation of the growth plate. Nature 423, 332–336 [DOI] [PubMed] [Google Scholar]
- 7. Miyazono K., Maeda S., Imamura T. (2005) BMP receptor signaling: transcriptional targets, regulation of signals, and signaling cross-talk. Cytokine Growth Factor Rev. 16, 251–263 [DOI] [PubMed] [Google Scholar]
- 8. Guo X., Wang X. F. (2009) Signaling cross-talk between TGF-β/BMP and other pathways. Cell Res. 19, 71–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pogue R., Lyons K. (2006) BMP signaling in the cartilage growth plate. Curr. Top. Dev. Biol. 76, 1–48 [DOI] [PubMed] [Google Scholar]
- 10. Hatakeyama Y., Tuan R. S., Shum L. (2004) Distinct functions of BMP4 and GDF5 in the regulation of chondrogenesis. J. Cell. Biochem. 91, 1204–1217 [DOI] [PubMed] [Google Scholar]
- 11. Haas A. R., Tuan R. S. (1999) Chondrogenic differentiation of murine C3H10T1/2 multipotential mesenchymal cells: II. Stimulation by bone morphogenetic protein-2 requires modulation of N-cadherin expression and function. Differentiation 64, 77–89 [DOI] [PubMed] [Google Scholar]
- 12. Fujii M., Takeda K., Imamura T., Aoki H., Sampath T. K., Enomoto S., Kawabata M., Kato M., Ichijo H., Miyazono K. (1999) Roles of bone morphogenetic protein type I receptors and Smad proteins in osteoblast and chondroblast differentiation. Mol. Biol. Cell 10, 3801–3813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Leboy P., Grasso-Knight G., D'Angelo M., Volk S. W., Lian J. V., Drissi H., Stein G. S., Adams S. L. (2001) Smad-Runx interactions during chondrocyte maturation. J. Bone Joint Surg. Am. 83, S15–S22 [PubMed] [Google Scholar]
- 14. Valcourt U., Gouttenoire J., Moustakas A., Herbage D., Mallein-Gerin F. (2002) Functions of transforming growth factor-β family type I receptors and Smad proteins in the hypertrophic maturation and osteoblastic differentiation of chondrocytes. J. Biol. Chem. 277, 33545–33558 [DOI] [PubMed] [Google Scholar]
- 15. Tsumaki N., Nakase T., Miyaji T., Kakiuchi M., Kimura T., Ochi T., Yoshikawa H. (2002) Bone morphogenetic protein signals are required for cartilage formation and differently regulate joint development during skeletogenesis. J. Bone Miner. Res. 17, 898–906 [DOI] [PubMed] [Google Scholar]
- 16. Yoon B. S., Ovchinnikov D. A., Yoshii I., Mishina Y., Behringer R. R., Lyons K. M. (2005) Bmpr1a and Bmpr1b have overlapping functions and are essential for chondrogenesis in vivo. Proc. Natl. Acad. Sci. U.S.A. 102, 5062–5067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Retting K. N., Song B., Yoon B. S., Lyons K. M. (2009) BMP canonical Smad signaling through Smad1 and Smad5 is required for endochondral bone formation. Development 136, 1093–1104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Volk S. W., Luvalle P., Leask T., Leboy P. S. (1998) A BMP-responsive transcriptional region in the chicken type X collagen gene. J. Bone Miner. Res. 13, 1521–1529 [DOI] [PubMed] [Google Scholar]
- 19. Kempf H., Ionescu A., Udager A. M., Lassar A. B. (2007) Prochondrogenic signals induce a competence for Runx2 to activate hypertrophic chondrocyte gene expression. Dev. Dyn. 236, 1954–1962 [DOI] [PubMed] [Google Scholar]
- 20. Kobayashi T., Lyons K. M., McMahon A. P., Kronenberg H. M. (2005) BMP signaling stimulates cellular differentiation at multiple steps during cartilage development. Proc. Natl. Acad. Sci. U.S.A. 102, 18023–18027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Scotti C., Tonnarelli B., Papadimitropoulos A., Scherberich A., Schaeren S., Schauerte A., Lopez-Rios J., Zeller R., Barbero A., Martin I. (2010) Recapitulation of endochondral bone formation using human adult mesenchymal stem cells as a paradigm for developmental engineering. Proc. Natl. Acad. Sci. U.S.A. 107, 7251–7256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pullig O., Weseloh G., Ronneberger D., Käkönen S., Swoboda B. (2000) Chondrocyte differentiation in human osteoarthritis: expression of osteocalcin in normal and osteoarthritic cartilage and bone. Calcif. Tissue Int. 67, 230–240 [DOI] [PubMed] [Google Scholar]
