

Background: Propionyl-CoA is a common metabolic intermediate that requires degradation to avoid intoxication of cellular metabolism.
Results: A key enzyme involved in a modified β-oxidation pathway in Candida albicans has been identified.
Conclusion: Although fungi generally use the methyl citrate cycle to degrade propionyl-CoA, CUG clade yeasts form an exception.
Significance: The modified β-oxidation pathway could provide a target for new antifungal compounds.
Keywords: Amino Acid, Candida albicans, Enzyme Kinetics, Fatty Acid Metabolism, Fatty Acid Oxidation, Pathogenesis, CUG clade, β-Hydroxypropionate, Odd-chain Fatty Acids
Abstract
Propionyl-CoA arises as a metabolic intermediate from the degradation of propionate, odd-chain fatty acids, and some amino acids. Thus, pathways for catabolism of this intermediate have evolved in all kingdoms of life, preventing the accumulation of toxic propionyl-CoA concentrations. Previous studies have shown that fungi generally use the methyl citrate cycle for propionyl-CoA degradation. Here, we show that this is not the case for the pathogenic fungus Candida albicans despite its ability to use propionate and valerate as carbon sources. Comparative proteome analyses suggested the presence of a modified β-oxidation pathway with the key intermediate 3-hydroxypropionate. Gene deletion analyses confirmed that the enoyl-CoA hydratase/dehydrogenase Fox2p, the putative 3-hydroxypropionyl-CoA hydrolase Ehd3p, the 3-hydroxypropionate dehydrogenase Hpd1p, and the putative malonate semialdehyde dehydrogenase Ald6p essentially contribute to propionyl-CoA degradation and its conversion to acetyl-CoA. The function of Hpd1p was further supported by the detection of accumulating 3-hydroxypropionate in the hpd1 mutant on propionyl-CoA-generating nutrients. Substrate specificity of Hpd1p was determined from recombinant purified enzyme, which revealed a preference for 3-hydroxypropionate, although serine and 3-hydroxyisobutyrate could also serve as substrates. Finally, virulence studies in a murine sepsis model revealed attenuated virulence of the hpd1 mutant, which indicates generation of propionyl-CoA from host-provided nutrients during infection.
Introduction
Candida albicans is an important opportunistic pathogen of humans (1), which is frequently found on mucosal surfaces such as the oral cavity, vaginal mucosa, or the digestive tract (2). Depending on the health status of its host, it can turn from a harmless commensal into a pathogen causing invasive mucosal or even life-threatening systemic infections. Epidemiologic studies have revealed that C. albicans is responsible for up to 15% of nosocomial bloodstream infections (3). Of note, nosocomial candidemia is connected with high mortality rates of about 40% (3). The ability to thrive in various host niches is the basis for the establishment of infections (4, 5). A prominent example is the metabolic switch from fermentative to nonfermentative growth upon phagocytosis by granulocytes (6–9). Although the bloodstream provides glucose as a preferred carbon source for the pathogen as indicated by increased expression of glycolytic genes (PFK2 and PYK1) (9), C. albicans is subjected to glucose starvation after phagocytosis (7, 9). To escape the hostile environment of phagocytes, the cells undergo a morphogenetic switch from yeast to hyphae (10) and invade the surrounding tissues. For this process, it has been shown that mutations in glycolysis, gluconeogenesis, and the glyoxylate cycle decrease virulence (9, 11, 12), indicating that the adaptation to the available nutrient sources is critical for a successful infection process.
Besides glucose, host-derived fatty acids, lipids, and proteins likely serve as additional nutrients for C. albicans. However, previous studies have shown that metabolism of fatty acids via β-oxidation seems dispensable for virulence (13, 14). Although a fox2 mutant, which is unable to utilize fatty acids as nutrient sources, revealed attenuated virulence, this effect was mainly attributed to the formation of giant peroxisomes causing transport defects into the peroxisomal compartment (15). Nevertheless, the efficient removal of propionyl-CoA as a side product from the degradation of some amino acids and odd-chain fatty acid can be assumed to be important for the proliferation within the host.
Accumulation of propionyl-CoA causes severe metabolic disorders not only in microorganisms but also in humans, where it is associated with life-threatening propionic aciduria and methylmalonic acidemia (16, 17). Investigations on mutants of the filamentous fungi Aspergillus nidulans (18, 19) and Aspergillus fumigatus (20) revealed that elevated propionyl-CoA concentrations interfere with the pyruvate dehydrogenase complex and the succinyl-CoA synthetase. Additionally, secondary metabolite production is affected under propionyl-CoA accumulation (21). Furthermore, A. fumigatus mutants, which are unable to metabolize propionyl-CoA, display strongly attenuated virulence in a murine infection model for pulmonary aspergillosis, indicating that propionyl-CoA is indeed formed from nutrients provided by the host (22). Thus, detoxification of propionyl-CoA appears of general importance to maintain normal cellular functions. However, it is worth noting that humans and fungi use strikingly different metabolic pathways for propionyl-CoA degradation.
In humans, propionyl-CoA is converted into the citric acid cycle intermediate succinyl-CoA, which requires a carboxylation to (R)-methylmalonyl-CoA, an isomerization to (S)-methylmalonyl-CoA, and finally, a rearrangement of the carbon skeleton to succinyl-CoA via coenzyme B12-dependent methylmalonyl-CoA mutase (23, 24). In contrast, most fungi seem to use the so-called methyl citrate cycle for the degradation of propionyl-CoA (24). This pathway, which is also present in several bacteria (25), results in the α-oxidation of propionate to pyruvate and is characterized by an initial condensation of propionyl-CoA and oxaloacetate to form methyl citrate (19), a dehydration to methyl-cis-aconitate (26), a rehydration to methyl isocitrate (26), and finally, a cleavage via methylisocitrate lyase into pyruvate and succinate (27–30). Although the reactions of this pathway resemble those of the citric acid and the glyoxylate cycle, the methyl citrate cycle uses its own specific set of enzymes.
Interestingly, a third pathway for the degradation of propionyl-CoA has been described but has not been studied in detail at the molecular level. This pathway seems to proceed via a modified β-oxidation pathway and is characterized by the formation of β-hydroxypropionate. β-Hydroxypropionate has been detected in plant seedlings incubated in the presence of 13C-labeled propionate (31), in insects (32), in some selected bacteria such as Rhodococcus erythropolis (33), and most strikingly, in the yeast Candida rugosa (34, 35). It has been assumed that propionyl-CoA first enters the β-oxidation pathway of fatty acid degradation. Here, a fatty acyl-CoA oxidase or dehydrogenase forms acrylyl-CoA, which is hydrated to β-hydroxypropionyl-CoA. In a complete β-oxidation cycle, the hydroxyacyl-CoA is oxidized to the ketoacyl-CoA and finally cleaved by ketoacyl-CoA thiolases under the release of an acetyl-CoA unit (14). This results in a residual acyl-CoA shortened by two carbon atoms which, in case of propionyl-CoA, would consist of the one carbon unit formyl-CoA. However, the detection of the key-metabolite β-hydroxypropionate suggests that the latter two reactions are not efficiently carried out. It has therefore been proposed that hydroxypropionyl-CoA exits the β-oxidation pathway and is converted by a series of reactions via hydroxypropionate, malonate semialdehyde and malonate to acetyl-CoA and CO2 (31, 32). However, evidence for enzymes performing these reactions still has only partially been provided.
Because previous studies on A. fumigatus demonstrated that the accumulation of intermediates from propionate degradation affects virulence (22), we were interested in elucidating the respective pathway in the dimorphic and pathogenic yeast C. albicans. To identify the responsible pathway, we performed growth analyses, two-dimensional proteomics, gene deletions, metabolite analyses, recombinant protein production, and enzyme characterizations. Additionally, virulence of a selected mutant was studied in a murine infection model to investigate the impact of propionyl-CoA degradation on host infection.
EXPERIMENTAL PROCEDURES
Media, Culture Conditions, and Growth Analyses
For standard cultivation, C. albicans strains were grown in 20 ml of YPD (per liter: 10 g of yeast extract, 20 g of peptone, and 20 g of glucose) or Candida minimal medium (CMM)2 (14). Liquid cultures were generally incubated at 30 °C and 200 rpm on a rotary shaker. Solid media were prepared by the addition of 2% agar. CMM was supplemented with one of the following carbon sources: 50 mm glucose, 50 mm sodium acetate, 50 mm sodium propionate, 10 mm sodium butyrate, 10 mm sodium valerate, or 10 mm sodium hexanoate. Amino acids as nutrient sources were generally added in a final concentration between 10 and 50 mm and replaced the nitrogen source (NH4)2SO4. For spot dilution growth analyses, C. albicans SC5314 wild type, mutants, and complemented strains were pre-grown in YPD, washed three times with ice-cold phosphate-buffered saline (PBS), adjusted to 3.3 × 105 cells/μl, and serially diluted (1:10 dilutions). From each dilution, 3 μl were spotted onto YPD and CMM agar plates containing the above mentioned carbon sources. When propionate or valerate were tested as substrates in spot dilution assays, ethanol from the vitamin solution was omitted to prevent initial growth of C. albicans by ethanol utilization. Plates were incubated at 30 and 37 °C. Plates were analyzed after 1 day on YPD, 2 days on glucose, or 4 days on all other nutrient sources. Growth analyses were carried out in triplicate with at least two independent deletion and complemented strains. Growth curves from liquid media were recorded by removing aliquots at different time points, dilution of samples in PBS, and determination of optical density at 600 nm on a Lambda25 UV-visible double beam spectrophotometer (PerkinElmer Life Sciences).
Sample Preparation for Two-dimensional Gel Analysis
C. albicans SC5314 wild-type cells were preincubated for 14 h in CMM + 50 mm glucose and collected by centrifugation at 3000 × g at 4 °C for 10 min and washed three times with PBS. CMM containing either 50 mm glucose, acetate, or propionate were inoculated at an OD600 of 0.2. Glucose cultures were incubated at 30 °C for 8 h and acetate and propionate cultures for 18 h. Cells were collected by centrifugation at 4000 × g at 20 °C for 10 min; the supernatant was discarded, and the pellet was resuspended in a minimal volume of trichloroacetic acid (TCA) solution (3.99 g of TCA, 0.09 g of DTT in 30 ml of acetone). Cells were disrupted using a speed mill (Analytik Jena, Jena, Germany) in the presence of zirconia beads (0.5–0.7 mm, Roth, Karlsruhe Germany). Cell lysis was performed by two intervals of 2 min with 2-min cooling on ice. Samples were stored at −20 °C for 16 h to allow for complete protein precipitation and centrifuged at 12,000 × g for 15 min. The precipitate was washed twice with 1 ml of washing solution (0.09 g of DTT in 30 ml of 90% acetone) and air-dried. After all residual acetone had evaporated, 300 μl of lysis buffer (7 m urea, 2 m thiourea, 2% CHAPS, 30 mm Tris, 1% Zwittergent, 0.8% Pharmalyte 3-10, 20 mm DTT) was added and mixed by vigorous shaking. Samples were incubated in an ultrasonic bath for 10 min and subsequently incubated at −80 °C for 1 h. Samples were thawed at room temperature and centrifuged at 12,000 × g and 4 °C for 20 min. The supernatant was collected, and protein concentration was determined by the protein assay kit from Bio-Rad using bovine serum albumin as standard.
First Dimension Isoelectric Focusing and SDS-PAGE for Second Dimension
Isoelectric focusing and second-dimension electrophoresis were performed with some modifications as described previously (36). In brief, for isoelectric focusing, 11-cm Immobilize dry strips, pH 3–11NL (GE Healthcare), were used. Strips were rehydrated in rehydration buffer as recommended by the manufacturer. Isoelectric focusing was performed using an Ettan II isoelectric focusing system (GE Healthcare). To a 100-μg protein sample in a maximum volume of 100 μl, 2.5 μl of DeStreak reagent (GE Healthcare) and 1 μl of IPG-buffer 3-11 NL (0.5%, GE Healthcare) were added, and the solution was applied to the strips by cup loading. The following program was used for isoelectric focusing: 0–3 h at 300 V; 3–7-h gradient to 600 V; 7–8-h gradient to 1000 V; 11–15-h gradient to 8000 V and hold for additional 24,000 V-h. Prior to second-dimension electrophoresis on 12.5% Criterion Precast gels (Tris/HCl, 1.0 mm for 11-cm IPG Strips, Bio-Rad), strips were equilibrated in reducing equilibration buffer containing iodoacetamide for protein acetylation (36). Gels were run for 1 h at constant 200 V. After fixation for at least 2 h in fixing solution (40% (v/v) methanol and 7% acetic acid), gels were washed in distilled water and stained with Coomassie Brilliant Blue G-250 (37). The gels were neutralized for 5 min in 100 mm Tris/o-phosphoric acid, pH 6.5, and kept at 4 °C in water until analyzed.
Protein Identification from Two-dimensional Gels
Stained gels were scanned and manually overlaid for spot selection. Protein spots of interest were manually excised from the gels and subjected to a tryptic digest (200 ng of trypsin; Promega, Mannheim, Germany). Protein fragments were extracted and prepared for MALDI-TOF/TOF analysis (38). An aliquot of the extracted peptides was mixed with the same amount of α-cyano-4-hydroxycinnamic acid matrix solved in TA30 (30% acetonitrile + 70% of 0.1% TFA). 2 μl were spotted onto a 800/384 anchor chip target (Bruker Daltonics, Bremen, Germany). Peptide mass analysis and peptide sequence determination were performed on a Bruker ultraflex TOF/TOF with Bruker Compass 1.2 software suite, including FlexControl (hardware control) and FlexAnalysis 3.0 (peak list creation). The following parameter settings for peak list generation were used: signal-to-noise ratio = 4; maximum number of peaks = 500; algorithm = SavitzkyGolay (width = 0.15 m/z, cycles = 4). The Proteinscape 1.3 software was used to collect peak lists that were sent to the Mascot in-house server 2.1.03 equipped with a monthly updated NCBI nonredundant database (taxonomy, fungi) for protein identification. For MS analysis, a peptide mass fingerprint search was performed with the following parameter settings: peptide mass tolerance ±50 ppm, peptide charge state = 1+; maximum of missed cleavages = 0–1. For further analysis, an MS/MS ion search was used. The parameters were set to peptide mass tolerance = ±300 ppm; fragment mass tolerance = ±1 Da; maximum of missed cleavages = 1. For statistical analysis, a based Mowse score was calculated automatically by the Proteinscape software.
