

Background: TDP-43 aggregates and mutations are observed in patients with ALS and FTLD.
Results: The equilibrium unfolding of the RRM domains reveals a highly populated intermediate in RRM2.
Conclusion: The stability of RRM2 may result from a large hydrophobic cluster, and the intermediate state may be essential for accessing the nuclear export sequence.
Significance: Accessing the RRM2 intermediate state can potentially propagate disease pathogenesis.
Keywords: Amyotrophic Lateral Sclerosis (Lou Gehrig's Disease), Circular Dichroism (CD), Fluorescence, Neurodegenerative Diseases, Protein Folding, Protein Misfolding, Protein-Nucleic Acid Interaction, RNA-binding Protein, Thermodynamics, Frontotemporal Lobar Degeneration (FTLD)
Abstract
Pathological alteration of TDP-43 (TAR DNA-binding protein-43), a protein involved in various RNA-mediated processes, is a hallmark feature of the neurodegenerative diseases amyotrophic lateral sclerosis and frontotemporal lobar degeneration. Fragments of TDP-43, composed of the second RNA recognition motif (RRM2) and the disordered C terminus, have been observed in cytoplasmic inclusions in sporadic amyotrophic lateral sclerosis cases, suggesting that conformational changes involving RRM2 together with the disordered C terminus play a role in aggregation and toxicity. The biophysical data collected by CD and fluorescence spectroscopies reveal a three-state equilibrium unfolding model for RRM2, with a partially folded intermediate state that is not observed in RRM1. Strikingly, a portion of RRM2 beginning at position 208, which mimics a cleavage site observed in patient tissues, increases the population of this intermediate state. Mutually stabilizing interactions between the domains in the tethered RRM1 and RRM2 construct reduce the population of the intermediate state and enhance DNA/RNA binding. Despite the high sequence homology of the two domains, a network of large hydrophobic residues in RRM2 provides a possible explanation for the increased stability of RRM2 compared with RRM1. The cluster analysis suggests that the intermediate state may play a functional role by enhancing access to the nuclear export signal contained within its sequence. The intermediate state may also serve as a molecular hazard linking productive folding and function with pathological misfolding and aggregation that may contribute to disease.
Introduction
Amyotrophic lateral sclerosis (ALS)2 is a highly debilitating and progressive motor neuron disease affecting approximately 1–2 of 100,000 new people each year, with death occurring 2–5 years after onset (1). Only 10% of ALS cases have been linked to genetic mutations in numerous genes (familial ALS), whereas the remaining 90% of cases result from an unknown cause (sporadic ALS) (2). The pathological hallmark of ALS is the presence of ubiquitinated inclusions in the cytoplasm of surviving spinal motor neurons. In 2006, biochemical and immunological approaches identified TAR DNA-binding protein 43 (TDP-43) as a major protein found in post-mortem brain inclusions of patients with both ALS and frontotemporal lobar degeneration with ubiquitinated inclusions (FTLD-U), providing a molecular connection between these diseases (3, 4). In the years since this initial discovery, 50 different TDP-43 mutations in familial and sporadic ALS patients have been identified (see the ALS Online Genetics Database), thereby underscoring a direct role for TDP-43 in ALS pathogenesis. Related research has been rapid (5), with numerous reports of mouse models, biomarkers, and assays for testing disease progression and cellular function. However, a molecular level understanding of how TDP-43 may lead to disease is still lacking (4), in part because of the poor solubility of the full-length protein and its tendency to fragment and aggregate.
TDP-43 is a 414-amino acid protein that contains two RNA recognition motifs (RRMs), a nuclear localization sequence in the N terminus, a nuclear export signal (NES) within RRM2, and a C-terminal glycine-rich domain (accession no. Q13148, UniProtKB/Swiss-Prot) (Fig. 1A). TDP-43 is ubiquitously expressed and has been implicated to play a functional role in many RNA processes, including gene transcription, splicing, mRNA processing, and mRNA stability (6–8). TDP-43 is localized in the nucleus in normal cells but redistributes to form cytoplasmic aggregates composed of hyperphosphorylated and ubiquitinated C-terminal fragments in diseased cells (9). As inferred from studies of other protein aggregates involved in neurodegenerative diseases (10), TDP-43 aggregates may arise from the population of non-native conformations that probably drive the neurodegeneration directly through a gain-of-function or loss-of-function mechanism. In these scenarios, the formation and accumulation of TDP-43 aggregates generate toxicity or impair normal TDP-43 cellular function, resulting in cell death. Mounting evidence supports a loss-of-function phenotype (11), where sequestration of functional protein into cytoplasmic aggregates would limit the pool of available functional nuclear TDP-43.
FIGURE 1.

TDP-43 contains both structured and disordered regions. A, domain architecture of TDP-43 and aggregation (16) and disorder (17) propensity predictions based on the amino acid sequence. The aggregation algorithms TANGO (green) and WALTZ (blue) predict an aggregation-prone stretch in RRM2 (residues 228–233), with WALTZ predicting a second region in the glycine-rich domain (residues 314–319) and TANGO predicting a region near the nuclear localization sequence (NLS; purple). The disorder algorithm PONDR (orange) predicts high amounts of disorder in the C-terminal glycine-rich region (orange), where the ALS-causing mutations are located. Regions of disorder are also predicted in the linker, which lies between the two RRM domains as well as within the nuclear localization sequence. By contrast, very little disorder is predicted within the putative NES (yellow). B, the two RRM domains show high sequence similarity by ClustalW2 sequence alignment (*, identical; :, similar). Residues in β-strands and α-helices are colored blue and red, respectively, with the residues removed in RRM2c boxed in cyan. The predicted aggregation prone sequence in β3 is highlighted in boldface type, and the putative NES sequence is underlined. C and D, RRM domains are structurally similar based on solution NMR (C, RRM1 (Protein Data Bank entry 2cqg); D, RRM2 (Protein Data Bank entry 1wf0)). β-Strands are colored blue, α-helices are red, and loops are gray. The key phenylalanine residues identified for RNA binding in β3 are shown as sticks (RRM1, Phe147 and Phe149; RRM2, Phe229 and Phe231), and intrinsic fluorophores for following tertiary structure folding are also shown (RRM1, Trp113 and Trp172; RRM2, Tyr214). The RRM2c construct, a model for the cleavage observed in disease, removes the first β-strand and half of the first α-helix (deleted regions shown in cyan) in RRM2. Note that the solution structure of RRMc is unknown and predicted to be much different from that mapped on RRM2 (Protein Data Bank entry 1wf0) due to removal of secondary structural elements in the protein core. E, NMR structure of the tethered RRMs with RNA (Protein Data Bank entry 4bs2) with RNA shown in gray (43). The secondary structure elements are colored as shown in C and D. The RNA-binding residues and intrinsic fluorophores are shown as sticks.
Several studies have demonstrated that both RRM2 and the disordered C terminus are required for aggregation and toxicity (12–15). Examination of the domain architecture of TDP-43 suggests a possible interplay between structured and disordered sequences that may play a key role in toggling between appropriate biological function and dysfunction leading to disease (Fig. 1A). Computer algorithms developed for predicting aggregation prone regions in unfolded polypeptide chains (16), WALTZ and TANGO, both show a high propensity for aggregation in RRM2 and the C terminus (Fig. 1A). The sequence-based disorder predictor algorithm, PONDR (17, 18), predicts a high degree of disorder in the C-terminal region of TDP-43. The two RRM domains share significant sequence (Fig. 1B) and structural (Fig. 1, C and D) homology. The NMR solution structures of the isolated RRM domains (Fig. 1, C and D) and the tethered RRM domains (Fig. 1E) show an α + β structure for each domain, composed of two repeating βαβ motifs with an extra β-strand (β4) inserted within the second βαβ motif. Together, these strands form an antiparallel β-sheet across one face of the RRM with the α-helices docked on the opposite face. Although RRM domains can bind a diverse set of targets, including RNA, DNA, and peptides and other proteins (19, 20), most studies on TDP-43 have focused on the role of the highly conserved phenylalanine side chains in β3 for RNA recognition (21–23).
To understand how the conformations populated by the RRM domains of TDP-43 may play a role in disease propagation, the equilibrium unfolding and RNA-binding properties of the isolated and tethered RRM domains were probed by a pair of complementary spectroscopic techniques. The results identified a novel intermediate state in the folding of the RRM2 domain. The population of this intermediate is increased in a cleavage fragment but is reduced upon tethering to RRM1. The intermediate state in RRM2 may serve as a molecular hazard that may partition between productive folding and function on the one hand and misfolding and aberrant protein-protein interactions that could lead to disease progression on the other.
EXPERIMENTAL PROCEDURES
Expression and Purification of TDP-43 Fragments
Gene fragments of TDP-43 encoding RRM1 (amino acids 102–181), RRM2 (amino acids 190–261), an RRM2 fragment mimicking the disease-relevant proteolytic cleavage site within RRM2 (9) (RRM2c; amino acids 208–261), and the tethered RRMs (tRRMs; amino acids 97–261) were purchased from Genescript with BamHI and EcoRI or NcoI and BamHI restriction enzyme digestion sites. The genes were inserted into a modified pGEX-6p1 (GE Healthcare) or pET-3d (Novagen) vector with a His6 tag and either a PreScission or TEV protease cleavage site, respectively, for His6 tag removal. The proteins were overexpressed in BL21 DE3 Escherichia coli cells (Stratagene) grown in LB medium until A600 = 0.8, followed by induction with 1 mm isopropyl 1-thio-β-d-galactopyranoside for 24 h at 20 °C (RRM1 and tRRMs) or for 16 h at 30 °C (RRM2 and RRM2c).
Cells were resuspended in lysis buffer (20 mm NaPi, pH 7.4, 300 mm NaCl, 30 mm imidazole) and lysed by sonication. RRM2 and RRM2c were only present in the insoluble fraction, and isolation from cell pellets was performed in the presence of 6 m urea. Each construct was bound to His60 resin (Clontech) overnight and washed with 10 column volumes of lysis buffer before elution with 300 mm imidazole. The eluted protein was dialyzed against protease cleavage buffer (50 mm Tris, pH 8.0, 1 mm EDTA, and 1 mm DTT), followed by subsequent cleavage with PreScission or TEV protease to remove the His6 tag. Minor contaminants were removed through ion exchange chromatography using S Sepharose (RRM1) or Q Sepharose (RRM2, tRRMs, and RRM2c) before dialysis into 10 mm KPi, pH 7.2, 150 mm KCl, 1 mm β-mercaptoethanol for subsequent studies. After cleavage, protein purity was >98%, as determined by both SDS-PAGE and reverse-phase MALDI-TOF mass spectroscopy carried out at the Proteomics and Mass Spectrometry Facility, University of Massachusetts Medical School.
