

Background: The extracellular Vibrio cholerae protease, VesB, is expressed in vitro and in stools of cholera patients.
Results: VesB is homologous to trypsin and thrombin and the C-terminal domain has an immunoglobulin-fold.
Conclusion: In contrast to thrombin, but similar to trypsin, VesB is not activated by sodium.
Significance: This is the first structure of a bacterial modular trypsin-like protease.
Keywords: Bacteria, Crystal Structure, Protein Domains, Protein Secretion, Proteolytic Enzymes, Serine Protease, Thrombin, Immunoglobulin-fold, Substrate Specificity, Trypsin
Abstract
The chymotrypsin subfamily A of serine proteases consists primarily of eukaryotic proteases, including only a few proteases of bacterial origin. VesB, a newly identified serine protease that is secreted by the type II secretion system in Vibrio cholerae, belongs to this subfamily. VesB is likely produced as a zymogen because sequence alignment with trypsinogen identified a putative cleavage site for activation and a catalytic triad, His-Asp-Ser. Using synthetic peptides, VesB efficiently cleaved a trypsin substrate, but not chymotrypsin and elastase substrates. The reversible serine protease inhibitor, benzamidine, inhibited VesB and served as an immobilized ligand for VesB affinity purification, further indicating its relationship with trypsin-like enzymes. Consistent with this family of serine proteases, N-terminal sequencing implied that the propeptide is removed in the secreted form of VesB. Separate mutagenesis of the activation site and catalytic serine rendered VesB inactive, confirming the importance of these features for activity, but not for secretion. Similar to trypsin but, in contrast to thrombin and other coagulation factors, Na+ did not stimulate the activity of VesB, despite containing the Tyr250 signature. The crystal structure of catalytically inactive pro-VesB revealed that the protease domain is structurally similar to trypsinogen. The C-terminal domain of VesB was found to adopt an immunoglobulin (Ig)-fold that is structurally homologous to Ig-folds of other extracellular Vibrio proteins. Possible roles of the Ig-fold domain in stability, substrate specificity, cell surface association, and type II secretion of VesB, the first bacterial multidomain trypsin-like protease with known structure, are discussed.
Introduction
Many bacteria require secretion of a variety of proteins for optimal adaptation to the diverse environments they encounter. One of the several bacterial protein secretion machineries is the type II secretion (T2S)5 system. This large two-membrane spanning assembly is remarkable in that it translocates multidomain and sometimes multimeric proteins in folded form across the outer membrane of Gram-negative bacteria (1–3). In Vibrio cholerae, the T2S system is responsible for the secretion of cholera toxin, the major virulence factor, and a large number of enzymes, including proteases into the extracellular milieu (4, 5).
Proteases are naturally occurring enzymes that are found in all domains of life. One of the largest classes of proteases, serine proteases, contains over 80 families. The S1, or chymotrypsin family consists of structurally homologous endoproteases with conserved His, Asp, and Ser residues that form the catalytic triad of the active site. Most members of this family are secreted as inactive precursors, or zymogens, with an N-terminal propeptide. The propeptide is proteolytically cleaved at a conserved activation site, which induces a conformational change that results in an active enzyme (6, 7).
Although structurally very similar, members of the chymotrypsin family are further subdivided into three groups based on substrate specificity. Trypsin-like proteases cleave at an arginine or lysine residue, chymotrypsin-like proteases act on tryptophan, tyrosine, or phenylalanine, and elastase-like proteases target small, uncharged residues (7). Eukaryotic proteases of the chymotrypsin family are important for many cellular processes. Functionally, they aid in digestion, blood coagulation, complement activation, inflammation, and development (7). Although the digestive enzymes, trypsin and chymotrypsin, are single domain proteases, proteins like tissue plasminogen activator, plasmin, and the complement component C1r contain multiple regulatory domains that are N-terminal to the protease domain (6, 8, 9). Furthermore, some chymotrypsin family proteases are anchored to membranes by either glycosylphosphatidylinositol, a single-pass transmembrane domain at the C terminus, or a signal anchor at the N terminus (10).
The S1A, or chymotrypsin A subfamily, consists primarily of eukaryotic proteases, however, a few bacterial serine proteases belong to this subfamily according to the peptidase database MEROPS (11). To date, proteases that are structurally related to trypsin have been identified in Streptomyces species (12, 13). These were shown to contain a single protease domain that has collagenolytic activity (12–14). In addition, we recently identified three related V. cholerae extracellular serine proteases, VesA, VesB, and VesC, in a proteomic screen designed to detect proteins secreted via the T2S system (5). Consistent with their reliance on the T2S system for extracellular secretion, when overexpressed, measurable activity of all three proteases has been detected in culture supernatants of V. cholerae (5, 15). Furthermore, these proteases have a predicted N-terminal signal peptide that allows them to enter the periplasm via the Sec pathway prior to engaging with the T2S system (16). In addition, they all consist of an N-terminal protease domain containing a putative activation site, residues that comprise the catalytic triad, and a C-terminal domain that varies in length and includes a Gly-Gly-CTERM extension of unknown function (Fig. 1) (17). The domain organization of VesA, VesB, and VesC differs from that of most bacterial and eukaryotic trypsin-like proteases in that the additional non-protease domain is positioned at the C terminus.