Generation of Gene Deletion and Complementation Constructs
The deletion cassettes were generated by procedures described previously (14, 39). For a list of oligonucleotides used for the different PCR amplifications refer to Table 1. In brief, about 200–500 bp of the 5′- and 3′-flanking regions from each gene of interest were amplified by PCR with phusion polymerase (Thermo Scientific, Schwerte, Germany) using a Speed Cycler (Analytik Jena, Jena, Germany). PCR products were cloned into the PCR cloning vector pJET1.2 (Thermo Scientific). Fragments were released from pJET1.2 by either ApaI + XhoI restriction for the 5′-flanking region or SacI + SacII restriction for the 3′-flanking region and cloned into the corresponding sites of vector pSSU1 comprising the SAT1 flipper cassette (39). The pSSU1 vector, including the flanking regions, was digested with ApaI + SacI to remove the vector backbone prior to transformation of C. albicans.
TABLE 1.
List of oligonucleotides used in this study
Pairs of oligonucleotides are indicated.

a Cleavage sites for the restriction enzymes ApaI, XhoI, SacI, or SacII and the InFusion recombination sites are highlighted in boldface.
b Description of the application for which the primer pairs were used.
c Two independent deletion cassettes were required to delete the first and second allele of HPD1.
For complementation of homozygous mutants, the gene of interest, including 200–500 bp of the upstream flanking region, was amplified from genomic DNA of the SC5314 wild-type strain. PCR products were subcloned into pJET1.2, released by ApaI + BglII restriction, and subcloned into the ApaI + BglII restricted plasmid pSAP2KS1, which contains the SAT1 resistance marker. Subsequently, the 3′-flanking region used for gene deletion (described above) was cloned downstream from the SAT1 cassette of pSAP2KS1. Complementation fragments were released by ApaI + SacI and used for transformation of the deletion mutants.
Transformation of C. albicans and Selection Marker Regeneration
Transformation of C. albicans was performed by electroporation similar to described procedures (39). After the pulse, 1 ml of 1 m sorbitol was added, and the suspension was divided in two aliquots (A and B) that were regenerated in YPD at 30 °C for at least 4 h. Aliquots were spread on YPD agar plates containing 100 μg/ml nourseothricin (Werner Bioagents, Jena, Germany). Transformants became visible after 48–72 h. To ensure that independent clones were subsequently investigated, transformants from both aliquots (A and B) were propagated. To regenerate nourseothricin sensitivity for subsequent transformations, the SAT1 selection marker of the flipper cassette was removed by cultivation of transformants in 20 ml of YPM medium (per liter: 10 g of yeast extract, 20 g of peptone, 20 g of maltose) (39). Cells were incubated for at least 18 h at 30 °C and 200 rpm. Cells from these cultures were screened for nourseothricin sensitivity. Homozygous deletion mutants were identified by PCR using the 5′-oligonucleotide from the amplification of the 5′-flanking region and a specifically designed 3′-deletion control oligonucleotide that hybridized to the corresponding gene of interest (Table 1). Selected transformants were additionally checked by Southern hybridization using digoxygenin-labeled probes (Roche Diagnostics).
Generation of EHD3-GFP and HPD1-GFP Fusion Constructs
To investigate the subcellular localization of Hpd1p and Ehd3p, fusions with the enhanced green fluorescent protein (GFP) were generated. Two kinds of independent constructs were generated as follows: (i) complete open reading frames together with their natural promoter fused with GFP and (ii) for Ehd3p a GFP fusion of an N-terminal fragment comprising the first 48 amino acids (144 bp) under control of the constitutively active actin promoter pACT1. All constructs contained the SAT1 resistance marker and replaced one wild-type allele in strain SC5314. Primer pairs for generating PCR fragments are listed in Table 1, and fusion of fragments was performed from 15-bp overlaps. To enable homologous integration into the respective locus, GFP constructs were flanked by the 5′- and 3′-flanking regions that were also used to delete the respective gene in the wild type. In brief, to generate the pEHD3-EHD3-GFP cassette, PCR fragments were amplified with primer pairs 22 and 23. The deletion plasmid for EHD3 was restricted with ApaI and XhoI, and the restriction fragment comprising the plasmid with the SAT1 cassette and the 3′-flanking region were gel-purified. The PCR products and the plasmid were fused using the In-Fusion® HD cloning kit (Takara Bio Europe/Clontech). Similarly, the pHPD1-HPD1-GFP cassette was generated with primer pairs 24 and 25 and fused with the respective ApaI/XhoI-restricted plasmid used for HPD1 deletion.
For constitutive expression of truncated N-terminal fragments, pACT1 was amplified with primer pair 19. The 144-bp EHD3 fragment was generated with primer pair 21. The PCR fragments were fused with the gel-purified XhoI-restricted EHD3 deletion plasmid resulting in construct pACT1-EHD3N-term-GFP. Transformation cassettes were released by ApaI/SacI restriction and used for transformation as described above. Transformants were checked by PCR for the integration of the fusion constructs with the respective primer pairs 26 and 27.
Fluorescence Intensity Levels, Staining of Mitochondria and Fluorescence Microscopy
Fluorescence intensity from pEHD3-EHD3-GFP and pHPD1-HPD1-GFP containing transformants was determined from cell-free extracts of cells grown exponentially on CMM with either glucose, acetate, valerate, or propionate as carbon sources. In brief, cells of the untransformed wild type and two independent transformants from each construct were harvested, resuspended in 50 mm Tris/HCl, pH 8.0, with 150 mm NaCl and 10% glycerol, and disrupted in a speed mill. Protein concentrations of cell-free extracts were determined, and serial 2-fold dilutions starting at 20 μg/100 μl were prepared in black 96-well plates with transparent bottom (Nunc/Thermo Fisher Scientific). GFP fluorescence was determined on a FLUOstar Omega Microplate reader (BMG Labtec) with the following settings: bottom reading, signal gain of 2100, excitation filter at 485 nm, emission filter at 520 nm, scan matrix 5 × 5 with 15 flashes per scan point. Fluorescence intensity values were exported to Microsoft Excel, and wild-type background fluorescence from the respective carbon sources and protein concentrations was subtracted from values of the transformants. Intensities were normalized to 1 μg/100 μl resulting in background-corrected relative fluorescence units/μg of protein. Mitochondria were stained by incubating cells for 30 min in the presence of MitoTracker Red 580 (Invitrogen) with subsequent washing in medium without MitoTracker. Cells were mounted in ProLong Gold antifade reagent (Invitrogen), and microscopy was performed on a Zeiss AXIO Imager. M1 under oil immersion with a Zeiss Plan-APOCHROMAT 100× objective. Mitochondria were visualized by using filter set 14, and GFP fluorescence was visualized by use of filter set 09. Images were adjusted within the MetaMorph software (Molecular Devices), and overlays were created by the “Overlay Images” tool.
Recombinant Production and Purification of Hpd1p in Escherichia coli
The sequence of the HPD1 gene (orf19.5565) was obtained from the CGD website, and the translated open reading frame was checked for a putative mitochondrial import sequence by Mitoprot. A cleavage site was detected at amino acid position 17 with a probability for mitochondrial import of 97%. Therefore, this sequence was omitted for recombinant production in E. coli. For generating a version with an N-terminal His tag, the gene was amplified from genomic DNA of SC5314 with oligonucleotides BamHI_HPD1_ATG_f (5′-GGA TCC ACC AAT TAC GGG TTT ATT GG-3′) and NotI_HPD1_TAA_re (5′-GCG GCC GCT TAT TTT CTT TTG ACA TCA ATT ACA TC-3′), cloned in pJET1.2, and excised by BamHI + NotI restriction. The fragment was subcloned in a modified pET43 vector in-frame with the His tag sequence (40, 41). For generation of a version with C-terminal His tag, the gene excluding the mitochondrial import sequence and the terminal stop codon was amplified with oligonucleotides IF_pet29HPD1_f (5′-aag gag ata tac ata tgA CCA ATT ACG GGT TTA TTG GTT TG-3′; overhang in lowercase letters) and IF_pet29HPD1_r (5′-aca ggt ttt cgg atc cTT TTC TTT TGA CAT CAA TTA CAT CAC-3′; overhang in lowercase letters) and directly cloned by In-Fusion® PCR cloning (Takara Bio Europe/Clontech) in an NdeI + BamHI-restricted pET29a vector (Merck). Both expression plasmids were used for transformation of E. coli BL21 Rosetta2 (DE3) cells (Merck). Protein production was induced either by cultivation in Overnight Express Instant TB medium or in minimal M9 medium by the addition of 0.5 mm isopropyl 1-thio-β-d-galactopyranoside at an OD550 of 0.8. Additionally, incubation temperatures between 18 and 30 °C were tested. Cell pellets were collected by centrifugation, resuspended in buffer A (50 mm Tris/HCl, 150 mm NaCl, 10% glycerol, pH 8.0), and disrupted by sonication. Lysates were cleared by centrifugation, filtered over a filter with 0.45-μm pore size, and loaded on a nickel-Sepharose 6 Fast Flow (GE Healthcare) gravity-flow column (1-ml bed volume). After a stringency wash with 6 column volumes of buffer B (buffer A + 30 mm imidazole), the protein was eluted in buffer C (buffer A + 200 mm imidazole). Purity of the proteins was checked by SDS-PAGE using NuPAGE BisTris 4–12% gradient gels (Invitrogen) in a MES-buffered running system. Fractions were combined, concentrated, and desalted by centrifugal filter devices (Merck). 50% glycerol was added, and the protein was stored at −20 °C without significant loss of activity. Protein concentrations were determined by use of the protein assay kit from Bio-Rad using bovine serum albumin as standard.
Enzyme Assays
All enzyme assays were performed at 22 °C using a Lambda25 UV-visible spectrophotometer (PerkinElmer Life Sciences). Citrate synthase and methylcitrate synthase activity was determined from C. albicans cell lysates by detection of CoASH release using 5,5′-dithiobis-(2-nitrobenzoic acid) as described previously (18). Hydroxypropionate dehydrogenase activity was determined by modification of a previously described procedure (42). The standard assay contained the following: 200 mm CHES, pH 9.5, 3 mm NAD, 36 mm 3-hydroxypropionate, purified enzyme and water to a final volume of 1 ml. The reaction was initiated by the addition of 3-hydroxypropionate. The following alternative substrates replaced 3-hydroxypropionate in the standard assay: (S)-3-hydroxyisobutyrate (10 mm), (R)-3-hydroxyisobutyrate (10 mm) and l-serine (40 mm). NADP replaced NAD in the standard assay to investigate the cofactor specificity. EDTA was added at 5 mm prior to addition of enzyme and substrate to test for metal dependence. For determination of pH and buffer dependence, the following buffers were used (all at 200 mm): Tris/HCl, pH 7.0, 8.0, and 9.0; CHES, pH 9.0, 9.5, 10.0; potassium carbonate, pH 9.5, 10.0, 10.5; glycine, pH 9.5, 10.0, 10.5; CAPS, pH 10.0, 10.5, 11.0. The Km values were determined by varying the concentration of one substrate, whereas that of the co-substrate was kept constant. At least five different substrate concentrations were tested. Data were plotted double reciprocally in Lineweaver-Burk diagrams, and Km values and maximum activities were calculated by use of the Microsoft Excel software from the interception points of y and x axis of a linear regression curve (R2 values for all regression curves were >0.95). Turnover numbers were calculated assuming one active site per enzyme subunit. 3-Hydroxypropionate dehydrogenase from cell lysates of C. albicans strains was determined in the standard assay with three replicates from each strain and growth condition.
Sample Preparation for HPLC and GC/MS Analysis
Cells of the C. albicans wild-type SC5314 and the hpd1 mutant were pre-grown at 30 °C and 200 rpm for 14 h in four cultures of 50 ml of YPD medium. Cells were collected by centrifugation for 5 min at 3000 × g and 4 °C, washed three times with PBS, and finally resuspended in 5 ml of PBS. Samples of each strain were pooled. For each strain, four cultures of 100 ml of CMM with 20 mm acetate and 20 mm propionate as carbon sources were inoculated with the respective cell suspension to give an OD600 of 15. One culture was directly collected by a 5-min centrifugation at 4000 × g at 4 °C. The other cultures were collected after 2, 4, and 6 h. The OD600 was again adjusted to 15 by dilution with fresh medium, and 100-ml aliquots were used for further processing. After centrifugation, the supernatants were discarded, and the cell pellets were directly frozen in liquid nitrogen and stored at −80 °C until analyzed. Cells were thawed on ice and resuspended in a minimal volume of 0.1 m HCl before they were transferred to microtubes equipped with zirconia beads (0.5–0.7 mm, Roth, Karlsruhe Germany). Cell disruption was carried out using a speed mill (Analytik Jena, Jena, Germany) with three disruption intervals of 2 min. Cells were cooled for 2 min on ice between the intervals. Samples were centrifuged for 30 min at 12,000 × g and at 4 °C, and supernatants were either directly used for HPLC analysis or further processed for GC/MS analysis. The same procedure of sample preparation was followed for the hpd1 mutant when samples were prepared for NMR analysis. However, only a 6-h time point was selected, and 20 mm 2-[13C]propionate replaced the unlabeled propionate from above. To process samples for GC/MS analysis, all chemical reagents were of analytical grade. Methoxyamine hydrochloride and pyridine were purchased from Sigma. N-Methyl-N-trimethysilyltrifluoroacetamide was purchased from Macherey-Nagel (Düren, Germany). 3-Hydroxypropionic acid was purchased from TCI (Zwijndrecht, Belgium). 50 μl of the supernatant was transferred in a GC/MS glass vial and evaporated to dryness under vacuum (Concentrator Plus, Eppendorf, Hamburg). The sample derivatization was performed according to the protocol of Roessner et al. (43) by adding 80 μl of methoxyamine hydrochloride (2 g/100 ml in pyridine) and 80 μl of N-methyl-N-trimethysilyltrifluoroacetamide to the dried sample. After each addition, a domestic microwave oven was used for heating (270 watts for 5 min) (44). Control standards were treated in an identical manner.
