

Background: The effects of glucocorticoids on the expression of negative feedback regulators of NF-κB are not well understood.
Results: A novel intronic enhancer for TNFAIP3 was synergistically induced by the glucocorticoid receptor and NF-κB.
Conclusion: The glucocorticoid receptor can cooperate with NF-κB to enhance the expression of anti-inflammatory genes such as TNFAIP3.
Significance: These results establish a novel mechanism for anti-inflammatory effects of glucocorticoids.
Keywords: Chromatin Immunoprecipitation (ChiP), Cooperativity, Glucocorticoid Receptor, NF-kappa B (NF-KB), Transcription Enhancers, TNFAIP3, Negative Feedback
Abstract
TNF expression is elevated in asthma and other inflammatory airway diseases that are commonly treated with glucocorticoid-based therapies, but the impact of glucocorticoids on negative feedback control of TNF is not well understood. We analyzed the effect of dexamethasone, a potent synthetic glucocorticoid, on TNF-regulated gene expression in cultured airway epithelial cells. Although dexamethasone-mediated activation of the glucocorticoid receptor (GR) potently repressed expression of IL1β, IL8, and several other pro-inflammatory TNF targets, the expression of anti-inflammatory TNF targets such as TNFAIP3 (A20) and NFKBIA was selectively spared or augmented by dexamethasone treatment. Despite divergent effects on gene expression, GR and NF-κB occupancy at the TNFAIP3 locus and GR-repressed targets was similar. A co-occupied intronic TNFAIP3 regulatory element mediated cooperative enhancement of transcription by GR and NF-κB that required the presence of a functional GR binding site (GBS). GBS exchanges between reporters for TNFAIP3 and FKBP5, a canonical GR-induced target, revealed substantial latitude in the GBS sequence requirements for GR/NF-κB cooperation, suggesting that the TNFAIP3 GBS acts primarily as a docking site in this context. Supporting this notion, a selective GR ligand with only weak agonist activity for induction of FKBP5 enabled robust GR/NF-κB cooperative induction of a mutant TNFAIP3 reporter harboring the FKBP5 GBS. Taken together, our data support a model in which the expression of anti-inflammatory targets of TNF is maintained during treatment with glucocorticoids through context-dependent cooperation between GR and NF-κB.
Introduction
Synthetic glucocorticoids continue to be a mainstay in treating immune-mediated disease (1). Glucocorticoids function primarily through binding to the glucocorticoid receptor (GR),2 which, in response to ligand, translocates to the nucleus and regulates gene expression, leading to both therapeutic inflammatory suppression and the development of deleterious side effects (1, 2). GR utilizes specific glucocorticoid binding sites and tethering interactions with other transcription factors to associate with DNA, leading to the assembly of activating or repressive complexes and alterations in polymerase II occupancy and processivity (3). GR-mediated gene regulation is itself subject to a wide range of regulatory mechanisms including alternate splicing and posttranslational modification of GR (4–6), restricted expression of co-regulators (7), cell type-specific chromatin architecture (8), and binding site-mediated effects on GR recruitment and activity (9, 10). Despite our growing understanding of the molecular basis for GR function, the precise targets and mechanisms through which GR orchestrates the resolution of inflammation across diverse cellular and disease contexts remain poorly understood.
It has recently become clear that normal termination of inflammatory responses requires the activity of negative feedback circuitry (11, 12). For example, in addition to provoking inflammation, tumor necrosis factor-α (TNF) and other cytokines induce potent anti-inflammatory genes such as TNFAIP3 (A20), a key inhibitor of NF-κB whose dysfunction is associated with inflammatory disorders ranging from rheumatoid arthritis to sepsis (13, 14). Repression of NF-κB function by GR has long been implicated as a crucial determinant in glucocorticoid-based therapeutics, but this activity has generally been attributed to GR associating directly with NF-κB to reduce the expression of specific pro-inflammatory targets (15, 16). Recent studies have suggested greater complexity to GR/NF-κB cross-talk (17–20), with co-occupancy by both factors implicated in driving a variety of effects on steady state target gene expression (21, 22). However, these studies have not directly addressed the role of GR in regulating the expression of negative feedback targets of NF-κB, such as TNFAIP3, nor have the mechanisms underpinning differential transcriptional consequences of GR/NF-κB cross-talk at specific loci been fully determined.
Elevated expression of TNF occurs in asthma, an inflammatory disorder of the airway that is frequently treated with glucocorticoid-based therapies (23). We therefore assayed expression of pro- and anti-inflammatory targets of TNF in airway epithelial cells after treatment with dexamethasone (dex), a potent glucocorticoid. We applied chromatin immunoprecipitation, reporter assays, binding site swaps, and varied GR ligand chemistries to probe the molecular basis for selectively maintained expression of negative feedback targets of TNF after dex treatment, with a primary focus on TNFAIP3 regulation. Our results suggest that context-dependent cooperation between GR and NF-κB enables glucocorticoids to preserve negative feedback control of inflammation, thus contributing to the potent effects of glucocorticoids in treating inflammatory disorders of the airway.
EXPERIMENTAL PROCEDURES
Cell Culture and Reagents
Beas-2B cells (ATCC CRL-9609) were cultured in Dulbecco's modified Eagle's medium (DMEM) containing 4.5 g/liter glucose, l-glutamine and supplemented with 1% penicillin-streptomycin and 10% fetal bovine serum (FBS; HyClone). dex (D1756) was purchased from Sigma and used at a concentration of 100 nm. TNF was purchased from Sigma (T6674) and Life Technologies (PHC3015L). TNF-neutralizing antibody (D1B4) was obtained from Cell Signaling Technology. MK-5932 was previously described (24) and was a generous gift from Merck and Co. Antibodies used for Western analyses were: anti-TNFAIP3 (ab13597), anti-HBEGF (ab92620), anti-β-actin (ab75186) from Abcam; anti-GR (H-300; sc-8992) and anti-NF-κB p65 (C-20; sc-372) from Santa Cruz Biotechnology; and enhanced chemiluminescence (ECL) donkey anti-rabbit IgG-horseradish peroxidase (HRP; NA9340V) and ECL sheep anti-mouse IgG-HRP (NA931V) from GE Healthcare. Antibodies used for chromatin immunoprecipitation (ChIP) were anti-GR (N-499, a generous gift from Dr. Keith Yamamoto, and 1A1, a generous gift from Dr. Miles Pufall) and anti-NFkB p65 (C-20; sc-372) from Santa Cruz Biotechnology. The GFP (Ad-GFP) and TNFAIP3 (Ad-TNFAIP3) adenoviruses were obtained from Welgen; Ad-TNFAIP3 was constructed using a previously described TNFAIP3 expression vector (25). siRNA studies were conducted using ON-TARGETplus SMARTpool against human GR (siNR3C1; NM-001020825), human NFkB (siRELA; L-003533-00-0020), and nontargeting control (siCtrl; D-001810-10-05) from Dharmacon.