FIGURE 1.

Schematic diagrams of VesA, VesB, and VesC. All three contain an N-terminal signal peptide in light blue, an N-terminal protease domain in purple, a C-terminal domain in dark blue with the highlighted Ig-fold domain in light blue, and the GlyGly-CTERM extension in yellow. The N-terminal protease domain contains the predicted activation site indicated by the arrow and the catalytic triad comprised of His, Asp, and Ser.
VesB, the focus of this study, shares ∼30% sequence identity with trypsin and other members of the S1A family of serine proteases and displays a similar positioning of its catalytic residues. Although the biological role of VesB has yet to be determined, VesB is produced both in vitro and in vivo. Besides its detection in laboratory-grown cultures, VesB has also been detected in V. cholerae that was isolated from stools of patients with clinical cholera, inferring that VesB may contribute to intestinal growth or pathogenesis (5, 18, 19). Also, the intestinal vesB gene expression was detected in V. cholerae cultivated in a rabbit ileum loop, an experimental model for cholera, further suggesting a possible role in survival or disease (20). Although VesB may contribute to intestinal growth of V. cholerae, it is not the only factor required for intestinal survival as vesB inactivation had no negative effect on infant mice colonization (5). Finally, VesB is capable of cleaving the A subunit of cholera toxin, a process important for cholera toxin activation (5, 21).
The rarity of characterized bacterial serine proteases in the chymotrypsin A subfamily, the unique domain arrangement, and the possible role of VesB in intestinal growth or pathogenesis led us to study VesB in detail. Here, we focus on the activity and structure of VesB. We show that the protease domain of pro-VesB contains a trypsin-like fold with an incomplete oxyanion hole, and an additional C-terminal domain with an Ig-fold containing seven β-strands and two short α-helices. Furthermore, we show that purified VesB has activity against synthetic peptides, cleaves after Arg independently of Na+ ions, can be inhibited by serine protease inhibitors, and is produced with a propeptide that is critical for activity but not for extracellular secretion by V. cholerae.
EXPERIMENTAL PROCEDURES
Growth Conditions
All strains were grown on Luria-Bertani (LB) agar (Fisher). Single colonies were inoculated in Luria-Bertani (LB) broth (Fisher) with 100 μg/ml of carbenicillin and 1 or 100 μm isopropyl β-d-thiogalactopyranoside (Sigma) and grown for 16 h at 37 °C.
Bacterial Strains and Plasmids
The V. cholerae strain N16961, an El Tor O1 biotype, and the isogenic ΔvesABC strain (5) were used for all experiments. VesB was expressed from pMMB67EH-vesB (22), a low copy vector with an isopropyl β-d-thiogalactopyranoside-inducible promoter and ampicillin cassette, and pMMB67EH served as a control plasmid or a cloning vector for additional constructs. The QuikChange Site-directed Mutagenesis Kit (Agilent) was used to create vesB-S221A and vesB-R32E with the following primers 5′-TCATGTCAGGGAGATGCTGGTGGCCCAATTGTA-3′ (Fwd), 5′-TACAATTGGGCCACCAGCATCTCCCTGACATGA-3′ (Rev) and 5′-TCCACAGCAGATATTTCATCTGAAATTATTAATGGTTCGAATGCA-3′ (Fwd), 5′-TGCATTCGAACCATTAATAATTTCAGATGAAATATCTGCTGTGGA-3′ (Rev), respectively. Primers were synthesized at IDT Technologies. Restriction enzymes, buffers, and T4 DNA ligase were purchased from New England Biolabs. Plasmid constructs were transformed into Escherichia coli MC1061 and a triparental conjugation protocol using the helper strain, MM294, was used to transfer the plasmids into N16961 and ΔvesABC strains (23).
Native VesB Purification for Protease Assays
VesB was overexpressed in V. cholerae and culture supernatant was isolated and precipitated with 60% ammonium sulfate for 1 h at 4 °C. The sample was centrifuged at 10,000 × g for 35 min at 4 °C. The pellet was suspended in 20 ml of 50 mm Tris-HCl, pH 8.0, 450 mm NaCl (Buffer A) and residual ammonium sulfate was removed through dialysis against Buffer A. The dialyzed sample was subjected to affinity chromatography on benzamidine-Sepharose (GE Healthcare). The flow-through fraction was collected and the column was washed with Buffer A. Then, VesB was eluted using 10 ml of 100 mm benzamidine, 50 mm Tris-HCl, pH 8.0, buffer. Fractions were pooled and the buffer was exchanged to 50 mm Tris-HCl, pH 8.0, using a PD10 column (GE Healthcare). VesB was concentrated by ultrafiltration (30 kDa cut-off; Millipore). Protein concentration was determined with the Bradford assay (Bio-Rad) using bovine serum albumin as a reference. Polyclonal antiserum against VesB was generated by Covance Inc.
