

Background: Monoamine oxidase A (MAO-A) catalyzes the degradation of neurotransmitters such as serotonin.
Results: Knockdown of MAO-A expression in embryos induces high serotonin levels and abnormal brain development, which can be rescued by inactivation of serotonin receptor-6 (5-Htr6).
Conclusion: 5-Htr6 activation is vital for early development of the embryonic brain.
Significance: Serotonin signaling and metabolism are important in early embryos.
Keywords: Eicosanoid, Embryo, Oxidase, Receptors, Serotonin, Signaling
Abstract
Monoamine oxidases A and B (MAO-A and MAO-B) are enzymes of the outer mitochondrial membrane that metabolize biogenic amines. In the adult central nervous system, MAOs have important functions for neurotransmitter homeostasis. Expression of MAO isoforms has been detected in the developing embryo. However, suppression of MAO-B does not induce developmental alterations. In contrast, targeted inhibition and knockdown of MAO-A expression (E7.5–E10.5) caused structural abnormalities in the brain. Here we explored the molecular mechanisms underlying defective brain development induced by MAO-A knockdown during in vitro embryogenesis. The developmental alterations were paralleled by diminished apoptotic activity in the affected neuronal structures. Moreover, dysfunctional MAO-A expression led to elevated levels of embryonic serotonin (5-hydroxytryptamine (5-HT)), and we found that knockdown of serotonin receptor-6 (5-Htr6) expression or pharmacologic inhibition of 5-Htr6 activity rescued the MAO-A knockdown phenotype and restored apoptotic activity in the developing brain. Our data suggest that excessive 5-Htr6 activation reduces activation of caspase-3 and -9 of the intrinsic apoptotic pathway and enhances expression of antiapoptotic proteins Bcl-2 and Bcl-XL. Moreover, we found that elevated 5-HT levels in MAO-A knockdown embryos coincided with an enhanced activation of extracellular signal-regulated kinase 1/2 (ERK1/2) and a reduction of proliferating cell numbers. In summary, our findings suggest that excessive 5-HT in MAO-A-deficient mouse embryos triggers cellular signaling cascades via 5-Htr6, which suppresses developmental apoptosis in the brain and thus induces developmental retardations.
Introduction
Monoamine oxidases (MAOs)2 constitute catalytic flavoproteins of the outer mitochondrial membrane that catalyze the oxidative deamination of monoamines to the corresponding aldehydes (1). Two genes encoding two distinct isoenzymes (MAO-A and MAO-B) occur in the human genome, and their expression is under the control of independent mechanisms (2). The two MAO isoforms can be distinguished by their substrate specificity and their sensitivity toward specific pharmacological inhibitors. MAO-A preferentially deaminates serotonin (5-hydroxytryptamine (5-HT)), whereas MAO-B has high affinity for phenylethylamine (1). Both isoenzymes are expressed in the central nervous system as well as in peripheral tissues, and their expression has also been detected during embryo development (3–5).
MAO isoforms are key enzymes of cerebral neurotransmitter homeostasis. The roles of these enzymes in aging brains and in the pathogenesis of neurodegenerative diseases have become a major focus of translational research (6). The biological roles of MAOs have been investigated using animals that carry dysfunctional Mao genes. Mice carrying a targeted knock-out of the Mao-b gene are viable and show no developmental defects (7). Similar observations were made when MAO-B expression was knocked down during in vitro embryogenesis using an siRNA silencing strategy (8). Unfortunately, no targeted animal knock-out models of the Mao-a gene are currently available. However, mouse strains with a naturally occurring mutation within the Mao-a gene or with an accidental insertion of an interferon β minigene have been reported (9, 10). Both mouse strains show impaired MAO-A expression but unfortunately have not yet been thoroughly characterized to rule out collateral alterations within the underlying genomes. MAO-A-deficient mice reproduce normally but show several behavioral abnormalities, which have been attributed to impaired postnatal maturation of the brain (9, 11–13). The prerequisites for postnatal brain development, however, are already laid out before birth, but little is known about the roles of the MAO isoenzymes during this stage of development. Both MAO isoenzymes are expressed early on in embryonic and extraembryonic tissues (5, 8). In the embryo, MAO-A is the dominant isoenzyme, and the time course of its expression persists during in vitro development of explanted mouse embryos (8). Recent loss-of-function experiments have indicated that MAO-A is an important factor during embryo development (8, 14, 15), whereas for MAO-B, no biological function during prenatal development has been identified so far (8). In humans, functional inactivation of MAO expression has been associated with a severe form of Norrie disease, which is characterized by various developmental retardations in particular of the eye (16). In adult humans, MAO activity has been implicated in the pathogenesis of neurological disorders such as depression and neurodegeneration (Alzheimer and Parkinson diseases) (6).
Monoamines are not only important neurotransmitters but also function as signaling molecules that control differentiation, proliferation, and cell death (17). Monoaminergic transmitter systems are present early on in the developing embryo, and disturbances of monoamine homeostasis lead to abnormal development or embryonic death (18, 19). We recently demonstrated that impaired MAO-A expression and/or activity during in vitro embryogenesis resulted in severe developmental retardation of the brain (8). Unfortunately, the underlying molecular mechanisms remained unclear. A hallmark of systemic MAO-A deficiency is the elevation of 5-HT levels, which was consistently observed in all models of MAO-A deficiency (8–10). However, it remains to be explored how such a 5-HT overload induces developmental abnormalities.
The present study was aimed at exploring the role of 5-HT signaling during in vitro embryogenesis. We found that 5-HT acts via serotonin receptor-6 (5-Htr6) to ameliorate developmental neuronal apoptosis, which contributes to the phenotypic alterations observed for MAO-A deficiency during in vitro embryogenesis.
EXPERIMENTAL PROCEDURES
Chemicals
The chemicals used were from the following sources. 5-HT, selective 5-HT transporter (5-HTT) antagonist fluoxetine, tryptophan hydroxylase (TPH) antagonist p-chlorophenylalanine (PCPA), and specific 5-Htr inhibitors methiothepin and ritanserin were from Sigma. 5-HT mimetic 5-carboxamidotryptamine and other specific 5-Htr inhibitors WAY1000635, SB224289, BRL15572, SB269970, and BRL54443 were from Tocris Bioscience. For quantitative real time RT-PCR (RT-qPCR), Superscript III reverse transcriptase and RNaseOUT from Invitrogen; BD Advantage 2 Polymerase Mix from BD Biosciences (Pharmingen); dNTPs from Carl Roth GmbH (Karlsruhe, Germany); QuantiTect SYBR Green PCR kit from Qiagen (Hilden, Germany); and PCR primers from BioTeZ (Berlin, Germany) were used.
RNA Extraction, Reverse Transcription, and RT-qPCR
Total RNA was extracted using the RNeasy Mini kit (Qiagen) and reverse transcribed according to standard protocols with oligo(dT)15 primers and SuperScript III reverse transcriptase (Invitrogen) according to the vendor's instructions. RT-qPCR was carried out with a Rotor Gene 3000 system (Corbett Research, Australia) using ImmoMix/SYBR Green (Bioline, Germany). TPH, MAO, and 5-Htr isoform-specific amplification primers were designed, and their sequences are as follows: Tph1, 5′-CAG-CAT-GAT-TGA-AGA-CAA-CAA-GGA-G-3′ and 5′-TCG-ATG-TGT-AAC-AGG-CTC-ACA-TGA-T-3′; Tph2, 5′-ATG-CAG-CCC-GCA-ATG-ATG-ATG-TTT-T-3′ and 5′-ACA-ACT-GCT-GTC-TTG-CTG-CTC-TCT-3′; Htr1a, 5′-TGG-ATA-TGT-TCA-GTC-TTG-GCC-AGG-3′ and 5′-ATT-GCC-GAG-CAC-CGC-GCA-GAA-AA-3′; Htr1b, 5′-TGG-AGG-AGC-AGG-GTA-TTC-AGT-GC-3′ and 5′-CAA-GGT-GAT-GAG-CGC-CAA-CAA-AGC-A-3′; Htr1d, 5′-ATG-TCT-CCT-CCA-AAC-CAG-TCC-CTA-3′ and 5′-AAG-GCA-TTG-GAG-AGG-ACA-GTG-GC-3′; Htr1f, 5′-CGC-ATC-AGA-CCA-AAA-CTT-GAC-CTC-3′ and 5′-AAT-AGT-TGG-CTG-GGT-GGT-GCA-GC-3′; Htr2a, 5′-ATG-ACT-CGC-TAG-TCT-CTC-CAC-ACT-3′ and 5′-TCC-CTG-GAG-TTG-AAG-TCA-TTA-GGG-3′; Htr2b, 5′-GAT-TAG-AGA-CAG-ACT-CAG-TAG-CAG-A-3′ and 5′-TAC-TGC-AGC-CTT-TTC-TCC-AGT-GCA-3′; Htr2c, 5′-ATG-GTG-AAC-CTG-GGC-ACT-GCG-G-3′ and 5′-CAC-GAC-TAT-TGA-AAG-TGC-TGG-CCA-3′; Hrtr3a, 5′-CTC-TGC-ATC-CCG-CAG-GTG-CTG-T-3′ and 5′-GTA-GGC-TTC-CTC-CAG-TCC-CGC-A-3′; Htr3b, 5′-ATG-ATT-CTT-CTG-TGG-TCC-TGC-CTC-3′ and 5′-GTG-GTG-GCC-TCA-GCC-CAG-TTG-T-3′; Htr4, 5′-TGA-TGC-TAA-TGT-GAG-TTC-CAA-CGA-G-3′ and 5′-GCA-AAG-GCG-AGA-GAC-ACA-ATG-AAA-T-3′; 5-Htr5a, 5′-GGA-TCT-GCC-TGT-AAA-CTT-GAC-CTC-3′ and 5′-CAG-CAC-CAG-CAG-GTT-CCA-AGT-GA-3′; 5-Htr5b, 5′-TGG-AAG-TTT-CTA-ACC-TCT-CAG-GCG-3′ and 5′-CAC-CAC-AAG-CAC-GGT-GAA-AGC-AG-3′; Htr6, 5′-TTC-CAG-AGC-CCG-GCC-CTG-TCA-A-3′ and 5′-AAG-TTA-GAC-GTG-TTG-CGC-AGC-GC-3′; Htr7, 5′-TGA-TGG-ACG-TTA-ACA-GCA-GCG-GC-3′ and 5′-TCT-GGG-AAG-CCG-CTC-AGC-AGG-T-3′; GAPDH, 5′-CCA-TCA-CCA-TCT-TCC-AGG-AGC-GA-3′ and 5′-GGA-TGA-CCT-TGC-CCA-CAG-CCT-TG-3′. RNA preparations were analyzed at least in triplicate. The experimental raw data were evaluated with Rotor-Gene Monitor software (version 4.6). To generate standard curves for quantification of expression levels, specific amplicons were used as external standards for each target gene (5 × 103–3 × 106 copy numbers). GAPDH mRNA was used as an internal standard to normalize expression of the target transcripts.