Plasmids
To generate pTNFAIP3I2, PCR primers to amplify +5670–6491 of the TNFAIP3 locus were designed based on visualizing published ChIP-seq peaks in the UCSC genome browser and identifying putative GR and NF-κB binding sites using MatInspector (Genomatix). A core binding sequence similarity of 95% was used as a cut-off to identify the consensus GR and NF-κB binding sequences demarcated in Fig. 4. Amplified PCR product was TA-cloned into the pCR 2.1-TOPO vector according to the manufacturer's protocol (Life Technologies) and subsequently introduced into the PGL3 promoter vector (Promega) as a KpnI/XhoI fragment to generate a luciferase reporter for TNFAIP3I2 enhancer activity. Site-directed mutagenesis of the putative GR binding site was accomplished using the QuikChange II site-directed mutagenesis kit from Agilent Technologies. Primer sequences are listed in the supplemental tables. pFKBP5, pIL8, and p3XNF-κB have been previously described (28, 31, 32) as indicated under “Results.”
FIGURE 4.
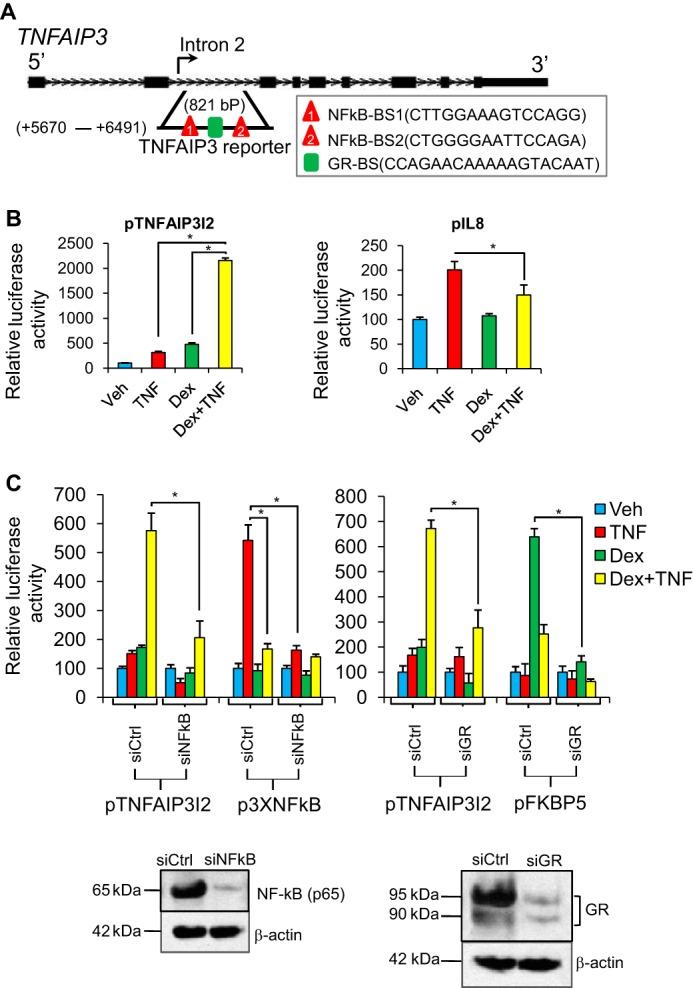
Synergistic induction of an intronic TNFAIP3 enhancer by NF-κB and GR. A, schematic diagram of the TNFAIP3 locus. The region from intron 2 used to generate the pTNFAIP3I2 reporter plasmid and the relative location of strong consensus GR (GR-BS) and NF-κB (NFkB-BS) binding sites are noted. B, relative luciferase activity of the indicated reporter constructs after transfection into Beas-2B cells and treatment with vehicle (Veh), TNF, dex, or TNF + dex for 8 h. C, relative luciferase activity of pTNFAIP3I2, p3xNFκB (NF-κB control reporter), and pFKBP5 (GR control reporter) in Beas-2B cells co-transfected with control nontargeting siRNA (siCtrl), NF-κB-targeting siRNA (siNFkB), or GR-targeting siRNA (siGR) and treated as indicated. For B and C, luciferase activity values for each reporter were normalized to a pSV40-Renilla control. Bars indicate means of biological quadruplicate + S.D. * indicates p ≤ 0.05 for relevant comparisons. D, Western blot analysis of NFkB, GR, and β-actin protein expression in Beas-2B cells transfected for 24 h with siCtrl, siNFkB, or siGR.
Transfections, Luciferase Assays, and qPCR
For luciferase assays, cells were plated in 250 μl of antibiotic-free DMEM supplemented with 10% FBS in 48-well plates in at a density of ∼4 × 104 cells/well and incubated overnight prior to plasmid transfection. The next day, a complex of Lipofectamine 2000 (1 μl) and total DNA (400 ng) diluted in 50 μl/well Opti-MEM (Life Technologies) was added to each well. DNA complexes were formed from firefly luciferase plasmids and the Renilla luciferase (RL) expression vector, pSV40-RL (Promega), at a ratio of 10:1 using Lipofectamine 2000 (Life Technologies) transfection reagent according to the manufacturer's protocol. 18 h after transfection, cells were treated with TNF and/or dex for 8 h. Cells were subsequently assayed for luciferase activity as described previously (26) in biologic quadruplicate. p values indicated in the figure legends were calculated using Student's t tests or nonparametric analysis. For siRNA transfection, Beas-2B cells were transfected with 25 nm siGR, siNFkB, or siCtrl using Lipofectamine RNAiMAX transfection reagent according to the manufacturer's protocol (Life Technologies). 24–48 h later cells were lysed and assayed for GR or NFkB knockdown using Western blot analysis. Cells were also co-transfected with the above siRNAs in combination with firefly and Renilla luciferase plasmids. 24 h later cells were treated as described above and subsequently assayed for luciferase activity. For gene expression analysis, cells were plated in 6-well plates at a density of 3 × 105 cells/well. The next day, cells were treated with TNF and/or dex, and RNA was subsequently prepared using TRIzol reagent and the Pure Link RNA mini kit, both from Life Technologies. RNA was reverse-transcribed, qPCR was performed, and gene expression was quantified as described previously (26). Primer sequences are in the supplemental tables. For adenoviral transduction, Beas 2B cells were transduced with adeno-TNFAIP3 (Ad-TNFAIP3) or control adeno-green fluorescent protein (Ad-GFP) at a multiplicity of infection of 100. After ∼17 h cells were treated as indicated under “Results” and assayed for gene expression by quantitative RT-PCR.
Western Analysis
To measure the expression of TNFAIP3 and HBEGF proteins or to assay for GR and NF-κB knockdown at the protein level upon siRNA transfection, treated Beas-2B cells were lysed with radioimmunoprecipitation assay buffer containing 1× protease inhibitor cocktail (Thermo Scientific). 50 μg of protein of each sample were separated by SDS-PAGE and transferred into PVDF membrane (Amersham Biosciences). Membranes were then immunoprobed for the detection of the corresponding proteins. Band visualization was carried out using ECL Plus Western blotting detection system (GE Healthcare).