- 23. Sandell L. J., Aigner T. (2001) Articular cartilage and changes in arthritis. An introduction: cell biology of osteoarthritis. Arthritis Res. 3, 107–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dreier R. (2010) Hypertrophic differentiation of chondrocytes in osteoarthritis: The developmental aspect of degenerative joint disorders. Arthritis Res. Ther. 12, 216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nelea V., Luo L., Demers C. N., Antoniou J., Petit A., Lerouge S., R Wertheimer M., Mwale F. (2005) Selective inhibition of type X collagen expression in human mesenchymal stem cell differentiation on polymer substrates surface-modified by glow discharge plasma. J. Biomed. Mater. Res. A 75, 216–223 [DOI] [PubMed] [Google Scholar]
- 26. Sekiya I., Vuoristo J. T., Larson B. L., Prockop D. J. (2002) In vitro cartilage formation by human adult stem cells from bone marrow stroma defines the sequence of cellular and molecular events during chondrogenesis. Proc. Natl. Acad. Sci. U.S.A. 99, 4397–4402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Imamura T., Takase M., Nishihara A., Oeda E., Hanai J., Kawabata M., Miyazono K. (1997) Smad6 inhibits signalling by the TGF-β superfamily. Nature 389, 622–626 [DOI] [PubMed] [Google Scholar]
- 28. Zhu H., Kavsak P., Abdollah S., Wrana J. L., Thomsen G. H. (1999) A SMAD ubiquitin ligase targets the BMP pathway and affects embryonic pattern formation. Nature 400, 687–693 [DOI] [PubMed] [Google Scholar]
- 29. Murakami G., Watabe T., Takaoka K., Miyazono K., Imamura T. (2003) Cooperative inhibition of bone morphogenetic protein signaling by Smurf1 and inhibitory Smads. Mol. Biol. Cell 14, 2809–2817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kawamura I., Maeda S., Imamura K., Setoguchi T., Yokouchi M., Ishidou Y., Komiya S. (2012) SnoN suppresses maturation of chondrocytes by mediating signal cross-talk between transforming growth factor-β and bone morphogenetic protein pathways. J. Biol. Chem. 287, 29101–29113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Obeid L. M., Linardic C. M., Karolak L. A., Hannun Y. A. (1993) Programmed cell death induced by ceramide. Science 259, 1769–1771 [DOI] [PubMed] [Google Scholar]
- 32. Sjöholm A. (1995) Ceramide inhibits pancreatic β-cell insulin production and mitogenesis and mimics the actions of interleukin-1 β. FEBS Lett. 367, 283–286 [DOI] [PubMed] [Google Scholar]
- 33. Mebarek S., Komati H., Naro F., Zeiller C., Alvisi M., Lagarde M., Prigent A. F., Némoz G. (2007) Inhibition of de novo ceramide synthesis upregulates phospholipase D and enhances myogenic differentiation. J. Cell Sci. 120, 407–416 [DOI] [PubMed] [Google Scholar]
- 34. Sharma K., Shi Y. (1999) The yins and yangs of ceramide. Cell Res. 9, 1–10 [DOI] [PubMed] [Google Scholar]
- 35. Aubin I., Adams C. P., Opsahl S., Septier D., Bishop C. E., Auge N., Salvayre R., Negre-Salvayre A., Goldberg M., Guénet J. L., Poirier C. (2005) A deletion in the gene encoding sphingomyelin phosphodiesterase 3 (Smpd3) results in osteogenesis and dentinogenesis imperfecta in the mouse. Nat. Genet. 37, 803–805 [DOI] [PubMed] [Google Scholar]
- 36. Khavandgar Z., Poirier C., Clarke C. J., Li J., Wang N., McKee M. D., Hannun Y. A., Murshed M. (2011) A cell-autonomous requirement for neutral sphingomyelinase 2 in bone mineralization. J. Cell Biol. 194, 277–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Stoffel W., Jenke B., Blöck B., Zumbansen M., Koebke J. (2005) Neutral sphingomyelinase 2 (smpd3) in the control of postnatal growth and development. Proc. Natl. Acad. Sci. U.S.A. 102, 4554–4559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Stoffel W., Jenke B., Holz B., Binczek E., Günter R. H., Knifka J., Koebke J., Niehoff A. (2007) Neutral sphingomyelinase (SMPD3) deficiency causes a novel form of chondrodysplasia and dwarfism that is rescued by Col2A1-driven smpd3 transgene expression. Am. J. Pathol. 171, 153–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chae Y. M., Heo S. H., Kim J. Y., Lee J. M., Ryoo H. M., Cho J. Y. (2009) Upregulation of smpd3 via BMP2 stimulation and Runx2. BMB Rep. 42, 86–90 [DOI] [PubMed] [Google Scholar]