Preparation of Mouse Embryos
Inbred imprinting control region pregnant mice were obtained from the animal house, and embryos at different developmental stages (gestational day 7.5 (E7.5) to E10.5) were prepared under a stereomicroscope (Olympus). For RT-qPCR, preparations were kept in PBS with 0.1% diethyl pyrocarbonate, and extraembryonic tissue was removed. For in vitro culture, the embryos were dissected in PB1 medium with 5% FBS. All of the animal research was approved by the Animal Experimentation Ethics Committee (08/011/ERG) and was performed in accordance with institutional guidelines.
Embryo in Vitro Culture
For in vitro embryo culture, developing mouse embryos were explanted from pregnant mothers at E7.5, and in vitro development of the embryos was permitted for up to 72 h. During this time window, embryos were manipulated by siRNA injection for targeted expression knockdown, by pharmacological intervention, or by ectopic overexpression. For our knockdown strategy, we designed isoform-specific MAO-A siRNAs, 5-Htr1a siRNAs, and 5-Htr6 siRNAs using the StealthTM RNAi program (BLOCK-iTTM RNAi Designer, Invitrogen): Mao-a: sense, 5′-GAG GAA UGA GCA UGU UAA AUG GGU A-3′; antisense, 5′-UAC CCA UUU AAC AUG CUC AUU CCU C-3′; 5-Htr1a: sense, 5′-AAU UAU GGC ACC CAA CAA CUC AGG C-3′; antisense, 5′-GCC UGA GUU GUU GGG UGC CAU AAU U-3′; 5-Htr6: sense, 5′-AAA GAA CAU GCU CAG CAG GAU GCC G-3′; antisense, 5′-CGG CAU CCU GCU GAG CAU GUU CUU U-3′. Among the designed probes, we selected those constructs exhibiting the highest estimated knockdown probability and a GC content below 40%. No sequence cross-homology was allowed. Scrambled siRNA duplexes with no homology to any vertebrate transcript were used as random control probes (Stealth RNAi Negative Controls, Invitrogen). For R1 overexpression, the R1 coding sequence was cloned into the mammalian pcDNA3.1 expression vector (8). R1 codes for a transcription factor that specifically represses Mao-a gene transcription (20, 21). Empty vector was used as a control vector. For knockdown experiments, murine embryos were explanted at E7.5 and transfected with siRNA constructs. 10 nl of annealed double-stranded siRNA (25–100 nm) or pcDNA3.1 cdca7l/R1 plasmid DNA (4 μg/μl) were mixed with 0.01% LipofectamineTM 2000 (Invitrogen) and then microinjected with an ASTP micromanipulator (Leica, Germany) into the amniotic cavity. After microinjection, the embryos were placed in a whole embryo culture roller incubator (BTC Engineering, United Kingdom). The embryos at early gastrulation stage (E7.5) were allowed to develop for up to 72 h until early organogenesis stages E9.5–E10.5 in 100% heat-inactivated rat serum with a continuous flow of gas mixtures (22).
Morphometric Scoring Assay
The impact of the treatments on embryonic growth (embryo size measured by the crown rump and head length) and on development of the brain, heart, and limbs was quantified by a microscopic scoring procedure. To quantify the developmental progress and judge cerebral embryogenesis, we assessed the degree of brain maturation by a number of morphological parameters (23). A score of 0 represented strong developmental retardations, whereas a score of 5 indicated normal development. The following morphological criteria were applied for the different parts of embryonic brain: forebrain: prosencephalon invisible, score 0; V-shape, score 1; U-shape, score 2; partially fused, score 3; completely fused, score 4; telencephalic evaginations, score 5; midbrain: mesencephalic invisible, score 0; V-shape, score 1; U-shape, score 2; partially fused, score 3; completely fused, score 4; discernible division between mesencephalon and diencephalons, score 5; hindbrain: rhombencephalon invisible, score 0; V-shape, score 1; U-shape, score 2; folds fused with anterior neuropore, score 3; anterior neuropore closure, score 4; dorsal flexion developed with fourth ventricle roof, score 5. Significances were calculated with the Students' t test.
Serotonin Assay
Serotonin concentration in the embryos was measured using the Serotonin EIA (Ultra Sensitive) kit (Immuno-Biological Laboratories, Inc.) according to the manufacturer's instructions. In brief, after treatments, in vitro cultured embryos were washed and homogenized, and aliquots of the lysates were applied for quantification. The lower detection limit of the kit was 1.0 nmol/liter, and a linear calibration plot was established between 1.45 and 100 nmol/liter (r2 = 0.9807 ± 0.0012). The intra- and interassay variation coefficients were <3.02%, and the recovery range varied between 100.4 and 102.3%.
Western Blotting
For immunoblotting, we prepared total protein extracts from whole embryo lysates after in vitro culture that were obtained by homogenizing embryos in lysis buffer (50 mm Tris-HCl, 0.3 m NaCl, 1 mm EDTA, 1 mm DTT, and 1 mm PMSF). Aliquots with equal amounts of total protein (100 μg) were separated by 12% SDS-PAGE, and separated proteins were transferred to nitrocellulose membranes (Amersham Biosciences). The membranes were blocked overnight with an ovalbumin solution (Sigma) and then incubated at 37 °C for 1 h with primary antibodies. Primary antibodies included active caspase-3 (p17) and active caspase-9 (p37) from Abcam (Cambridge, UK) and anti-β-actin, anti-MAO-A, anti-5-Hrt1a, anti-5-Hrt6, anti-procaspase-3 (p35), anti-procaspase-9 (p46), anti-extracellular signal-regulated kinase (ERK), anti-Bcl-XL, anti-Bcl-2, and anti-Ki-67 from Santa Cruz Biotechnology, Inc.; and anti-phospho-ERK from Cell Signaling Technology (Hertfordshire, UK). The membranes were then washed three times with TBS and incubated with HRP-labeled secondary antibody (Dako, Carpinteria, CA) at 1:1000 dilution (30 min at room temperature). Blots were finally developed using the ECL kit (GE Healthcare). Immunoreactive protein bands were imaged using the Quantity One quantification software (Bio-Rad) and normalized to β-actin expression. For ERK/phospho-ERK Western blotting, antibody binding was revealed with enhanced chemiluminescence Western blotting detection reagent (Pierce). Digital images were captured using an LAS-3000 image analyzer (Fuji Film Co. Ltd., Tokyo, Japan), and band intensity was quantified using Aida software (Raytest GmbH, Straubenhardt, Germany).
TUNEL Staining
Apoptotic cells were stained with the standard TUNEL technique on the sagittal sections of cultured embryos at the E10.5 stage using a Chemicon (Temecula, CA) kit according to the vendor's instructions. Nuclear DNA was counterstained with 0.5% (w/v) methyl green for microscopic examination.
RESULTS
Excessive Serotonin Induces Abnormal Development of the Brain
5-HT is the dominant physiological substrate of MAO-A, and defective MAO-A expression leads to elevated 5-HT levels (8–10). We demonstrated previously that similar results were obtained when we specifically knocked down MAO-A expression in an isoform-specific manner (8). To test whether increased 5-HT levels might contribute to the developmental abnormalities observed as a consequence of MAO-A expression knockdown (8), mouse embryos were exposed to elevated 5-HT concentrations during in vitro maturation studies. We found that exogenous 5-HT induced similar developmental alterations compared with siRNA-induced expression silencing of MAO-A (Fig. 1A). The retardations were most prominent in the head region and the developing brain (Fig. 1B). In contrast, other embryonic regions remained unaffected by both MAO-A siRNA and 5-HT (data not shown). To further confirm this finding, we mimicked 5-HT elevation by exposing the embryos to the 5-HT mimetic 5-carboxamidotryptamine and to the selective 5-HTT antagonist fluoxetine. We found that 5-carboxamidotryptamine and fluoxetine induced similar morphological alterations as 5-HT and MAO-A siRNA (Fig. 1, A and B). These data suggest that 5-HT is a dominant factor causing abnormal brain development induced by MAO-A inactivation.