SDS-PAGE and Immunoblotting
Samples were prepared and analyzed via SDS-PAGE and immunoblotting as described previously (23) with the following modifications. VesB antiserum was incubated in culture supernatants from the ΔvesABC strain for 1 h to pre-absorb cross-reactive antibodies prior to incubating with the membrane for 2 h (1:5,000 dilution) and horseradish peroxidase-conjugated goat anti-rabbit IgG (Bio-Rad) was used at 1:20,000 dilution. Membranes were developed using ECL 2 Western blotting reagent (Thermo Scientific) and protein was visualized using a Typhoon Trio variable mode imager system and ImageQuant software. SDS-PAGE gels were stained with GelCode Blue (Thermo Scientific) or Silver Stain Kit reagents (Invitrogen).
Protease Assay
Protease activity in V. cholerae culture supernatants was measured using Boc-Gln-Ala-Arg-7-amino-4-methylcoumarin as described previously (24). In other assays, supernatants were preincubated with benzamidine (Thermo Scientific) at the various concentrations listed for 10 min at 37 °C prior to measuring protease activity. Purified VesB was measured for protease activity with various methylcoumarin-conjugated peptides (Peptides International) listed in Table 1 in a final reaction volume of 100 μl. The peptides, Suc-Ala-Ala-Ala-AMC, Suc-(OMe)-Ala-Ala-Pro-Val-AMC, Z-Leu-Arg-Gly-Gly-AMC, Leu-AMC, Suc-Ala-Ala-Pro-Phe-AMC, Boc-Glu-Lys-Lys-AMC, and Boc-Gln-Ala-Arg-AMC are substrates that have been successfully used for elastase, pancreatic elastase, isopeptidase, aminopeptidase, chymotrypsin, plasmin, and trypsin, respectively. All assays were done with 50 mm Tris-HCl, pH 8.0. Where stated, assays were performed in the presence of different concentrations of NaCl. Inhibition assays were done with purified VesB that was preincubated for 10 min at 37 °C with 50 μm leupeptin (Thermo Scientific), 10 mm EDTA, or 1 mm benzamidine. A change in fluorescence per minute was converted to moles of liberated 7-amino-4-methylcoumarin (AMC) per minute via a standard curve with known amounts of AMC. The results were then normalized by the amount of purified VesB or A600 of the bacterial cultures. Standard errors were generated from three independent experiments each performed in three technical replicates.
TABLE 1.
Proteolytic activity of VesB
| Peptide sequencea | VesB activityb |
|---|---|
| nmol AMC/min/nmol VesB | |
| Suc-Ala-Ala-Ala-AMC | 0 |
| Suc (OMe)-Ala-Ala-Pro-Val-AMC | 0 |
| Z-Leu-Arg-Gly-Gly-AMC | 0 |
| Leu-AMC | 0.518 ± 0.124 |
| Suc-Ala-Ala-Pro-Phe-AMC | 0 |
| Boc-Glu-Lys-Lys-AMC | 0.449 ± 0.060 |
| Boc-Gln-Ala-Arg-AMC | 286 ± 34.0 |
| Boc-Gln-Ala-Arg-AMC + 200 mm NaCl | 122 ± 21.9c |
| Boc-Gln-Ala-Arg-AMC + 400 mm NaCl | 137 ± 31.3c |
| Boc-Gln-Ala-Arg-AMC + 600 mm NaCl | 110 ± 18.9c |
| Boc-Gln-Ala-Arg-AMC + 800 mm NaCl | 97.2 ± 21.6c |
a Peptide sequence of the substrates all conjugated to AMC.
b Substrate concentration was 0.05 mm. VesB concentration was 0.079 μg/ml when Boc-Gln-Ala-Arg-AMC was analyzed and 7.9 μg/ml for all other substrates. Experiments were done in triplicates and the mean ± S.E. are shown.
c p values < 0.01: all compared to the sample with no NaCl.
N-terminal Sequencing
Purified VesB secreted by V. cholerae was subjected to SDS-PAGE and transferred to a PVDF membrane using 10 mm CAPS, 10% methanol, pH 11. The membrane was stained with 0.025% Coomassie Brilliant Blue R-250, 40% methanol, 5% acetic acid solution, destained using 40% methanol, 5% acetic acid, washed with water, and then air-dried. The VesB band was cut and subjected to automated Edman degradation at the protein facility core at University of Michigan, Ann Arbor, MI.
Statistical Analysis
Student's t tests were done on samples as indicated. Results yielding a p value of <0.05 were considered statistically significant.