GC/MS, HPLC, and NMR Analyses
GC/MS analysis was performed using a GC-QqQ-MS (Agilent 7890A, 7000 MS). One μl of the sample was injected into a VF 5-ms column (30 m with 5 m of EZguard, 0.25 mm inner diameter, and 0.25-μm film thickness, Agilent, Waldbronn, Germany). The injection was performed in split mode (15:1), and the temperature of the injector was set to 290 °C. The oven program started at 50 °C (2 min) and rose up with a rate of 10 °C/min to 320 °C (11 min). Electron ionization was conducted at 70 eV, and data were collected in full scan mode (m/z 30–600).
Quantification of 3-hydroxypropionate was performed using an HPLC system (Jasco, Japan) equipped with a diode array detector and a refractive index detector). Samples were chromatographed over an Aminex HPX-87H column (9 μm, 300 × 7.8 mm, Bio-Rad) with 0.005 m H2SO4 as mobile phase. The column temperature was 50 °C. The flow rate of the mobile phase was set to 0.5 ml/min, and 50 μl of the sample was injected. Quantification was achieved using external standard calibration method.
NMR spectra were recorded at 300 K on a Bruker Avance III 500 spectrometer with methanol-d4 as solvent and internal standard. The 1H NMR spectrum of 3-[2-13C]hydroxypropionic acid showed two signals at 2.50 ppm (2H, dt, J = 127.8, 6.3 Hz, H-2) and 3.80 ppm (2H, dt, J = 6.3, 2.3 Hz, H-3), respectively. The 1H-decoupled 13C NMR spectrum included resonances at 38.2 ppm (carbon 2) and 58.9 ppm (carbon 3). A signal for the quaternary carbon of the carboxylic acid function (carbon 1) was not detected, which is likely due to signal dispersion caused by the homonuclear coupling to carbon 2.
Murine Model of Hematogenously Disseminated Candidiasis
8–10-Week-old female BALB/c mice (18–20 g; Charles River, Germany) were used for the experiments. The animals were housed in groups of five in individually ventilated cages and cared for in strict accordance with the principles outlined in the European Convention for the Protection of Vertebrate Animals Used for Experimental and Other Scientific Purposes in compliance with the German Animal Welfare Act. Protocols were approved by the responsible Federal State authority (Thüringer Landesamt für Lebensmittelsicherheit und Verbraucherschutz) and its ethics committee. C. albicans strains were grown for 14 h at 30 °C, washed three times with PBS, and adjusted to 2.5 × 106 cells/ml. Mice were challenged intravenously on day 0 with 2.5 × 104 cfu/g body weight via the lateral tail vein. The health status of the mice was examined at least twice a day by a veterinarian. Body surface temperature and body weight were recorded daily. Mice showing severe signs of illness, like isolation from the group, apathy, hypothermia, and drastic weight loss, were euthanized by application of 200 μl of ketamine hydrochloride (50 mg/ml). Gross pathological alterations were recorded during necropsy.
RESULTS
Propionate Consumption by C. albicans
To test for the ability of C. albicans to utilize propionate, growth analyses were performed. Of note, Saccharomyces cerevisiae is unable to utilize propionate as the sole carbon source but metabolizes it via the methyl citrate cycle in the presence of glucose (45). Because of the phylogenetic relation between S. cerevisiae and C. albicans, both belonging to the Saccharomycetales, we initially monitored growth of the C. albicans wild-type strain SC5314 on glucose, glucose/acetate, and glucose/propionate medium and simultaneously determined the consumption of glucose (Fig. 1). As long as glucose was present in the culture media, no difference among the three conditions was observed. After glucose consumption, cells continued to grow in the presence of acetate with only a short lag phase. In contrast, the adaptation to propionate caused an interruption of cell growth for ∼20 h, after which an additional slight increase in biomass formation was observed. This result shows that glucose is the preferred carbon source and suppresses the use of alternative carbon sources. However, after glucose depletion, both acetate and propionate support growth, although the latter supports it only to a limited extent. To confirm the utilization of propionate, we inoculated C. albicans in a medium with propionate as the sole carbon source and monitored growth and propionate consumption. As indicated in Fig. 1, the slow increase in biomass was accompanied by the consumption of propionate. Thus, C. albicans utilizes, albeit at low rates, propionate as sole carbon source and does not require the co-metabolism of glucose.
FIGURE 1.

Propionate utilization by C. albicans. A, growth analysis of C. albicans wild type on 5 mm glucose, 5 mm glucose + 50 mm acetate, and 5 mm glucose + 50 mm propionate media. An arrow indicates the time point of total glucose consumption in all cultures. In medium supplemented with acetate, growth continues after glucose consumption. On medium supplemented with propionate, a 20-h lag phase follows glucose consumption after which biomass starts to increase. No further increase is observed from cultures growing without acetate or propionate supplementation. Data points show mean values with standard deviations from three independent cultures. B, growth of C. albicans on 20 mm propionate as sole carbon source. Cells were inoculated at high density, and aliquots were removed for propionate detection by HPLC analysis. C, HPLC-based determination of propionate consumption from cultures shown in B. After 30 h, no residual propionate is detected from the culture medium.
Utilization of Methyl Citrate Cycle or Methylmalonyl-CoA Pathway in C. albicans
To identify genes involved in propionyl-CoA degradation, we first screened the genome for genes coding for enzymes of the methyl citrate cycle. For this analysis, the methylcitrate synthases Cit3p from S. cerevisiae (46) and McsA from A. fumigatus (20) served as templates. Analysis revealed the sole presence of the putative citrate synthase Cit1p (orf19.4393), but no additional methylcitrate synthase. To check for a specific methylisocitrate lyase, Icl2p from S. cerevisiae (30) and MclA from A. fumigatus (25) were used as templates, but they only revealed the phylogenetically closely related isocitrate lyase Icl1p (orf19.6844) from C. albicans (4, 11, 25). When the putative methylcitrate dehydratase from S. cerevisiae (accession number NP_015326) or A. fumigatus (accession number EDP47611) was used for BLASTP analyses, no homologue was detected in the C. albicans genome. To verify the absence of methyl citrate cycle activity in C. albicans, strain SC5314 was grown on glucose or propionate containing media and tested for citrate and methylcitrate synthase activity. Specific citrate synthase activity on glucose was 0.5 units/mg, whereas methylcitrate synthase activity was near the background level with ∼1 milliunit/mg. On propionate, citrate synthase activity increased to 5.1 units/mg, which is in agreement with glyoxylate cycle induction on nonfermentable nutrient sources (4, 9). However, methylcitrate synthase remained low with ∼6 milliunits/mg. Thus, the absence of genes specifically contributing to a methyl citrate cycle in combination with the lack of methylcitrate synthase activity on propionate indicates that C. albicans does not utilize the methyl citrate cycle for the degradation of propionyl-CoA.
To identify genes required for a functional methylmalonyl-CoA pathway, we selected the essential coenzyme B12-dependent methylmalonyl-CoA mutase (23) for genome analyses. Methylmalonyl-CoA mutases from different species are highly conserved, and the enzymes from Homo sapiens (accession number AAA59569), the bacterium Rhodobacter sphaeroides (accession number ACJ71672), and the nematode Caenorhabditis elegans (accession number CAA84676) display between 63 and 75% amino acid identity. However, no methylmalonyl-CoA mutase was found in the C. albicans genome. It is therefore unlikely that C. albicans uses the methylmalonyl-CoA pathway for propionate degradation.
Two-dimensional Gel Proteomic Analyses for Identification of Proteins Involved in Propionate Degradation
Because C. albicans neither uses the methyl citrate cycle nor the methylmalonyl-CoA pathway, an alternative pathway for propionyl-CoA degradation was required. Because of the very long adaptation phase of C. albicans to utilize propionate, we decided to perform proteomic analyses on cells actively growing on this carbon source. To allow for a selection of proteins of interest, we compared the protein spot pattern of propionate-grown cells with those from cells grown on glucose and acetate medium. We excised about 80 major protein spots from each condition and focused especially on those proteins that were apparently unique or highly abundant under the respective conditions (Fig. 2).
FIGURE 2.
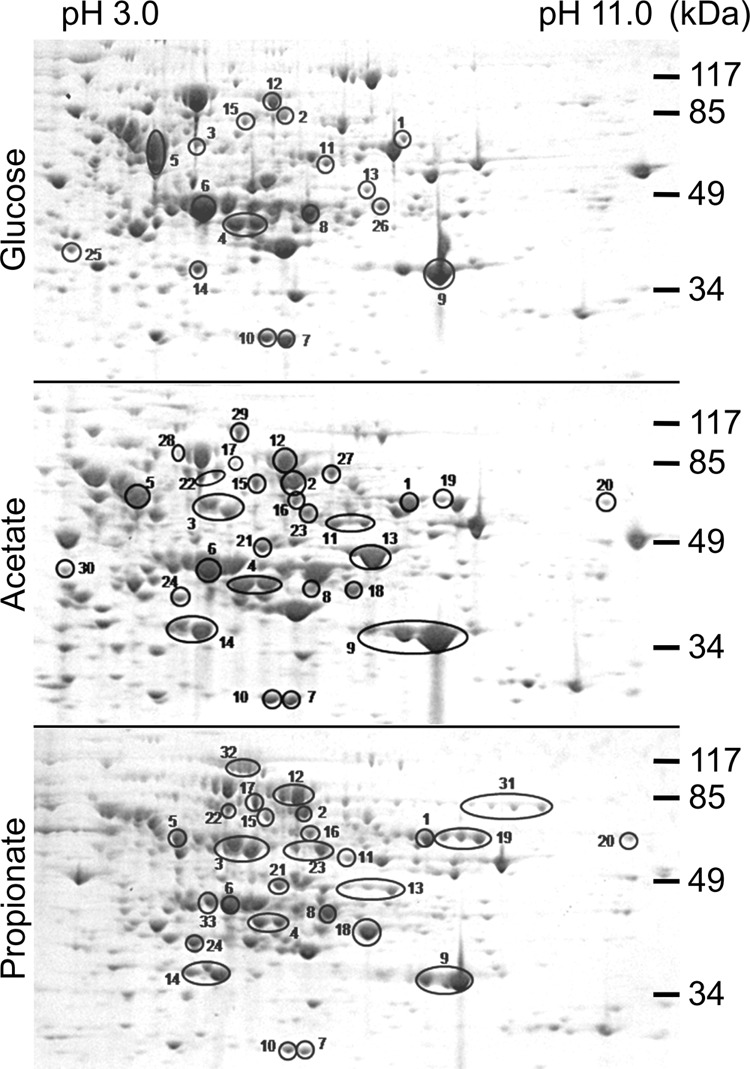
Proteomic analysis of protein extracts from C. albicans wild type cultivated on glucose, acetate, or propionate medium. Protein extracts were prepared from cells in the exponential growth phase and separated by two-dimensional gel electrophoresis. Major or unique spots from different conditions were analyzed by MALDI-TOF-MS analysis. Proteins assigned to catabolic processes by GO term analyses (see also Table 2) are highlighted and marked by numbered 1–33. Details on all proteins identified can be found in supplemental Table S1.