FIGURE 1.
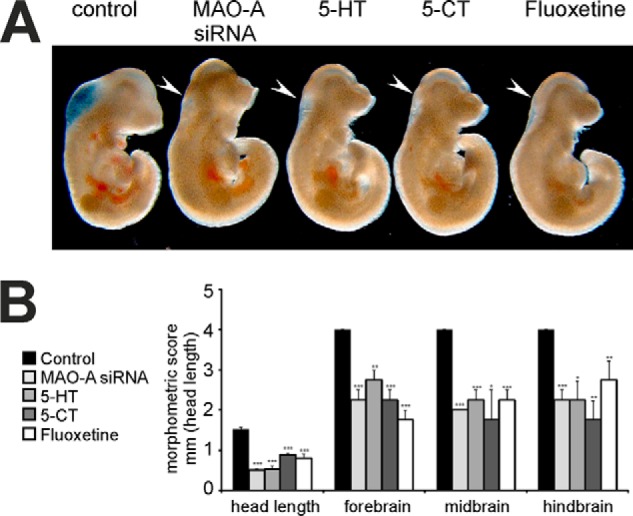
Developmental retardations induced by elevated levels of 5-HT or 5-HT analogues. Mouse embryos were explanted at E7.5 and transfected with MAO-A siRNA or stimulated with 10 μm 5-HT, 10 μm 5-carboxamidotryptamine (5-CT), or 10 μm fluoxetine. After 72 h of in vitro culture, embryos were subjected to microscopic inspection (A) and morphometric scoring (B). Figures are representative of four independent experiments, which were used for morphometric scoring. Arrows indicate brain regions with defective development. Significances were calculated using Student's t test; n = 4. ***, p < 0.001; **, p < 0.01; *, p < 0.05. Error bars represent S.D. If no error bars are visible in the diagrams, no developmental variations between embryos of one experiment could be detected.
Inhibition of Serotonin Biosynthesis Rescues Abnormal Brain Development
Systemic 5-HT concentrations are not only controlled by its cellular uptake and its intracellular degradation but also by the rate of biosynthesis. The key step of 5-HT biosynthesis is catalyzed by TPHs (24). To investigate whether the two murine TPH isoforms (TPH1 and TPH2) are expressed in developing mouse embryos, we first monitored their expression by RT-qPCR. Using this approach, we observed dominant expression of TPH1 in whole embryos (Fig. 2A). We then analyzed TPH1 expression in the developing brain in more detail. Our RT-qPCR data indicate that before E9.5 TPH1 is also the dominant isoform in all parts of the brain (Fig. 2A). However, from E10.5 onward, TPH2 expression dramatically increases, and TPH2 remains the dominant isoform in the brain from then until birth (data not shown) and into adulthood (25).
FIGURE 2.

Inhibition of embryonic 5-HT biosynthesis rescues developmental retardations induced by impaired MAO-A expression. A, mouse embryos were dissected (whole embryos and embryonic brain) at different embryonic stages (E7.5–E10.5). Embryonic brains from E8.5 onward were further dissected into fore-, mid- and hindbrain. RNA was extracted, and mRNA levels of TPH isoforms (TPH1 and TPH2) were quantified by qRT-PCR. B–E, mouse embryos were explanted at E7.5 and subjected to in vitro culture. Transfection complexes containing 100 nm siRNA (scrambled siRNA or MAO-A siRNA) or 1 μg/μl plasmid DNA (pcDNA3.1 backbone or pcDNA3.1 R1) were injected into the amniotic cavity. 1 μm PCPA or vehicle was added to the growth medium. After 72 of in vitro culture, embryos were collected and analyzed. B, serotonin levels were quantified as outlined under “Experimental Procedures.” Significances were calculated using Student's t test; n = 6. *, p < 0.001. C, representative images of differently treated mouse embryos are shown. Arrows indicate brain regions with defective development. D and E, developmental progress of the embryos following different treatments was quantified by morphometric scoring. Significances were calculated using Student's t test; n = 4. *, p < 0.001. Error bars represent S.D. If no error bars are visible in the diagrams, no developmental variations between embryos of one experiment could be detected.
TPH activity can be inhibited by the pharmacological antagonist PCPA (9, 19). PCPA has been shown previously to effectively rescue post- and perinatal phenotypic alterations observed in MAO-A-deficient mice by lowering abnormally elevated 5-HT levels (11, 14). When we exposed developing embryos in the absence of other stimuli to PCPA, embryonic 5-HT levels were almost unaffected, suggesting a low rate of 5-HT biosynthesis during this stage of embryo development (Fig. 2B). However, when 5-HT levels were elevated either by siRNA-mediated expression knockdown or by overexpression of the specific transcriptional MAO-A repressor R1 (8, 20), PCPA normalized the elevated 5-HT levels (Fig. 2B). Moreover, PCPA treatment rescued the phenotypes of MAO-A-deficient mouse embryos (Fig. 2C). In fact, PCPA normalized the developmental scores in MAO-A-deficient embryos (Fig. 2, D and E). These data suggest that elevated 5-HT levels contribute to the developmental defects induced by MAO-A expression silencing.
Inhibition of 5-HT Receptor-6 Rescues MAO-A-deficient Phenotypes of Embryo Development
5-HT exerts its biological functions by binding to a class of 5-Htrs, and 5-Htr-1a, -1b, and -2 subtypes have been implicated previously in abnormal perinatal maturation of neuronal structures of MAO-A-deficient mice (26–28). However, the roles of these receptor isoforms during early embryo development have not been explored in detail. Initially, we screened for expression of 5-Htr subtypes using conventional RT-PCR and observed expression of all known 5-Htr subtypes during our experimental time window (E7–E10) (data not shown). Functionality of the various 5-Htr isoforms can be blocked by isoform-specific antagonists. Methiothepin, an antagonist with a broad specificity toward 5-Htr1a, -1b, -1d, -1f, -2a, -2b, -2c, -6, and -7 rescued the MAO-A knockdown phenotype in our in vitro embryogenesis model and restored normal developmental scores (Fig. 3, A and B). In contrast, ritanserin, which antagonizes 5-Htr2 subtypes, did not prevent the developmental defects induced by MAO-A expression silencing. These data suggest that 5-Htr1s, 5-Htr6, and/or 5-Htr7 but not 5-Htr2 subtypes may be involved in the developmental defects. We then monitored in more detail the expression of these receptor subtypes using an RT-qPCR approach (Fig. 4). Here we found that most receptors were expressed at similar levels of about 0.2–1 mRNA copies/1000 mRNA copies of GAPDH. Only 5-Htr7 showed higher expression levels of up to 4 mRNA molecules/1000 copies of GAPDH mRNA. These data indicate a similar expression of most receptor subtypes. To further narrow down the 5-Htr subtypes involved in developmental retardations of MAO-A-deficient embryos, we consecutively inhibited individual 5-Htrs by using specific sets of receptor antagonists in MAO-A knockdown embryos. Neither WAY1000635 (antagonist of 5-Htr1a), SB224289 (antagonist of 5-Htr1b), BRL15572 (antagonist of 5-Htr1d), nor SB269970 (antagonist of 5-Htr7) was able to completely rescue the MAO-A knockdown phenotype (Fig. 3, A and B). Although SB269970 did not restore the developmental scores, it positively affected the head length, implicating at least a partial contribution of 5-Htr7. Because of the lack of specific inhibitors for 5-Htr1e and 5-Htr1f, we used the dual receptor agonist BRL54443 to mimic 5-HT-induced developmental alterations. When embryos were treated with BRL54443 in the absence of additional stimuli, normal embryonic development was observed (Fig. 3, C and D), suggesting that these receptor subtypes may not be of major relevance for 5-HT-induced developmental defects. In summary, our antagonist/agonist studies suggest that 5-Htr1, -2, and -7 subtypes do not contribute to embryonic brain development during this experimental time window. In contrast, the 5-Htr6 may be the most likely receptor candidate. Moreover, when we monitored 5-Htr6 expression in different regions of the embryonic brain by our RT-qPCR strategy, we found expression of 5-Htr6 in all parts of the developing brain (fore-, mid-, and hindbrain; data not shown).
FIGURE 3.