Protein Expression and Purification for Crystallization
A DNA fragment corresponding to residues 24–373 of VesB was PCR amplified and cloned into a modified pCDF-Duet-1 vector (EMD Millipore). The construct contains an N-terminal pelB signal sequence and a C-terminal tobacco etch virus protease cleavage site followed by a His6 tag. To overcome possible difficulties when overexpressing WT pro-VesB in E. coli, the Ser221 residue was replaced with Ala using QuikChange mutagenesis protocol (Stratagene). The S221A variant of pro-VesB was expressed in E. coli Rosetta2(DE3) cells at 18 °C for 4 h after induction with 0.5 mm isopropyl β-d-thiogalactopyranoside. The cells were harvested by centrifugation and resuspended in buffer containing 20 mm Tris-HCl, pH 7.4, 0.5 m NaCl, 1 mm PMSF, 1 mm benzamidine, and 15 mm imidazole. Pro-VesB was purified via a nickel-nitrilotriacetic acid-agarose (Qiagen) column followed by His6 tag cleavage with tobacco etch virus protease and a second nickel-nitrilotriacetic acid-agarose chromatography step. The yield of pro-VesB was improved from <0.5 to ∼1.5 mg/6 liters of culture after addition of 2 mm Fe2+ ions to the clarified cell lysate (25). The final size-exclusion chromatography was performed using a Superose 6 column (GE Healthcare) in buffer containing 20 mm Tris-HCl, pH 7.4, and 0.5 m NaCl. Pro-VesB was concentrated to 3 mg ml−1 and flash frozen in liquid nitrogen (26).
Crystallization, Data Collection, and Structure Solution
Screening for crystallization conditions was performed using Index (Hampton Research) and Wizard I and II (Emerald Bio) screens. Crystallization trials were set-up in vapor diffusion sitting drop format (0.2 μl of protein + 0.2 μl of crystallization solution) using a Phoenix crystallization robot (Art Robbins Instruments). The initial crystals grew with 0.1 m HEPES, pH 7.5, 3.0 m NaCl. The optimized crystals were obtained using a crystallization solution containing 0.1 m Tris-HCl, pH 9.0, 3.0 m NaCl (1.0 μl of protein + 1.0 μl of crystallization solution + 0.5 μl of water). Crystals were cryoprotected in crystallization solution supplemented with 20% glycerol and flash-cooled in liquid nitrogen.
A 2.4-Å native dataset was collected at the Advanced Light Source beamline 8.2.2. In addition, a derivative dataset was obtained in-house using a Saturn 94 CCD detector on a Rigaku Micromax HF-7 rotating anode from a crystal incubated in 0.1 m Tris-HCl, pH 9.0, 2.0 m NaCl, 1.0 m NaI. Data were processed using HKL2000 and XDS (27, 28). The pro-VesB structure was solved by a combination of molecular replacement and single wavelength diffraction methods as implemented in Phaser (29). The initial molecular replacement solution was found by using the Balbes server (30) and the structure of MT-SP1/matriptase (PDB code 1EAX) as a search model for the protease domain of pro-VesB (30, 31). 12 iodide sites were found by Phaser during the first round of phasing. After density modification using Parrot, model building using Buccaneer, and iterative model improvement using Coot (32–34), the preliminary model was submitted for the second round of phasing, which led to identification of 5 additional (17 total) iodide sites. Subsequent iterative density modification and automated model building led to an almost complete model that included the second domain of VesB. The model was improved using Coot and refined using REFMAC5 and 9 translation/libration/screw (TLS) groups identified by the TLSMD server (35, 36). The quality of the model was assessed using Coot and the MolProbity server (37). Residues 32–372 are included in the final model, whereas residues 166–172, 214–217, and 244–247 had poorly defined density and were not modeled. The final model includes 93 water molecules and has good geometry with 97.5% residues in the favorable areas of the Ramachandran plot (Table 2). The atomic coordinates and structure factors for pro-VesB (PDB code 4LK4) have been deposited in the Protein Data Bank.
TABLE 2.
Data collection and refinement statistics
| Native (PDB 4LK4) | NaI derivativea | |
|---|---|---|
| Data collection | ||
| Wavelength (Å) | 0.9999 | 1.5418 |
| Space group | P42212 | P42212 |
| Cell dimensions | ||
| a, b, c (Å) | 121.57, 121.57, 71.31 | 121.44, 121.44, 71.86 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 |
| Resolution (Å) | 46.3–2.40 (2.53–2.40)b | 33.9–3.20 (3.37–3.20) |
| Rsym | 0.065 (0.985) | 0.154 (0.683) |
| I/σI | 20.4 (2.5) | 14.9 (3.6) |
| Completeness (%) | 99.5 (99.9) | 99.9 (100) |
| Multiplicity | 6.3 (6.5) | 7.7 (7.6) |
| Refinement | ||
| Resolution (Å) | 46.3–2.40 | |
| No. reflections (total/free) | 21376/1070 | |
| Rwork/Rfree | 0.187/0.236 | |
| No. atoms | ||
| Protein | 2465 | |
| Ligand/ion | 0 | |
| Water | 93 | |
| B-factors | ||
| Protein | 60.2 | |
| Water | 48.0 | |
| Wilson B | 59.0 | |
| Root mean square deviations | ||
| Bond lengths (Å) | 0.012 | |
| Bond angles (°) | 1.466 | |
| Ramachandran distribution (%)c | ||
| Favored | 97.5 | |
| Outliers | 0.0 | |
a Friedel pairs are treated as different reflections.
b Values in parentheses are for the highest-resolution shell.
c Calculated using the MolProbity server (37).