ChIP
Beas-2B cells were grown to confluence in 100-mm dishes and treated with vehicle (ethanol), 100 nm dex, 20 ng/ml TNF or with a combination of dex and TNF for 1 h. 16% methanol-free formaldehyde (Thermo Scientific) was added directly to the culture medium to a final concentration of 1%, and ChIP was subsequently performed as described (26). Resulting DNA was analyzed using qPCR. Relative occupancy was calculated on a log2 scale based on comparison with the geometric mean of CT values for two or three negative control regions. Validity of negative controls was established through demonstrating that amplification of dilute input DNA generated similar CT values for primers to control and putative occupied regions. Primer sequences are in the supplemental tables.
RESULTS
Glucocorticoids Selectively Spare the Expression of Anti-inflammatory Targets of TNF
TNF is implicated as a driver in inflammatory airway diseases that are treated with glucocorticoids, but the effects of glucocorticoids on negative feedback control of TNF signaling have not been evaluated in airway epithelial cells. We therefore used Beas-2B cells, a human airway epithelial cell line, to analyze the expression of a set of well described pro- and anti-inflammatory targets of TNF signaling. Cells were treated with TNF, dex, which is a potent GR agonist, or a combination of both agents for a total of 4 h. As expected, the expression of several typical pro-inflammatory targets of TNF, such as IL8 and HBEGF, was strongly induced by TNF (Fig. 1A, red bars). The inductive effect of TNF on each of these genes was abrogated significantly (p < 0.05) by co-treatment with dex (Fig. 1A, yellow bars). In contrast, but concordant with findings from recent studies in other cell types (17, 19, 22), the expression of anti-inflammatory targets of TNF such as TNFAIP3 (A20), TNIP1, and NFKBIA was spared or augmented by TNF + dex co-treatment in comparison with treatment with TNF alone (Fig. 1B, compare red and yellow bars). The differential effects of dex on the expression of pro- and anti-inflammatory TNF target genes were confirmed at the protein level for HBEGF and TNFAIP3 (Fig. 1C). Thus, induction of GR signaling by dex spares or augments the expression of anti-inflammatory targets of TNF, whereas it potently represses the expression of inflammatory mediators.
FIGURE 1.
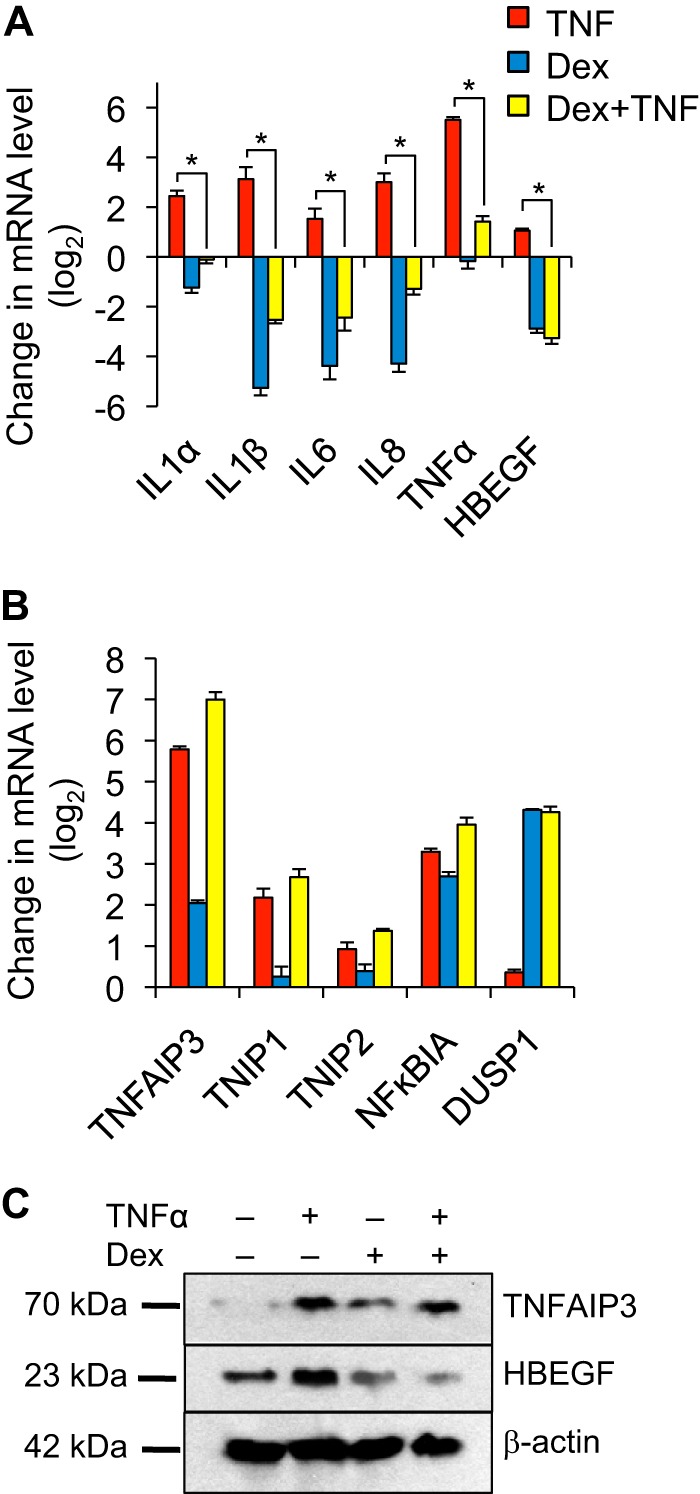
Glucocorticoids selectively repress pro-inflammatory responses to TNF. A and B, qPCR analysis of pro-inflammatory (A) and anti-inflammatory (B) gene expression in Beas-2B cells treated for 4 h with vehicle, TNF (20 ng/ml), dex (100 nm), or TNF + dex as indicated. For both A and B, bar graphs indicate mean normalized CT values of biological quadruplicate samples relative to vehicle control + S.D., * indicates p ≤ 0.05 for relevant comparisons. C, Western analysis of TNFAIP3, HBEGF, and β-actin (loading control) proteins using lysates prepared from Beas-2B cells treated as indicated for 24 h.
Next we asked whether differential regulation of pro- and anti-inflammatory gene expression is specific to GR-based inhibition of TNF signaling. We took two approaches to address this question. First, we used a TNF-neutralizing antibody to block upstream TNF signaling. We incubated increasing concentrations of TNF-neutralizing antibody with recombinant TNF for 2 h and treated cells with the resultant complexes for 4 h prior to analyzing gene expression via qPCR. In contrast to the effects of dex treatment, antibody-based blockade of TNF repressed the induction of both pro-inflammatory and anti-inflammatory genes by TNF in a dose-dependent manner (Fig. 2A). Second, we used an adenoviral system to overexpress TNFAIP3 (Ad-TNFAIP3) as a mechanism to abrogate TNF signaling through reducing NF-κB activity. Here, gene regulation by TNF, dex, or TNF + dex was analyzed after infection with Ad-TNFAIP3 in comparison with infection with a control adenovirus (Ad-GFP). Although there were some differences in sensitivity of different targets, forced TNFAIP3 expression generally reduced the expression of both pro-inflammatory and anti-inflammatory targets (Fig. 2B). Primers that amplify a region of the TNFAIP3 5′-UTR were used in this experiment to measure endogenous TNFAIP3 expression (Fig. 2B), whereas overexpression of TNFAIP3 driven by the adenovirus was confirmed both by qPCR using primers for TNFAIP3 exon 5 (Fig. 2B) and by Western blot (Fig. 2C). Taken together, these data indicate that, unlike the selective regulatory effects of GR activation, restraining TNF signaling through either upstream blockade or inhibition of NF-κB activity decreases both pro-inflammatory and anti-inflammatory gene expression.