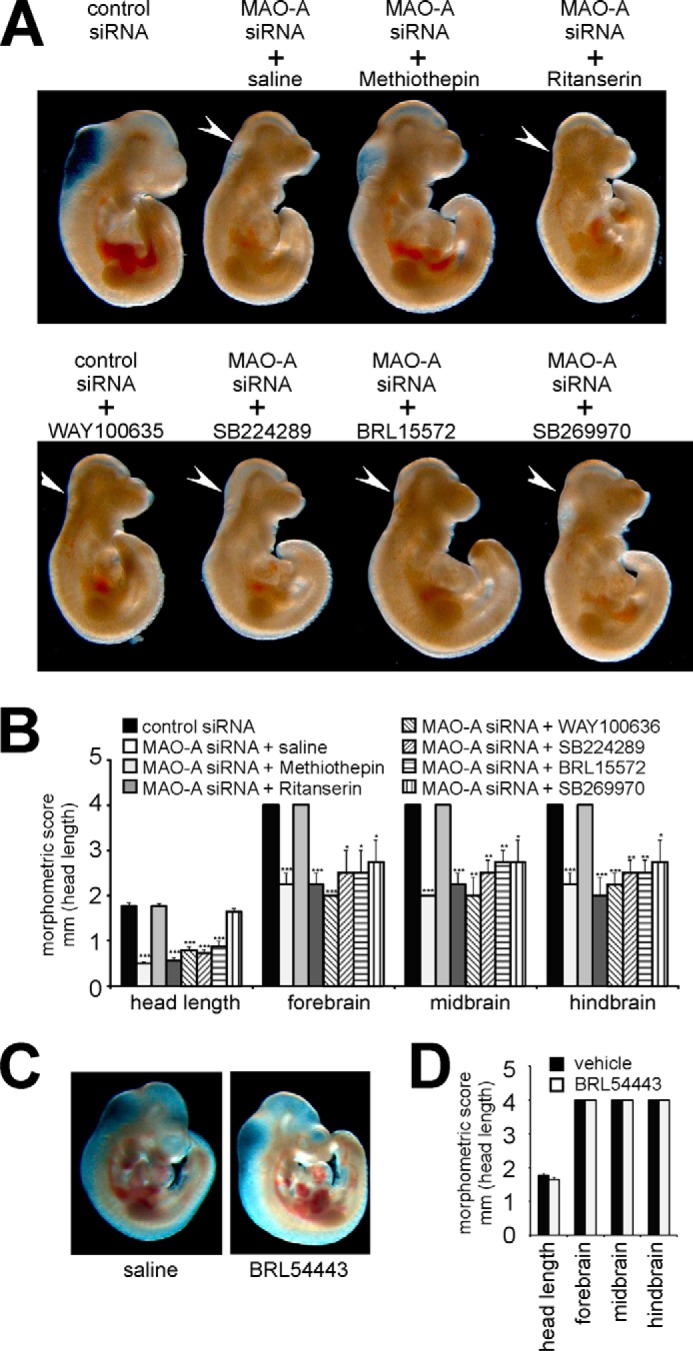
Selective inhibition and activation of 5-HT receptor isoforms. Mouse embryos were explanted at E7.5 and subjected to in vitro culture for 72 h. Transfection complexes containing 100 nm siRNA (scrambled siRNA or MAO-A siRNA) were injected into the amniotic cavity. 5-HT receptor antagonists (5 μm methiothepin, 5 μm ritanserin, 0.5 μm WAY100635, 2.5 μm SB224289, 2.5 μm BRL15572, and 0.1 μm SB269970), 5-HT receptor agonist (1.25 μm BRL54443), or vehicle was added to the growth medium at E7.5. A and C, representative images of differently treated mouse embryos are shown. Arrows indicate brain regions with defective development. B and D, developmental progress of the embryos following different treatments was quantified by morphometric scoring. Significances were calculated using Student's t test; n = 4. ***, p < 0.001; **, p < 0.01; *, p < 0.05. Error bars represent S.D. If no error bars are visible in the diagrams, no developmental variations between embryos of one experiment could be detected.
FIGURE 4.
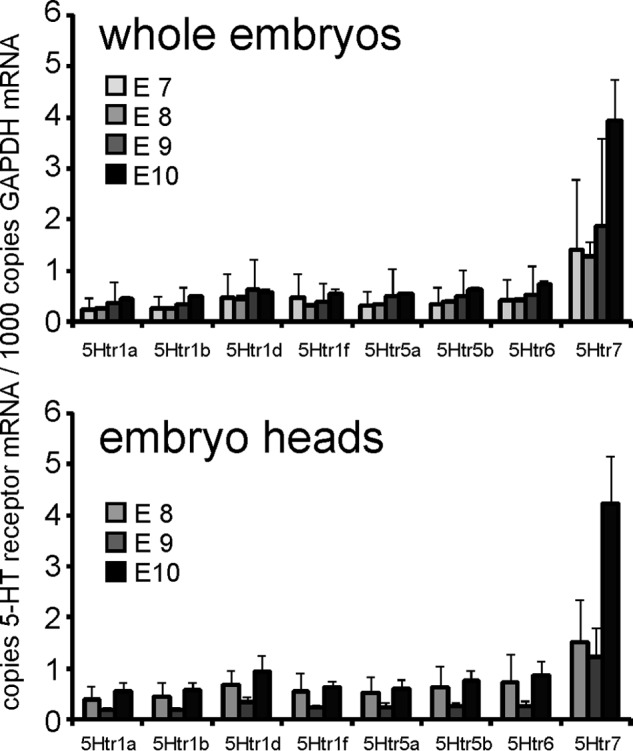
Expression of 5-HT receptor isoforms in whole embryos and in the embryo head region. Mouse embryos were dissected at different embryonic stages (E8, E9, and E10) and pooled (n = 10), RNA was extracted, and mRNA levels of 5-HT receptor isoforms (5-Htr1a, 5-Htr1b, 5-Htr1d, 5-Htr1f, 5-Htr5a, 5-Htr5b, 5-Htr6, and 5-Htr7) were quantified by qRT-PCR. Error bars represent S.D.
5-Htr6 Confers Developmental Signals in MAO-A Knockdown Embryos
Our agonist/antagonist data suggest the involvement of 5-Htr6 in the abnormal development of MAO-A knockdown embryos. To confirm these findings, we used a genetic loss-of-function strategy. For this purpose, we co-transfected the embryos with specific siRNA constructs directed against 5-Htr6 and MAO-A. Successful knockdown of target gene expression was confirmed by Western blotting (Fig. 5A). All siRNA experiments indicated strong and specific suppression of target protein expression when compared with control experiments (scrambled siRNA). In developing embryos, scrambled siRNA (Fig. 5B, panel b) did not affect the phenotype of the MAO-A knockdown embryos (Fig. 5B, panel a). Moreover, siRNA directed against 5-Htr1a was not able to rescue the MAO-A knockdown phenotype (Fig. 5B, panel c). These results are consistent with the data obtained for MAO-A knockdown embryos cultured in the presence of the 5-Htr1a antagonist WAY100636 (Fig. 3). We then applied two independent 5-Htr6-specific siRNA probes and were able to rescue the MAO-A knockdown phenotype (Fig. 5B, panels d and e). In fact, both 5-Htr6 siRNA probes normalized developmental scores (Fig. 5C), whereas scrambled siRNA and 5-Htr1a siRNA probes were ineffective.
FIGURE 5.
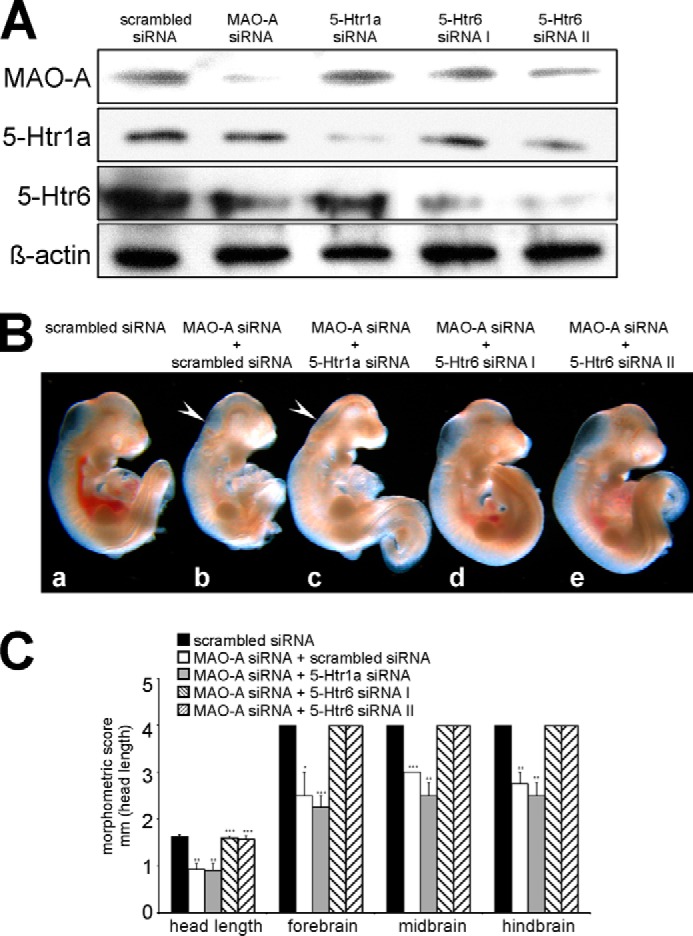
Knockdown of 5-Htr6 expression rescues developmental retardations induced by MAO-A knockdown. Mouse embryos were explanted at E7.5 and subjected to in vitro culture. Transfection complexes containing 100 nm siRNA (scrambled siRNA, MAO-A siRNA, 5-Htr1a siRNA, or different 5-Htr6 siRNAs) were injected into the amniotic cavity. After 72 of in vitro culture, embryos were collected and analyzed. A, expression of MAO-A, 5-Htr1a, 5-Htr6, and the housekeeping protein β-actin was analyzed by Western blotting. B, panels a–e, representative images of differently treated mouse embryos are shown. Arrows indicate brain regions with defective development. C, developmental progress of the embryos following different treatments was quantified by morphometric scoring. Significances were calculated using Student's t test; n = 4. ***, p < 0.001; **, p < 0.01; *, p < 0.05. Error bars represent S.D. If no error bars are visible in the diagrams, no developmental variations between embryos of one experiment could be detected.