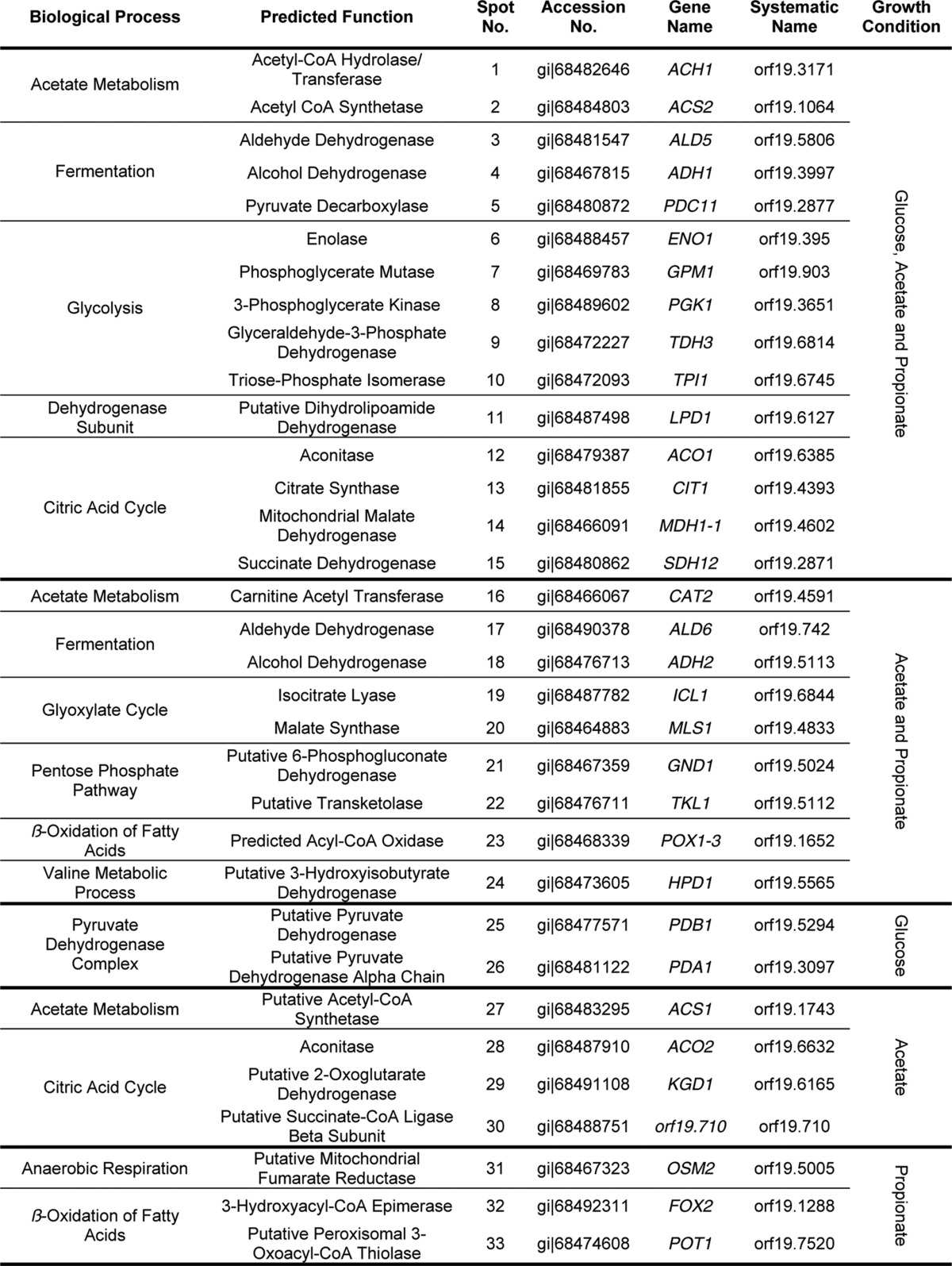
Among the ∼250 spots analyzed, we identified 73 different proteins (supplemental Table S1). This low number was mainly due to post-translational modifications that led to multiple identifications of the same protein from different spots on one gel. 33 of these proteins were assigned to catabolic processes of primary carbon metabolism by GO-term analyses and are shown in Table 2. Among these, 15 proteins were identified from all three growth conditions and were associated with central metabolic pathways such as acetyl-CoA metabolism, fermentation, glycolysis, and the citric acid cycle. Additionally, a dihydrolipoamide-containing dehydrogenase subunit was found, which is part of several dehydrogenase complexes in central metabolic pathways. There were nine proteins that were detected from acetate and propionate but not from glucose-grown cells. These proteins were specific for acetate metabolism, the glyoxylate cycle, the pentose phosphate pathway, and β-oxidation. Moreover, a protein, which is proposed to be associated with valine metabolism, was identified. Finally, we also detected proteins that were unique to specific growth conditions. On glucose, two subunits of the pyruvate dehydrogenase complex were identified, indicating a higher abundance of this complex during growth on glucose. On acetate, the acetyl-CoA synthetase Acs1p and enzymes of the citric acid cycle were identified, which indicates an increased turnover of metabolites via this pathway and is in agreement with activity of the glyoxylate cycle for anaplerosis of oxaloacetate. On propionate, a putative fumarate reductase was identified that is difficult to assign a specific function without further investigation. Additionally, two enzymes of the β-oxidation, namely the bifunctional enoyl-CoA hydratase/hydroxyacyl-CoA dehydrogenase Fox2p and the main 3-ketoacyl-CoA thiolase Pot1p (14), were identified. Taken together, enzymes involved in β-oxidation of fatty acids were highly abundant in cells grown on propionate, which implies that propionyl-CoA might enter the β-oxidation pathway for further degradation.
TABLE 2.
Proteins with the GO-term “carbon metabolism” from proteomic analyses of glucose-, acetate-, or propionate-grown cells
For a complete list of all proteins identified and their respective scores from analysis refer to supplemental Table S1.
Selection of Target Genes for Knock-out Experiments
As mentioned previously, a pathway for propionyl-CoA degradation via a modified β-oxidation pathway has been suggested for plants (31), some insects (32), some bacteria (33, 47), and a relative of C. albicans, which is C. rugosa (34, 35). Based upon substrate labeling studies, possible reactions leading to the key intermediate 3-hydroxypropionate and, eventually, to acetate or acetyl-CoA formation have been postulated. Additional information on the enzymes involved in these reactions is, however, lacking, although candidate genes have been proposed for the plant Arabidopsis thaliana (31) as indicated in the scheme in Fig. 3. Based on our proteomics experiment and genome analyses, we speculated on the respective candidates in C. albicans.
FIGURE 3.
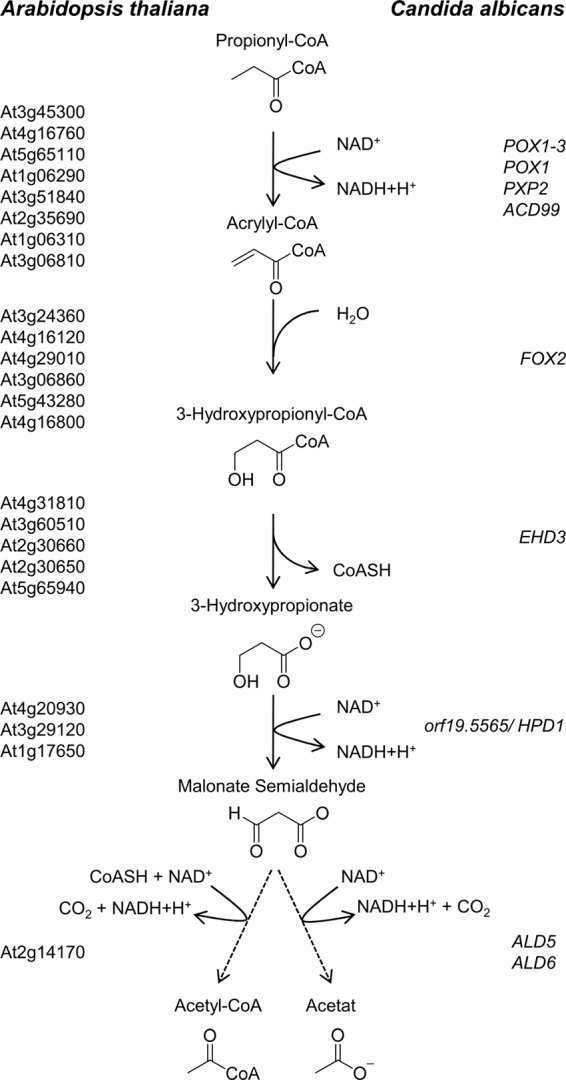
Scheme of the modified β-oxidation pathway via 3-hydroxypropionate. Candidate genes predicted for the plant A. thaliana (31) are shown on the left side and candidate genes for C. albicans on the right side of the pathway. Propionyl-CoA enters the β-oxidation pathway and is oxidized to 3-hydroxypropionyl-CoA. This intermediate exits the β-oxidation pathway by hydrolysis of the CoA ester and is further oxidized to either acetate or acetyl-CoA.
While entering the β-oxidation pathway, propionyl-CoA becomes oxidized to acrylyl-CoA, which could be performed by one of the fatty acyl-CoA oxidases. A likely candidate from our analyses was Pox1–3p, because it was identified from acetate- and propionate-grown cells. However, an involvement of other isoenzymes cannot be excluded. Subsequently, a hydration of acrylyl-CoA leads to the formation of 3-hydroxypropionyl-CoA. Because there is only a single peroxisomal bifunctional enoyl-CoA hydratase/hydroxyacyl-CoA dehydrogenase present in the genome of C. albicans (13–15, 48), it is extremely likely that this step is performed by Fox2p. In addition, Fox2p was identified as a highly abundant protein in propionate-grown cells. However, if Fox2p is involved in this reaction, it needs to release 3-hydroxypropionyl-CoA rather than converting it to 3-ketopropionyl-CoA, which does not belong to the assumed intermediates of a 3-hydroxypropionate pathway (31, 32). In contrast, removal of hydroxypropionyl-CoA to a different compartment such as the mitochondria followed by the action of a thioester hydrolase would lead to the formation of 3-hydroxypropionate. Although we did not identify a respective protein in our proteomic approach, we used the proposed A. thaliana candidate proteins (31) as template for genome analyses. Indeed, we were able to identify Ehd3p as a putative candidate in C. albicans, which has been predicted as a hydroxyisobutyryl-CoA hydrolase with putative mitochondrial localization. 3-Hydroxypropionate could then undergo a dehydrogenase reaction to malonate semialdehyde, and a candidate from our analyses was the protein encoded by orf19.5565 that had been annotated as a putative 3-hydroxyisobutyrate dehydrogenase from valine metabolism. In agreement with a putative mitochondrial localization of Ehd3p, the protein encoded by orf19.5565 also contains a putative mitochondrial import sequence and was detected from acetate and propionate grown cells. Finally, it has been assumed that additional dehydrogenase reactions convert malonate semialdehyde to either malonate with subsequent decarboxylation to acetate or via a coenzyme A-acylating aldehyde dehydrogenase directly to acetyl-CoA. At least one additional dehydrogenase would be required for these reactions, and we identified Ald6p, an enzyme with a putative mitochondrial localization, from acetate- and propionate-grown cells. Furthermore, Ald5p that does not display a sequence for mitochondrial import was present under all conditions. Both aldehyde dehydrogenase candidates displayed 49.6 and 51.6% identity to the putative A. thaliana candidate At2g14170. The conversion of propionyl-CoA to acetate or acetyl-CoA would subsequently require the glyoxylate bypass for gluconeogenesis, and in agreement, isocitrate lyase and malate synthase were found in both acetate- and propionate-grown cells.
In summary, a complete set of enzymes required for propionyl-CoA degradation via a modified β-oxidation pathway is present in C. albicans, and most of the candidates were identified from propionate-grown cells. However, because several of the proteins were not specifically produced in the presence of propionate, but also on acetate, their in vivo contribution to propionyl-CoA degradation remained to be elucidated. For this purpose, we investigated previously generated fox2 and 3-ketoacyl-CoA thiolase mutants (14) for their contribution to propionate metabolism. Furthermore, we generated homozygous mutants of ehd3, orf19.5565, ald5, ald6, and a double knock-out of ald5 and ald6 for phenotypic characterization.
Selection of Carbon Sources for Characterization of Mutant Strains
The genes from the selection above were deleted in the background of the C. albicans SC5314 wild-type strain utilizing the re-usable SAT1 flipper cassette (39). To characterize these mutants, seven different solid media were used in a screening approach (Fig. 4). All strains were analyzed for their growth behavior on YPD and glucose medium as positive controls. Furthermore, we tested growth on the carboxylic acids acetate, propionate, butyrate, valerate, and hexanoate. Acetate does not require the β-oxidation pathway, because its activation directly results in a single acetyl-CoA unit. Butyrate and hexanoate require one or two rounds of β-oxidation to yield two and three units of acetyl-CoA, respectively. Finally, although propionate might not require a complete round of β-oxidation, but branches into a modified β-oxidation pathway, valerate contains five carbon atoms and could first undergo one complete cycle of “normal” β-oxidation that leads to the formation of acetyl-CoA and propionyl-CoA. For the breakdown of the latter, the modified β-oxidation pathway would also be required.
FIGURE 4.

Growth analysis of C. albicans wild type and deletion mutants on solid media containing different carbon sources. Serial dilutions (106 to 101 cells/spot) were used for inoculation, and plates were incubated at either 30 or 37 °C. The scheme on top denotes the order of strains on all plates. Lane 1, SC5314 wild-type strain; lane 2, homozygous fox2 deletion strain; lane 3, homozygous ehd3 deletion strain; lane 4, homozygous hpd1 (orf19.5565) deletion strain; lane 5, homozygous ald5 deletion strain; lane 6, homozygous ald6 deletion strain; lane 7, homozygous mutant with ald5 and ald6 deletions; lane 8, homozygous triple mutant with deletion of the ketoacyl-CoA thiolases pot1, fox3, and pot13. Strains complemented on one allele showed no altered phenotype in comparison with the wild type (data not shown).
Phenotypic Characterization of the β-Oxidation Mutants fox2 and pot1/pot13/fox3
As expected, a fox2 mutant (lane 2 in Fig. 4) and the triple 3-ketoacyl-CoA thiolase pot1/pot13/fox3 mutant (lane 8 in Fig. 4) displayed no growth defects on YPD and glucose medium (14) but showed reduced growth on the nonfermentable carbon source acetate. Although a β-oxidation pathway is not required on acetate, studies on a fox2 mutant showed that peroxisomal morphology is altered, which negatively affects the glyoxylate cycle (15). The same could be true for the 3-ketoacyl-CoA thiolase triple mutant. Both mutants were also defective in utilization of butyrate, valerate, and hexanoate that require the β-oxidation pathway. However, the fox2 mutant was completely unable to grow on propionate, whereas growth of the triple 3-ketoacyl-CoA thiolase mutant was similar to that on acetate. This indicates that Fox2p is an essential enzyme required for propionyl-CoA degradation, which is in agreement with the modified β-oxidation pathway. Furthermore, a branch in the β-oxidation pathway at the level of Fox2p would also avoid action of the 3-ketoacyl-CoA thiolases, which is in agreement with the observed phenotype. Thus, Fox2p but none of the 3-ketoacyl-CoA thiolases appears essential for propionyl-CoA degradation.