FIGURE 2.

TNF blockade and TNFAIP3 overexpression repress both pro-inflammatory and anti-inflammatory gene expression in Beas-2B cells. A, relative mRNA levels of pro-inflammatory (left) and anti-inflammatory (right) genes in Beas-2B cells treated with a combination of TNF (20 ng/ml) and increasing concentrations of anti-TNF-neutralizing antibody for 4 h; TNF was allowed to complex with antibody for 2 h prior to addition to the cells. B, effect of adenoviral mediated TNFAIP3 (Ad-TNFAIP3) overexpression on mRNA levels relative to Ad-GFP control of the indicated pro-inflammatory (top) and anti-inflammatory (bottom) genes in Beas-2B cells treated with TNF (20 ng/ml), dex (100 nm), or both as indicated. For both A and B, bar graphs indicate mean normalized CT values of biological quadruplicate samples relative to vehicle-treated control + S.D., * indicates p ≤ 0.05 for relevant comparisons.
Patterns of GR and NF-κB Occupancy Are Similar at Pro- and Anti-inflammatory Regulatory Loci
GR is generally believed to mediate therapeutic effects via transrepression of inflammatory transcription factors such as NF-κB and AP-1, whereas gene induction by GR has been associated with side effects (27). However, our gene expression analysis suggested that GR actively regulates the expression of anti-inflammatory genes that are induced by TNF. To explore the mechanistic basis for regulation of TNF responses by GR, we applied ChIP to compare GR and NF-κB occupancy at selected pro- and anti-inflammatory loci. For this analysis, factor binding peaks from a published ChIP-seq study of GR and NF-κB occupancy in HeLa cells (22), visualized in the UCSC genome browser, were used to identify putative GR and NF-κB binding regions in Beas-2B cells within the TNFAIP3, NFKBIA, IL8, and IL1β loci; a well characterized GR binding site within FKBP5 was used as a control (28, 29). Occupancy for GR and the p65 subunit of NF-κB was determined relative to non-occupied control regions after exposure to each of four conditions for 1 h: vehicle (ethanol), TNF (20 ng/ml), dex (100 nm), or TNF + dex co-treatment. Occupancy data for both factors in each condition at the interrogated regions are shown in composite in Fig. 3 on a log2 scale. Even with vehicle treatment, both factors exhibited a significant (p < 0.05 and greater than 2-fold) increase in occupancy at each of the interrogated regions (with the exception of NF-κB occupancy of the TNFAIP3 promoter) in comparison with occupancy of control sites, likely reflecting basal activity of both GR and NF-κB signaling in Beas-2B cells under standard culture conditions. Dex treatment significantly increased GR binding above baseline at tested regions associated with FKBP5, IL8, TNFAIP3 (+5936–6047), and NFKBIA, whereas basal binding at IL1β for GR was increased by TNF treatment, consistent with studies indicating that inflammatory signals can modulate GR localization independent of ligand (30). Across treatment conditions, GR exhibited similar occupancy patterns at the TNFAIP3 and IL8 loci despite the dichotomous effects of GR activation on TNFAIP3 and IL8 gene expression. Likewise, NF-κB (p65) occupancy at the TNFAIP3 +5.5 region and the IL8 promoter was similar across treatment conditions. In aggregate, these data indicate that occupancy of GR and NF-κB in Beas-2B cells does not correlate with a specific transcriptional outcome of the associated target genes.
FIGURE 3.
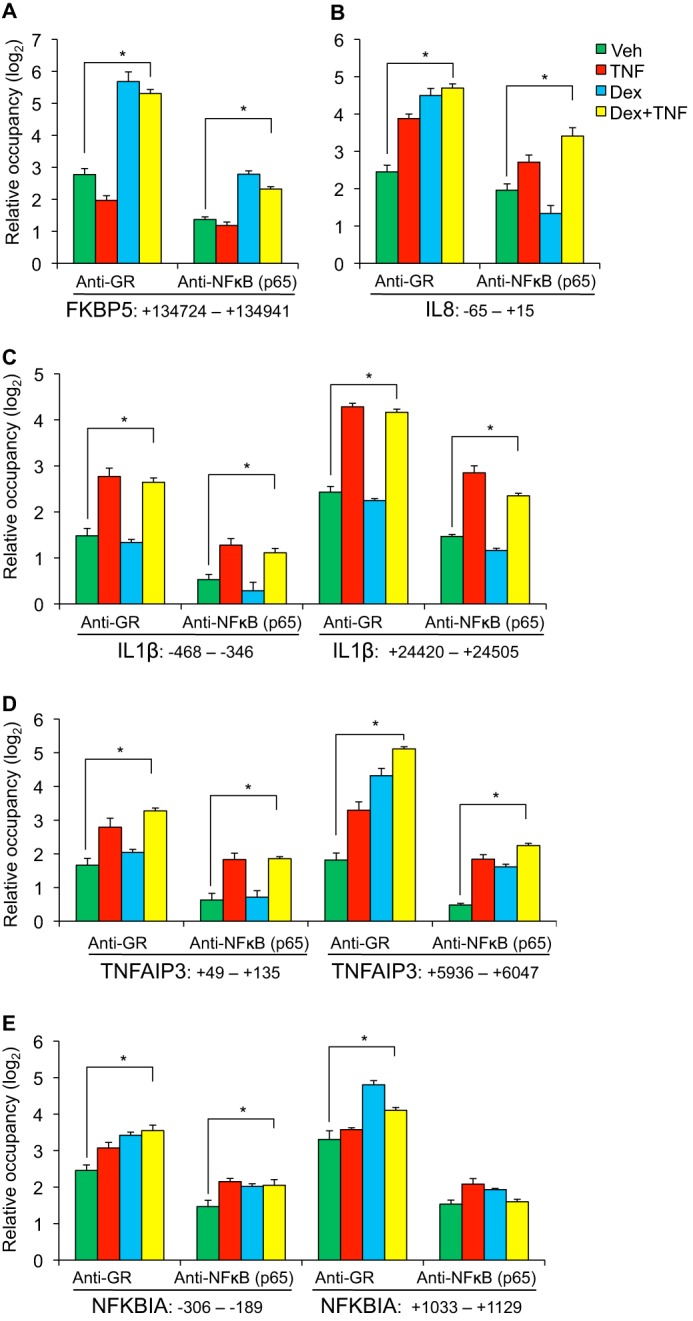
GR and NF-κB occupancy patterns do not distinguish between pro- and anti-inflammatory targets of TNF. A–E, ChIP-qPCR analysis of GR and NFkB (p65) occupancy within the FKBP5 (A), IL8 (B), IL1β (C), TNFAIP3 (D), and NFKBIA (E) loci in Beas-2B cells treated with TNF (20 ng/ml), dex (100 nm), or both for 1 h. Regions that were interrogated for factor occupancy are identified in relationship to the transcriptional start site of the associated gene, as indicated below the x axes. Relative factor occupancy was calculated as a difference between CT values for each target as compared with the geometric mean of CT values of three control regions that are not occupied by either GR or NFkB (p65). ChIP experiments were conducted in biological quadruplicate, bars indicate means + S.D. * indicates p ≤ 0.05 for relevant comparisons. Veh, vehicle.