RRM2 contains a second TEV protease-like cleavage site (246EDLIIKG252), as determined by mass spectrometry, preventing isolation of the intact domain. Thus, all RRM2-containing constructs were expressed instead with a PreScission protease site after the N-terminal His tag. Cleavage with TEV protease (24) results in an N-terminal Gly residue before the RRM amino acid sequence, and cleavage with PreScission protease leaves an N-terminal GPLGS sequence, with the LGS sequence being required for cloning.
Size Exclusion Chromatography to Determine Oligomerization State
Size exclusion chromatography was performed on all constructs using a 24-ml Superdex 200 10/300 GL column run at a flow rate of 0.2 ml min−1. Oligomerization status of the constructs was monitored as a function of loaded protein concentration by comparison with protein molecular weight standards (GE Healthcare). All size exclusion chromatography was performed at 4 °C in 10 mm KPi, pH 7.2, 150 mm KCl, and 1 mm β-mercaptoethanol.
Equilibrium Unfolding Experiments
The native state circular dichroism (CD) spectrum of each construct was collected from 190 to 280 nm on a Jasco-810 spectropolarimeter with a thermoelectric temperature control system in a 0.1-cm cuvette (Hellma). Guanidine hydrochloride (GdnHCl)-induced denaturation spectra were collected from 260 to 215 nm at a scan rate of 50 nm min−1 and a response time of 8 s. Samples were prepared from native and unfolded stock solutions mixed in precise amounts by in-house software and a Hamilton series 500 titrator. The resulting solutions were incubated overnight at room temperature or at 37 °C before recording CD spectra. All GdnHCl concentrations were measured using an ABBE refractometer, and all CD measurements were baseline-corrected for buffer contributions. Protein concentration was measured by A280 absorbance (25), using an extinction coefficient of 15,470 m−1 cm−1 for tRRMs, 13,980 m−1 cm−1 for RRM1, and 1490 m−1 cm−1 for RRM2 and RRM2c. The protein concentration was varied from 5 to 60 μm for CD experiments. Each CD spectra was normalized for protein concentration and number of amino acids and reported as mean residue ellipticity (26). Reversibility was confirmed to be >95% by the coincidence of equilibrium profiles for samples prepared from initial protein stocks in either buffer or denaturant.
Steady-state fluorescence (FL) measurements were performed on a Spex Fluorolog-3 equipped with a wavelength electronics temperature controller. For RRM2 and RRM2c, each GdnHCl titration sample was excited at 274 nm, and tyrosine emission spectra were collected from 280 to 400 nm at 20 or 37 °C with 5-nm slit widths. For the tryptophan-containing RRM1 and tRRMs proteins, excitation was at 295 nm, and tryptophan emission spectra were collected from 300 to 500 nm at 20 °C with 5-nm slit widths.
Denaturation experiments by both CD and FL were performed in replicates of three for each construct to ensure reproducibility. The equilibrium folding data for each construct were analyzed using an appropriate equilibrium folding model with the in-house data analysis software Savuka (27). Each data set was subjected to a global analysis, where the baselines were local parameters, and the free energy of folding in the absence of denaturant (ΔG0H2O) and the m value were globally linked between data sets. All of the Trp and Tyr fluorescence equilibrium profiles as well as the CD equilibrium profile for RRM1 were best fit to a two-state model, N ⇆ U. For these experiments, the change in free energy between the native and unfolded states is assumed to depend linearly on the denaturant concentration as shown in Equation 1 (28).
The change in free energy in the absence of denaturant, ΔG0H2O, can be used to determine the equilibrium constant, Keq, from Equation 2.
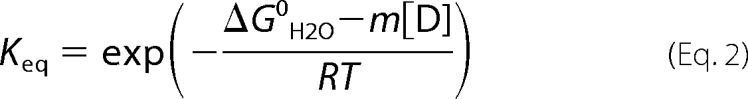 |
For the CD equilibrium unfolding profiles of RRM2 and RRM2c, a three-state model best described the transition between the native and unfolded forms of the protein with the population of a stable intermediate state (I), N ⇆ I ⇆ U. Tethering the two RRM domains by the natural 15-amino acid residue linker sequence (tRRMs) results in the population of multiple stable intermediates at equilibrium by CD. In this case, the data were best fit with a four-state equilibrium model, N ⇆ I1 ⇆ I2 ⇆ U, with the population of two stable intermediate states, I1 and I2.
The fractional population of the intermediate state for RRM2 and RRM2c at a given denaturant concentration was determined from the partition function according to Equation 3 (29),
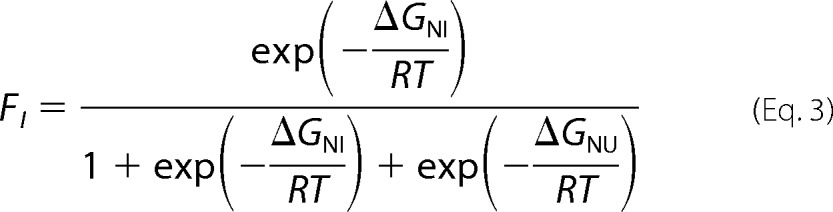 |
where ΔGNI = ΔG0NIH2O − mNI[D], and ΔGNU = ΔG0NUH2O − mNU[D]. The I2 intermediate in the four-state model for tRRMs corresponds to the single intermediate for RRM2, and its fractional population was calculated as defined in Equation 4,
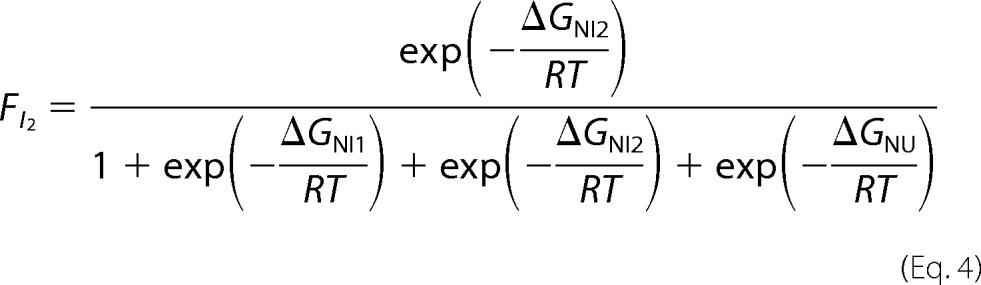 |
where ΔGNI1 = ΔG0NI1 − mNI1[D], ΔGNI2 = ΔG0NI2 − mNI2[D], and ΔGNU = ΔG0NU − mNU[D].
Nucleic Acid Binding Assays
EMSAs and tryptophan (Trp) lifetimes were used to determine the apparent binding affinity of the TDP-43 RRM domains to UGUGUGUGUGUG ((UG)6), TGTGTGTGTGTG ((TG)6), and TTTTTTTTTTTT (T12) 12-mer oligonucleotides (IDT Technologies). For EMSA, the oligonucleotides were fluorescently labeled at the 5′-end by 5-carboxyfluorescein (IDT Technologies).
A typical EMSA consists of 50-μl reactions of 3 nm nucleotides incubated with increasing protein concentrations up to 8 μm. Binding reactions were performed in binding buffer, 10 mm KPi, pH 7.2, 150 mm KCl, 2 mm DTT, 10 μg ml−1 tRNA, and 0.01% IGEPAL CA-630 (Sigma-Aldrich) (30) and incubated for 2 h at room temperature. Prior to loading on an 8% polyacrylamide gel, 5 μl of bromocresol blue in 30% glycerol was added to each reaction, and 45 μl of the reaction mixture was added to the acrylamide gel. The samples were run for 1 h at 140 V in 1× Tris-boric acid buffer followed by subsequent imaging with a Fujifilm FLA-5000 system using a 473-nm excitation wavelength. The fraction of bound DNA or RNA, θ, was measured using Multi Gauge software (Fujifilm) to quantify the bound fractions (upper bands) and free DNA fractions (lower bands) from the polyacrylamide gel.
 |
Trp lifetime assays were performed in 500-μl reactions consisting of 2 μm protein incubated with increasing amounts of nucleic acid in the EMSA binding buffer described above. The samples were excited at 295 nm in a 50-μl cuvette (Hellma) using an autosampler configuration to prevent Trp photobleaching. The laser intensity was adjusted using 4 μm N-acetyl-tryptophanamide as a standard to ensure a count rate between 8 × 104 and 1 × 105 per second prior to sample acquisition. Trp lifetime decays were measured for 2 min in 30-s intervals for each sample with ∼60,000 counts total in the peak channel. The Trp lifetime decays were corrected for buffer contributions and subsequently fit to two exponential decays, which differed slightly, depending on protein construct and DNA/RNA. In comparison with samples containing only protein, the amplitudes of the ∼3.8 and ∼5.8 ns components decreased and increased, respectively, with increased DNA/RNA concentration. Thus, the amplitude of the faster phase represented the unbound protein, and the slower phase resulted from the DNA/RNA-bound protein.
 |
The percentage bound was determined as a function of DNA/RNA concentration and modeled to the quadratic binding equation (Equation 7) using Igor Pro (Wavemetrics, Inc.) to determine the apparent dissociation constant, Kd,app, (31),
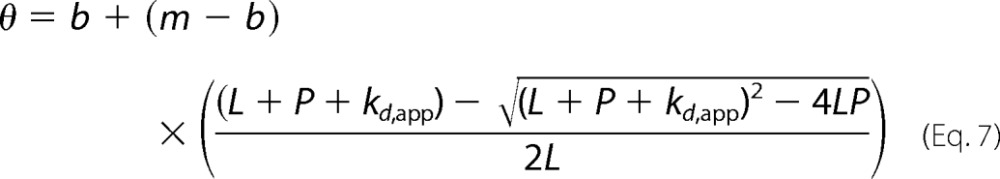 |
where L is the fixed ligand concentration (for EMSA, DNA/RNA; for Trp lifetime assays, RRM1 or tRRMs), P is the independent variable (for EMSA, protein concentration; for Trp lifetime assays, DNA or RNA concentration), Kd,app is the apparent dissociation constant, and b and m are the baseline and maximum percentage bound used to normalize the data sets.