RESULTS
Purification of Secreted VesB
To determine whether benzamidine-Sepharose could be utilized for affinity purification of native VesB, the reversible serine protease inhibitor benzamidine was analyzed for its ability to inhibit VesB activity. Specifically, VesB was overexpressed from pMMB67EH-vesB in ΔvesABC, a V. cholerae strain lacking all three serine protease genes vesA, vesB, and vesC (5). Culture supernatant was isolated and incubated with increasing amounts of benzamidine, and then subjected to a kinetic protease activity assay using the synthetic peptide Boc-Gln-Ala-Arg-7-amino-4-methylcoumarin as a substrate. The results showed that as the concentration of benzamidine increased the activity of VesB decreased, suggesting that benzamidine is binding to VesB and that benzamidine-Sepharose can be used for the purification of VesB (Fig. 2A). To purify VesB, culture supernatant was isolated, concentrated, and applied to benzamidine-Sepharose chromatography. VesB was eluted with benzamidine and all of the fractions were analyzed by SDS-PAGE followed by GelCode blue and silver staining (Fig. 2, B and C). The GelCode blue and silver-stained gels showed that some VesB was found in the flow-through fraction (lane 4), whereas there was no additional loss of VesB during washing (lanes 5–10). Bound VesB eluted efficiently with 100 mm benzamidine (lanes 11–14) and once benzamidine was removed the activity of VesB could be measured (Fig. 3). Antibodies were then raised against the purified material and the saved fractions were analyzed by SDS-PAGE and immunoblotting with anti-VesB antiserum (Fig. 2D).
FIGURE 2.

VesB purification by benzamidine-Sepharose affinity chromatography. A, the supernatants of ΔvesABC strains with pMMB67EH (p) or pVesB were incubated for 10 min at 37 °C with different concentrations of the serine protease inhibitor, benzamidine, and the protease activity was measured (experiments were done in triplicates and S.E. bars are shown). B–D, the supernatant from ΔvesABC overexpressing VesB (lane 1) was added to 60% saturation of ammonium sulfate and the precipitated material (Lane 2) and supernatant (Lane 3) were separated by centrifugation. The pellet was resuspended and dialyzed in 50 mm Tris-HCl, pH 8.0, 450 mm NaCl. The sample was used for affinity chromatography using a benzamidine-Sepharose column. Flow-through fraction was discarded (lane 4) and the column was washed with 50 mm Tris-HCl, pH 8.0, 450 mm NaCl (lanes 5–10). VesB was eluted using 100 mm benzamidine (lanes 11–14). The samples were analyzed by SDS-PAGE and GelCode blue staining (B), silver staining (C), or Western blotting with anti-VesB antibodies (D).
FIGURE 3.

Protease activity of purified VesB. A, VesB activity was measured with different concentrations of Boc-Gln-Ala-Arg-7-amino-4-AMC in 5 mm HEPES, pH 7.5, at 37 °C. B, purified VesB (0.08 μg/ml) was incubated with 50 μm leupeptin, 1 mm benzamidine, or 10 mm EDTA for 10 min at 37 °C. The Boc-Gln-Ala-Arg-7-amino-4-AMC (0.05 mm final concentration) was added and VesB activity was measured. All experiments were done in triplicates and S.E. bars are shown.
Characterization of Purified Secreted VesB
Previously, we have shown that Boc-Gln-Ala-Arg-7-amino-4-methylcoumarin is cleaved when incubated with supernatant from V. cholerae overexpressing VesB and that this activity is inhibited by common serine protease inhibitors (5) (Fig. 2A). To further determine the substrate specificity and verify that purified VesB is inhibited by serine protease inhibitors, we added purified VesB to different commercially available synthetic peptides that are conjugated to AMC (Table 1 and Fig. 3A) and preincubated VesB with different inhibitors (Fig. 3B). VesB efficiently cleaved the trypsin substrate Boc-Gln-Ala-Arg-AMC, whereas the plasmin and aminopeptidase substrates, Boc-Glu-Lys-Lys-AMC and Leu-AMC, respectively, were cleaved with greatly reduced efficiency, and no cleavage was observed for the chymotrypsin or elastase substrates (Table 1). Measurement of kinetic parameters yielded Vmax and Km values of 0.137 ± 0.0030 nmol/min and 0.0327 ± 0.0025 mm, respectively, for Boc-Gln-Ala-Arg-AMC (Fig. 3A). We were unable to determine Vmax and Km for Boc-Glu-Lys-Lys-AMC and Boc-Leu-AMC as the reactions never approached maximum rates due to the lack of solubility of the substrates at higher concentrations. In addition, we observed inhibition of purified VesB activity by the serine protease inhibitors benzamidine and leupeptin, whereas VesB was not affected by the presence of the metal chelator, EDTA (Fig. 3B). Taken together, our results suggest that VesB is an extracellular trypsin-like protease that may have a preference for arginine at the P1 position of substrate(s) (using the commonly accepted nomenclature (38)) and does not require divalent metal ions for activity.