A Novel Enhancer in the TNFAIP3 Locus Mediates Transcriptional Synergism between GR and NF-κB
Although our ChIP data demonstrate that GR and NF-κB occupancy at binding regions within repressed and induced genes is similar, these results do not directly establish the transcriptional consequences of factor occupancy within these regions. To explore this question, we designed primers to amplify a region in the second intron of TNFAIP3 that was occupied by both GR and NF-κB (Fig. 4A). This specific region was selected for further analysis because it contains a strong consensus GR binding sequence and two consensus NF-κB binding sequences, all three of which are highly conserved across most mammalian species, as well as in lizard and chicken (supplemental Fig. S1). We introduced a roughly 900-bp fragment spanning these binding sequences and conserved flanking sequence into the PGL3 promoter vector to generate a firefly luciferase reporter, pTNAFIP3I2. We transfected pTNFAIP3I2 into Beas-2B cells and subsequently treated cells with vehicle (ethanol), TNF (20 ng/ml), dex (100 nm), or TNF + dex co-treatment for 8 h. We found that pTNFAIP3I2 was induced by both TNF (3.1 ± 0.02) and dex (4.7 ± 0.03-fold) as compared with vehicle treatment. Remarkably, TNF + dex treatment led to robust induction (21.5 ± 0.53-fold) relative to vehicle, which was greater than the product of the individual inductive effects of dex and TNF on pTNFAIP3I2 activity. In contrast, as reported previously (31), TNF-mediated induction of an IL8 reporter, pIL8, which encompasses the IL8 GR/NF-κB binding region we assayed by ChIP, was significantly reduced by treatment with dex (Fig. 4B). Thus, dex and TNF induce dichotomous effects on transcription driven from regulatory elements within the IL8 and TNFAIP3 loci despite similar occupancy of the corresponding genomic regions by GR and NF-κB.
To confirm that cooperative induction of pTNFAIP3I2 by dex and TNF was due to direct effects of GR and NF-κB we transfected Beas-2B cells with pTNFAIP3I2 and siRNA directed against either GR or the p65 subunit of NF-κB. A reporter with three multimerized NF-κB binding sites, p3XNF-κB, was used as a positive control for NF-κB activity (32), whereas an established GR-responsive reporter for FKBP5 (28), here termed pFKBP5, served as a positive control for GR. Knockdown of p65 eliminated the induction of both p3XNF-κB and pTNFAIP3I2 by TNF. The combinatorial effect of TNF and dex on pTNFAIP3I2 was also reduced to ∼35% of levels obtained with control siRNA transfection. Similarly, knockdown of GR prevented the inductive effects of dex on the pTNFAIP3I2 and pFKBP5 reporters and substantially reduced cooperative induction of pTNFAIP3I2 by TNF + dex co-treatment. Western blots confirmed that transfection with siNF-κB and siGR reduced the protein levels of p65 and GR, respectively. Thus, induction of pTNFAIP3I2 by TNF and dex is mediated through NF-κB and GR.
GR Binding Site Context Controls GR/NF-κB Synergism
GR can modulate NF-κB activity both through glucocorticoid binding sites and also in the context of regulatory regions that lack a strong consensus GR binding sequence (21, 33, 34). Therefore, to test whether the GR binding sequence within the second intron of TNFAIP3 was required for GR/NF-κB cooperativity, we disrupted this sequence using site-directed mutagenesis (Fig. 5A) and tested the resultant plasmid, pTNFAIP3I2 (mutGBS), for inducibility by dex and TNF. The activity of pTNFAIP3I2 was entirely dependent on the presence of this GBS (Fig. 5B). Next, to determine whether specific sequence features of this GBS regulate transcriptional outcome, we replaced the native TNFAIP3I2 GBS with a distinct GBS from the FBKP5 locus that is required for regulation of the pFKBP5 reporter by GR (28). This plasmid, pTNFAIP3I2/FKBP5 GBS, exhibited similar induction to the wild type pTNFAIP3I2 plasmid both with dex treatment and after combined treatment with dex and TNF. Moreover, dex-mediated induction of pTNFAIP3/FKBP5 GBS was substantially less than induction of pFKBP5 despite the two plasmids harboring the identical GBS. Taken together, these data indicate that regulation of pTNFAIP3I2 by GR depends on the presence of a single GBS and that the surrounding context of this GBS is a major determinant of the transcriptional response to dex and TNF.
FIGURE 5.
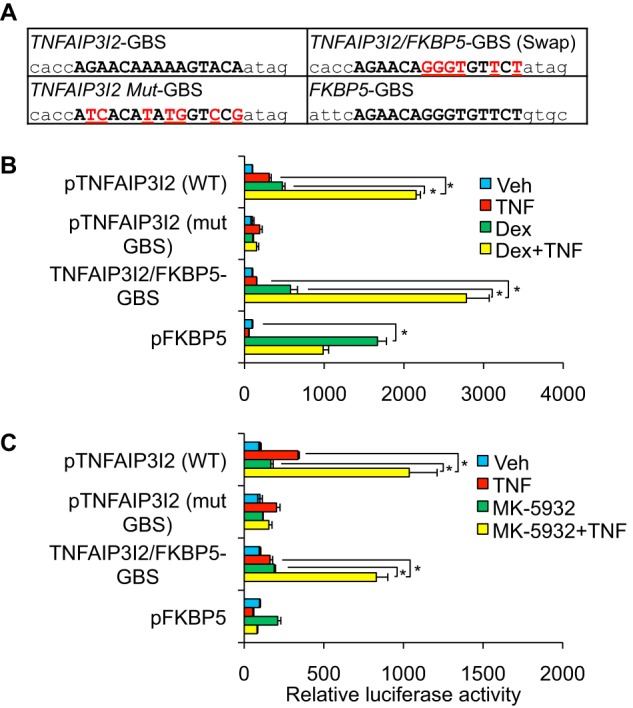
Glucocorticoid receptor binding site context mediates GR/NF-κB synergism. A, sequences of GR binding sites from TNFAIP3 intron 2, FKBP5, and mutations. B, relative luciferase activity of the indicated plasmids after transfection into Beas-2B cells and treatment for 8 h with vehicle, TNF (20 ng/ml), dex (100 nm), or TNF + dex. C, relative luciferase activity of the indicated plasmids after transfection into Beas-2B cells and treatment for 8 h with vehicle, TNF (20 ng/ml), MK-5932 (100 nm), or TNF+ MK-5932. For B and C, bars represent mean luciferase activity normalized to that of the control reporter (SV40-Renilla) +S.D. * indicates p ≤ 0.05 for relevant comparisons.