5-Htr6 Confers the Developmental Signals Triggered by Excess 5-HT
Next we tested whether the developmental defects induced by 5-HT can be rescued by silencing the expression of 5-Htr6. As indicated in Fig. 6A, the developmental retardations induced by 5-HT (Fig. 6A, panel b) were reversed by pharmacological 5-Htr6 inhibition and siRNA-mediated expression knockdown of 5-Htr6 (panels c and d). This observation was confirmed by the morphometric scores (Fig. 6B), suggesting that MAO-A knockdown and exogenous 5-HT trigger similar signaling events.
FIGURE 6.
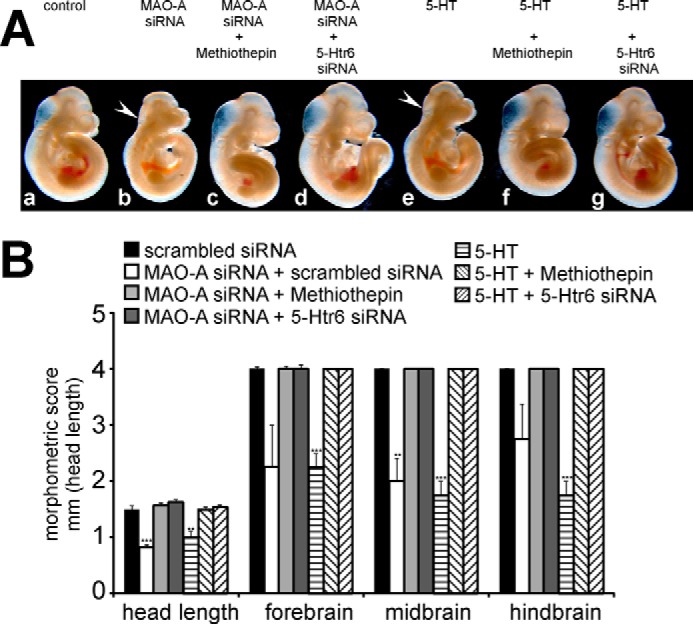
Invalidation of 5-Htr6 activity rescues developmental retardations induced by exogenous 5-HT. Mouse embryos were explanted at E7.5 and subjected to in vitro culture for 72 h. 10 μm 5-HT, 5 μm methiothepin, and/or vehicle was added to the growth medium at E7.5. A, panels a–d, representative images of differently treated mouse embryos are shown. Arrows indicate brain regions with defective development. B, developmental progress of the embryos following different treatments was quantified by morphometric scoring. Significances were calculated using Student's t test; n = 4. ***, p < 0.001; **, p < 0.01. Error bars represent S.D. If no error bars are visible in the diagrams, no developmental variations between embryos of one experiment could be detected.
5-Htr6 Activates Developmental Apoptosis and Suppresses Proliferation
Besides controlling amine levels, MAO-A functions as a modulator of apoptotic cell death (21, 29). Thus, we speculated that 5-Htr6 may be involved in 5-HT-induced developmental apoptosis. If this is the case, inactivation and/or expression silencing of 5-Htr6 should not only rescue the phenotypic alterations induced by MAO-A knockdown but should also normalize the degree of developmental apoptosis. We found that MAO-A knockdown reduced developmental apoptosis in the neuroepithelium (Fig. 7B, panel b versus panel a). A similar decrease in apoptotic cells was found when embryos were cultured in the presence of exogenous 5-HT (Fig. 7A, panel e versus panel a). We then explored the role of 5-Htr6 in this experimental setup and found that methiothepin and 5-Htr6 expression knockdown normalized the impaired apoptotic activity in MAO-A knockdown embryos (Fig. 7B).
FIGURE 7.

Activation of apoptosis and cell proliferation signaling in mouse embryos following in vitro culture. Mouse embryos were explanted at E7.5 and subjected to in vitro culture. Transfection complexes containing 100 nm siRNA (scrambled siRNA, MAO-A siRNA, or 5-Htr6 siRNA) were injected into the amniotic cavity. 10 μm 5-HT, 5 μm methiothepin, and/or vehicle was added to the growth medium at E7.5. After 72 of in vitro culture, embryos were collected and analyzed by TUNEL staining (A and B) and Ki-67 immunostaining (C). A, TUNEL staining was performed on microscopic cross-sections to stain (dark brown) apoptotic cells. The highlighted area indicates the embryonic region shown in B and C. Scale bar, 0.5 mm. B, panels a–g, apoptotic cells with dominant staining are indicated by red arrows in the neuroepithelial lining of the developing midbrain. Images are representative for three independent experiments. Scale bar, 0.1 mm. C, panels a–g, Ki-67 immunostaining was performed on microscopic cross-sections. Strong Ki-67-positive staining (dark brown) is indicated by red arrows. Scale bar, 0.1 mm.
We demonstrated previously that MAO-A knockdown coincides with ameliorated cell proliferation as indicated by reduced cyclin D levels in developing embryos (8). To investigate whether proliferative activity is also affected by 5-Htr6 signaling, we monitored Ki-67 expression in our experimental system (Fig. 7C). Ki-67 is a nuclear marker protein of proliferating cells and is absent from resting cells (30). Indeed, MAO-A siRNA and exogenous 5-HT reduced the number of Ki-67-positive cells (Fig. 7C, panels b and e versus panel a), and this was reversed by inhibition or knockdown of 5-Htr6 (Fig. 7C).
To further explore the underlying mechanisms that affect apoptotic signaling, we monitored expression and activation states of various regulatory components of the apoptotic cascade. MAO-A inactivation has previously been associated with increased protein levels of Bcl-2 and Bcl-XL (6, 31, 32). Here we found that MAO-A expression knockdown as well as exogenous 5-HT ameliorated the activation of caspase-3 and caspase-9 (Fig. 8, lanes 2 and 5 versus lane 1). Moreover, the protein levels of the antiapoptotic proteins Bcl-2 and Bcl-XL were moderately but consistently elevated under these conditions (Fig. 8, lanes 2 and 5 versus lane 1). We then inhibited 5-Htr6 activity by methiothepin or 5-Htr6 siRNA in MAO-A knockdown embryos and in embryos exposed to excess 5-HT. Here we found that activation of caspase-3 and -9 was reversed, whereas total caspase protein levels remained unaffected (Fig. 8, lanes 3 and 4 versus lane 2 and lanes 6 and 7 versus lane 5). Similarly, expression of the antiapoptotic proteins Bcl-2 and Bcl-XL was reduced when 5-Htr6 activity was abrogated (Fig. 8, lanes 3 and 4 versus lane 2 and lanes 6 and 7 versus lane 5). The results suggest that developmental apoptosis during early embryonic MAO-A deficiency may be conferred by 5-Htr6.
FIGURE 8.
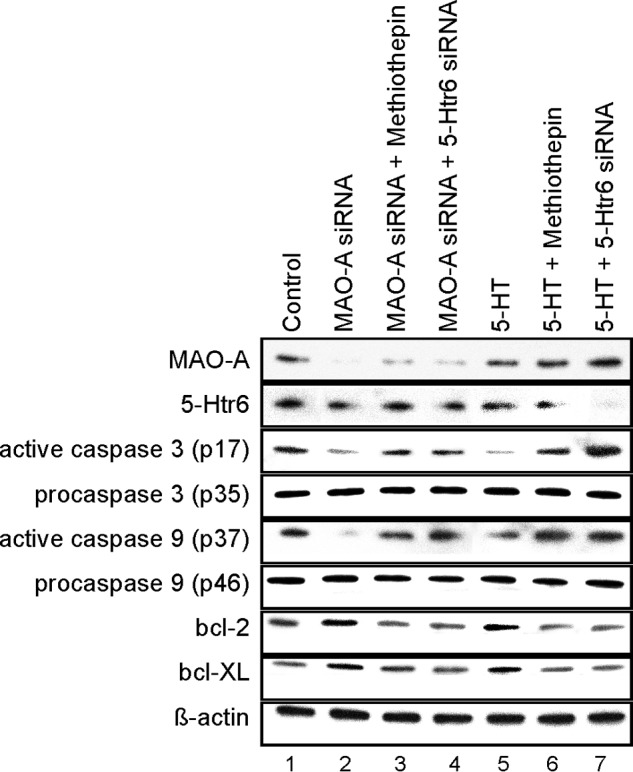
Activation of apoptosis in mouse embryos following in vitro culture. Mouse embryos were explanted at E7.5 and subjected to in vitro culture. Transfection complexes containing 100 nm siRNA (scrambled siRNA, MAO-A siRNA, or 5-Htr6 siRNA) were injected into the amniotic cavity. 10 μm 5-HT, 5 μm methiothepin, and/or vehicle was added to the growth medium at E7.5. After 72 of in vitro culture, embryos were collected, and expression of MAO-A, 5-Htr6, and apoptotic marker proteins was analyzed by Western blotting. β-Actin served as an internal standard, which was not affected by our treatments.
5-HT Signaling Affects ERKs
5-Htr activation triggers a number of intracellular signaling cascades (33) including the mitogen-activated kinase ERK (34–36), and 5-Htr6 has been suggested to activate ERK1/2 via Fyn kinase (37). ERK1/2 activates a number of downstream targets including Bcl-2 (38) and may therefore constitute a link between 5-HT signaling and the observed developmental defects. Thus, we explored the activation state of ERK following MAO-A inactivation and 5-HT stimulation. Indeed, elevated levels of phosphorylated (activated) ERK were detected in mouse embryos treated with MAO-A siRNA and in embryos cultured in the presence of exogenous 5-HT (Fig. 9, A and D).
FIGURE 9.