Phenotypic Characterization of the ehd3 Mutant
Ehd3p displays 33.2% identity and 53% similarity to the putative hydroxyisobutyryl-CoA hydrolase CHY1 from A. thaliana (accession number Q9LKJ1) and was therefore selected as a candidate gene involved in a modified β-oxidation pathway. Furthermore, Ehd3p is also 37.7% identical (54.4% similarity) to the human hydroxyisobutyryl-CoA hydrolase (accession number NP_055177), which has been shown to be highly active with 3-hydroxypropionyl-CoA as substrate (49). Therefore, we speculated that Ehd3p might perform the hydrolysis of 3-hydroxypropionyl-CoA, and we speculated for a growth defect on propionate and valerate but not on the other carbon sources tested. In agreement, the ehd3 mutant displayed no growth defect on YPD, glucose, butyrate (lane 3 in Fig. 4) or the amino acid valine (Fig. 5C). The latter finding was unexpected, because classic valine degradation produces 3-hydroxyisbutyrate and propionyl-CoA as intermediates (50, 51). Some growth inhibition was observed on acetate and hexanoate, but this was more pronounced at elevated temperatures than at 30 °C. Interestingly, reduced growth on hexanoate could be complemented by carnitine supplementation (data not shown). The most striking effects of the ehd3 mutant were observed on valerate and propionate, where growth was nearly completely abolished and could not be restored by the addition of carnitine (data not shown). Additionally, growth of the ehd3 mutant was also completely abolished in liquid medium (Fig. 5A). This is in accordance with an essential role of Ehd3p in the hydrolysis of 3-hydroxypropionyl-CoA to provide 3-hydroxypropionate for subsequent enzymatic reactions. Furthermore, from the inability to grow on valerate, we speculated that accumulating 3-hydroxypropionyl-CoA could cause toxic effects on Fox2p.
FIGURE 5.
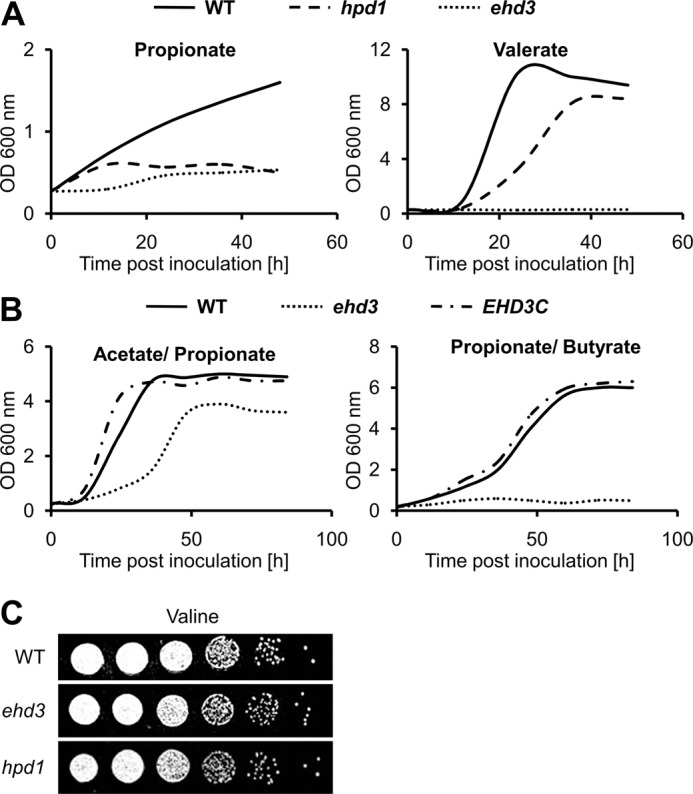
Growth analysis of wild type, Δ/Δhpd1 (hpd1) mutant, and Δ/Δehd3 (ehd3) mutant on liquid media and on valine-containing solid media. A, growth of wild type, hpd1 deletion mutant, and ehd3 deletion mutant on propionate and valerate medium. The hpd1 mutant shows some initial biomass formation on propionate, but growth is completely abolished at later time points. On valerate, the mutant shows a decreased growth rate and reduced final biomass. The ehd3 deletion mutant only reveals very limited growth after a 15–20-h lag phase and is unable to grow on valerate. Mean values from three independent parallel cultures are shown. B, growth of the ehd3 deletion mutant and its complemented strain in comparison with the wild type on mixed carbon sources. Although the mutant is able to grow at a reduced rate in the presence of acetate with propionate, no growth is observed when butyrate is supplemented with propionate. Mean values from three independent parallel cultures are shown. C, growth analysis on valine-containing solid media. The homozygous ehd3 and hpd1 deletion mutants show no altered growth phenotype in comparison with the wild type.
Growth of the ehd3 mutant on butyrate was not affected and on acetate and hexanoate only slightly affected. This indicates that Ehd3p is not essential for β-oxidation or growth on acetyl-CoA units. In contrast, valerate must first undergo one β-oxidation cycle leading to acetyl-CoA and propionyl-CoA. Although this acetyl-CoA could be sufficient for energy metabolism and biomass formation, the ehd3 mutant was completely blocked on valerate, implying that accumulating hydroxypropionyl-CoA could specifically inhibit subsequent β-oxidation cycles at the level of Fox2p. To test this hypothesis, we incubated the ehd3 mutant and the wild type on acetate/propionate and on butyrate/propionate medium. As expected from an inhibition of Fox2p, no growth was observed for the ehd3 mutant on butyrate/propionate medium (Fig. 5B), whereas growth on acetate/propionate medium was only slightly inhibited (Fig. 5B). This agrees with a specific inhibitory effect of 3-hydroxypropionyl-CoA on fatty acid degradation and its minor toxicity toward general acetyl-CoA metabolism. In summary, Ehd3p seems responsible for hydroxypropionyl-CoA hydrolysis, and the protein appears essential for the proposed modified β-oxidation pathway. Furthermore, the inability to efficiently remove hydroxypropionyl-CoA seems to cause a metabolic intoxication toward fatty acid degradation.
Phenotypic Characterization of the orf19.5565 Mutant
The protein encoded by orf19.5565 was detected by proteomic analyses and had been annotated as a putative 3-hydroxyisobutyrate dehydrogenase from valine catabolism, whereby enzymatic characteristics had not yet been determined. However, 3-hydroxyisobutyrate dehydrogenases from Pseudomonas putida (42) and Bacillus cereus (52) had been shown to be also active as 3-hydroxypropionate dehydrogenases. Thus, we expected that a deletion of orf19.5565 might affect growth on propionate and valerate but not on any of the other carbon sources tested. Indeed, growth of the mutant (lane 4 in Fig. 4) on YPD, glucose, acetate, butyrate, and hexanoate was indistinguishable from that of the wild type. Furthermore, growth on valine (Fig. 5C) was not affected, implying a different pathway for valine degradation in C. albicans. However, consistent with a contribution in propionyl-CoA degradation, colony formation on propionate was no longer observed, and growth on valerate was strongly reduced. Additionally, when tested in liquid cultures, the mutant only revealed some initial growth on propionate that was due to minor amounts of ethanol from the vitamin mix. After this consumption, further growth was completely abolished. Additionally, although the mutant was able to grow on valerate, growth and total biomass formation were reduced. This indicates that orf19.5565 is essential for propionyl-CoA degradation and might indeed act as a 3-hydroxypropionate dehydrogenase.
Phenotypic Characterization of ald5 and ald6 Mutants
The two aldehyde dehydrogenases Ald6p and Ald5p were identified from our proteomic approach (Table 2). Because 3-hydroxypropionate dehydrogenase would lead to the formation of malonate semialdehyde, further degradation requires at least one additional oxidation to form either malonate with a subsequent decarboxylation to acetate or a one-step reaction, in which acetyl-CoA is produced. The latter reaction has been described for methylmalonate-semialdehyde dehydrogenases (acylating CoA) from Bacillus subtilis (53, 54) and, most strikingly, from rat liver (EC 1.2.1.18, accession number Q02253) (55, 56) to which Ald6p displays 50.6% identity. In contrast, identity of Ald5p to the rat liver enzyme was only 29.1%. This made Ald6p a likely candidate for the proposed enzymatic reaction.
A deletion of ALD5 (Fig. 4, lane 5) reduced growth on propionate especially at elevated temperatures, but growth on valerate was similar to that of the wild type. This indicates that Ald5p might contribute to but is not essential for degradation of propionyl-CoA. Further analyses revealed that Ald5p seems generally required for growth on amino acids, but this was not followed in detail. In contrast to the ald5 mutant, the ald6 mutant (Fig. 4, lane 6) revealed growth defects on propionate and valerate similar to the ehd3 and orf19.5565 mutant, whereas colony formation on all other media tested appeared virtually unaffected. Thus, Ald6p might indeed act as an aldehyde dehydrogenase converting malonate semialdehyde to acetyl-CoA. Additionally, as indicated for Ehd3p and the protein encoded by orf19.5565, Ald6p also possesses a putative mitochondrial localization signal. Therefore, production of mitochondrial acetyl-CoA allows further oxidation via the citric acid cycle. Interestingly, a double mutant deleted in ald5 and ald6 (Fig. 4, lane 7) also showed growth defects on glucose, acetate, and butyrate medium, implying that at least one of these dehydrogenases is also required for other metabolic processes independent of propionyl-CoA degradation.
Subcellular Localization of Ehd3p and Hpd1p
In silico analyses by Mitoprot detected putative signal peptides for Ehd3p and Hpd1p comprising the first 24 (Ehd3p) or 16 amino acids (Hpd1p). For both proteins the probability for import was predicted as >95%. Thus, we assumed a transport of 3-hydroxypropionyl-CoA to the mitochondrial compartment, which might relieve inhibition of Fox2p by this CoA ester. To confirm the subcellular localization of Ehd3p and Hpd1p and, additionally, to analyze the induction of gene expression on propionate, we generated HPD1 and EHD3 fusions with enhanced GFP under control of the respective natural promoter. In both cases, the reporter constructs replaced one allele of the respective gene in the wild type. Gene expression was analyzed by determination of background-corrected fluorescence intensities from cell-free extracts of transformants grown on glucose, acetate, valerate, or propionate medium (Fig. 6). An increase in fluorescence was clearly detected for the HPD1-GFP fusion on propionate and, to a lesser extent, on valerate and acetate but not on glucose (Fig. 6A). Differential staining of mitochondria from propionate-grown HPD1-GFP fusion cells with MitoTracker confirmed a co-localization of GFP fluorescence with the mitochondrial network (Fig. 6B). In contrast, fluorescence intensities of EHD3-GFP fusions did not significantly exceed the wild-type background levels regardless of the applied growth condition (data not shown). This implies a general low expression of EHD3 under its natural promoter, which is in agreement with the lack of Ehd3p from our proteomics analyses. However, low abundance of this putative 3-hydroxypropionyl-CoA hydrolase might be required to avoid unspecific hydrolysis of related CoA esters such as acetyl-CoA or malonyl-CoA, which is in agreement with an inability to produce a full-length Ehd3p under control of the actin promoter (data not shown). To confirm that not only Hpd1p but also Ehd3p localizes to mitochondria, we selected the first 48 amino acids of Ehd3p for a GFP fusion under the control of the actin promoter pACT1. The construct replaced one wild-type allele in SC5314 and revealed GFP fluorescence co-localizing with the MitoTracker (Fig. 6C). Thus, Ehd3p and Hpd1p contain functional mitochondrial import signals supporting a relocation of the modified β-oxidation from peroxisomes to mitochondria.
FIGURE 6.
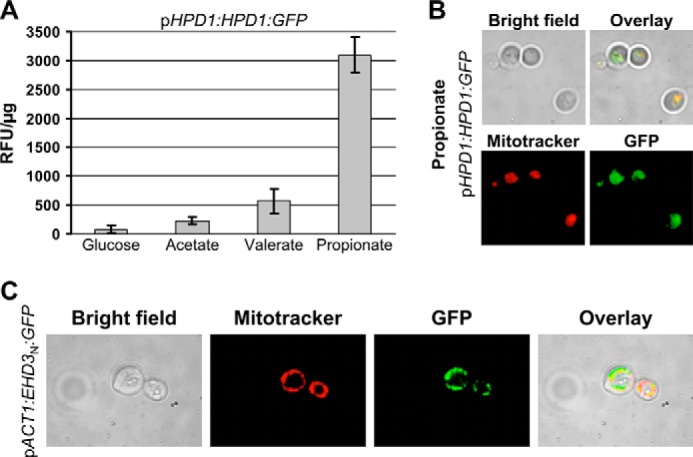
Analysis of HPD1-GFP and EHD3-GFP fusion strains. A, fluorescence intensity of the HPD1-GFP fusion under control of its natural HPD1 promoter (pHPD1). Background fluorescence from the wild type has been subtracted, and values are given as relative fluorescence units/μg of total protein (RFU/μg). Data represent mean values from two independent transformants measured in at least three serial dilutions. Error bars indicate standard deviations. EHD3-GFP fusion strains showed no fluorescence above background levels and are not depicted. B, fluorescence microscopy of a representative pHPD1-HPD1-GFP fusion strain grown in the presence of propionate. Mitochondria were stained with MitoTracker Red. C, subcellular localization of GFP fused with an N-terminal fragment of Ehd3p under the control of the constitutively expressed actin promoter pACT1 (pACT1-EHD3N-GFP). Mitochondria were stained with MitoTracker Red.
Detection of 3-Hydroxypropionate
All our mutant analyses corroborated the proposed metabolism of propionyl-CoA via a modified β-oxidation pathway in which 3-hydroxypropionate is formed as a key intermediate. Therefore, we aimed at the identification of 3-hydroxypropionate from the orf19.5565 mutant. We first cultivated the wild type and the orf19.5565 mutant on a medium containing acetate and propionate to allow for growth of both strains. After 6 h, cells were harvested and lysed, and extracts were analyzed by GC/MS analysis using authentic 3-hydroxypropionate as internal standard. Indeed, 3-hydroxypropionate was detected from the extract of the mutant (Fig. 7). To determine the production of 3-hydroxypropionate in more detail, we quantified the production of 3-hydroxypropionate from a defined number of mutant and wild-type cells. For this purpose, both strains were pre-grown on YPD medium and shifted to acetate/propionate medium at high cell density. Samples were removed after 0, 2, 4, and 6 h, adjusted to the same optical density, and applied to the preparation of cell-free extracts. These extracts were subjected to analytical HPLC. The amount of 3-hydroxypropionate was quantified from the peak area around 15.5 min with authentic 3-hydroxypropionate as standard. Results revealed that up to 4 h no 3-hydroxypropionate was detectable in the wild type, whereas after 6 h a total amount of 2 μmol of 3-hydroxypropionate was detected from the cell extract. For the mutant, 3-hydroxypropionate was undetectable directly after the shift at 0 h, but levels strongly increased to 16 μmol at 2 h, 27 μmol at 4 h, and 41 μmol at 6 h (Fig. 7). Similarly, when wild type and mutant were incubated on valerate, the mutant, but not the wild type, showed a time-dependent accumulation of 3-hydroxypropionate as determined by GC/MS analysis (Fig. 7). These data confirmed the following: (i) 3-hydroxypropionate is produced during growth on nutrient sources that support propionyl-CoA formation, and (ii) the protein encoded by orf19.5565 most likely acts as a 3-hydroxypropionate dehydrogenase. Subsequently, orf19.5565 was therefore referred to as Hpd1p.