RESULTS
RRM Domains Are Monomeric and Well Folded
Each isolated RRM domain (RRM1 and RRM2) could be expressed at high levels, was soluble to concentrations exceeding 5 mg ml−1 (∼0.5 mm), and was monomeric by size exclusion chromatography (Fig. 2A). Despite their structural similarity by NMR (Fig. 1, C and D), the CD spectra of the two isolated RRM domains were strikingly different from one another. Indeed, CD spectra can vary widely between RRM domains, including RNA-binding proteins that contain a single RRM domain, such as FUS/TLS (32), or multiple RRM domain-containing proteins, such as Musashi-1 (33) and U1A (34). RRM1 contains prominent minima at 208 and 218 nm, suggesting significant α-helical propensity for this domain. RRM1 contains a dramatically increased CD signal compared with RRM2 (Fig. 2B), consistent in part with the enhanced α-helical structure content of RRM1 (25%) compared with RRM2 (20%), as predicted by DSSP (35, 36), based on the NMR structures (Fig. 1B). The CD spectrum of RRM2 was approximately half as intense as that of RRM1, with a minimum at 210 nm and a shoulder at 230 nm. Although the two RRM domains have identical β-strand content (30%), the NMR structures indicate that RRM1 has more twist to its β-sheet compared with RRM2, consistent also with the increased intensity of the CD signal for RRM1 (37).
FIGURE 2.
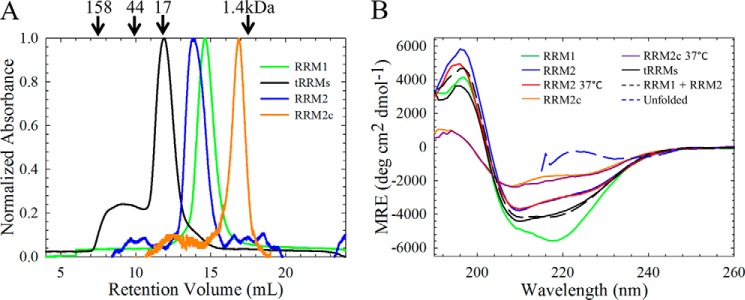
The isolated RRM domains are monomeric and contain significant secondary structure under physiological salt conditions. A, analytical size exclusion chromatography of the isolated (green, RRM1; blue, RRM2; orange, RRM2c) RRM domains reveals that each isolated domain is predominantly monomeric. The tethered domains (tRRMs, black) are also predominantly monomeric; however, some higher order species are also present, probably due to domain swapping. The arrows indicate retention times of molecular weight standards. B, RRM1 contains significant secondary structure compared with RRM2. RRM1 (green) has a minimum at 218 nm with a shoulder at 208 nm, whereas RRM2 (blue) has reduced ellipticity with a minimum at 210 nm and a shoulder at 220 nm. The cleavage fragment, RRM2c (orange) has further reduced ellipticity. All CD spectra were taken at 20 °C unless otherwise noted; physiological temperature (37 °C) does not alter the secondary structures of RRM2 (red) or RRM2c (purple). Tethering the RRMs (tRRMs; black) results in a CD spectrum resembling the sum of the isolated domains (black dashed line). The CD spectrum for each protein, reported as mean residue ellipticity as a function of wavelength, did not vary in the range of 5–60 μm, and all proteins showed similar unfolded spectra when denatured in 7 m GdnHCl (blue dashed line).
RRM2 Populates an Intermediate State
To probe the equilibrium folding mechanism of the RRM domains and its potential role in ALS, denaturant-induced unfolding was used to sample other protein conformations. The loss in secondary and tertiary structure was monitored by CD and FL, respectively (Fig. 3, A and B). Initial equilibrium folding studies performed in urea (data not shown) showed that RRM2 remains partially folded at high urea concentrations (>8 m). By contrast, both RRM1 and RRM2 were fully unfolded in the presence of 7.5 m GdnHCl.
FIGURE 3.

The isolated and tethered RRM domains have complex equilibrium unfolding profiles at 20 °C. A, equilibrium unfolding of RRM1 unfolds by a two-state mechanism (N ⇆ U) by both CD (filled circles, solid line) and Trp FL (open circles, dashed line). B, RRM2 unfolds through the population of an equilibrium intermediate (N ⇆ I ⇆ U) by CD (filled triangles, solid line). By contrast, the Tyr FL (open triangles, dashed line) unfolds in a two-state process that coincides with the N ⇆ I transition observed by CD. C, tethering the two RRMs stabilizes the RRM1 transition, resulting in a complex equilibrium unfolding profile with two stable intermediate states (N ⇆ I1 ⇆ I2 ⇆U) by CD (closed diamonds, solid line). Trp FL (open diamonds, dashed line) follows the denaturation of RRM1 only and coincides with the first transition (N ⇆ I1) by CD. D, a cleavage fragment of RRM2 (RRM2c) unfolds through a three-state mechanism by CD (filled squares, solid line) with destabilization of both transitions compared with RRM2. Similar to RRM2, the two-state transition observed by Tyr FL (open squares, dashed lines) coincides with the N ⇆ I transition by CD. Each experiment was consistently reproduced three times.
RRM1 has a CD spectrum with two prominent minima (Fig. 2B) characteristic of an α + β protein. The two tryptophan residues, unique to RRM1 (Fig. 1C), monitor the tertiary structure within this domain upon unfolding. For RRM1 at 20 °C, the CD and Trp FL reveal a single cooperative transition between the native folded state and the unfolded state (Fig. 3A) that is well described by a two-state model for the free energy of folding in the absence of denaturant; ΔG0H2O = 3.7 kcal mol−1 (Table 1). The midpoints (Cm) of the transition between these two states are coincident between the two spectroscopic techniques (Fig. 4, A and B), consistent with a two-state mechanism of folding for the RRM1 domain.
TABLE 1.
Thermodynamic parameters of the isolated and tethered RRM domains
The CD or FL experiments for the appropriate RRM construct were fit to an appropriate model: 1) two-state (N ⇆ U), 2) three-state (N ⇆ I ⇆ U), or 3) four-state (N ⇆ I1 ⇆ I2 ⇆ U) with the corresponding free energy in the absence of denaturant (ΔG0), m value (m), and midpoint (Cm) for each transition. The m value correlates to the buried surface area of each domain, and the Cm is the denaturant concentration where the domains populate two states equally. The total ΔG0 and m value for each construct are reported in the right-most column. Units: ΔG0 (kcal mol−1), m(kcal mol−1 m−1), and Cm (m). Each experiment is an average of three (n = 3) independent trials.
FIGURE 4.

Cleavage and physiological temperatures destabilize RRM2 and increase the population of the intermediate state. A, fraction apparent plot of the Trp FL of RRM1 (open circles, green dashed line) and tRRMs (open triangles, black dashed line) at 20 °C reveals a shift toward higher denaturant for RRM1 unfolding upon tethering to RRM2. B, fraction apparent plots of all TDP-43 RRM constructs by CD. RRM1 displays a two-state equilibrium profile (green), whereas RRM2 (20 °C, blue; 37 °C, red) and the cleavage fragment, RRM2c (20 °C, orange; 37 °C, purple) display a three-state profile with the population of an intermediate state. tRRMs (black) display a complex unfolding profile with the population of two stable intermediates. C, RRM2 intermediate population as a function of denaturant using Equation 3 for a three-state profile (RRM2 and RRM2c) or Equation 4 for a four-state profile (tRRMs). Increasing temperature (red) or cleavage (orange and purple) within the RRM2 domain (blue) results in an increased intermediate population under native conditions (0 m GdnHCl) with a shift to lower denaturant for the maximum intermediate population. Tethering RRM2 to RRM1 (black) reduces the RRM2 intermediate state under native conditions and shifts the maximum population to higher denaturant. D, physiological temperatures destabilize the N ⇆ I transition by CD (filled inverted triangles, solid line) and Tyr FL transition (open inverted triangles, dashed line) with a shift in the Cm to lower denaturant compared with RRM2. E, physiological temperature results in a similar unfolding profile for RRM2c to RRM2 at 37 °C (compare filled inverted triangles with filled hexagons). Tyr FL revealed little change between the folded and unfolded states (open hexagons), suggesting that Tyr214 resembles the unfolded state at this temperature. Each experiment was consistently reproduced three times.
In comparison with RRM1, RRM2 contains reduced ellipticity and a single tyrosine as a fluorescence probe. The unfolding profile of RRM2 at 20 °C (Fig. 3B) monitored by CD is significantly different from that of RRM1. A change in the cooperativity observed at 4 m GdnHCl indicates a three-state unfolding process with contributions from a stably populated intermediate state. The transitions in the N ⇆ I ⇆ U three-state mechanism contribute 3.6 kcal mol−1 (N ⇆ I) and 3.8 kcal mol−1 (I ⇆ U) to the total stability of RRM2 (Table 1; 7.4 kcal mol−1). The CD and FL transitions are not coincident, further supporting the presence of an intermediate state on the RRM2 unfolding pathway. The Tyr fluorescence becomes insensitive to denaturant in the second transition (>4 m GdnHCl), suggesting that the region surrounding this residue is unfolded in the intermediate state. Using the thermodynamic parameters for the individual transitions (N ⇆ I and I ⇆ U), the population of the intermediate state was calculated as a function of denaturant by Equation 3. As shown in Fig. 4C, the intermediate state is maximally populated (80%) at 4 m GdnHCl, and a small proportion (<1%) of RRM2 populates this partially unfolded intermediate state under native conditions (0 m GdnHCl).