A Selective GR Ligand Distinguishes between GR/NF-κB Cooperation and Activation of a Simple GR Response Element
Selective GR ligands have been developed that alter GR-mediated transcriptional regulation differentially in comparison with classical synthetic GR agonists such as dexamethasone. A design goal for such ligands has been to enable GR-mediated repression of NF-κB function, while reducing GR-mediated gene induction (16). However, it is unknown whether selective ligands enable GR to cooperate with NF-κB to enhance transcription, an activity that our data implicates as regulating the expression of TNFAIP3, a potent anti-inflammatory protein. We therefore tested the ability of one such ligand developed by Merck and Co., MK-5932 (24), to regulate pTNFAIP3I2. Treatment with MK-5932 and TNF resulted in cooperative induction of pTNFAIP3I2 that was ∼40% of the level achieved with dex and TNF treatment (Fig. 5). In contrast, pFKBP5 was only weakly induced by MK-5932 to ∼10% of the level obtained with dex. These data indicate that MK-5932 selectively maintains cooperation between GR and NF-κB to induce pTNFAIP3I2 in comparison with induction of pFKBP5. Moreover, although pFKBP5 was only weakly induced by MK-5932, combined treatment with MK-5932 and TNF resulted in induction of the pTNFAIP3/FKBP5 swap that was comparable with the effect of MK-5932 + TNF on wild type pTNFAIP3I2 (Fig. 5C). Taken together, these data indicate that GR/NF-κB cooperation can be pharmacologically distinguished from classical GR-mediated induction of an isolated GBS. Moreover, the context of the composite GR/NF-κB regulatory element within TNFAIP3, rather than the sequence of the GBS, is the major determinant of combinatorial induction by GR and NF-κB.
DISCUSSION
Glucocorticoids are extremely effective anti-inflammatory agents, but the mechanisms that underpin their potency are not fully understood. Here we show that in comparison with antibody-based blockade of TNF, glucocorticoids selectively spare or augment the expression of negative feedback targets of TNF such as TNFAIP3, TNIP1, and NFKBIA. In contrast to well described repression of NF-κB by GR, co-treatment with dex + TNF caused GR and NF-κB to synergistically induce transcriptional responses through an intronic enhancer for TNFAIP3. GR/NF-κB cooperation depended on the context of the GR binding site, rather than the specific binding site sequence, and could be dissociated from induction of a simple glucocorticoid response element by GR through altered ligand chemistry. Our data support a model (Fig. 6) in which context-dependent cooperation between GR and NF-κB alters the balance between positive and negative feedback control of inflammation to bias termination of inflammatory responses.
FIGURE 6.
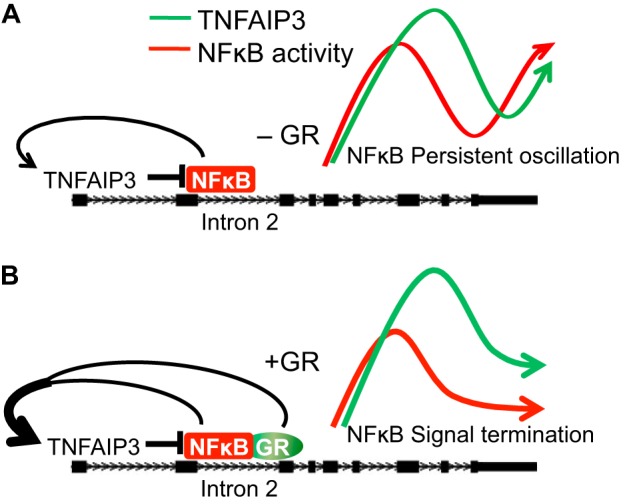
A model of cooperative regulation of TNFAIP3 by GR and NF-κB leading to termination of NF-κB signaling. A, due to negative feedback regulation of NF-κB, in the absence of activated GR, the level of NF-κB transcriptional activity oscillates (41). As NF-κB drives TNFAIP3 expression, TNFAIP3 levels oscillate in phase with NF-κB activity levels. B, activation of GR signaling maintains TNFAIP3 expression through cooperating with NF-κB, resulting in a shift of TNFAIP3 expression levels relative to NF-κB activity. TNFAIP3-mediated reduction of NF-κB activity below a threshold can lead to signal termination.
Although GR inhibits numerous NF-κB-regulated genes, two earlier studies described promoters with three NF-κB sites in proximity to a GBS that were co-induced by GR and NF-κB (35, 36). GR occupancy has also been observed to have a neutral effect on NF-κB activity at the IkB promoter (31). More recent genome-wide studies in HeLa cells and murine macrophages have indicated that GR and NF-κB co-occupy numerous loci (21, 22) and have associated factor occupancy with both induction and repression of steady state gene expression, including activated GR augmenting the induction of TNFAIP3 by TNF in HeLa cells. Our study extends on these findings through establishing strongly cooperative enhancement of transcription as a possible outcome of GR/NF-κB co-occupancy. Whether GR/NF-κB cooperation occurs across a range of cell types and encompasses other negative feedback targets of NF-κB remains to be determined.
The cooperative interaction between NF-κB and GR at the TNFAIP3 enhancer we observed in reporter assays was more than multiplicative. This transcriptional synergism correlated with an ∼2-fold increase in endogenous TNFAIP3 expression after TNF + dex treatment in comparison with treatment with TNF alone. The difference between reporter activity and endogenous TNFAIP3 expression levels is likely secondary to combinatorial effects of dex and TNF on both positive and negative control of NF-κB, a primary driver of TNFAIP3 transcription that binds to several sites in the TNFAIP3 locus. In that regard, as a consequence of negative feedback, NF-κB signaling induces oscillatory cycling of downstream target gene expression that is subject to late-phase damping by TNFAIP3 (37). It remains to be determined experimentally whether the cooperative induction of TNFAIP3 enhancer activity by GR and NF-κB decouples oscillation of TNFAIP3 expression relative to NF-κB activity, as we propose in our model (Fig. 6).
The role of GR binding site sequence in determining transcriptional outcomes is currently controversial. Published analyses of the GR cistrome have yielded contradictory conclusions with respect to whether specific GBS sequences mediate transcriptional repression by GR (21, 34). Similarly, although recent studies have indicated that DNA can serve as an allosteric effector that modulates the magnitude of GR-mediated gene induction and influences GR dimerization (9, 38), others have argued that binding site affinity is a primary determinant of GR activity (39). Although our data do not address the role of GBS sequence in transcriptional outcomes on a genome-wide level, they strongly suggest that GR/NF-κB cooperation within the TNFAIP3 intron 2 enhancer is determined primarily by the context of the binding site, rather than specific GBS sequence features that enable GR/NF-κB cooperativity. Whether the context and positioning of the GR and NF-κB binding sites within the TNFAIP3 locus result in transcriptional synergism through mediating cooperative binding to DNA by both factors, or through facilitating the engagement of co-activators, has yet to be established.