FIGURE 7.

Identification and quantification of 3-hydroxypropionic acid by HPLC and GC-MS analysis. Silylation of 3-hydroxypropionate leads to the 3-[(trimethylsilyl)oxy]-trimethylsilyl ester of propionic acid. A, GC-MS spectrum of silylated 3-hydroxypropionate from cell-free extracts of propionate/acetate-cultivated hpd1 mutant cells (upper panel), authentic 3-hydroxypropionate (middle panel), and a reference spectrum from a GC-MS database (lower panel). B, HPLC quantification of 3-hydroxypropionate from cell-free extracts of the hpd1 mutant (left) and wild type (right) cultivated on acetate + propionate medium for the indicated time points. C, GC-MS analysis of 3-hydroxypropionate accumulation from cell-free extracts of the hpd1 mutant (left) and wild type (right) cultivated for the indicated time points on valerate.
To further confirm that that the observed 3-hydroxypropionate directly derives from propionate, we shifted the mutant to a medium containing 20 mm acetate and 20 mm 2-[13C]propionate, harvested cells after 6 h, prepared cell-free extracts, and isolated 3-hydroxypropionate by HPLC. The concentrated fraction was subsequently analyzed by NMR spectroscopy. Its 1H NMR spectrum (Fig. 8) revealed the presence of an AX system consisting of two methylene groups. The corresponding protons were vicinally coupled as indicated by their coupling constants and homonuclear correlation spectroscopy. Furthermore, it was evident that the methylene signal in the higher field was significantly split due to heteronuclear coupling to an adjacent 13C atom (J = 127.8 Hz).
FIGURE 8.

NMR analysis of 3-hydroxypropionate isolated from homozygous hpd1 mutant cultivated for 6 h in the presence of acetate and 2-[13C]propionate. A, 1H NMR spectrum of 3-[2-13C]hydroxypropionic acid recorded at 500 Hz in methanol-d4. The signal of the methylene protons resonating at 2.50 ppm is split into two discrete triplets because of 1J coupling to 13C. B, 1H-decoupled 13C NMR spectrum of 3-[2-13C]hydroxypropionic acid recorded at 125 MHz in methanol-d4. The α-carbon in 3-hydroxypropionic acid is strongly enriched in the 1H-decoupled 13C NMR spectrum, supporting the metabolic origin from 2-[13C]propionate.
Recombinant Production and Characterization of Hpd1p
Accumulation of 3-hydroxypropionate in the hpd1 (orf19.5565) mutant showed that the protein is directly involved in the conversion of this intermediate. Still, substrate specificity and turnover rate of 3-hydroxypropionate remained unknown. Thus, we produced a recombinant protein for biochemical characterization. To this end, we first replaced the N-terminal mitochondrial import sequence with a His tag and used a pET-vector expression system. Unfortunately, most of the protein produced in E. coli ended up in inclusion bodies. The soluble fraction purified by nickel-chelate chromatography yielded an enzyme of only about 20% purity with co-purification of several fragments that may have resulted from premature transcription termination. Therefore, a version with the C-terminal His tag was constructed. Again, a significant proportion of the protein was found in inclusion bodies, but a larger fraction remained soluble, and purification via nickel-chelate affinity chromatography resulted in a protein of about 90–95% purity as judged by SDS-PAGE analysis (Fig. 9A).
FIGURE 9.

Purification and pH dependence of recombinant purified Hpd1p from C. albicans. A, SDS-PAGE analysis. Lane M, molecular mass marker; lane 1, cell-free extract; lane 2, column flow-through; lane 3, wash fraction; lanes 4–6, elution fractions. B, buffer and pH dependence of recombinant Hpd1p. Highest activity is observed in a range between pH 9.5 and 10.0 with CHES buffer.
For biochemical characterization, we studied pH and buffer dependence of the enzymatic reaction, tested several different substrates, and determined Km values for the assumed main substrates. First, we evaluated the effect of different buffers on enzymatic activity using 3-hydroxypropionate as a substrate (Fig. 9B). Similar to bacterial β-hydroxyisobutyrate dehydrogenases (42), an alkaline pH was strongly required for activity. However, a strong dependence on the buffer system was also observed. Thus, CHES buffer at pH 9.5 turned out to be the optimal buffer with only slightly decreased activities at the surrounding pH values of 9.0 and 10.0, and all subsequent analyses were performed using this buffer. Investigations on substrate specificity and specific activity (Table 3) revealed that 3-hydroxypropionate served as substrate supporting highest turnover rates, followed by l-serine, (S)-β-hydroxyisobutyrate, and (R)-β-hydroxyisobutyrate. Although the additional methyl group in the hydroxyisobutyrate isomers negatively affects the maximum turnover rate, it may enhance the substrate binding. The Km values were lowest for (S)-β-hydroxyisobutyrate followed by (R)-β-hydroxyisobutyrate, 3-hydroxypropionate, and l-serine. Furthermore, the Km value for NAD was 10 times lower with (S)-β-hydroxyisobutyrate than with 3-hydroxypropionate. Notwithstanding that the catalytic efficiency was highest with (S)-β-hydroxyisobutyrate, the activity as 3-hydroxypropionate dehydrogenase must play an essential role under in vivo conditions, because deletion of hpd1 led to an accumulation of 3-hydroxypropionate. Further characterization also showed that the enzyme was strictly dependent on the cofactor NAD, because no activity was observed when NADP was used as acceptor for reducing equivalents. EDTA added at concentrations of up to 5 mm did not affect enzymatic activity, indicating that catalysis does not require metal ions.
TABLE 3.
Biochemical characteristics of recombinant purified C. albicans Hpd1p
All values were determined at 22 °C in CHES buffer, pH 9.5.
| Substrate | Aspec (exp)a | Aspec (theo)b | Km | Km (NAD) | kcatc | Keff |
|---|---|---|---|---|---|---|
| units/mg | units/mg | mm | mm | s−1 | m−1 s−1 | |
| 3-Hydroxy-propionate | 40.7 (@36 mm) | 47.40 | 5.37 | 0.22 | 30.97 | 5.77 × 103 |
| (S)-β-Hydroxyisobutyrate | 3.62 (@10 mm) | 3.79 | 0.13 | 0.02 | 2.48 | 1.91 × 104 |
| (R)-β-Hydroxyisobutyrate | 1.85 (@10 mm) | 2.00 | 0.58 | NDd | 1.31 | 2.26 × 103 |
| l-Serine | 5.16 (@30 mm) | 14.40 | 46.7 | NDd | 9.41 | 2.02 × 102 |
a Maximum-specific activities as determined experimentally at given substrate concentrations are shown in parentheses.
b Theoretic maximum-specific activities as deduced from Lineweaver-Burk diagrams.
c Turnover numbers were calculated assuming one active site per subunit, a molecular mass of 39.2 kDa for the recombinant enzyme, and using the theoretical maximum activity value.
d ND = not determined.
Contribution of Hpd1p to 3-Hydroxypropionate Dehydrogenase Activity in C. albicans
To confirm that Hpd1p is the major enzyme catalyzing the oxidation of 3-hydroxypropionate in C. albicans, we cultivated the wild type, the hpd1 deletion mutant, and a strain complemented on one HPD1 allele on glucose, acetate, propionate, and valerate medium. To avoid substrate limitation or the production of acetate during growth on glucose, the strains were pre-cultivated on YPD medium, washed, and transferred for 6 h to glucose and 8 h to the other carbon sources. Activity determinations from cell-free extracts (Table 4) revealed that no activity was detected when cells were shifted to glucose, which is in agreement with results from the GFP fusion constructs and our proteome analyses, in which Hpd1p was not detected. Furthermore, although the homozygous deletion mutant showed no activity above background levels under any of the applied conditions, the wild type and the complemented mutant revealed activity in the order propionate > acetate > valerate, similar to the results obtained from the GFP fusion construct of Hpd1p (Fig. 6A). The induction on acetate and propionate additionally agrees with the identification of Hpd1p from these carbon sources by proteome analyses. Additionally, a gene dosage effect was observed for the complemented mutant that revealed lower Hpd1p activities compared with the wild type. These data clearly indicate that Hpd1p is the major 3-hydroxypropionate dehydrogenase in C. albicans, and accumulation of 3-hydroxypropionate in hpd1 mutants is due to lack of this activity.
TABLE 4.
3-Hydroxypropionate dehydrogenase activity of wild type, Δ/Δhpd1 mutant, and complemented strain (Δhpd1/HPD1)
Cultures were pregrown on YPD and shifted for 6 h to glucose and 8 h to acetate, valerate, or propionate medium. Activities are given in milliunits/mg, and mean values from three determinations with standard deviation are shown.
| Strain | Glucose | Acetate | Propionate | Valerate |
|---|---|---|---|---|
| Wild type | 1.2 ± 1.3 | 95.0 ± 9.7 | 230.1 ± 31.9 | 61.0 ± 8.9 |
| Δ/Δhpd1 | 0 ± 0 | 0.2 ± 0.3 | 1.1 ± 1.0 | 0 ± 0 |
| Δhpd1/HPD1 | 0.1 ± 0.2 | 36.9 ± 3.4 | 120.7 ± 6.3 | 52.3 ± 11.1 |
Contribution of Hpd1p to Virulence in a Murine Model of Systemic Candidiasis
Although deletion of HPD1 only caused minor growth defects under in vitro conditions, the inability to yield energy and building blocks from propionyl-CoA degradation reduced growth speed and maximum biomass yield in the presence of nutrient sources that support propionyl-CoA formation. Therefore, we were interested whether deletion of HPD1 reduces virulence in a murine model of systemic candidiasis, which could indicate that propionyl-CoA is an intermediate from the utilization of host-provided nutrients. Hence, we selected the wild-type SC5314, the homozygous hpd1 deletion strain, and a strain complemented on one allele for systemic mouse infection. As shown in Fig. 10, virulence of the hpd1 mutant was strongly attenuated. Wild type and complemented mutant showed similar virulence, in which 90–100% of mice succumbed to infection within 14 days. In contrast, mice infected with the hpd1 mutant showed a strongly delayed progression of infection and only 30% of mice succumbed to infection within the 21-day observation period. This indicates that, at least in a systemic bloodstream infection model, C. albicans utilizes nutrients that lead to the production of propionyl-CoA, which requires the modified β-oxidation pathway for further utilization.
FIGURE 10.

Murine infection model of disseminated candidiasis. BALB/C mice (n = 10 mice per strain) were infected intravenously with 2.5 × 104 cfu/g of body weight of the hpd1 mutant, the wild type (WT), or the reconstituted mutant (HPD1C). Survival was monitored over a period of 21 days, and data are shown as Kaplan-Meyer plots. The hpd1 mutant reveals a significantly attenuated virulence (p < 0.01) compared with the wild type and the reconstituted mutant as calculated by the Log-rank (Mantel-Cox) test of the GraphPad Prism 5 software.
DISCUSSION
In this study, we aimed at the identification of the metabolic processes that enable C. albicans to utilize propionyl-CoA. This was of special interest, because studies on other pathogenic fungi, e.g. A. fumigatus, have shown that accumulating propionyl-CoA disturbs primary metabolism (20) and attenuates virulence (22). Therefore, it had been assumed that interruption of propionyl-CoA metabolism might provide a suitable target for new antifungal compounds. The methyl citrate cycle is common in Ascomycota such as Aspergillus species (19, 20), Fusarium species (57, 58), and the yeast S. cerevisiae (30, 46). Furthermore, genes encoding enzymes of the methyl citrate cycle have also been detected in Basidiomycota (25), which led to the assumption that this pathway is the general mechanism by which fungi degrade propionyl-CoA. However, exceptions for use of a modified β-oxidation pathway had been proposed for C. rugosa and Candida catenulata (34). However, although 3-hydroxypropionate accumulation had been described for C. rugosa (35), the enzymes involved in propionate metabolism had not been characterized.
As shown by our analyses, C. albicans neither possesses genes of a methyl citrate cycle nor of the methylmalonyl-CoA pathway. It thus appeared likely that propionyl-CoA metabolism in C. albicans could occur via a modified β-oxidation pathway. To test this hypothesis, we initially conducted microarray analyses (data not shown) to identify metabolic genes induced in the presence of propionate compared with those on acetate. Because of the long adaptation phase of C. albicans toward propionate, it was difficult to identify suitable time points for comparative analyses. Early time points of up to 4 h mainly revealed responses pointing to nutrient starvation and stress adaptation without a clear picture on the induction of genes from primary metabolism (data not shown). Therefore, we performed proteome analyses and focused on the major proteins and proteins that were found on one but not the other carbon source. This approach allowed the identification of several proteins that were responsible for propionyl-CoA degradation, underlining the power of proteomics in the analysis of new metabolic pathways.
Propionyl-CoA first enters the β-oxidation pathway as shown by the deletion of the marker gene FOX2 that encodes the sole peroxisomal enoyl-CoA hydratase/hydroxyacyl-CoA dehydrogenase in C. albicans (13, 14, 48). Fox2p performs two consecutive reactions in the oxidation of fatty acids, namely the hydration of an enoyl-CoA and a subsequent dehydrogenase reaction. Because a mutant defective in all ketoacyl-CoA thiolases only showed a moderate growth defect on propionate similar to that observed on acetate (14), we conclude that propionyl-CoA degradation branches at the first part of the Fox2p reaction, which is the formation of 3-hydroxypropionyl-CoA from acrylyl-CoA.