Intermediate Population Is Enhanced by Increased Temperature or Cleavage of RRM2
In ALS patient tissues, fragments of TDP-43 comprising the C terminus and regions of RRM2 are present in cytoplasmic inclusions (9). One of these fragments of RRM2, starting at position Arg208, results in the removal of β1 and a region of α1 and leads to severe aggregation and toxicity in cell models (12, 13, 15). The disruption of the native fold in RRM2c could enhance the population of partially unfolded states. A construct consisting of residues 208–261 (RRM2c) of RRM2 was designed, cloned, and expressed for denaturation studies. RRM2c rapidly precipitates from solution upon the addition of salt, suggesting that RRM2c adopts a structure that facilitates aggregation under cellular ionic strength conditions (38). The fragment was predominantly monomeric by size exclusion chromatography (Fig. 2A); however, to enhance the solubility of RRM2c in the absence of denaturation, the 150 mm KCl was eliminated from the folding buffer.
The CD spectrum of RRM2c reveals that cleavage within the domain greatly reduces but does not obliterate its secondary structure (Fig. 2B). The fragment retains the three-state behavior of the RRM2 domain upon denaturation at 20 °C (Fig. 3D) and significant stability (Table 1; 4.1 kcal mol−1). The transitions (N ⇆ I and I ⇆ U) are destabilized compared with the intact domain (Table 1) and display non-coincident CD and tyrosine FL transitions that support a three-state unfolding model for RRM2c. The FL results suggest that the fragment can fold to a conformation that decreases the solvent accessibility of the Tyr, although the Tyr is close to the new N terminus. Comparing the fraction species plot of the intermediate state as a function of denaturant reveals that 2% of the RRM2c conformational ensemble samples the intermediate state under native conditions at 20 °C (Fig. 4B, compare orange and blue traces). These results indicate that fragments of RRM2 sample partially folded states to a greater extent than the intact domain and may provide a platform for subsequent TDP-43 aggregation (38).
Incubation of RRM1 at physiological temperatures (37 °C) resulted in aggregation (39, 40), whereas RRM2 and RRM2c remain in native conformations (Fig. 2B) with little change in secondary structure. Denaturation of the intact RRM2 at 37 °C (Fig. 4C) also follows a three-state unfolding process, albeit with a reduction in the overall stability (5.7 versus 7.4 kcal mol−1). As with denaturation of RRM2 at 20 °C, the Tyr FL data were not coincident with the CD data (Table 1), supporting the presence of an intermediate in RRM2 even at elevated temperatures. Surprisingly, denaturation of RRM2c at 37 °C revealed a three-state unfolding profile coincident with the intact domain (Fig. 4B) but with a slight increase in total stability upon increasing temperature (4.1 versus 4.8 kcal mol−1). The increase in overall free energy suggests either a self-association of RRM2c or that the folding of this fragment is driven by the hydrophobic effect, which is stronger at higher temperatures (41, 42). Self-association is concentration-dependent, and thus higher concentration would be expected to drive the association reaction and stabilize the protein against denaturation. However, our experiments revealed no concentration dependence for the unfolding transition by CD in the range from 15 to 60 μm RRM2c. Further, the lack of denaturant dependence of the tyrosine emission spectrum for RRM2c shows that the Tyr is exposed to solvent in this RRM2 fragment at physiological temperatures (Fig. 4E). This conformation differs from the Tyr FL at 20 °C, where the Tyr is at least partially buried in the native RRM2c structure. The destabilized native conformations of both RRM2 and RRM2c at physiological temperature suggest that the intermediate state becomes more populated under native conditions at increased temperatures. Indeed, the intermediate state is 5–10-fold more populated at 37 °C compared with 20 °C for both the intact RRM2 and the fragment of RRM2. Together, these results show that either a cleavage event within the RRM2 domain or physiological temperature increases the population of a potentially pathological intermediate state that may contribute to possible misfolding and aggregation.
Tethering the RRM Domains Results in Stabilization of RRM1 and Decreased Access to the RRM2 Intermediate
Under normal cellular conditions, the RNA binding domain of TDP-43 consists of RRM2 tethered to RRM1 by a short 15-amino acid linker. As such, the presence of RRM1 may influence the folding of RRM2 through mutual stabilizing interactions and decrease the population of the RRM2 intermediate state. The tRRM construct, which comprises RRM1, RRM2, and its natural linker, exhibits a CD spectrum that is very similar to the additive sum of the individual RRM domain spectra (Fig. 2B). This observation suggests that tethering the two RRM domains does not significantly affect the overall secondary structure of each RRM. The slight differences may arise through a small decrease in α-helical content of the RRM1 domain when tethered, as suggested by the decrease in the 190 nm band and the shift in the ratio of the CD signal at 222 nm to 208 nm for tRRMs compared with the additive spectra (Fig. 2B).
However, upon denaturation with GdnHCl, tRRMs displayed a complex equilibrium unfolding profile when monitored by CD that was best described by N ⇆ I1 ⇆ I2 ⇆ U, suggesting the population of two discrete intermediate states (Fig. 3C). To elucidate the extent that each RRM domain contributes to the intermediates, the Trp FL data were used as a constraint to define the first transition. Because Trp residues are only present within RRM1, the Trp FL provides structural insights into the unfolding of only RRM1 when tethered to RRM2. Interestingly, comparison of the Trp FL of the isolated RRM1 with the tethered RRMs (Fig. 4A) reveals a shift in the transition midpoint (Cm) to higher denaturant (from 1.5 to 2.3 m) upon tethering. These results suggest that RRM1 and RRM2 interact in the absence of RNA, with interdomain interactions stabilizing the native state of each RRM domain by 0.9 kcal mol−1 (Table 1). Indeed, a recent NMR study on the tethered RRM domain revealed that in the absence of RNA, the two RRMs do not tumble independently of one another (43), further supporting mutual stabilizing interactions between the domains. The Trp FL also suggests that RRM1 is completely unfolded at the first intermediate, I1, of the CD equilibrium unfolding profile. The extent of the RRM2 folding at the I1 intermediate is unclear because this domain lacks Trp residues, and the contributions from the single tyrosine residue are masked by RRM1. However, the thermodynamic parameters derived from the CD profile suggest that RRM2 is at least partially unfolded because the I1 ⇆ I2 transition of tRRMs is not coincident with N ⇆ I of RRM2 (Fig. 4B).
The third transition in the equilibrium profile of tRRMs (I2 ⇆ U) corresponds to the I ⇆ U transition of RRM2 (Table 1) because both transitions provide similar stability and midpoints. Therefore, the I2 species in the tethered domains probably correspond to the same RRM2 intermediate state. Using Equation 4 to identify the population of the RRM2 intermediate as a function of denaturant reveals that tethering shifts the maximal population of RRM2 to higher denaturant (5.0 m GdnHCl) compared with the individual RRM2 domain (4.0 m Gdn; Fig. 4C), suggesting that RRM1 inhibits the formation of the RRM2 intermediate. Indeed, based on the stability measurements from the four-state model, RRM1 contributes ∼0.7 kcal mol−1 of stability to the RRM2 native state, which would shift the N ⇆ I transition to higher denaturant concentration and reduce the intermediate state population under native conditions. These results suggest that in the intact TDP-43 with RRM2 tethered to RRM1, mutual interactions between these domains can serve two purposes: 1) stabilizing RRM1 against populating unfolded conformations and 2) decreasing the RRM2 intermediate to almost negligible populations under native conditions (<0.1%). Isolating or fragmenting RRM2 removes stabilizing contributions from RRM1 and allows this region of TDP-43 to sample a potentially pathogenic partially folded state.
RRM2 Binds Weakly to RNA and Enhances RRM1 Affinity
TDP-43 is involved in multiple RNA processes, and thus, RNA probably plays a role in the folding and stability of the RRM domains. As shown in Fig. 4A, RRM1 becomes stabilized against denaturation when the two RRMs are tethered, suggesting that this interaction could also enhance the affinity of RRM1 and possibly RRM2 for RNA. Two complementary nucleic acid binding assays, EMSA (Fig. 5A) and the tryptophan lifetime-based assay (Fig. 5B), were used to determine the dissociation constant, Kd,app, for each individual and tethered RRM domain. Based on the results of previous binding affinity measurements on various TDP-43 constructs (21), the oligonucleotides (UG)6 and (TG)6 were selected for comparing the affinities of each domain, and T12 was selected as a control oligonucleotide.
FIGURE 5.
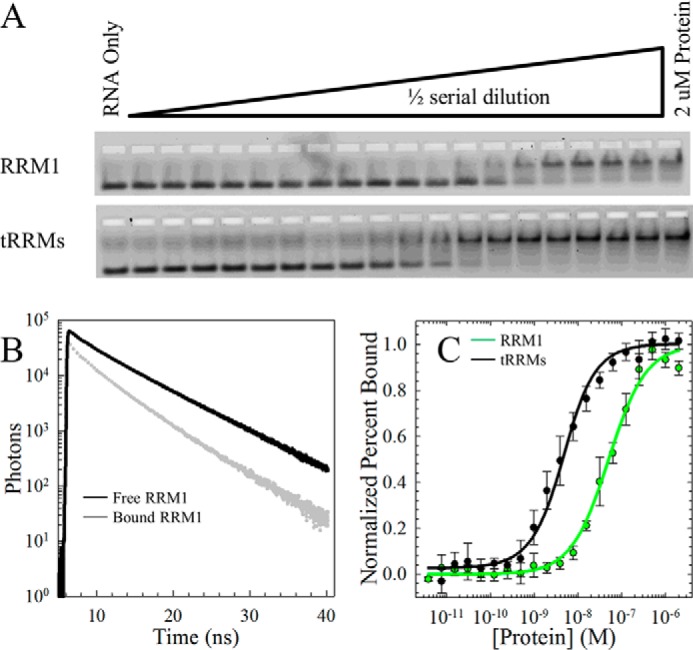
Tethering the RRM domains enhances the RNA binding affinity compared with RRM1. A, EMSA of increasing concentrations of RRM1 (top) and tRRMs (bottom) to a fixed concentration of 5′-carboxyfluorescein (FAM)-labeled (UG)6. The upper and lower bands on the native polyacrylamide gel represent the bound and free fractions of labeled RNA, respectively. B, Trp lifetimes of RRM1 alone (black) or bound by (UG)6 (gray) reveal a decrease in lifetime in the Trp on RRM1 upon binding. Trp lifetimes are insensitive to RRM2 due to a lack of Trp in this domain. Trp lifetimes were performed as a function of nucleic acid to a fixed concentration of RRM1 and tRRMs. C, quantification of the EMSA. Each data set is an average of three independent runs and modeled to the quadratic binding equation (Equation 6). A complete summary of binding affinities for tRRMs, RRM1, RRM2, and RRM2c is provided in Table 2.