Dysregulation of TNFAIP3 transcription is strongly associated with autoimmune disease (13); however, the role of TNFAIP3 regulation by glucocorticoids in immune-mediated disease pathogenesis and therapeutics has not been established. A recent study demonstrated that LPS-mediated induction of TNFAIP3 involves looping of both a distal 3′ enhancer and intron 2 to the TNFAIP3 promoter region (40); looping was disrupted in cells harboring genomic variants associated with the development of lupus. In that system, the recruitment of intron 2 to the TNFAIP3 promoter was relatively modest, and enhancer properties for intron 2 were not established. Our results raise the possibility that activation of GR signaling will increase LPS-driven association of intron 2 with the TNFAIP3 promoter/3′ enhancer complex. Long range association of GR-bound intron 2 with other regulatory elements could thus provide a mechanism through which distal SNPs modulate GR responsiveness without directly disrupting a GBS.
Negative feedback is known to play a central role in terminating inflammatory responses and is dysregulated in a number of immune-mediated diseases. Although comparatively little is known about the role of negative feedback in airway inflammation, we propose that cooperation between GR and NF-κB contributes to the efficacy of GR agonists in treating inflammatory airway disorders through augmenting the expression of endogenous anti-inflammatory pathways. A logical extension of this notion is that disruption of GR/NF-κB cooperation may contribute to corticosteroid-resistant airway disease. The availability of clinical samples from patients with airway disease, and murine models in which negative feedback regulators such as TNFAIP3 are disrupted in the airway, will facilitate future testing of these hypotheses.
Supplementary Material
Acknowledgments
We thank Jennifer Richer for providing the p3XNF-κB plasmid. We thank Keith Yamamoto and Miles Pufall for generous gifts of GR antibodies. Qian Wang provided valuable technical assistance. We thank Monica Gerber for critical review of the manuscript.
This work was supported in part by National Institutes of Health Grant R01HL109557 (to A. N. G.) and with institutional funds from National Jewish Health.

This article contains supplemental Tables S1–S4 and Fig. S1.
- GR
- glucocorticoid receptor
- GBS
- glucocorticoid binding site
- dex
- dexamethasone
- qPCR
- quantitative PCR
- Ad
- adenovirus.
REFERENCES
- 1. Flammer J. R., Rogatsky I. (2011) Minireview: Glucocorticoids in autoimmunity: unexpected targets and mechanisms. Mol. Endocrinol. 25, 1075–1086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. McMaster A., Ray D. W. (2008) Drug insight: selective agonists and antagonists of the glucocorticoid receptor. Nat. Clin. Pract. Endocrinol. Metab. 4, 91–101 [DOI] [PubMed] [Google Scholar]
- 3. Nicolaides N. C., Galata Z., Kino T., Chrousos G. P., Charmandari E. (2010) The human glucocorticoid receptor: molecular basis of biologic function. Steroids 75, 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Galliher-Beckley A. J., Williams J. G., Collins J. B., Cidlowski J. A. (2008) Glycogen synthase kinase 3β-mediated serine phosphorylation of the human glucocorticoid receptor redirects gene expression profiles. Mol. Cell. Biol. 28, 7309–7322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Oakley R. H., Cidlowski J. A. (2013) The biology of the glucocorticoid receptor: New signaling mechanisms in health and disease. J. Allergy Clin. Immunol. 132, 1033–1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chen W., Dang T., Blind R. D., Wang Z., Cavasotto C. N., Hittelman A. B., Rogatsky I., Logan S. K., Garabedian M. J. (2008) Glucocorticoid receptor phosphorylation differentially affects target gene expression. Mol. Endocrinol. 22, 1754–1766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Grøntved L., John S., Baek S., Liu Y., Buckley J. R., Vinson C., Aguilera G., Hager G. L. (2013) C/EBP maintains chromatin accessibility in liver and facilitates glucocorticoid receptor recruitment to steroid response elements. EMBO J. 32, 1568–1583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. John S., Sabo P. J., Thurman R. E., Sung M. H., Biddie S. C., Johnson T. A., Hager G. L., Stamatoyannopoulos J. A. (2011) Chromatin accessibility pre-determines glucocorticoid receptor binding patterns. Nat. Genet. 43, 264–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Meijsing S. H., Pufall M. A., So A. Y., Bates D. L., Chen L., Yamamoto K. R. (2009) DNA binding site sequence directs glucocorticoid receptor structure and activity. Science 324, 407–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. So A. Y., Cooper S. B., Feldman B. J., Manuchehri M., Yamamoto K. R. (2008) Conservation analysis predicts in vivo occupancy of glucocorticoid receptor-binding sequences at glucocorticoid-induced genes. Proc. Natl. Acad. Sci. U.S.A. 105, 5745–5749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Weber A., Wasiliew P., Kracht M. (2010) Interleukin-1 (IL-1) pathway. Sci. Signal. 3, cm1. [DOI] [PubMed] [Google Scholar]
- 12. Ruland J. (2011) Return to homeostasis: downregulation of NF-κB responses. Nat. Immunol. 12, 709–714 [DOI] [PubMed] [Google Scholar]
- 13. Ma A., Malynn B. A. (2012) A20: linking a complex regulator of ubiquitylation to immunity and human disease. Nat. Rev. Immunol. 12, 774–785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Boone D. L., Turer E. E., Lee E. G., Ahmad R. C., Wheeler M. T., Tsui C., Hurley P., Chien M., Chai S., Hitotsumatsu O., McNally E., Pickart C., Ma A. (2004) The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat. Immunol. 5, 1052–1060 [DOI] [PubMed] [Google Scholar]
- 15. De Bosscher K., Haegeman G., Elewaut D. (2010) Targeting inflammation using selective glucocorticoid receptor modulators. Curr. Opin. Pharmacol. 10, 497–504 [DOI] [PubMed] [Google Scholar]
- 16. Clark A. R., Belvisi M. G. (2012) Maps and legends: the quest for dissociated ligands of the glucocorticoid receptor. Pharmacol. Ther. 134, 54–67 [DOI] [PubMed] [Google Scholar]
- 17. Joanny E., Ding Q., Gong L., Kong P., Saklatvala J., Clark A. R. (2012) Anti-inflammatory effects of selective glucocorticoid receptor modulators are partially dependent on up-regulation of dual specificity phosphatase 1. Br. J. Pharmacol. 165, 1124–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vandevyver S., Dejager L., Tuckermann J., Libert C. (2013) New insights into the anti-inflammatory mechanisms of glucocorticoids: an emerging role for glucocorticoid-receptor-mediated transactivation. Endocrinology 154, 993–1007 [DOI] [PubMed] [Google Scholar]