VesB Is Produced as a Zymogen
The alignment of trypsin, thrombin, and the protease domain of VesB revealed two important features of the chymotrypsin family of serine proteases: the catalytic triad and an activation site, with the latter corresponding to residues Arg32–Ile33 in pro-VesB (Fig. 1). To verify cleavage at the predicted activation site, purified VesB was subjected to N-terminal sequencing by Edman degradation. The sequence of the first five residues, IINGS, indicated that the propeptide had been removed from VesB and that VesB is cleaved at Arg32–Ile33 similar to other trypsin-like proteases. To determine whether cleavage of the propeptide is necessary for VesB activity, we mutated the vesB gene to replace the activation site residue Arg32 with Glu. In another variant, the putative catalytic residue Ser221 was replaced with Ala. The mutant proteins were expressed in V. cholerae strain ΔvesABC and supernatants and cells were isolated and subjected to analysis of protease activity (Fig. 4A) and immunoblotting with VesB antiserum (Fig. 4B). Both VesB-S221A and VesB-R32E were expressed and secreted in amounts similar to WT VesB (Fig. 4B). However, VesB-S221A and VesB-R32E were deficient in cleaving the synthetic peptide Boc-Gln-Ala-Arg-AMC (Fig. 4A). Taken together, the activation site and catalytic triad are vitally important for the activity of VesB, whereas they play no role in VesB secretion.
FIGURE 4.

Protease activity and secretion of VesB-S221A and VesB-R32E. The vesB gene was subjected to site-directed mutagenesis and the obtained vesB-S221A and vesB-R32E constructs were cloned into pMMB67EH and expression was induced with 1 μm isopropyl β-d-thiogalactopyranoside in the ΔvesABC strain. A, the culture supernatants of ΔvesABC strains with pMMB67EH (p), pVesB, pVesB-S221A, and pVesB-R32E were analyzed for protease activity. The data of VesB were compared with the data of the three strains to generate p values. B, the supernatants (S) and cell extracts (C) were isolated from the bacterial cultures of ΔvesABC strains with pMMB67EH, VesB, VesB-S221A, and VesB-R32E and subjected to SDS-PAGE and Western blot analysis using anti-VesB antibodies.
Structure of Pro-VesB
As the yield of VesB purified from V. cholerae culture supernatant was not sufficiently high for crystallization, a soluble variant of VesB comprising residues 24–373, but lacking C-terminal residues 374–403, which contain the C-terminal Gly-Gly-CTERM extension, was expressed in E. coli and purified for crystallization and structure determination. To overcome a putative overexpression toxicity, the catalytic Ser221 of VesB was replaced with Ala. Crystallization trials produced crystals that yielded x-ray diffraction data to 2.4 Å (Table 2). The structure was solved by a combination of molecular replacement and single wavelength diffraction methods using a sodium iodide derivative crystal. The structure was refined to Rwork/Rfree of 0.187/0.236 with good geometry (Table 2).
The crystal structure revealed that pro-VesB consists of two domains: an N-terminal protease domain with a typical trypsin/chymotrypsin-fold and a C-terminal domain with an Ig-fold (Fig. 5). The structure of the protease domain of pro-VesB can be superimposed onto the bovine trypsinogen structure (PDB 1TGB (39)) with a root mean square deviation of 1.7 Å with 34% sequence identity for 198 residues (Fig. 6). Although there are two conserved disulfide bonds, Cys63–Cys79 and Cys190–Cys208, that are clearly present in the crystal structure, a putative Cys217–Cys244 disulfide is not visible as both Cys residues are part of disordered regions in pro-VesB. The protease domain of pro-VesB was captured in an inactive conformation in our structure either because E. coli does not express a protease that is capable of cleaving VesB and/or VesB is unable to undergo autocatalysis due to the S221A substitution. Residues 24–32, corresponding to the N-terminal propeptide sequence, were present in the crystallized protein, although most of the propeptide is disordered in the crystal. Due to the presence of the intact propeptide, the side chain of residue Ile33, which would be the N-terminal residue in the active form of VesB, cannot occupy the hydrophobic pocket lined by residues Val159 and Val180. Instead, this hydrophobic pocket in the current structure is occupied by Ile164, which prevents the correct orientation of Asp220 that, based on homology with trypsin and trypsinogen (PDB 2PTC and 1TGB (39, 40)), should coordinate the N-terminal amino group of Ile33 in active VesB (compare Fig. 7, A and B). These structural features probably contribute to the disorder of three loops in the vicinity of the active site, which include residues 166–172, 214–217, and 244–247 (Fig. 5A). Consistent with VesB being a trypsin-like protease, the disordered loops in pro-VesB are reminiscent of the disordered loops in the crystal structure of bovine trypsinogen (39).
FIGURE 5.