In the EMSA, RRM2 bound to the UG repeat sequence very weakly with a dissociation constant of 17.0 μm (Table 2). Strikingly, RRM2c had a binding affinity for UG repeats enhanced by ∼5-fold compared with the intact domain. This enhancement indicates that RRM2c may access conformations that favor RNA binding compared with the intact RRM2 domain. Interactions with DNA were much weaker than for RNA (>100 μm) and unmeasurable by EMSA. This weak binding of RRM2 and RRM2c to RNA suggests that these domains may serve to stabilize RRM1 (Fig. 4A) rather than provide a major contribution to RNA binding. In fact, RRM1 binds with a Kd,app in the nanomolar range compared with the micromolar range of RRM2 (Table 2), supporting RRM1 as the major contributor to RNA binding. Notably, tethering the two RRM domains enhances binding affinity by an order of magnitude to result in dissociation constants for UG and TG repeat sequences in the low nanomolar range (Fig. 5C and Table 2). Trp lifetimes on the RRM domain reveal a reduction of Trp intensity and lifetime upon binding RNA (Fig. 5B) and provide structural insights into the binding interaction because the Trp residues are located on the β-sheet of RRM1. The RNA binding results suggest that the presence of RNA will play a role in defining the populations of all species on the folding free energy surface of TDP-43 by stabilizing the RRM domains and modulating access to the partially folded state in RRM2.
TABLE 2.
Dissociation constants of the isolated and tethered RRM domains
Shown are the dissociation constants of each construct by an EMSA and Trp lifetimes (FL) for RRM1, RRM2, RRM2c, and tRRMs to (UG)6, (TG)6, and T12. FL assays were only performed on RRM1 and tRRMs, whereas an EMSA was suitable for all constructs. Each experiment is an average of three independent experiments.
| RRM domain | Assay | Dissociation constant |
||
|---|---|---|---|---|
| (UG)6 | (TG)6 | T12 | ||
| nm | nm | nn | ||
| RRM1 | EMSA | 49.3 ± 12.0 | 232 ± 48 | >8000 |
| FL | 34.8 ± 14.1 | 630 ± 75 | >100,000 | |
| RRM2 | EMSA | 17,000 ± 3700 | >100,000 | >100,000 |
| RRM2c | EMSA | 2750 ± 480 | >100,000 | >100,000 |
| tRRMs | EMSA | 3.1 ± 1.3 | 20.2 ± 1.8 | >8000 |
| FL | 3.2 ± 2.2 | 279 ± 90 | >100,000 | |
DISCUSSION
Although the ALS-linked proteins, such as SOD1 (44), TDP-43 (45, 46), FUS/TLS (47), profilin 1 (48), and C9ORF72 (49, 50) among others (51), are functionally different, common mechanisms for cellular toxicity and aggregation may govern the folding pathways of these proteins to result in disease. Indeed, the RNA-binding proteins, TDP-43 and FUS/TLS, have been linked not only to ALS but also to other neurodegenerative diseases, including FTLD-U (1), Alzheimer disease (52), and Parkinson disease (53), suggesting a potential common mechanism of disease pathogenesis between these proteins. Here, we performed denaturation and RNA binding studies to probe the equilibrium unfolding pathways of the isolated and tethered RRM domains of TDP-43. The data revealed a highly populated stable folding intermediate within RRM2 (Figs. 3B and 4C) that may link the pathways governing TDP-43 folding and function with those of misfolding and aggregation. Specifically, the intermediate state may have a normal cellular function in nuclear export, but at the same time, populating this intermediate state may sow the seeds for misfolding and aggregation in disease.
A major focus of the TDP-43 field has been to investigate the potential impacts of TDP-43 loss-of-function and aggregation propensity on the propagation of ALS and FTLD (1, 8, 11, 54, 55). Aggregation studies on RRM2 revealed increased formation of fibrillar aggregates with truncated RRM2 and peptide fragments consisting of β3 and β5 (56). These results suggest that the core sequences within RRM2 may directly participate in driving the aggregation of TDP-43. TANGO and WALTZ (16) predict an aggregation-prone region, β3 (Fig. 1A), localized within RRM2 that may serve as a template for aggregation propagation. In combination with the C terminus, intact or fragmented RRM2 was shown to severely enhance cellular aggregation and toxicity in cell models (12, 13, 15, 38, 57). The C terminus, which also contains a particularly aggregation-prone stretch (Met311–Met323), was shown to have increased β-strand propensity and aggregation tendency (38), whereas a Gln/Asn-rich region that is critical for proper TDP-43 protein-protein interactions was also sufficient for aggregation of GFP fused with multiple Gln/Asn repeats (58). Taken together, these results suggest that the population of the RRM2 intermediate state could expose aggregation-prone residues, such as β3 and β5 (56), either through the intact domain, cleavage by caspases (13, 59–61), or increased temperatures. This partially folded state could aberrantly interact with RRM1 (39, 62), the aggregation-prone C-terminal peptides (56, 63), or potential sequences within other proteins, such as heterogeneous nuclear ribonucleoprotein A1 and A2/B1 (64, 65), to promote misfolding and propagate the aggregation observed in ALS and FTLD patients.
Biophysical studies on TDP-43 have been hindered by the poor solubility and aggregation tendencies of the full-length protein. Thermal denaturation studies have shown that RRM2 remains well folded and resistant to denaturation beyond 85 °C, whereas RRM1 undergoes a conformational change at 50 °C (38, 66). These results suggest an inherent stability difference between the two domains, which are 30% identical in sequence by ClustalW2 (Fig. 1B). Similarly, in our study, the GdnHCl-induced denaturation of these domains revealed markedly different equilibrium unfolding profiles with significantly different stabilities (Table 1). GdnHCl denaturation fortuitously revealed a partially folded state that was not evident by thermal denaturation. Thus, chemical denaturation provides access to a partially folded state that may be relevant for both function and aggregation.
Hydrogen exchange experiments on the TIM barrel protein family have shown that clusters of isoleucine, leucine, and valine residues can accurately predict cores of stability (67) and early folding events (68, 69). Using an in-house algorithm (see the Clusters of Branched Aliphatic Side Chains in Globular Proteins (BASiC) Web site) (70, 71)), clusters of the branched aliphatic side chain residues in RRM1 (Fig. 6D) and RRM2 (Fig. 6A) were identified using the NMR structures. As shown in Fig. 6, RRM2 contains one large cluster with 12 ILV residues, eight of which provide multiple contacts (Fig. 6C). RRM1 (Fig. 6E), on the other hand, contains three clusters of ILV residues with two single-contact clusters and a remaining cluster that consists of eight loosely connected residues. Upon tethering to RRM2 and the addition of RNA, the Leu106-Leu177 contact (Fig. 6, D and E, purple) and Val161 in α2 are incorporated into the RRM1 cluster and possibly enhance the stability of RRM1. In RRM2c (Fig. 6B), three ILV residues (Val193, Val195, and Leu207) (Fig. 5D, cyan) are removed, and the remaining contacts may provide a significantly different hydrophobic core for folding (Fig. 2A). This networked cluster of ILV residues in RRM2 and RRM2c (Fig. 6C) probably contributes to the presence of a stable intermediate and increased stability of RRM2 compared with RRM1 (Fig. 6E).
FIGURE 6.

Extensive and networked ILV cluster in RRM2. A, solution NMR structure of RRM2 (Protein Data Bank code 1wf0) with isoleucine (I), leucine (L), and valine (V) residues highlighted that contact other ILV residues (blue). B, residues from the large cluster in RRM2 that are removed in RRM2c are highlighted in cyan, and the NES (residues 239–250), which is still part of the large cluster in RRM2c, is highlighted in yellow. C, ILV cluster contact map for RRM2 displayed on the secondary structure elements of RRM2. The RRM2 ILV cluster contains a single highly connected cluster (blue) spanning all α-helices and β-strands, and the cluster is unaffected by tethering to RRM1. The residues removed in RRM2c (cyan) are marked with an asterisk. The NES (yellow) contributes three residues to the large ILV cluster (Leu243, Leu248, and Ile250), which may contribute to the increased stability of RRM2 compared with RRM1. D, RRM1 (Protein Data Bank code 2cqg) contains three small clusters (purple, red, and green), two of which were single surface residue contacts (red and purple). The remaining cluster (green) is small and less connected compared with RRM2. E, the RRM1 cluster contains a small ILV cluster with single contacts centered in β1–β3. Upon tethering to RRM2 and RNA binding, the cluster exhibits a few contact changes that include the addition of Val161 and the purple cluster, resulting in a more networked RRM1 compared with the isolated RRM1.
Alternatively, the RRM2 intermediate may serve a functional role in RNA processing. Normal TDP-43 shuttling to the cytoplasm is probably governed by a leucine-rich NES (72) located within RRM2 (residues 239–250). Alanine mutation of any of the hydrophobic residues within this sequence resulted in nuclear mutants and endogenous aggregates (73). The NES forms α2 and β4 within RRM2 to possibly sequester the hydrophobic residues (Leu243, Leu248, and Ile250) and contribute to the ILV cluster (Fig. 6B). Another ALS-linked protein, FUS/TLS, also contains a NES within an RRM domain (74) with low affinity for RNA (75). Strikingly, we have observed that the RRM domain of FUS/TLS also unfolds through an equilibrium intermediate state.3 The presence of an intermediate state in the RRM unfolding pathways of TDP-43 and FUS/TLS suggests that these RRMs may serve to stabilize the other RNA-binding domain and sequester the NES within the hydrophobic core to control cytoplasmic shuttling or to prevent aggregation. RRM2 must partially unfold to expose the NES for recognition by exportin (76), which mediates the transport to the cytoplasm. This hypothesis suggests that cleavage within RRM2, loss of RRM1, or external stressors would increase the population of the intermediate state and disrupt the normal cellular distribution. Up-regulated export to the cytoplasm could increase the formation of dysfunctional complexes with other proteins and cytoplasmic aggregates that would ultimately decrease the population of functional nuclear TDP-43. Further experiments to investigate the role of the intermediate state on TDP-43 nuclear export would delineate the consequences of populating this partially folded state.