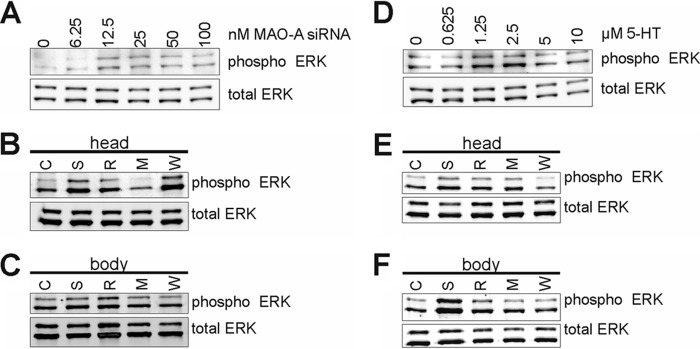
Activation of ERK1/2 in mouse embryos following in vitro culture. Mouse embryos were explanted at E7.5 and subjected to in vitro culture. After 72 h, embryos were collected as whole embryos (A and D) or dissected into head region (B and E) or body region (C and F) and subjected to Western blotting for total ERK or phospho-ERK. A, transfection complexes containing varying amounts of MAO-A siRNA (complemented to 100 nm with scrambled siRNA) were injected into the amniotic cavity. D, varying concentrations of 5-HT or saline were added to the growth medium at E7.5. B and C, mouse embryos transfected with 100 nm MAO-A siRNA (S, R, M, and W) or scrambled siRNA (C) were concomitantly treated with 5 μm methiothepin (M), 5 μm ritanserin (R), 0.5 μm WAY100635 (W), or saline (S) added to the growth medium at E7.5. E and F, mouse embryos cultured in the presence of 5-HT (S, R, M, and W) or vehicle (C) were concomitantly treated with 5 μm methiothepin (M), 5 μm ritanserin (R), 0.5 μm WAY100635 (W), or saline (S) added to the growth medium at E7.5. Western blotting was performed on lysates from at least three pooled embryos.
Because the developmental defects of MAO-A knockdown were most pronounced in the head region of the embryos, we prepared this region and repeated the experiments with 5-Htr antagonists. Here we found that in MAO-A knockdown embryos ERK activation was strongly reduced when 5-HT signaling was blocked by methiothepin. In contrast, ritanserin and WAY100635 were less effective (Fig. 9B). In the caudal body region, activation of ERK following MAO-A knockdown was less evident, and neither ritanserin nor methiothepin altered ERK activity. Only WAY100635 somewhat attenuated ERK activation (Fig. 9C). When embryos were exposed to exogenous 5-HT, all 5-Htr antagonists impaired ERK activation in both the head and caudal body regions when compared with corresponding controls (Fig. 9, E and F).
DISCUSSION
In addition to its contribution to neurotransmitter homeostasis, MAO-A activity has been implicated as a regulator of apoptotic cell death, cell proliferation, and differentiation, but the molecular mechanisms have not been explored in detail (15, 21, 29). Most of our knowledge on MAO-A functions has been obtained in adult mammals. However, recently a number of reports have aimed at investigating the biological roles of MAOs during embryogenesis. For instance, embryonic MAO-A-deficient stem cells obtained at E3.5 show a reduced capacity to differentiate into neuronal cells (15). When MAO-A expression was knocked down during in vitro embryogenesis (E7.5–E10.5), apoptotic activity as well as the proliferative activity in particular within the developing brain was reduced (8). In contrast, later on in development (E12.5–E17.5), MAO-A expression appears to mainly affect proliferation of cells of embryonic brains, but its effect on apoptosis appears to be negligible (14). This suggests that MAO-A activity very likely affects different cellular mechanisms depending on the developmental stage. In addition to cell proliferation and apoptosis, differentiation and cell size may also contribute to the developmental retardations induced by MAO-A knockdown (39). However, these pathways were beyond the scope of the present study. Finally, it must be noted that the genetic background of the animal model appears to have an impact on 5-HT levels and on the cellular responses to 5-HT (40). Moreover, many data on MAO-A function are derived from stem cell-based knock-out approaches, which might be impacted by genetic drift and epigenetic counter-regulations (41–43). This is not the case in our in vitro model of murine embryogenesis, but there are other limitations with this system (in vitro model and limited time window). Thus, each experimental model has its advantages and limitations. In conclusion, multiple factors including genetic background of the experimental mouse strain, methodological limitations of the experimental setup, and the experimental time window affect the outcome of knock-out and knockdown studies, and this may lead to partially deviating observations. In the present study, we aimed at exploring the molecular mechanisms that induce the developmental retardations of MAO-A-deficient mouse embryos between E7.5 and E10.5. For this purpose, we used an in vitro embryogenesis model and dissected the signaling cascades triggered by MAO-A deficiency. Although our experimental model is based on the explantation of embryos and their culture in vitro, it was designed to adequately mirror embryonic in utero development during the selected time window (23). Because siRNA-dependent expression silencing may have off-target effects (44), we complement such genetic approaches with pharmacologic intervention studies using inhibitors and receptor antagonists.
One of the primary biological functions of MAO-A is the breakdown of 5-HT. However, the biological activity of 5-HT is not only regulated by its breakdown but also by its biosynthesis and intracellular handling (33). In 5-HT-synthesizing cells, the transmitter is packed into vesicles with the help of the vesicular monoamine transporter (VMAT) and is secreted if needed. Conversely, 5-HT is cleared from the extracellular space via 5-HTT and is subsequently catabolized intracellularly by the catalytic activity of MAO-A. In all established in vitro and in vivo models, MAO-A deficiency is hallmarked by increased 5-HT levels, and thus, 5-HT is the most likely effector of MAO-A deficiency. This conclusion is supported by our finding that retarded progression of brain development observed in MAO-A knockdown embryos can be mimicked by addition of exogenous 5-HT, with the 5-HT mimetic 5-carboxamidotryptamine, or when extracellular 5-HT levels are elevated by inhibiting 5-HT clearance. Despite ongoing discussion about the source of 5-HT in embryos, several reports suggested the embryonic capacity of 5-HT biosynthesis (25, 45, 46). In our in vitro embryogenesis model, we found that pharmacologic inhibition of 5-HT biosynthesis reduced embryonic 5-HT levels and restored normal development of MAO-A knockdown embryos. In conclusion, our data suggest that the developmental retardations observed in MAO-A knockdown embryos are conferred by 5-HT. This is in line with observations made during postnatal development of MAO-A-deficient mice and of 5-Htt knock-out mice, which are also characterized by elevated 5-HT levels (11, 47).
5-HT acts via a family of membrane-bound cell surface receptors (5-Htrs) that trigger intracellular signaling cascades (48). To identify the receptors that mediate the developmental aberrations induced by 5-HT, we initially screened for the expression of the various receptor isoforms and found that most 5-Htrs are expressed early during embryogenesis (49). In MAO-A-deficient neonates, abnormal maturation of neuronal structures of the brain was shown to be dependent on 5-Htr1b and 5-Htr2a activation (27, 28). Indeed, abnormal maturation of the retinogeniculate and somatosensory thalamocortical systems observed in mice that were MAO-A-deficient as well as 5-HTT−/− could be rescued when 5-Htr1b expression was also inhibited (27, 28). Interestingly, however, 5-Htr1b knock-out mice show no abnormal thalamocortical or retinogeniculate projections (28), suggesting compensatory mechanisms conferred by other 5-Htr subtypes. We found that most of the developmental retardations induced by MAO-A knockdown can be attributed to 5-Htr6 activity. 5-Htr6 has only recently been identified (37). It is coupled to adenylate cyclases, and its expression can be detected in various neuronal cells (37). No targeted knock-out model for 5-Htr6 is currently available. However, 5-Htr6 inhibition studies indicated that 5-Htr6 activity modulates interneuron migration in the cerebral cortex during perinatal development (50). Activation of 5-Htr6 triggers the activation of protein kinase A (PKA) and ERK1/2, which then stimulate further downstream signaling events such as the activation of transcription factors. Indeed, excess 5-HT caused activation of ERK1/2 in our in vitro model of embryo development. Moreover, ERK1/2 and cAMP activate transcription factors that stimulate transcription of the Bcl-2 gene (51, 52). Activation of Bcl-2 expression has frequently been observed when MAO-A activity is inhibited (29, 31, 32, 51). Consistent with these findings, we also found that Bcl-2 protein levels were increased following siRNA-dependent knockdown of MAO-A expression, and this effect was reversed when 5-Htr6 signaling was shut off. However, our data indicate that when 5-HT levels are artificially elevated in wild type embryos, which express normal levels of MAO-A, additional signaling events may be triggered. For instance, the MAO reaction releases hydrogen peroxide and aldehydes, which subsequently may activate/inactivate signaling events (29, 53). In fact, aldehydes produced by MAO activity have been suggested to cause neuronal cell death (54, 55). In addition, hydrogen peroxide is a potent signaling molecule, and the precise control of its production and clearance appears to be vital for normal embryo development (56).