Crystal structure of pro-VesB. A, the crystal structure is shown in ribbon representation with α-helices in purple and β-strands in green. The inset on the right shows the catalytic triad residues Asp125, His78, and Ser221. Note that Ser221 was substituted to Ala to alleviate possible toxicity during protein expression. The disordered loops are indicated by dotted lines. B, the structure of VesB in surface representation. The protease domain of VesB is shown in light green and the Ig domain in dark green. The catalytic triad residues are colored in orange. Note that the catalytic site is not blocked by the Ig-fold domain.
FIGURE 6.

Similarities between pro-VesB, trypsinogen, thrombin, and S. griseus trypsin. A, stereo view of superposition of the protease domain of pro-VesB and trypsinogen (PDB 2PTC (40)). VesB is shown in the same colors as described in the legend to Fig. 5A, i.e. with α-helices in purple and β-strands in green; trypsinogen is shown in cyan. The catalytic triad residues of both proteases are shown as sticks. B, structure-based sequence alignment of VesB, S. griseus trypsin (SGT), bovine trypsinogen and human thrombin (trypsin template numbering for trypsinogen and thrombin (39)). Catalytic triad residues are highlighted in blue. Tyr250 of VesB, at a position equivalent to that of Tyr225 in thrombin is highlighted in cyan. In thrombin, this residue is involved in Na+ binding (but neither in S. griseus trypsin, trypsinogen or VesB, see text). Conserved Cys residues that form disulfide bonds are highlighted in yellow, note that the disulfide Cys217–Cys244 was invisible in the pro-VesB structure due to flexibility. The three disordered loops in the pro-VesB crystal structure are indicated with dotted lines above the top sequence. The sequence numbers on top are those of VesB, at the bottom those of thrombin. The alignment was rendered using the ESPript server (64).
FIGURE 7.
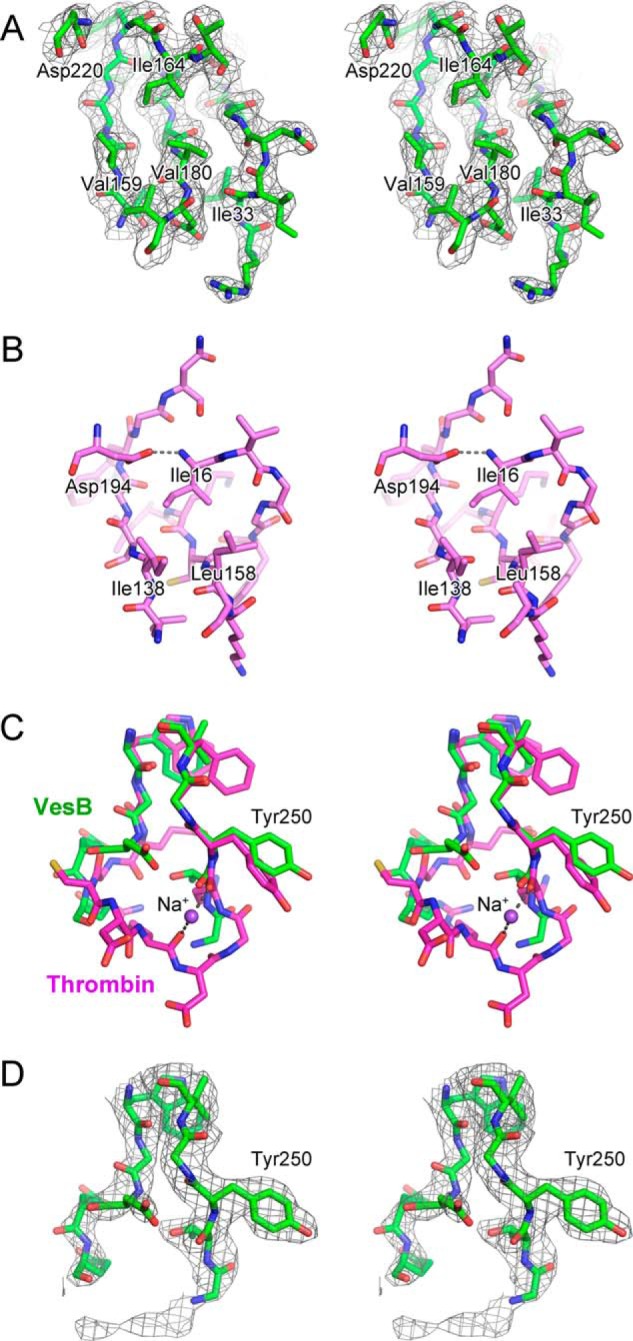
Close-up views of the activation site and the Tyr250 region of pro-VesB. A, stereo view of the activation site of VesB. Selected residues including Ile33 are shown in stick representation and labeled. σA-Weighted 2Fo − Fc map is shown as gray mesh (contoured at 1σ). B, stereo view of the equivalent region in trypsin is shown (PDB code 2PTC). Note that the N-terminal Ile16 residue occupies the hydrophobic pocket formed by Ile138 and Ile158, and coordinates Asp194. C, stereo view of the superposition of the Na+ binding loop of thrombin (magenta) and the equivalent loop of pro-VesB (green). Na+ ion is shown as a purple sphere. D, the same view as in C showing the σA-weighted 2Fo − Fc map as gray mesh (contoured at 1σ) of pro-VesB.