RRM2 may also enhance the function of TDP-43 through interactions with RRM1 and RNA. Tethering RRM2 to RRM1 increases the stability of RRM1 (Fig. 4C) and the affinity to UG-rich sequences (Fig. 5C). Studies on RNA binding in disease progression have indicated that removal of RRM1 or mutational analysis of key phenylalanine residues in RRM1 results in non-functional TDP-43 with neurotoxicity and aggregation phenotypes (21–23, 77, 78). A cell-free system revealed that incubation with (UG)6 or (TG)6 resulted in an increase in TDP-43 solubility and reduced the tendency to aggregate compared with mutant RRM1 TDP-43 (22). These results suggest that RNA binding may be playing a critical role in limiting access to the partially folded intermediate in RRM2 and reducing aggregation. RNA binding assays by our group and others (21, 66) suggest that RRM2 binds weakly to RNA compared with RRM1 (Table 2). RRM2 could contribute to RNA binding through two potential mechanisms: 1) RRM2 may contribute to binding through allosteric interactions with RRM1, and 2) RRM2 may indirectly contribute to binding by stabilizing RRM1. In either scenario, mutual interactions between RRM1 and RRM2 would reduce access to the RRM2 intermediate state. Structural information on the DNA/RNA-bound tethered domains would provide critical insights into the RNA-binding modes for each of these domains and could further delineate the role of RNA in the RRM folding pathway.
The thermodynamic and DNA/RNA binding experiments suggest a mechanism for potential TDP-43 dysfunction and aggregation through the RRM2 intermediate (Fig. 7). Normal cellular TDP-43 is involved in multiple RNA processes with protein partners in the nucleus. TDP-43 probably exists in equilibrium between two states: the folded TDP-43 and functional TDP-43 that populates the RRM2 intermediate. This intermediate could serve two potential roles: 1) a productive functional intermediate allowing for NES recognition by the cellular export machinery or 2) a non-productive misfolding intermediate with exposed aggregation-prone peptides, such as β3 and β5, that may aberrantly interact with other intact and fragmented TDP-43 as well as other known protein partners. Normally, only a small fraction of TDP-43 populates the intermediate state, and the equilibrium heavily favors the folded TDP-43 with a properly folded RRM2. The NES remains sequestered in the hydrophobic core of the molecule for the nuclear localization of TDP-43. However, cellular stresses, such as oxidative damage, could result in TDP-43 cleavage and loss in RNA binding or protein partners. Such events could potentially shift the equilibrium toward the misfolding of the intermediate state. These misfolded, non-functional proteins may increase transport to the cytoplasm, where exposure of the hydrophobic residues in the NES or aggregation-prone peptides in RRM2 results in the formation of dysfunctional complexes and aggregates of TDP-43. In any case, the reduced functional pool of TDP-43 in the nucleus would influence neuronal death. Thus, the RRM2 intermediate state may link the productive folding and function of TDP-43 with non-productive misfolding and aggregation that leads to disease progression. Therapeutic intervention strategies can be developed that take into account approaches that limit pathological access to this intermediate state, potentially offering a viable drug target for treating ALS and FTLD.
FIGURE 7.
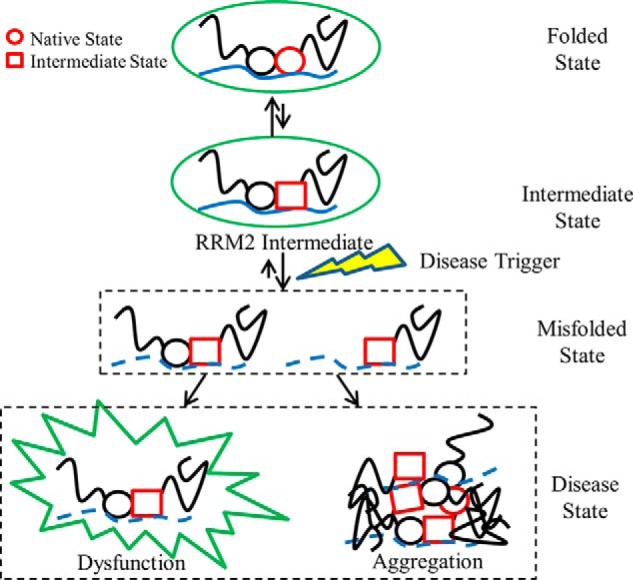
TDP-43 aggregation model through the RRM2 intermediate state. For simplicity, TDP-43 is represented with two circles for the RRM domains (black, RRM1; red, RRM2) and black lines for the disordered N and C terminus. TDP-43 can interact with other protein partners to form functional complexes represented by a green circle. An equilibrium exists where a small percentage of RRM2 can populate its intermediate state (RRM2 conversion from red circle to red square) but remain in functional complexes. Disease triggers (lightning bolt) can result in loss of RNA binding (dashed blue line), protein-protein interactions, or cleavage (removal of RRM1 and N terminus) that produces misfolded states (dashed boxes) where the population of the RRM2 intermediate increases. These misfolded states can result in dysfunctional complexes by aberrant protein-protein interactions (spiked green circle) or aggregates with other proteins (overlaid TDP-43 molecules). These disease states reduce the available pool of functional TDP-43 and lead to the propagation of ALS and FTLD.
Acknowledgements—We thank Muska Hassan, Daniel Lindstrom, and Amanda Winn for help with molecular biology and/or protein purification and Dr. Sagar Kathuria for assistance with the hydrophobic cluster analysis program. We are grateful to Drs. Francesca Massi and Sean Ryder and the members of their respective laboratories for many helpful suggestions. We also thank Drs. Selma Avendano-Vazquez, Osman Bilsel, Daryl Bosco, Larry Hayward, Bill Royer, Zuoshang Xu, and Chunxing Yang for many insightful discussions.
This work was supported, in whole or in part, by National Institutes of Health Grant GM54836 (to C. R. M.). This work was also supported by the ALS Therapy Alliance, Inc. (to J. A. Z.), the ALS Association (to J. A. Z.), and a University of Massachusetts Medical School Faculty Scholar Award (to J. A. Z.).
B. C. Mackness, M. T. Tran, S. P. McClain, C. R. Matthews, and J. A. Zitzewitz, unpublished data.
- ALS
- amyotrophic lateral sclerosis
- FL
- fluorescence
- FTLD
- frontotemporal lobar degeneration
- GdnHCl
- guanidine hydrochloride
- RRM
- RNA recognition motif
- RRM1
- first RRM domain, comprising residues 102–181
- RRM2
- second RRM domain, comprising residues 190–261
- RRM2c
- fragment of RRM2, comprising residues 208–261
- tRRM
- tethered RRM domain construct, comprising residues 97–261
- NES
- nuclear export signal
- TEV
- tobacco etch virus.
REFERENCES
- 1. Da Cruz S., Cleveland D. W. (2011) Understanding the role of TDP-43 and FUS/TLS in ALS and beyond. Curr. Opin. Neurobiol. 21, 904–919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Turner M. R., Hardiman O., Benatar M., Brooks B. R., Chio A., de Carvalho M., Ince P. G., Lin C., Miller R. G., Mitsumoto H., Nicholson G., Ravits J., Shaw P. J., Swash M., Talbot K., Traynor B. J., Van den Berg L. H., Veldink J. H., Vucic S., Kiernan M. C. (2013) Controversies and priorities in amyotrophic lateral sclerosis. Lancet Neurol. 12, 310–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Neumann M., Sampathu D. M., Kwong L. K., Truax A. C., Micsenyi M. C., Chou T. T., Bruce J., Schuck T., Grossman M., Clark C. M., McCluskey L. F., Miller B. L., Masliah E., Mackenzie I. R., Feldman H., Feiden W., Kretzschmar H. A., Trojanowski J. Q., Lee V. M. (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133 [DOI] [PubMed] [Google Scholar]
- 4. Arai T., Hasegawa M., Akiyama H., Ikeda K., Nonaka T., Mori H., Mann D., Tsuchiya K., Yoshida M., Hashizume Y., Oda T. (2006) TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 351, 602–611 [DOI] [PubMed] [Google Scholar]
- 5. Chen-Plotkin A. S., Lee V. M., Trojanowski J. Q. (2010) TAR DNA-binding protein 43 in neurodegenerative disease. Nat. Rev. Neurol. 6, 211–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lee E. B., Lee V. M., Trojanowski J. Q. (2012) Gains or losses: molecular mechanisms of TDP43-mediated neurodegeneration. Nat. Rev. Neurosci. 13, 38–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Buratti E., Baralle F. E. (2008) Multiple roles of TDP-43 in gene expression, splicing regulation, and human disease. Front. Biosci. 13, 867–878 [DOI] [PubMed] [Google Scholar]
- 8. Lagier-Tourenne C., Polymenidou M., Cleveland D. W. (2010) TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum. Mol. Genet. 19, R46–R64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Igaz L. M., Kwong L. K., Xu Y., Truax A. C., Uryu K., Neumann M., Clark C. M., Elman L. B., Miller B. L., Grossman M., McCluskey L. F., Trojanowski J. Q., Lee V. M. (2008) Enrichment of C-terminal fragments in TAR DNA-binding protein-43 cytoplasmic inclusions in brain but not in spinal cord of frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Am. J. Pathol. 173, 182–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ross C. A., Poirier M. A. (2004) Protein aggregation and neurodegenerative disease. Nat. Med. 10, S10–S17 [DOI] [PubMed] [Google Scholar]