- 40. Atsumi T., Miwa Y., Kimata K., Ikawa Y. (1990) A chondrogenic cell line derived from a differentiating culture of AT805 teratocarcinoma cells. Cell Differ. Dev. 30, 109–116 [DOI] [PubMed] [Google Scholar]
- 41. Goldring M. B., Birkhead J. R., Suen L. F., Yamin R., Mizuno S., Glowacki J., Arbiser J. L., Apperley J. F. (1994) Interleukin-1 β-modulated gene expression in immortalized human chondrocytes. J. Clin. Invest. 94, 2307–2316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Alvarez J., Sohn P., Zeng X., Doetschman T., Robbins D. J., Serra R. (2002) TGFβ2 mediates the effects of hedgehog on hypertrophic differentiation and PTHrP expression. Development 129, 1913–1924 [DOI] [PubMed] [Google Scholar]
- 43. Shukunami C., Ohta Y., Sakuda M., Hiraki Y. (1998) Sequential progression of the differentiation program by bone morphogenetic protein-2 in chondrogenic cell line ATDC5. Exp. Cell Res. 241, 1–11 [DOI] [PubMed] [Google Scholar]
- 44. Yao Y., Wang Y. (2013) ATDC5: an excellent in vitro model cell line for skeletal development. J. Cell. Biochem. 114, 1223–1229 [DOI] [PubMed] [Google Scholar]
- 45. Origuchi N., Ishidou Y., Nagamine T., Onishi T., Matsunaga S., Yoshida H., Sakou T. (1998) The spatial and temporal immunolocalization of TGF-β 1 and bone morphogenetic protein-2/-4 in phallic bone formation in inbred Sprague Dawley male rats. In Vivo 12, 473–480 [PubMed] [Google Scholar]
- 46. Heinonen J., Taipaleenmäki H., Roering P., Takatalo M., Harkness L., Sandholm J., Uusitalo-Järvinen H., Kassem M., Kiviranta I., Laitala-Leinonen T., Säämänen A. M. (2011) Snorc is a novel cartilage specific small membrane proteoglycan expressed in differentiating and articular chondrocytes. Osteoarthritis Cartilage 19, 1026–1035 [DOI] [PubMed] [Google Scholar]
- 47. Korchynskyi O., ten Dijke P. (2002) Identification and functional characterization of distinct critically important bone morphogenetic protein-specific response elements in the Id1 promoter. J. Biol. Chem. 277, 4883–4891 [DOI] [PubMed] [Google Scholar]
- 48. Lefebvre V., Li P., de Crombrugghe B. (1998) A new long form of Sox5 (L-Sox5), Sox6 and Sox9 are coexpressed in chondrogenesis and cooperatively activate the type II collagen gene. EMBO J. 17, 5718–5733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Marchesini N., Luberto C., Hannun Y. A. (2003) Biochemical properties of mammalian neutral sphingomyelinase 2 and its role in sphingolipid metabolism. J. Biol. Chem. 278, 13775–13783 [DOI] [PubMed] [Google Scholar]
- 50. Qin J., Berdyshev E., Poirer C., Schwartz N. B., Dawson G. (2012) Neutral sphingomyelinase 2 deficiency increases hyaluronan synthesis by up-regulation of hyaluronan synthase 2 through decreased ceramide production and activation of Akt. J. Biol. Chem. 287, 13620–13632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rokutanda S., Fujita T., Kanatani N., Yoshida C. A., Komori H., Liu W., Mizuno A., Komori T. (2009) Akt regulates skeletal development through GSK3, mTOR, and FoxOs. Dev. Biol. 328, 78–93 [DOI] [PubMed] [Google Scholar]
- 52. Shukunami C., Shigeno C., Atsumi T., Ishizeki K., Suzuki F., Hiraki Y. (1996) Chondrogenic differentiation of clonal mouse embryonic cell line ATDC5 in vitro: differentiation-dependent gene expression of parathyroid hormone (PTH)/PTH-related peptide receptor. J. Cell Biol. 133, 457–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Matsumoto K., Li Y., Jakuba C., Sugiyama Y., Sayo T., Okuno M., Dealy C. N., Toole B. P., Takeda J., Yamaguchi Y., Kosher R. A. (2009) Conditional inactivation of Has2 reveals a crucial role for hyaluronan in skeletal growth, patterning, chondrocyte maturation and joint formation in the developing