To avoid accumulation of toxic 3-hydroxypropionyl-CoA, it has been proposed that the CoA ester is hydrolyzed (31, 32). Interestingly, our analyses showed that this reaction seems to occur within mitochondria and not in peroxisomes as proposed for plants (31), because our candidate protein Ehd3p localized to mitochondria. For the transport of 3-hydroxypropionyl-CoA to mitochondria, a carnitine shuttle system would be required, because the inner mitochondrial membrane is impermeable for peroxisomal/cytoplasmic CoA esters (60). This transport could be performed by the putative carnitine:acylcarnitine antiporter Crc1p, which is very similar to the acyl-carnitine transporter AcuH (61) from A. nidulans (55.6% identity) which was previously shown to be present in the mitochondrial membrane (62). Furthermore, Cat2p (also called Ctn2p), a peroxisomal and mitochondrial carnitine acyltransferase (63, 64), was detected from acetate- and propionate-grown cells (supplemental Table S1), indicating that the required machinery for the transport of 3-hydroxypropionyl-CoA is present. Carnitine acyltransferases perform reversible reactions. Although acyl-CoA esters are transformed into carnitine esters in the cytoplasm and peroxisomes, the back-reaction is performed by the same enzyme in mitochondria (60). Interestingly, C. albicans contains three carnitine acyltransferase isoenzymes with overlapping function, and virulence studies showed no defects of single or double deletion mutants (64). However, no triple mutant or a mutant defective in the transporter gene CRC1 has been constructed and tested for virulence as yet. Thus, a single carnitine acyltransferase might be sufficient for hydroxypropionyl-CoA transport.
Accumulation of 3-hydroxypropionyl-CoA seems to cause toxic effects as deduced from analyses of the ehd3 mutant. Propionate addition to the ehd3 mutant inhibited growth on all fatty acids, but not on acetate, leading to the speculation that 3-hydroxypropionyl-CoA competitively blocks the dehydrogenase function of Fox2p. However, despite its contribution to propionyl-CoA degradation, it should also be noted that Ehd3p was annotated as an enzyme with 3-hydroxyisobutyryl-CoA hydrolase activity, which is required, at least in mammals, for the degradation of the amino acid valine (65). Studies on purified 3-hydroxyisobutyryl-CoA hydrolase from rat liver have shown that the enzyme is also highly active with 3-hydroxypropionate (66), which made it conceivable that C. albicans Ehd3p could act on valine and propionyl-CoA catabolism. However, deletion of EHD3 did not affect growth on valine. This implies that either a second 3-hydroxyisobutyryl-CoA hydrolase is specifically involved in valine metabolism or that valine catabolism in C. albicans proceeds via a different pathway. Our preliminary results point to a mechanism of valine metabolism that is reminiscent of that in the plant A. thaliana. Here, mutations in the hydroxyisobutyryl-CoA hydrolases (chy1 mutant) showed increased sensitivity toward propionate and isobutyrate but not to valine. In contrast, feeding studies with labeled valine revealed that the labeling pattern of valine was subsequently found in leucine, which indicates that valine is first converted to leucine prior to its degradation (31). Analyses on a C. albicans leu1 mutant, which is defective in the putative 3-isopropylmalate dehydratase, confirmed the expected leucine auxotrophy, but the mutant was also unable to grow in the presence of valine.3 Further studies will be performed to analyze valine degradation in C. albicans in more detail.
The identification of the key intermediate 3-hydroxypropionate in C. albicans provided substantial support for the presence of a modified β-oxidation pathway. In this study, we were able to show that the previously uncharacterized orf19.5565 (now named HPD1) encodes a 3-hydroxypropionate dehydrogenase, which accumulates 3-hydroxypropionate from propionate and the odd-chain fatty acid valerate. Interestingly, similar to Ehd3p, Hpd1p was also annotated as a putative enzyme from valine metabolism by acting as a 3-hydroxyisobutyrate dehydrogenase. Our analyses on recombinant Hpd1p confirmed a maximum substrate turnover with 3-hydroxypropionate. The accumulation of 3-hydroxypropionate in the hpd1 mutant and the lacking dehydrogenase activity in this mutant under inducing conditions confirms its essential contribution to propionyl-CoA degradation. Furthermore, as observed for the ehd3 mutant, the hpd1 mutant showed no growth defect in the presence of valine implying the following: (i) Hpd1p may have specifically adapted from valine degradation to serve for a modified β-oxidation pathway, and (ii) valine degradation in C. albicans does not proceed via the classical valine degradation pathway. Therefore, our analysis describes the first characterization of a eukaryotic 3-hydroxypropionate dehydrogenase that is specifically adapted to a modified β-oxidation pathway for propionyl-CoA degradation. Nevertheless, we cannot exclude that a homologue from C. rugosa had been purified in a previous investigation (67). C. rugosa had been cultivated on isobutyrate with the aim to identify a 3-hydroxyisobutyrate dehydrogenase. A respective enzyme was purified, but activity was only tested with the substrates (S)- and (R)-3-hydroxyisobutyrate. Protein or gene sequences had not been determined. Thus, it cannot be excluded that the C. rugosa enzyme also acts as a 3-hydroxypropionate dehydrogenase. In agreement with this assumption, it should be noted that Hpd1p is conserved in all yeasts of the CUG clade. However, an orthologue is not found in the genome of S. cerevisiae. Bakers' yeast metabolizes propionyl-CoA via the methyl citrate cycle (30, 46) and does not require Hpd1p for a modified β-oxidation pathway. Furthermore, the valine degradation pathway in this yeast has not been explored, but it could also proceed via leucine. This would make Hpd1p dispensable.
The putative aldehyde dehydrogenase Ald6p seems to perform the final step in the conversion of propionyl-CoA to acetyl-CoA for several reasons. (i) Deletion of ALD6 caused growth defects on carbon sources that lead to propionyl-CoA. (ii) Ald6p is >40% identical to bacterial methylmalonate semialdehyde dehydrogenase and >50% identical to the methylmalonate semialdehyde dehydrogenase from rat liver, and these enzymes have been shown to produce acetyl-CoA from malonate semialdehyde (53–56). (iii) Similar to other enzymes of the modified β-oxidation pathway, Ald6p also seems to localize to the mitochondria. Thus, acetyl-CoA is formed in mitochondria and allows direct oxidation via the citric acid cycle for energy production. This mitochondrial localization also causes a problem that may explain the very long adaptation time and low growth rate of C. albicans on propionate, whereas a good growth support is observed on valerate. Genome analyses imply that C. albicans neither possesses an ATP:citrate lyase, nor a citrate lyase. Mitochondrial acetyl-CoA is hence trapped in this compartment and cannot be used for gluconeogenic and biosynthetic purposes. In contrast, metabolism of valerate produces one peroxisomal (by normal β-oxidation) and one mitochondrial (by modified β-oxidation) acetyl-CoA. Because the peroxisomal glyoxylate cycle can generate oxaloacetate, energy gain in mitochondria and biomass production from peroxisomes is balanced. At this point, it remains open to which mechanism cytoplasmic acetyl-CoA or oxaloacetate can be produced during growth on propionate as the sole carbon source. Probably, the putative fumarate reductase is involved in this process which was found to be highly abundant only in propionate-grown cells. Further studies are required to elucidate its contribution in detail. Nevertheless, concerning the conversion of propionyl-CoA to acetyl-CoA, candidates for all key enzymes required for the branch in the modified β-oxidation pathway (Ehd3p, Hpd1p, and Ald6p) have been identified by our analyses.
Finally, we performed virulence studies using the hpd1 mutant as a marker for propionyl-CoA degradation. Our analyses clearly indicate a contribution of this enzyme to virulence implying that propionyl-CoA is generated during the infection process. However, it needs to be investigated whether the sources of propionyl-CoA are amino acids or other compounds, such as cholesterol, as speculated for the pathogenic bacterium Mycobacterium tuberculosis (59, 68). However, the reduced virulence of a C. albicans mutant defective in propionyl-CoA metabolism implies that the modified β-oxidation pathway could provide a target for new antifungal compounds.
Supplementary Material
Acknowledgments
We thank Daniela Hildebrandt and Armin Siering for excellent technical support in gene cloning and metabolite detection by HPLC and GC/MS.
This work was supported by funds from the Hans-Knoell-Institute and the Jena School for Microbial Communication.

This article contains supplemental Table S1.
C. Otzen, B. Bardl, I. D. Jacobsen, M. Nett, and M. Brock, unpublished data.
- CMM
- Candida minimal medium
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- CHES
- 2-(cyclohexylamino)ethanesulfonic acid
- CAPS
- 3-(cyclohexylamino)propanesulfonic acid.
REFERENCES
- 1. Kim J., Sudbery P. (2011) Candida albicans, a major human fungal pathogen. J. Microbiol. 49, 171–177 [DOI] [PubMed] [Google Scholar]
- 2. Gow N. A., Hube B. (2012) Importance of the Candida albicans cell wall during commensalism and infection. Curr. Opin. Microbiol. 15, 406–412 [DOI] [PubMed] [Google Scholar]
- 3. Moran C., Grussemeyer C. A., Spalding J. R., Benjamin D. K., Jr., Reed S. D. (2009) Candida albicans and non-albicans bloodstream infections in adult and pediatric patients: comparison of mortality and costs. Pediatr. Infect Dis. J. 28, 433–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brock M. (2009) Fungal metabolism in host niches. Curr. Opin. Microbiol. 12, 371–376 [DOI] [PubMed] [Google Scholar]
- 5. Fleck C. B., Schöbel F., Brock M. (2011) Nutrient acquisition by pathogenic fungi: nutrient availability, pathway regulation, and differences in substrate utilization. Int. J. Med. Microbiol. 301, 400–407 [DOI] [PubMed] [Google Scholar]
- 6. Rubin-Bejerano I., Fraser I., Grisafi P., Fink G. R. (2003) Phagocytosis by neutrophils induces an amino acid deprivation response in Saccharomyces cerevisiae and Candida albicans. Proc. Natl. Acad. Sci. U.S.A. 100, 11007–11012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lorenz M. C., Bender J. A., Fink G. R. (2004) Transcriptional response of Candida albicans upon internalization by macrophages. Eukaryot. Cell 3, 1076–1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fradin C., De Groot P., MacCallum D., Schaller M., Klis F., Odds F. C., Hube B. (2005) Granulocytes govern the transcriptional response, morphology and proliferation of Candida albicans in human blood. Mol. Microbiol. 56, 397–415 [DOI] [PubMed] [Google Scholar]
- 9. Barelle C. J., Priest C. L., Maccallum D. M., Gow N. A., Odds F. C., Brown A. J. (2006) Niche-specific regulation of central metabolic pathways in a fungal pathogen. Cell. Microbiol. 8, 961–971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shareck J., Belhumeur P. (2011) Modulation of morphogenesis in Candida albicans by various small molecules. Eukaryot. Cell 10, 1004–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lorenz M. C., Fink G. R. (2001) The glyoxylate cycle is required for fungal virulence. Nature 412, 83–86 [DOI] [PubMed] [Google Scholar]
- 12. Lorenz M. C., Fink G. R. (2002) Life and death in a macrophage: role of the glyoxylate cycle in virulence. Eukaryot. Cell 1, 657–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Piekarska K., Mol E., van den Berg M., Hardy G., van den Burg J., van Roermund C., MacCallum D., Odds F., Distel B. (2006) Peroxisomal fatty acid β-oxidation is not essential for virulence of Candida albicans. Eukaryot. Cell 5, 1847–1856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Otzen C., Müller S., Jacobsen I. D., Brock M. (2013) Phylogenetic and phenotypic characterisation of the 3-ketoacyl-CoA thiolase gene family from the opportunistic human pathogenic fungus Candida albicans. FEMS Yeast Res. 13, 553–564 [DOI] [PubMed] [Google Scholar]
- 15. Piekarska K., Hardy G., Mol E., van den Burg J., Strijbis K., van Roermund C., van den Berg M., Distel B. (2008) The activity of the glyoxylate cycle in peroxisomes of Candida albicans depends on a functional β-oxidation pathway: evidence for reduced metabolite transport across the peroxisomal membrane. Microbiology 154, 3061–3072 [DOI] [PubMed] [Google Scholar]
- 16. Schwab M. A., Sauer S. W., Okun J. G., Nijtmans L. G., Rodenburg R. J., van den Heuvel L. P., Dröse S., Brandt U., Hoffmann G. F., Ter Laak H., Kölker S., Smeitink J. A. (2006) Secondary mitochondrial dysfunction in propionic aciduria: a pathogenic role for endogenous mitochondrial toxins. Biochem. J. 398, 107–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Morath M. A., Okun J. G., Müller I. B., Sauer S. W., Hörster F., Hoffmann G. F., Kölker S. (2008) Neurodegeneration and chronic renal failure in methylmalonic aciduria–a pathophysiological approach. J. Inherit. Metab. Dis. 31, 35–43 [DOI] [PubMed] [Google Scholar]