MAO-A is located at the outer surface of the outer mitochondrial membrane and has been shown to be a constituent of the intrinsic apoptotic signaling cascade (21, 29, 31). The intrinsic pathway of apoptosis relies on the equilibrium of pro- and antiapoptotic proteins associated to the mitochondrial membranes (57). Activation of the intrinsic apoptotic cascade proceeds via caspase-9 and effector caspase-3. MAO-A inactivation is generally associated with decreased apoptotic activity, and thus, MAO-A is thought to be a proapoptotic factor. Developmental apoptosis is a physiological process that ensures proper maturation of tissues and organs (58–60), and disturbances of the equilibrium of apoptotic regulators may cause abnormal development (61). We have shown previously that MAO-A knockdown ameliorates apoptotic activity in the embryo (8), and the data shown here indicate 5-HT as a major signaling molecule for this. Similar observations were made in neonatal MAO-A-deficient mice, which also showed enhanced 5-HT levels and reduced activation of developmental apoptosis (62). In contrast, mice with targeted deletion of the Vmat2 gene show reduced 5-HT levels because 5-HT cannot be packed into vesicles, and this causes increased apoptosis in the cerebral cortex. When VMAT−/− mice were crossed with MAO-A-deficient mice, apoptotic activity in the cortex was normalized (62). Moreover, in that study, a 5-Htr2 antagonist also normalized apoptotic activity in the brains of neonates, which suggests that in neonates 5-Htr2 receptors are responsible for the apoptotic effects conferred by 5-HT. However, our data indicate that the 5-Htr2 antagonist ritanserin affects neither embryo morphology nor the activation state of apoptosis during early embryogenesis. Instead, our data point to an involvement of the 5-HT receptor subtype 6. In fact, 5-Htr6 inhibition or expression knockdown normalized apoptotic activities and caspase activation in both MAO-A knockdown and 5-HT-stimulated embryos. Moreover, inactivation of 5-Htr6 activity normalized embryonic development, reduced Bcl-2 and Bcl-XL levels, and elevated the activity state of caspases-3 and -9 in the brain. Different activities of the various 5-Htr subtypes during prenatal and postnatal development may explain the differing findings of our study and investigations obtained in neonatal mice. In conclusion, our data highlight the vital role of MAO-A during early embryo development and shed light on the underlying molecular mechanisms causing the developmental retardations in MAO-A-deficient mouse embryos that are mediated by 5-Htr6.
Footnotes
- MAO
- monoamine oxidase
- E
- gestational day
- 5-HT
- 5-hydroxytryptamine; serotonin
- 5-Htr
- serotonin receptor
- TPH
- tryptophan hydroxylase
- PCPA
- p-chlorophenylalanine
- qPCR
- quantitative PCR
- VMAT
- vesicular monoamine transporter
- 5-HTT
- 5-HT transporter.
REFERENCES
- 1. Edmondson D. E., Binda C., Wang J., Upadhyay A. K., Mattevi A. (2009) Molecular and mechanistic properties of the membrane-bound mitochondrial monoamine oxidases. Biochemistry 48, 4220–4230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chen K. (2004) Organization of MAO A and MAO B promoters and regulation of gene expression. Neurotoxicology 25, 31–36 [DOI] [PubMed] [Google Scholar]
- 3. Billett E. E. (2004) Monoamine oxidase (MAO) in human peripheral tissues. Neurotoxicology 25, 139–148 [DOI] [PubMed] [Google Scholar]
- 4. Nicotra A., Pierucci F., Parvez H., Senatori O. (2004) Monoamine oxidase expression during development and aging. Neurotoxicology 25, 155–165 [DOI] [PubMed] [Google Scholar]
- 5. Wang C. C., Billett E., Borchert A., Kuhn H., Ufer C. (2013) Monoamine oxidases in development. Cell. Mol. Life Sci. 70, 599–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Youdim M. B., Edmondson D., Tipton K. F. (2006) The therapeutic potential of monoamine oxidase inhibitors. Nat. Rev. Neurosci. 7, 295–309 [DOI] [PubMed] [Google Scholar]
- 7. Grimsby J., Toth M., Chen K., Kumazawa T., Klaidman L., Adams J. D., Karoum F., Gal J., Shih J. C. (1997) Increased stress response and beta-phenylethylamine in MAOB-deficient mice. Nat. Genet. 17, 206–210 [DOI] [PubMed] [Google Scholar]
- 8. Wang C. C., Borchert A., Ugun-Klusek A., Tang L. Y., Lui W. T., Chu C. Y., Billett E., Kuhn H., Ufer C. (2011) Monoamine oxidase a expression is vital for embryonic brain development by modulating developmental apoptosis. J. Biol. Chem. 286, 28322–28330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cases O., Seif I., Grimsby J., Gaspar P., Chen K., Pournin S., Müller U., Aguet M., Babinet C., Shih J. C., De Maeyer E. (1995) Aggressive behavior and altered amounts of brain serotonin and norepinephrine in mice lacking MAOA. Science 268, 1763–1766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Scott A. L., Bortolato M., Chen K., Shih J. C. (2008) Novel monoamine oxidase A knock out mice with human-like spontaneous mutation. Neuroreport 19, 739–743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cases O., Vitalis T., Seif I., De Maeyer E., Sotelo C., Gaspar P. (1996) Lack of barrels in the somatosensory cortex of monoamine oxidase A-deficient mice: role of a serotonin excess during the critical period. Neuron 16, 297–307 [DOI] [PubMed] [Google Scholar]
- 12. Upton A. L., Salichon N., Lebrand C., Ravary A., Blakely R., Seif I., Gaspar P. (1999) Excess of serotonin (5-HT) alters the segregation of ipsilateral and contralateral retinal projections in monoamine oxidase A knock-out mice: possible role of 5-HT uptake in retinal ganglion cells during development. J. Neurosci. 19, 7007–7024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bou-Flores C., Lajard A. M., Monteau R., De Maeyer E., Seif I., Lanoir J., Hilaire G. (2000) Abnormal phrenic motoneuron activity and morphology in neonatal monoamine oxidase A-deficient transgenic mice: possible role of a serotonin excess. J. Neurosci. 20, 4646–4656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cheng A., Scott A. L., Ladenheim B., Chen K., Ouyang X., Lathia J. D., Mughal M., Cadet J. L., Mattson M. P., Shih J. C. (2010) Monoamine oxidases regulate telencephalic neural progenitors in late embryonic and early postnatal development. J. Neurosci. 30, 10752–10762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang Z. Q., Chen K., Ying Q. L., Li P., Shih J. C. (2011) Monoamine oxidase A regulates neural differentiation of murine embryonic stem cells. J. Neural. Transm. 118, 997–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Saito M., Yamagata T., Matsumoto A., Shiba Y., Nagashima M., Taniguchi S., Jimbo E., Momoi M. Y. (2014) MAOA/B deletion syndrome in male siblings with severe developmental delay and sudden loss of muscle tonus. Brain Dev. 36, 64–69 [DOI] [PubMed] [Google Scholar]
- 17. Azmitia E. C. (2001) Modern views on an ancient chemical: serotonin effects on cell proliferation, maturation, and apoptosis. Brain Res. Bull. 56, 413–424 [DOI] [PubMed] [Google Scholar]
- 18. Herlenius E., Lagercrantz H. (2004) Development of neurotransmitter systems during critical periods. Exp. Neurol. 190, Suppl. 1, S8–S21 [DOI] [PubMed] [Google Scholar]
- 19. Vitalis T., Cases O., Passemard S., Callebert J., Parnavelas J. G. (2007) Embryonic depletion of serotonin affects cortical development. Eur. J. Neurosci. 26, 331–344 [DOI] [PubMed] [Google Scholar]
- 20. Chen K., Ou X. M., Chen G., Choi S. H., Shih J. C. (2005) R1, a novel repressor of the human monoamine oxidase A. J. Biol. Chem. 280, 11552–11559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ou X.-M., Chen K., Shih J. C. (2006) Monoamine oxidase A and repressor R1 are involved in apoptotic signaling pathway. Proc. Natl. Acad. Sci. U.S.A. 103, 10923–10928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ufer C., Wang C. C., Fähling M., Schiebel H., Thiele B. J., Billett E. E., Kuhn H., Borchert A. (2008) Translational regulation of glutathione peroxidase 4 expression through guanine-rich sequence-binding factor 1 is essential for embryonic brain development. Genes Dev. 22, 1838–1850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Van Maele-Fabry G., Delhaise F., Picard J. J. (1990) Morphogenesis and quantification of the development of post-implantation mouse embryos. Toxicol. in Vitro 4, 149–156 [DOI] [PubMed] [Google Scholar]
- 24. Nakamura K., Hasegawa H. (2007) Developmental role of tryptophan hydroxylase in the nervous system. Mol. Neurobiol. 35, 45–54 [DOI] [PubMed] [Google Scholar]
- 25. Amireault P., Sibon D., Côté F. (2013) Life without peripheral serotonin: insights from tryptophan hydroxylase 1 knockout mice reveal the existence of paracrine/autocrine serotonergic networks. ACS Chem. Neurosci. 4, 64–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shih J. C., Ridd M. J., Chen K., Meehan W. P., Kung M. P., Seif I., De Maeyer E. (1999) Ketanserin and tetrabenazine abolish aggression in mice lacking monoamine oxidase A. Brain Res. 835, 104–112 [DOI] [PubMed] [Google Scholar]