limb. Development 136, 2825–2835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ulici V., Hoenselaar K. D., Gillespie J. R., Beier F. (2008) The PI3K pathway regulates endochondral bone growth through control of hypertrophic chondrocyte differentiation. BMC Dev. Biol. 8, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Fujita T., Azuma Y., Fukuyama R., Hattori Y., Yoshida C., Koida M., Ogita K., Komori T. (2004) Runx2 induces osteoblast and chondrocyte differentiation and enhances their migration by coupling with PI3K-Akt signaling. J. Cell Biol. 166, 85–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kang M. H., Kim J. S., Seo J. E., Oh S. C., Yoo Y. A. (2010) BMP2 accelerates the motility and invasiveness of gastric cancer cells via activation of the phosphatidylinositol 3-kinase (PI3K)/Akt pathway. Exp. Cell Res. 316, 24–37 [DOI] [PubMed] [Google Scholar]
- 57. Bartke N., Hannun Y. A. (2009) Bioactive sphingolipids: metabolism and function. J. Lipid Res. 50, S91–S96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Mora A., Sabio G., Risco A. M., Cuenda A., Alonso J. C., Soler G., Centeno F. (2002) Lithium blocks the PKB and GSK3 dephosphorylation induced by ceramide through protein phosphatase-2A. Cell. Signal. 14, 557–562 [DOI] [PubMed] [Google Scholar]
- 59. Kolesnick R., Hannun Y. A. (1999) Ceramide and apoptosis. Trends Biochem. Sci. 24, 224–225 [DOI] [PubMed] [Google Scholar]
- 60. Wiesner D. A., Kilkus J. P., Gottschalk A. R., Quintáns J., Dawson G. (1997) Anti-immunoglobulin-induced apoptosis in WEHI 231 cells involves the slow formation of ceramide from sphingomyelin and is blocked by bcl-XL. J. Biol. Chem. 272, 9868–9876 [DOI] [PubMed] [Google Scholar]
- 61. Lee J. T., Xu J., Lee J. M., Ku G., Han X., Yang D. I., Chen S., Hsu C. Y. (2004) Amyloid-β peptide induces oligodendrocyte death by activating the neutral sphingomyelinase-ceramide pathway. J. Cell Biol. 164, 123–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Grassme H., Jekle A., Riehle A., Schwarz H., Berger J., Sandhoff K., Kolesnick R., Gulbins E. (2001) CD95 signaling via ceramide-rich membrane rafts. J. Biol. Chem. 276, 20589–20596 [DOI] [PubMed] [Google Scholar]
- 63. Dumitru C. A., Gulbins E. (2006) TRAIL activates acid sphingomyelinase via a redox mechanism and releases ceramide to trigger apoptosis. Oncogene 25, 5612–5625 [DOI] [PubMed] [Google Scholar]
- 64. Kultti A., Kärnä R., Rilla K., Nurminen P., Koli E., Makkonen K. M., Si J., Tammi M. I., Tammi R. H. (2010) Methyl-β-cyclodextrin suppresses hyaluronan synthesis by down-regulation of hyaluronan synthase 2 through inhibition of Akt. J. Biol. Chem. 285, 22901–22910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sabatini M., Rolland G., Léonce S., Thomas M., Lesur C., Pérez V., de Nanteuil G., Bonnet J. (2000) Effects of ceramide on apoptosis, proteoglycan degradation, and matrix metalloproteinase expression in rabbit articular cartilage. Biochem. Biophys. Res. Commun. 267, 438–444 [DOI] [PubMed] [Google Scholar]
- 66. Gilbert S. J., Blain E. J., Jones P., Duance V. C., Mason D. J. (2006) Exogenous sphingomyelinase increases collagen and sulphated glycosaminoglycan production by primary articular chondrocytes: an in vitro study. Arthritis Res. Ther. 8, R89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ehlert K., Frosch M., Fehse N., Zander A., Roth J., Vormoor J. (2007) Farber disease: clinical presentation, pathogenesis and a new approach to treatment. Pediatr. Rheumatol. Online J. 5, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Goldring M. B. (1999) The role of cytokines as inflammatory mediators in osteoarthritis: lessons from animal models. Connect. Tissue Res. 40, 1–11 [DOI] [PubMed] [Google Scholar]
- 69. Wiegmann K., Schütze S., Machleidt T., Witte D., Krönke M. (1994) Functional dichotomy of neutral and acidic sphingomyelinases in tumor necrosis factor signaling. Cell 78, 1005–1015 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.