- 11. Xu Z.-S. (2012) Does a loss of TDP-43 function cause neurodegeneration? Mol. Neurodegener. 7, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Johnson B. S., McCaffery J. M., Lindquist S., Gitler A. D. (2008) A yeast TDP-43 proteinopathy model: exploring the molecular determinants of TDP-43 aggregation and cellular toxicity. Proc. Natl. Acad. Sci. U.S.A. 105, 6439–6444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Igaz L. M., Kwong L. K., Chen-Plotkin A., Winton M. J., Unger T. L., Xu Y., Neumann M., Trojanowski J. Q., Lee V. M. (2009) Expression of TDP-43 C-terminal fragments in vitro recapitulates pathological features of TDP-43 proteinopathies. J. Biol. Chem. 284, 8516–8524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang Y.-J., Xu Y.-F., Cook C., Gendron T. F., Roettges P., Link C. D., Lin W.-L., Tong J., Castanedes-Casey M., Ash P., Gass J., Rangachari V., Buratti E., Baralle F., Golde T. E., Dickson D. W., Petrucelli L. (2009) Aberrant cleavage of TDP-43 enhances aggregation and cellular toxicity. Proc. Natl. Acad. Sci. U.S.A. 106, 7607–7612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yang C., Tan W., Whittle C., Qiu L., Cao L., Akbarian S., Xu Z. (2010) The C-terminal TDP-43 fragments have a high aggregation propensity and harm neurons by a dominant-negative mechanism. PLoS One 5, e15878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fernandez-Escamilla A. M., Rousseau F., Schymkowitz J., Serrano L. (2004) Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nat. Biotechnol. 22, 1302–1306 [DOI] [PubMed] [Google Scholar]
- 17. Romero P., Obradovic Z., Li X., Garner E. C., Brown C. J., Dunker A. K. (2001) Sequence complexity of disordered protein. Proteins 42, 38–48 [DOI] [PubMed] [Google Scholar]
- 18. Uversky V. N., Oldfield C. J., Dunker A. K. (2008) Intrinsically disordered proteins in human diseases: introducing the D2 concept. Annu. Rev. Biophys. 37, 215–246 [DOI] [PubMed] [Google Scholar]
- 19. Cléry A., Blatter M., Allain F. H. (2008) RNA recognition motifs. Boring? Not quite. Curr. Opin. Struct. Biol. 18, 290–298 [DOI] [PubMed] [Google Scholar]
- 20. Daubner G. M., Cléry A., Allain F. H. (2013) RRM-RNA recognition. NMR or crystallography … and new findings. Curr. Opin. Struct. Biol. 23, 100–108 [DOI] [PubMed] [Google Scholar]
- 21. Buratti E., Baralle F. E. (2001) Characterization and functional implications of the RNA binding properties of nuclear factor TDP-43, a novel splicing regulator of CFTR exon 9. J. Biol. Chem. 276, 36337–36343 [DOI] [PubMed] [Google Scholar]
- 22. Huang Y.-C., Lin K.-F., He R.-Y., Tu P.-H., Koubek J., Hsu Y.-C., Huang J. J. (2013) Inhibition of TDP-43 aggregation by nucleic acid binding. PLoS One 8, e64002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ayala Y. M., De Conti L., Avendaño-Vázquez S. E., Dhir A., Romano M., D'Ambrogio A., Tollervey J., Ule J., Baralle M., Buratti E., Baralle F. E. (2011) TDP-43 regulates its mRNA levels through a negative feedback loop. EMBO J. 30, 277–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lucast L. J., Batey R. T., Doudna J. A. (2001) Large-scale purification of a stable form of recombinant tobacco etch virus protease. BioTechniques 30, 544–546, 548, 550 passim [DOI] [PubMed] [Google Scholar]
- 25. Gill S. C., von Hippel P. H. (1989) Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 182, 319–326 [DOI] [PubMed] [Google Scholar]
- 26. Greenfield N. J. (2006) Using circular dichroism spectra to estimate protein secondary structure. Nat. Protoc. 1, 2876–2890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bilsel O., Zitzewitz J. A., Bowers K. E., Matthews C. R. (1999) Folding mechanism of the α-subunit of tryptophan synthase, an α/β barrel protein: global analysis highlights the interconversion of multiple native, intermediate, and unfolded forms through parallel channels. Biochemistry 38, 1018–1029 [DOI] [PubMed] [Google Scholar]
- 28. Pace C. N. (1986) Determination and analysis of urea and guanidine hydrochloride denaturation curves. Methods Enzymol. 131, 266–280 [DOI] [PubMed] [Google Scholar]
- 29. Finn B. E., Chen X., Jennings P., Saalau-Bethell S. M., Matthews C. R. (1992) in Protein Engineering. A Practical Approach (Rees A. R., Sternberg M. J. E., Wetzel R., eds) pp. 167–189, Oxford University Press, London [Google Scholar]
- 30. Pagano J. M., Farley B. M., McCoig L. M., Ryder S. P. (2007) Molecular basis of RNA recognition by the embryonic polarity determinant MEX-5. J. Biol. Chem. 282, 8883–8894 [DOI] [PubMed] [Google Scholar]
- 31. Pagano J. M., Clingman C. C., Ryder S. P. (2011) Quantitative approaches to monitor protein-nucleic acid interactions using fluorescent probes. RNA 17, 14–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Iko Y., Kodama T. S., Kasai N., Oyama T., Morita E. H., Muto T., Okumura M., Fujii R., Takumi T., Tate S., Morikawa K. (2004) Domain architectures and characterization of an RNA-binding protein, TLS. J. Biol. Chem. 279, 44834–44840 [DOI] [PubMed] [Google Scholar]
- 33. Kurihara Y., Nagata T., Imai T., Hiwatashi A., Horiuchi M., Sakakibara S., Katahira M., Okano H., Uesugi S. (1997) Structural properties and RNA-binding activities of two RNA recognition motifs of a mouse neural RNA-binding protein, mouse-Musashi-1. Gene 186, 21–27 [DOI] [PubMed] [Google Scholar]
- 34. Lu J., Hall K. B. (1995) An RBD that does not bind RNA: NMR secondary structure determination and biochemical properties of the C-terminal RNA binding domain from the human U1A protein. J. Mol. Biol. 247, 739–752 [DOI] [PubMed] [Google Scholar]
- 35. Kabsch W., Sander C. (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577–2637 [DOI] [PubMed] [Google Scholar]
- 36. Joosten R. P., te Beek T. A., Krieger E., Hekkelman M. L., Hooft R. W., Schneider R., Sander C., Vriend G. (2011) A series of PDB related databases for everyday needs. Nucleic Acids Res. 39, D411–D419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Berova N., Nakanishi K., Woody R. W. (eds) (2000) Circular Dichroism. Principles and Applications, 2nd Ed., pp. 601–620, Wiley-VCH, Inc., New York [Google Scholar]
- 38. Wang Y.-T., Kuo P.-H., Chiang C.-H., Liang J.-R., Chen Y.-R., Wang S., Shen J. C., Yuan H. S. (2013) The truncated C-terminal RNA recognition motif of TDP-43 protein plays a key role in forming proteinaceous aggregates. J. Biol. Chem. 288, 9049–9057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shodai A., Morimura T., Ido A., Uchida T., Ayaki T., Takahashi R., Kitazawa S., Suzuki S., Shirouzu M., Kigawa T., Muto Y., Yokoyama S., Takahashi R., Kitahara R., Ito H., Fujiwara N., Urushitani M. (2013) Aberrant assembly of RNA recognition motif 1 links to pathogenic conversion of TAR DNA-binding protein of 43 kDa (TDP-43). J. Biol. Chem. 288, 14886–14905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chang C.-K., Chiang M.-H., Toh E. K.-W., Chang C.-F., Huang T.-H. (2013) Molecular mechanism of oxidation-induced TDP-43 RRM1 aggregation and loss of function. FEBS Lett. 587, 575–582 [DOI] [PubMed] [Google Scholar]
- 41. Baldwin R. L. (1986) Temperature dependence of the hydrophobic interaction in protein folding. Proc. Natl. Acad. Sci. U.S.A. 83, 8069–8072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schellman J. A. (1997) Temperature, stability, and the hydrophobic interaction. Biophys. J. 73, 2960–2964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lukavsky P. J., Daujotyte D., Tollervey J. R., Ule J., Stuani C., Buratti E., Baralle F. E., Damberger F. F., Allain F. H. (2013) Molecular basis of UG-rich RNA recognition by the human splicing factor TDP-43. Nat. Struct. Mol. Biol. 20, 1443–1449 [DOI] [PubMed] [Google Scholar]
- 44. Rosen D. R., Siddique T., Patterson D., Figlewicz D. A., Sapp P., Hentati A., Donaldson D., Goto J., O'Regan J. P., Deng H. X. (1993) Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362, 59–62 [DOI] [PubMed] [Google Scholar]
- 45. Kabashi E., Valdmanis P. N., Dion P., Spiegelman D., McConkey B. J., Vande Velde C., Bouchard J.-P., Lacomblez L., Pochigaeva K., Salachas F., Pradat P.-F., Camu W., Meininger V., Dupre N., Rouleau G. A. (2008) TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet. 40, 572–574 [DOI] [PubMed] [Google Scholar]
- 46. Sreedharan J., Blair I. P., Tripathi V. B., Hu X., Vance C., Rogelj B., Ackerley S., Durnall J. C., Williams K. L., Buratti E., Baralle F., de Belleroche J., Mitchell J. D., Leigh P. N., Al-Chalabi A., Miller C. C., Nicholson G., Shaw C. E. (2008) TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319, 1668–1672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kwiatkowski T. J., Jr., Bosco D. A., Leclerc A. L., Tamrazian E., Vanderburg C. R., Russ C., Davis A., Gilchrist J., Kasarskis E. J., Munsat T., Valdmanis P., Rouleau G. A., Hosler B. A., Cortelli P., de Jong P. J., Yoshinaga Y., Haines J. L., Pericak-Vance M. A., Yan J., Ticozzi N., Siddique T., McKenna-Yasek D., Sapp P. C., Horvitz H. R., Landers J. E., Brown R. H., Jr. (2009) Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323, 1205–1208 [DOI] [PubMed] [Google Scholar]