- 27. Bras H., Gaytán S. P., Portalier P., Zanella S., Pásaro R., Coulon P., Hilaire G. (2008) Prenatal activation of 5-HT2A receptor induces expression of 5-HT1B receptor in phrenic motoneurons and alters the organization of their premotor network in newborn mice. Eur. J. Neurosci. 28, 1097–1107 [DOI] [PubMed] [Google Scholar]
- 28. Salichon N., Gaspar P., Upton A. L., Picaud S., Hanoun N., Hamon M., De Maeyer E., Murphy D. L., Mossner R., Lesch K. P., Hen R., Seif I. (2001) Excessive activation of serotonin (5-HT) 1B receptors disrupts the formation of sensory maps in monoamine oxidase a and 5-ht transporter knock-out mice. J. Neurosci. 21, 884–896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fitzgerald J. C., Ufer C., De Girolamo L. A., Kuhn H., Billett E. E. (2007) Monoamine oxidase-A modulates apoptotic cell death induced by staurosporine in human neuroblastoma cells. J. Neurochem. 103, 2189–2199 [DOI] [PubMed] [Google Scholar]
- 30. Yerushalmi R., Woods R., Ravdin P. M., Hayes M. M., Gelmon K. A. (2010) Ki67 in breast cancer: prognostic and predictive potential. Lancet Oncol. 11, 174–183 [DOI] [PubMed] [Google Scholar]
- 31. Yi H., Maruyama W., Akao Y., Takahashi T., Iwasa K., Youdim M. B., Naoi M. (2006) N-Propargylamine protects SH-SY5Y cells from apoptosis induced by an endogenous neurotoxin, N-methyl(R)salsolinol, through stabilization of mitochondrial membrane and induction of anti-apoptotic Bcl-2. J. Neural. Transm. 113, 21–32 [DOI] [PubMed] [Google Scholar]
- 32. Inaba-Hasegawa K., Akao Y., Maruyama W., Naoi M. (2012) Type A monoamine oxidase is associated with induction of neuroprotective Bcl-2 by rasagiline, an inhibitor of type B monoamine oxidase. J. Neural. Transm. 119, 405–414 [DOI] [PubMed] [Google Scholar]
- 33. Fidalgo S., Ivanov D. K., Wood S. H. (2013) Serotonin: from top to bottom. Biogerontology 14, 21–45 [DOI] [PubMed] [Google Scholar]
- 34. Watts S. W., Yang P., Banes A. K., Baez M. (2001) Activation of Erk mitogen-activated protein kinase proteins by vascular serotonin receptors. J. Cardiovasc. Pharmacol. 38, 539–551 [DOI] [PubMed] [Google Scholar]
- 35. Pearson G. W., Earnest S., Cobb M. H. (2006) Cyclic AMP selectively uncouples mitogen-activated protein kinase cascades from activating signals. Mol. Cell. Biol. 26, 3039–3047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Brown P., Gerfen C. R. (2006) Plasticity within striatal direct pathway neurons after neonatal dopamine depletion is mediated through a novel functional coupling of serotonin 5-HT2 receptors to the ERK 1/2 map kinase pathway. J. Comp. Neurol. 498, 415–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Marazziti D., Baroni S., Borsini F., Picchetti M., Vatteroni E., Falaschi V., Catena-Dell'Osso M. (2013) Serotonin receptors of type 6 (5-HT6): from neuroscience to clinical pharmacology. Curr. Med. Chem. 20, 371–377 [PubMed] [Google Scholar]
- 38. Désiré L., Courtois Y., Jeanny J. C. (2000) Endogenous and exogenous fibroblast growth factor 2 support survival of chick retinal neurons by control of neuronal bcl-xL and bcl-2 expression through a fibroblast growth factor receptor 1- and ERK-dependent pathway. J. Neurochem. 75, 151–163 [DOI] [PubMed] [Google Scholar]
- 39. Franke T. F., Hornik C. P., Segev L., Shostak G. A., Sugimoto C. (2003) PI3K/Akt and apoptosis: size matters. Oncogene 22, 8983–8998 [DOI] [PubMed] [Google Scholar]
- 40. Bortolato M., Chen K., Godar S. C., Chen G., Wu W., Rebrin I., Farrell M. R., Scott A. L., Wellman C. L., Shih J. C. (2011) Social deficits and perseverative behaviors, but not overt aggression, in MAO-A hypomorphic mice. Neuropsychopharmacology 36, 2674–2688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Thyagarajan T., Totey S., Danton M. J., Kulkarni A. B. (2003) Genetically altered mouse models: the good, the bad, and the ugly. Crit. Rev. Oral Biol. Med. 14, 154–174 [DOI] [PubMed] [Google Scholar]
- 42. Rivera J., Tessarollo L. (2008) Genetic background and the dilemma of translating mouse studies to humans. Immunity 28, 1–4 [DOI] [PubMed] [Google Scholar]
- 43. Weiss K. M., Fullerton S. M. (2000) Phenogenetic drift and the evolution of genotype-phenotype relationships. Theor. Popul. Biol. 57, 187–195 [DOI] [PubMed] [Google Scholar]
- 44. Jackson A. L., Linsley P. S. (2010) Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat. Rev. Drug Discov. 9, 57–67 [DOI] [PubMed] [Google Scholar]
- 45. Walther D. J., Bader M. (1999) Serotonin synthesis in murine embryonic stem cells. Brain Res. Mol. Brain Res. 68, 55–63 [DOI] [PubMed] [Google Scholar]
- 46. Basu B., Desai R., Balaji J., Chaerkady R., Sriram V., Maiti S., Panicker M. M. (2008) Serotonin in pre-implantation mouse embryos is localized to the mitochondria and can modulate mitochondrial potential. Reproduction 135, 657–669 [DOI] [PubMed] [Google Scholar]
- 47. Persico A. M., Baldi A., Dell'Acqua M. L., Moessner R., Murphy D. L., Lesch K. P., Keller F. (2003) Reduced programmed cell death in brains of serotonin transporter knockout mice. Neuroreport 14, 341–344 [DOI] [PubMed] [Google Scholar]
- 48. Berger M., Gray J. A., Roth B. L. (2009) The expanded biology of serotonin. Annu. Rev. Med. 60, 355–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Amireault P., Dubé F. (2005) Intracellular cAMP and calcium signaling by serotonin in mouse cumulus-oocyte complexes. Mol. Pharmacol. 68, 1678–1687 [DOI] [PubMed] [Google Scholar]
- 50. Riccio O., Potter G., Walzer C., Vallet P., Szabó G., Vutskits L., Kiss J. Z., Dayer A. G. (2009) Excess of serotonin affects embryonic interneuron migration through activation of the serotonin receptor 6. Mol. Psychiatry 14, 280–290 [DOI] [PubMed] [Google Scholar]
- 51. Chiou S. H., Ku H. H., Tsai T. H., Lin H. L., Chen L. H., Chien C. S., Ho L. L., Lee C. H., Chang Y. L. (2006) Moclobemide upregulated Bcl-2 expression and induced neural stem cell differentiation into serotoninergic neuron via extracellular-regulated kinase pathway. Br. J. Pharmacol. 148, 587–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Jaworski J., Mioduszewska B., Sánchez-Capelo A., Figiel I., Habas A., Gozdz A., Proszynski T., Hetman M., Mallet J., Kaczmarek L. (2003) Inducible cAMP early repressor, an endogenous antagonist of cAMP responsive element-binding protein, evokes neuronal apoptosis in vitro. J. Neurosci. 23, 4519–4526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Naoi M., Maruyama W., Yi H., Inaba K., Akao Y., Shamoto-Nagai M. (2009) Mitochondria in neurodegenerative disorders: regulation of the redox state and death signaling leading to neuronal death and survival. J. Neural Transm. 116, 1371–1381 [DOI] [PubMed] [Google Scholar]
- 54. Burke W. J., Li S. W., Schmitt C. A., Xia P., Chung H. D., Gillespie K. N. (1999) Accumulation of 3,4-dihydroxyphenylglycolaldehyde, the neurotoxic monoamine oxidase A metabolite of norepinephrine, in locus ceruleus cell bodies in Alzheimer's disease: mechanism of neuron death. Brain Res. 816, 633–637 [DOI] [PubMed] [Google Scholar]
- 55. Anderson D. G., Mariappan S. V., Buettner G. R., Doorn J. A. (2011) Oxidation of 3,4-dihydroxyphenylacetaldehyde, a toxic dopaminergic metabolite, to a semiquinone radical and an ortho-quinone. J. Biol. Chem. 286, 26978–26986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ufer C., Wang C. C., Borchert A., Heydeck D., Kuhn H. (2010) Redox control in mammalian embryo development. Antioxid. Redox Signal. 13, 833–875 [DOI] [PubMed] [Google Scholar]
- 57. Danial N. N., Korsmeyer S. J. (2004) Cell death: critical control points. Cell 116, 205–219 [DOI] [PubMed] [Google Scholar]
- 58. Boumela I., Assou S., Aouacheria A., Haouzi D., Dechaud H., De Vos J., Handyside A., Hamamah S. (2011) Involvement of BCL2 family members in the regulation of human oocyte and early embryo survival and death: gene expression and beyond. Reproduction 141, 549–561 [DOI] [PubMed] [Google Scholar]
- 59. Blaschke A. J., Staley K., Chun J. (1996) Widespread programmed cell death in proliferative and postmitotic regions of the fetal cerebral cortex. Development 122, 1165–1174 [DOI] [PubMed] [Google Scholar]
- 60. Fuchs Y., Steller H. (2011) Programmed cell death in animal development and disease. Cell 147, 742–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Copp A. J. (2005) Neurulation in the cranial region—normal and abnormal. J. Anat. 207, 623–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Stankovski L., Alvarez C., Ouimet T., Vitalis T., El-Hachimi K. H., Price D., Deneris E., Gaspar P., Cases O. (2007) Developmental cell death is enhanced in the cerebral cortex of mice lacking the brain vesicular monoamine transporter. J. Neurosci. 27, 1315–1324 [DOI] [PMC free article] [PubMed] [Google Scholar]