- 18. Brock M., Buckel W. (2004) On the mechanism of action of the antifungal agent propionate. Eur. J. Biochem. 271, 3227–3241 [DOI] [PubMed] [Google Scholar]
- 19. Brock M., Fischer R., Linder D., Buckel W. (2000) Methylcitrate synthase from Aspergillus nidulans: implications for propionate as an antifungal agent. Mol. Microbiol. 35, 961–973 [DOI] [PubMed] [Google Scholar]
- 20. Maerker C., Rohde M., Brakhage A. A., Brock M. (2005) Methylcitrate synthase from Aspergillus fumigatus. Propionyl-CoA affects polyketide synthesis, growth and morphology of conidia. FEBS J. 272, 3615–3630 [DOI] [PubMed] [Google Scholar]
- 21. Zhang Y. Q., Brock M., Keller N. P. (2004) Connection of propionyl-CoA metabolism to polyketide biosynthesis in Aspergillus nidulans. Genetics 168, 785–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ibrahim-Granet O., Dubourdeau M., Latgé J. P., Ave P., Huerre M., Brakhage A. A., Brock M. (2008) Methylcitrate synthase from Aspergillus fumigatus is essential for manifestation of invasive aspergillosis. Cell. Microbiol. 10, 134–148 [DOI] [PubMed] [Google Scholar]
- 23. Chandler R. J., Venditti C. P. (2005) Genetic and genomic systems to study methylmalonic acidemia. Mol. Genet. Metab. 86, 34–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Brock M. (2010) in Handbook of Hydrocarbon and Lipid Microbiology (Timmis K. N., ed) pp. 3279–3291, Springer, Berlin [Google Scholar]
- 25. Müller S., Fleck C. B., Wilson D., Hummert C., Hube B., Brock M. (2011) Gene acquisition, duplication and metabolic specification: the evolution of fungal methylisocitrate lyases. Environ. Microbiol. 13, 1534–1548 [DOI] [PubMed] [Google Scholar]
- 26. Brock M., Maerker C., Schütz A., Völker U., Buckel W. (2002) Oxidation of propionate to pyruvate in Escherichia coli. Involvement of methylcitrate dehydratase and aconitase. Eur. J. Biochem. 269, 6184–6194 [DOI] [PubMed] [Google Scholar]
- 27. Brock M., Darley D., Textor S., Buckel W. (2001) 2-Methylisocitrate lyases from the bacterium Escherichia coli and the filamentous fungus Aspergillus nidulans: characterization and comparison of both enzymes. Eur. J. Biochem. 268, 3577–3586 [DOI] [PubMed] [Google Scholar]
- 28. Grimm C., Evers A., Brock M., Maerker C., Klebe G., Buckel W., Reuter K. (2003) Crystal structure of 2-methylisocitrate lyase (PrpB) from Escherichia coli and modelling of its ligand bound active centre. J. Mol. Biol. 328, 609–621 [DOI] [PubMed] [Google Scholar]
- 29. Brock M. (2005) Generation and phenotypic characterization of Aspergillus nidulans methylisocitrate lyase deletion mutants: methylisocitrate inhibits growth and conidiation. Appl. Environ. Microbiol. 71, 5465–5475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Luttik M. A., Kötter P., Salomons F. A., van der Klei I. J., van Dijken J. P., Pronk J. T. (2000) The Saccharomyces cerevisiae ICL2 gene encodes a mitochondrial 2-methylisocitrate lyase involved in propionyl-coenzyme A metabolism. J. Bacteriol. 182, 7007–7013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lucas K. A., Filley J. R., Erb J. M., Graybill E. R., Hawes J. W. (2007) Peroxisomal metabolism of propionic acid and isobutyric acid in plants. J. Biol. Chem. 282, 24980–24989 [DOI] [PubMed] [Google Scholar]
- 32. Halarnkar P. P., Blomquist G. J. (1989) Comparative aspects of propionate metabolism. Comp. Biochem. Physiol. B 92, 227–231 [DOI] [PubMed] [Google Scholar]
- 33. Lee S. H., Park S. J., Park O. J., Cho J., Rhee J. W. (2009) Production of 3-hydroxypropionic acid from acrylic acid by newly isolated Rhodococcus erythropolis LG12. J. Microbiol. Biotechnol. 19, 474–481 [DOI] [PubMed] [Google Scholar]
- 34. Miyakoshi S., Uchiyama H., Someya T., Satoh T., Tabuchi T. (1987) Distribution of the methylcitric acid cycle and β-oxidation pathway for propionate catabolism in fungi. Agric. Biol. Chem. 51, 2381–2387 [Google Scholar]
- 35. Hasegawa J., Ogura M., Kanema H., Kawaharada H., Watanabe K. (1982) Production of 3-hydroxypropionic acid from propionic acid by a Candida rugosa mutant unable to assimilate propionic acid. J. Ferment. Technol. 60, 591–594 [Google Scholar]
- 36. Kniemeyer O., Lessing F., Scheibner O., Hertweck C., Brakhage A. A. (2006) Optimisation of a 2-D gel electrophoresis protocol for the human-pathogenic fungus Aspergillus fumigatus. Curr. Genet. 49, 178–189 [DOI] [PubMed] [Google Scholar]
- 37. Neuhoff V., Arold N., Taube D., Ehrhardt W. (1988) Improved staining of proteins in polyacrylamide gels including isoelectric focusing gels with clear background at nanogram sensitivity using Coomassie Brilliant Blue G-250 and R-250. Electrophoresis 9, 255–262 [DOI] [PubMed] [Google Scholar]
- 38. Vödisch M., Albrecht D., Lessing F., Schmidt A. D., Winkler R., Guthke R., Brakhage A. A., Kniemeyer O. (2009) Two-dimensional proteome reference maps for the human pathogenic filamentous fungus Aspergillus fumigatus. Proteomics 9, 1407–1415 [DOI] [PubMed] [Google Scholar]
- 39. Reuss O., Vik A., Kolter R., Morschhäuser J. (2004) The SAT1 flipper, an optimized tool for gene disruption in Candida albicans. Gene 341, 119–127 [DOI] [PubMed] [Google Scholar]
- 40. Hortschansky P., Eisendle M., Al-Abdallah Q., Schmidt A. D., Bergmann S., Thön M., Kniemeyer O., Abt B., Seeber B., Werner E. R., Kato M., Brakhage A. A., Haas H. (2007) Interaction of HapX with the CCAAT-binding complex–a novel mechanism of gene regulation by iron. EMBO J. 26, 3157–3168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fazius F., Shelest E., Gebhardt P., Brock M. (2012) The fungal α-aminoadipate pathway for lysine biosynthesis requires two enzymes of the aconitase family for the isomerization of homocitrate to homoisocitrate. Mol. Microbiol. 86, 1508–1530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chowdhury E. K., Nagata S., Misono H. (1996) 3-Hydroxyisobutyrate dehydrogenase from Pseudomonas putida E23: purification and characterization. Biosci. Biotechnol. Biochem. 60, 2043–2047 [DOI] [PubMed] [Google Scholar]
- 43. Roessner U., Wagner C., Kopka J., Trethewey R. N., Willmitzer L. (2000) Technical advance: simultaneous analysis of metabolites in potato tuber by gas chromatography-mass spectrometry. Plant J. 23, 131–142 [DOI] [PubMed] [Google Scholar]
- 44. Villas-Bôas S. G., Noel S., Lane G. A., Attwood G., Cookson A. (2006) Extracellular metabolomics: a metabolic footprinting approach to assess fiber degradation in complex media. Anal. Biochem. 349, 297–305 [DOI] [PubMed] [Google Scholar]
- 45. Pronk J. T., van der Linden-Beuman A., Verduyn C., Scheffers W. A., van Dijken J. P. (1994) Propionate metabolism in Saccharomyces cerevisiae: implications for the metabolon hypothesis. Microbiology 140, 717–722 [DOI] [PubMed] [Google Scholar]
- 46. Graybill E. R., Rouhier M. F., Kirby C. E., Hawes J. W. (2007) Functional comparison of citrate synthase isoforms from S. cerevisiae. Arch. Biochem. Biophys. 465, 26–37 [DOI] [PubMed] [Google Scholar]
- 47. Ansede J. H., Pellechia P. J., Yoch D. C. (1999) Metabolism of acrylate to β-hydroxypropionate and its role in dimethylsulfoniopropionate lyase induction by a salt marsh sediment bacterium, Alcaligenes faecalis M3A. Appl. Environ. Microbiol. 65, 5075–5081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Shen Y. Q., Burger G. (2009) Plasticity of a key metabolic pathway in fungi. Funct. Integr. Genomics 9, 145–151 [DOI] [PubMed] [Google Scholar]
- 49. Hawes J. W., Jaskiewicz J., Shimomura Y., Huang B., Bunting J., Harper E. T., Harris R. A. (1996) Primary structure and tissue-specific expression of human β-hydroxyisobutyryl-coenzyme A hydrolase. J. Biol. Chem. 271, 26430–26434 [DOI] [PubMed] [Google Scholar]
- 50. Ooiwa T., Goto H., Tsukamoto Y., Hayakawa T., Sugiyama S., Fujitsuka N., Shimomura Y. (1995) Regulation of valine catabolism in canine tissues: tissue distributions of branched-chain aminotransferase and 2-oxo acid dehydrogenase complex, methacrylyl-CoA hydratase and 3-hydroxyisobutyryl-CoA hydrolase. Biochim. Biophys. Acta 1243, 216–220 [DOI] [PubMed] [Google Scholar]
- 51. Steele M. I., Lorenz D., Hatter K., Park A., Sokatch J. R. (1992) Characterization of the mmsAB operon of Pseudomonas aeruginosa PAO encoding methylmalonate-semialdehyde dehydrogenase and 3-hydroxyisobutyrate dehydrogenase. J. Biol. Chem. 267, 13585–13592 [PubMed] [Google Scholar]
- 52. Yao T., Xu L., Ying H., Huang H., Yan M. (2010) The catalytic property of 3-hydroxyisobutyrate dehydrogenase from Bacillus cereus on 3-hydroxypropionate. Appl. Biochem. Biotechnol. 160, 694–703 [DOI] [PubMed] [Google Scholar]
- 53. Stines-Chaumeil C., Talfournier F., Branlant G. (2006) Mechanistic characterization of the MSDH (methylmalonate semialdehyde dehydrogenase) from Bacillus subtilis. Biochem. J. 395, 107–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Talfournier F., Stines-Chaumeil C., Branlant G. (2011) Methylmalonate-semialdehyde dehydrogenase from Bacillus subtilis: substrate specificity and coenzyme A binding. J. Biol. Chem. 286, 21971–21981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Goodwin G. W., Rougraff P. M., Davis E. J., Harris R. A. (1989) Purification and characterization of methylmalonate-semialdehyde dehydrogenase from rat liver. Identity to malonate-semialdehyde dehydrogenase. J. Biol. Chem. 264, 14965–14971 [PubMed] [Google Scholar]
- 56. Popov K. M., Kedishvili N. Y., Harris R. A. (1992) Coenzyme A- and NADH-dependent esterase activity of methylmalonate semialdehyde dehydrogenase. Biochim. Biophys. Acta 1119, 69–73 [DOI] [PubMed] [Google Scholar]
- 57. Domin N., Wilson D., Brock M. (2009) Methylcitrate cycle activation during adaptation of Fusarium solani and Fusarium verticillioides to propionyl-CoA-generating carbon sources. Microbiology 155, 3903–3912 [DOI] [PubMed] [Google Scholar]
- 58. Lee S. H., Han Y. K., Yun S. H., Lee Y. W. (2009) Roles of the glyoxylate and methylcitrate cycles in sexual development and virulence in the cereal pathogen Gibberella zeae. Eukaryot. Cell 8, 1155–1164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Griffin J. E., Pandey A. K., Gilmore S. A., Mizrahi V., McKinney J. D., Bertozzi C. R., Sassetti C. M. (2012) Cholesterol catabolism by Mycobacterium tuberculosis requires transcriptional and metabolic adaptations. Chem. Biol. 19, 218–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hynes M. J., Murray S. L., Andrianopoulos A., Davis M. A. (2011) Role of carnitine acetyltransferases in acetyl coenzyme A metabolism in Aspergillus nidulans. Eukaryot. Cell 10, 547–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. De Lucas J. R., Domínguez A. I., Valenciano S., Turner G., Laborda F. (1999) The acuH gene of Aspergillus nidulans, required for growth on acetate and long-chain fatty acids, encodes a putative homologue of the mammalian carnitine/acylcarnitine carrier. Arch. Microbiol. 171, 386–396 [DOI] [PubMed] [Google Scholar]
- 62. Ramón De Lucas J., Martínez O., Pérez P., Isabel López M., Valenciano S., Laborda F. (2001) The Aspergillus nidulans carnitine carrier encoded by the acuH gene is exclusively located in the mitochondria. FEMS Microbiol. Lett. 201, 193–198 [DOI] [PubMed] [Google Scholar]
- 63. Strijbis K., van Roermund C. W., Visser W. F., Mol E. C., van den Burg J., MacCallum D. M., Odds F. C., Paramonova E., Krom B. P., Distel B. (2008) Carnitine-dependent transport of acetyl coenzyme A in Candida albicans is essential for growth on nonfermentable carbon sources and contributes to biofilm formation. Eukaryot. Cell 7, 610–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zhou H., Lorenz M. C. (2008) Carnitine acetyltransferases are required for growth on non-fermentable carbon sources but not for pathogenesis in Candida albicans. Microbiology 154, 500–509 [DOI] [PubMed] [Google Scholar]
- 65. Ishigure K., Shimomura Y., Murakami T., Kaneko T., Takeda S., Inoue S., Nomoto S., Koshikawa K., Nonami T., Nakao A. (2001) Human liver disease decreases methacrylyl-CoA hydratase and β-hydroxyisobutyryl-CoA hydrolase activities in valine catabolism. Clin. Chim. Acta 312, 115–121 [DOI] [PubMed] [Google Scholar]
- 66. Shimomura Y., Murakami T., Fujitsuka N., Nakai N., Sato Y., Sugiyama S., Shimomura N., Irwin J., Hawes J. W., Harris R. A. (1994) Purification and partial characterization of 3-hydroxyisobutyryl-coenzyme A hydrolase of rat liver. J. Biol. Chem. 269, 14248–14253 [PubMed] [Google Scholar]
- 67. Hasegawa J. (1981) Purification, crystallization and some properties of β-hydroxyisobutyrate dehydrogenase from Candida rugosa IFO 0750. Agric. Biol. Chem. 45, 2805–2814 [Google Scholar]
- 68. Brzostek A., Pawelczyk J., Rumijowska-Galewicz A., Dziadek B., Dziadek J. (2009) Mycobacterium tuberculosis is able to accumulate and utilize cholesterol. J. Bacteriol. 191, 6584–6591 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.