- 48. Wu C.-H., Fallini C., Ticozzi N., Keagle P. J., Sapp P. C., Piotrowska K., Lowe P., Koppers M., McKenna-Yasek D., Baron D. M., Kost J. E., Gonzalez-Perez P., Fox A. D., Adams J., Taroni F., Tiloca C., Leclerc A. L., Chafe S. C., Mangroo D., Moore M. J., Zitzewitz J. A., Xu Z.-S., van den Berg L. H., Glass J. D., Siciliano G., Cirulli E. T., Goldstein D. B., Salachas F., Meininger V., Rossoll W., Ratti A., Gellera C., Bosco D. A., Bassell G. J., Silani V., Drory V. E., Brown R. H., Jr., Landers J. E. (2012) Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature 488, 499–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. DeJesus-Hernandez M., Mackenzie I. R., Boeve B. F., Boxer A. L., Baker M., Rutherford N. J., Nicholson A. M., Finch N. A., Flynn H., Adamson J., Kouri N., Wojtas A., Sengdy P., Hsiung G.-Y., Karydas A., Seeley W. W., Josephs K. A., Coppola G., Geschwind D. H., Wszolek Z. K., Feldman H., Knopman D. S., Petersen R. C., Miller B. L., Dickson D. W., Boylan K. B., Graff-Radford N. R., Rademakers R. (2011) Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Renton A. E., Majounie E., Waite A., Simón-Sánchez J., Rollinson S., Gibbs J. R., Schymick J. C., Laaksovirta H., van Swieten J. C., Myllykangas L., Kalimo H., Paetau A., Abramzon Y., Remes A. M., Kaganovich A., Scholz S. W., Duckworth J., Ding J., Harmer D. W., Hernandez D. G., Johnson J. O., Mok K., Ryten M., Trabzuni D., Guerreiro R. J., Orrell R. W., Neal J., Murray A., Pearson J., Jansen I. E., Sondervan D., Seelaar H., Blake D., Young K., Halliwell N., Callister J. B., Toulson G., Richardson A., Gerhard A., Snowden J., Mann D., Neary D., Nalls M. A., Peuralinna T., Jansson L., Isoviita V.-M., Kaivorinne A.-L., Hölttä-Vuori M., Ikonen E., Sulkava R., Benatar M., Wuu J., Chiò A., Restagno G., Borghero G., Sabatelli M., ITALSGEN Consortium, Heckerman D., Rogaeva E., Zinman L., Rothstein J. D., Sendtner M., Drepper C., Eichler E. E., Alkan C., Abdullaev Z., Pack S. D., Dutra A., Pak E., Hardy J., Singleton A., Williams N. M., Heutink P., Pickering-Brown S., Morris H. R., Tienari P. J., Traynor B. J. (2011) A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chen S., Sayana P., Zhang X., Le W. (2013) Genetics of amyotrophic lateral sclerosis: an update. Mol. Neurodegener. 8, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Youmans K. L., Wolozin B. (2012) TDP-43. A new player on the AD field? Exp. Neurol. 237, 90–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Rayaprolu S., Fujioka S., Traynor S., Soto-Ortolaza A. I., Petrucelli L., Dickson D. W., Rademakers R., Boylan K. B., Graff-Radford N. R., Uitti R. J., Wszolek Z. K., Ross O. A. (2013) TARDBP mutations in Parkinson's disease. Parkinsonism Relat. Disord. 19, 312–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Janssens J., Van Broeckhoven C. (2013) Pathological mechanisms underlying TDP-43 driven neurodegeneration in FTLD-ALS spectrum disorders. Hum. Mol. Genet. 22, R77–R87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Geser F., Lee V. M., Trojanowski J. Q. (2010) Amyotrophic lateral sclerosis and frontotemporal lobar degeneration: a spectrum of TDP-43 proteinopathies. Neuropathology 30, 103–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Saini A., Chauhan V. S. (2011) Delineation of the core aggregation sequences of TDP-43 C-terminal fragment. Chembiochem 12, 2495–2501 [DOI] [PubMed] [Google Scholar]
- 57. Furukawa Y., Kaneko K., Nukina N. (2011) Molecular properties of TAR DNA binding protein-43 fragments are dependent upon its cleavage site. Biochim. Biophys. Acta 1812, 1577–1583 [DOI] [PubMed] [Google Scholar]
- 58. Budini M., Romano V., Avendaño-Vázquez S. E., Bembich S., Buratti E., Baralle F. E. (2012) Role of selected mutations in the Q/N-rich region of TDP-43 in EGFP-12xQ/N-induced aggregate formation. Brain Res. 1462, 139–150 [DOI] [PubMed] [Google Scholar]
- 59. Dormann D., Capell A., Carlson A. M., Shankaran S. S., Rodde R., Neumann M., Kremmer E., Matsuwaki T., Yamanouchi K., Nishihara M., Haass C. (2009) Proteolytic processing of TAR DNA binding protein-43 by caspases produces C-terminal fragments with disease defining properties independent of progranulin. J. Neurochem. 110, 1082–1094 [DOI] [PubMed] [Google Scholar]
- 60. Nonaka T., Kametani F., Arai T., Akiyama H., Hasegawa M. (2009) Truncation and pathogenic mutations facilitate the formation of intracellular aggregates of TDP-43. Hum. Mol. Genet. 18, 3353–3364 [DOI] [PubMed] [Google Scholar]
- 61. Zhang Y.-J., Xu Y.-F., Dickey C. A., Buratti E., Baralle F., Bailey R., Pickering-Brown S., Dickson D., Petrucelli L. (2007) Progranulin mediates caspase-dependent cleavage of TAR DNA binding protein-43. J. Neurosci. 27, 10530–10534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Takagi S., Iguchi Y., Katsuno M., Ishigaki S., Ikenaka K., Fujioka Y., Honda D., Niwa J., Tanaka F., Watanabe H., Adachi H., Sobue G. (2013) RNP2 of RNA recognition motif 1 plays a central role in the aberrant modification of TDP-43. PLoS ONE 8, e66966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Chen A. K., Lin R. Y., Hsieh E. Z., Tu P.-H., Chen R. P., Liao T.-Y., Chen W., Wang C.-H., Huang J. J. (2010) Induction of amyloid fibrils by the C-terminal fragments of TDP-43 in amyotrophic lateral sclerosis. J. Am. Chem. Soc. 132, 1186–1187 [DOI] [PubMed] [Google Scholar]
- 64. Buratti E., Brindisi A., Giombi M., Tisminetzky S., Ayala Y. M., Baralle F. E. (2005) TDP-43 binds heterogeneous nuclear ribonucleoprotein A/B through its C-terminal tail: an important region for the inhibition of cystic fibrosis transmembrane conductance regulator exon 9 splicing. J. Biol. Chem. 280, 37572–37584 [DOI] [PubMed] [Google Scholar]
- 65. D'Ambrogio A., Buratti E., Stuani C., Guarnaccia C., Romano M., Ayala Y. M., Baralle F. E. (2009) Functional mapping of the interaction between TDP-43 and hnRNP A2 in vivo. Nucleic Acids Res. 37, 4116–4126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kuo P.-H., Doudeva L. G., Wang Y.-T., Shen C.-K., Yuan H. S. (2009) Structural insights into TDP-43 in nucleic-acid binding and domain interactions. Nucleic Acids Res. 37, 1799–1808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Gu Z., Zitzewitz J. A., Matthews C. R. (2007) Mapping the structure of folding cores in TIM barrel proteins by hydrogen exchange mass spectrometry: the roles of motif and sequence for the indole-3-glycerol phosphate synthase from Sulfolobus solfataricus. J. Mol. Biol. 368, 582–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Gu Z., Rao M. K., Forsyth W. R., Finke J. M., Matthews C. R. (2007) Structural analysis of kinetic folding intermediates for a TIM barrel protein, indole-3-glycerol phosphate synthase, by hydrogen exchange mass spectrometry and Gō model simulation. J. Mol. Biol. 374, 528–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wu Y., Vadrevu R., Kathuria S., Yang X., Matthews C. R. (2007) A tightly packed hydrophobic cluster directs the formation of an off-pathway sub-millisecond folding intermediate in the α subunit of tryptophan synthase, a TIM barrel protein. J. Mol. Biol. 366, 1624–1638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kathuria S. V., Day I. J., Wallace L. A., Matthews C. R. (2008) Kinetic traps in the folding of beta alpha-repeat proteins: CheY initially misfolds before accessing the native conformation. J. Mol. Biol. 382, 467–484 [DOI] [PubMed] [Google Scholar]
- 71. Sobolev V., Sorokine A., Prilusky J., Abola E. E., Edelman M. (1999) Automated analysis of interatomic contacts in proteins. Bioinformatics 15, 327–332 [DOI] [PubMed] [Google Scholar]
- 72. la Cour T., Kiemer L., Mølgaard A., Gupta R., Skriver K., Brunak S. (2004) Analysis and prediction of leucine-rich nuclear export signals. Protein Eng. Des. Sel. 17, 527–536 [DOI] [PubMed] [Google Scholar]
- 73. Winton M. J., Igaz L. M., Wong M. M., Kwong L. K., Trojanowski J. Q., Lee V. M. (2008) Disturbance of nuclear and cytoplasmic TAR DNA-binding protein (TDP-43) induces disease-like redistribution, sequestration, and aggregate formation. J. Biol. Chem. 283, 13302–13309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ito D., Seki M., Tsunoda Y., Uchiyama H., Suzuki N. (2011) Nuclear transport impairment of amyotrophic lateral sclerosis-linked mutations in FUS/TLS. Ann. Neurol. 69, 152–162 [DOI] [PubMed] [Google Scholar]
- 75. Liu X., Niu C., Ren J., Zhang J., Xie X., Zhu H., Feng W., Gong W. (2013) The RRM domain of human fused in sarcoma protein reveals a non-canonical nucleic acid binding site. Biochim. Biophys. Acta 1832, 375–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Kawahara Y., Mieda-Sato A. (2012) TDP-43 promotes microRNA biogenesis as a component of the Drosha and Dicer complexes. Proc. Natl. Acad. Sci. U.S.A. 109, 3347–3352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Ayala Y. M., Zago P., D'Ambrogio A., Xu Y.-F., Petrucelli L., Buratti E., Baralle F. E. (2008) Structural determinants of the cellular localization and shuttling of TDP-43. J. Cell Sci. 121, 3778–3785 [DOI] [PubMed] [Google Scholar]
- 78. Ihara R., Matsukawa K., Nagata Y., Kunugi H., Tsuji S., Chihara T., Kuranaga E., Miura M., Wakabayashi T., Hashimoto T., Iwatsubo T. (2013) RNA binding mediates neurotoxicity in the transgenic Drosophila model of TDP-43 proteinopathy. Hum. Mol. Genet. 22, 4474–4484 [DOI] [PubMed] [Google Scholar]