- 19. King E. M., Chivers J. E., Rider C. F., Minnich A., Giembycz M. A., Newton R. (2013) Glucocorticoid repression of inflammatory gene expression shows differential responsiveness by transactivation- and transrepression-dependent mechanisms. PLoS One 8, e53936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lannan E. A., Galliher-Beckley A. J., Scoltock A. B., Cidlowski J. A. (2012) Proinflammatory actions of glucocorticoids: glucocorticoids and TNFα coregulate gene expression in vitro and in vivo. Endocrinology 153, 3701–3712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Uhlenhaut N. H., Barish G. D., Yu R. T., Downes M., Karunasiri M., Liddle C., Schwalie P., Hübner N., Evans R. M. (2013) Insights into negative regulation by the glucocorticoid receptor from genome-wide profiling of inflammatory cistromes. Mol. Cell 49, 158–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rao N. A., McCalman M. T., Moulos P., Francoijs K. J., Chatziioannou A., Kolisis F. N., Alexis M. N., Mitsiou D. J., Stunnenberg H. G. (2011) Coactivation of GR and NFKB alters the repertoire of their binding sites and target genes. Genome Res. 21, 1404–1416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Morjaria J. B., Babu K. S., Polosa R., Holgate S. T. (2007) Tumor necrosis factor-α in severe corticosteroid-refractory asthma. Expert Rev. Respir. Med. 1, 51–63 [DOI] [PubMed] [Google Scholar]
- 24. Bungard C. J., Hartman G. D., Manikowski J. J., Perkins J. J., Bai C., Brandish P. E., Euler D. H., Hershey J. C., Schmidt A., Fang Y., Norcross R. T., Rushmore T. H., Thompson C. D., Meissner R. S. (2011) Discovery of selective glucocorticoid receptor modulator MK-5932. Bioorg. Med. Chem. 19, 7374–7386 [DOI] [PubMed] [Google Scholar]
- 25. Wang Y. Y., Li L., Han K. J., Zhai Z., Shu H. B. (2004) A20 is a potent inhibitor of TLR3- and Sendai virus-induced activation of NF-κB and ISRE and IFN-β promoter. FEBS Lett. 576, 86–90 [DOI] [PubMed] [Google Scholar]
- 26. Sasse S. K., Mailloux C. M., Barczak A. J., Wang Q., Altonsy M. O., Jain M. K., Haldar S. M., Gerber A. N. (2013) The glucocorticoid receptor and KLF15 regulate gene expression dynamics and integrate signals through feed-forward circuitry. Mol. Cell. Biol. 33, 2104–2115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. De Bosscher K., Van Craenenbroeck K., Meijer O. C., Haegeman G. (2008) Selective transrepression versus transactivation mechanisms by glucocorticoid receptor modulators in stress and immune systems. Eur. J. Pharmacol. 583, 290–302 [DOI] [PubMed] [Google Scholar]
- 28. Hubler T. R., Scammell J. G. (2004) Intronic hormone response elements mediate regulation of FKBP5 by progestins and glucocorticoids. Cell Stress Chaperones 9, 243–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Reddy T. E., Pauli F., Sprouse R. O., Neff N. F., Newberry K. M., Garabedian M. J., Myers R. M. (2009) Genomic determination of the glucocorticoid response reveals unexpected mechanisms of gene regulation. Genome Res. 19, 2163–2171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hu A., Josephson M. B., Diener B. L., Nino G., Xu S., Paranjape C., Orange J. S., Grunstein M. M. (2013) Pro-asthmatic cytokines regulate unliganded and ligand-dependent glucocorticoid receptor signaling in airway smooth muscle. PLoS One 8, e60452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Luecke H. F., Yamamoto K. R. (2005) The glucocorticoid receptor blocks P-TEFb recruitment by NFκB to effect promoter-specific transcriptional repression. Genes Dev. 19, 1116–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dan H. C., Cooper M. J., Cogswell P. C., Duncan J. A., Ting J. P., Baldwin A. S. (2008) Akt-dependent regulation of NF-κB is controlled by mTOR and Raptor in association with IKK. Genes Dev. 22, 1490–1500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chinenov Y., Gupte R., Dobrovolna J., Flammer J. R., Liu B., Michelassi F. E., Rogatsky I. (2012) Role of transcriptional coregulator GRIP1 in the anti-inflammatory actions of glucocorticoids. Proc. Natl. Acad. Sci. U.S.A. 109, 11776–11781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Surjit M., Ganti K. P., Mukherji A., Ye T., Hua G., Metzger D., Li M., Chambon P. (2011) Widespread negative response elements mediate direct repression by agonist-liganded glucocorticoid receptor. Cell 145, 224–241 [DOI] [PubMed] [Google Scholar]
- 35. Webster J. C., Huber R. M., Hanson R. L., Collier P. M., Haws T. F., Mills J. K., Burn T. C., Allegretto E. A. (2002) Dexamethasone and tumor necrosis factor-α act together to induce the cellular inhibitor of apoptosis-2 gene and prevent apoptosis in a variety of cell types. Endocrinology 143, 3866–3874 [DOI] [PubMed] [Google Scholar]
- 36. Hofmann T. G., Schmitz M. L. (2002) The promoter context determines mutual repression or synergism between NF-κB and the glucocorticoid receptor. Biol. Chem. 383, 1947–1951 [DOI] [PubMed] [Google Scholar]
- 37. Werner S. L., Kearns J. D., Zadorozhnaya V., Lynch C., O'Dea E., Boldin M. P., Ma A., Baltimore D., Hoffmann A. (2008) Encoding NF-κB temporal control in response to TNF: distinct roles for the negative regulators IκBα and A20. Genes Dev. 22, 2093–2101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hudson W. H., Youn C., Ortlund E. A. (2013) The structural basis of direct glucocorticoid-mediated transrepression. Nat. Struct. Mol. Biol. 20, 53–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bain D. L., Yang Q., Connaghan K. D., Robblee J. P., Miura M. T., Degala G. D., Lambert J. R., Maluf N. K. (2012) Glucocorticoid receptor-DNA interactions: binding energetics are the primary determinant of sequence-specific transcriptional activity. J. Mol. Biol. 422, 18–32 [DOI] [PubMed] [Google Scholar]
- 40. Wang S., Wen F., Wiley G. B., Kinter M. T., Gaffney P. M. (2013) An enhancer element harboring variants associated with systemic lupus erythematosus engages the TNFAIP3 promoter to influence A20 expression. PLoS Genet. 9, e1003750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nelson D. E., Ihekwaba A. E., Elliott M., Johnson J. R., Gibney C. A., Foreman B. E., Nelson G., See V., Horton C. A., Spiller D. G., Edwards S. W., McDowell H. P., Unitt J. F., Sullivan E., Grimley R., Benson N., Broomhead D., Kell D. B., White M. R. (2004) Oscillations in NF-κB signaling control the dynamics of gene expression. Science 306, 704–708 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.