In thrombin, residues Tyr184, Arg221, and Lys224 were shown to coordinate a Na+ ion that binds to and allosterically enhances thrombin activity (41, 42). Although the catalytic activity of several other coagulation and complement factors is also enhanced by sodium, trypsin is not. In the Na+-activated members of the trypsin-like family, Tyr225 (thrombin numbering) plays a critical role. A proline residue at this position changes the orientation of the nearby Lys224 such that its carbonyl O atom can no longer coordinate Na+ (41). When thrombin residue Tyr225 was replaced with a Pro, as occurs in several trypsin-like proteases that are not Na+-sensitive, there was a loss in Na+ activation (41). The sequence alignment of VesB with thrombin shows that the residue corresponding to Tyr225 in thrombin is Tyr250 in VesB (Fig. 6B), i.e. a residue that is compatible with Na+ activation. The structural superposition of pro-VesB and thrombin is shown in Fig. 7C, specifically highlighting the position of Tyr225 and surrounding residues in thrombin and the corresponding residues in pro-VesB. However, no sodium ion is apparent in the pro-VesB electron density (Fig. 7D). The absence of sodium may be due to the inability of pro-VesB to coordinate sodium. Similarly, sodium is not present in the zymogen forms of thrombin (43). Thus, the structure of pro-VesB does not provide insight into the presence of a putative Na+ binding site in active VesB. To establish whether active VesB can be stimulated by Na+, we determined the activity of purified VesB in the absence and presence of increasing concentrations of NaCl using Boc-Gln-Ala-Arg-AMC (Table 1). The presence of Na+ did not stimulate VesB, instead we observed a decrease in activity (Table 1). When a different substrate with Arg at the P1 position (Z-Gly-Gly-Arg-AMC) was tested, a similar negative effect on the VesB activity was observed (not shown). In addition, we isolated culture supernatant from a V. cholerae culture grown in a medium without NaCl and determined the activity of VesB in the absence or presence of 200 mm NaCl. There was no change in VesB activity when 200 mm NaCl was included (data not shown). Taken together, VesB does not appear to be activated by Na+ in a similar fashion to thrombin.
The crystal structure of the C-terminal domain of VesB revealed a domain rich in β-strands that adopts an Ig-fold (Fig. 5). A structural similarity search with the Dali server identified a number of distant homologs with Ig-folds (Table 3). Although the best hit was a domain from a Porphyromonas gingivalis protein of unknown function (Z factor = 9.3 and 12% amino acid sequence identity), homologs of possibly greater interest are domains from bacterial PapD-like chaperones such as SfaE (Z factor = 8.2) and subtilisin-like proteases like cucumisin (Z factor = 7.7). Superpositions of the C-terminal domain of VesB with SfaE and cucumisin domains are shown in Fig. 8. Interestingly, VesB contains two additional short α-helices compared with the other proteins, and two extra β-strands compared with SfaE. We also observed a degree of structural homology, albeit less closely than with the previous three homologs, between the Ig domain of VesB and domains from several extracellular Vibrio proteins, including chitin-binding protein (GbpA), chitinase (ChiA), rugose and biofilm structure modifier (RbmA), and another protease of V. cholerae (PrtV) (Table 3). The Ig domain of GbpA has been reported to support its V. cholerae surface association (44), whereas the Ig domain of ChiA is a carbohydrate binding module (45). Although we were unable to detect binding of purified VesB to chitin, suggesting that VesB is not a chitin-binding protein (data not shown), we have observed that a large fraction of extracellular VesB is surface associated.6 We have not yet confirmed a role for the Ig domain in this interaction as the removal of the Ig domain of VesB results in an unstable protein that cannot be detected when expressed in V. cholerae (data not shown).
TABLE 3.
Structural homologs of the C-terminal domain of VesB
| Protein | Z-scorea | Root mean square deviation | Aligned aa | Sequence identity | PDB ID | Reference |
|---|---|---|---|---|---|---|
| % | ||||||
| Unknown function | 9.3 | 2.5 | 94 | 12 | 2QSV | NAb |
| SfaE | 8.2 | 2.7 | 92 | 5 | 1L4I | 65 |
| Cucumisin | 7.7 | 2.7 | 91 | 11 | 3VTA | 57 |
| RbmA | 7.0 | 2.6 | 88 | 10 | 4KKQ | 61 |
| GbpA | 5.8 | 3.4 | 84 | 11 | 2XWX | 44 |
| ChiA | 4.5 | 3.7 | 73 | 11 | 3B8S | 45 |
| PrtV | 3.7 | 2.8 | 63 | 17 | 4L9D | 60 |
| KP-43 | 2.3 | 2.9 | 60 | 7 | 1WMD | 59 |
| ASP | 2.0 | 2.9 | 59 | 10 | 3HJR | 58 |
a Z-score from the Dali server.
b NA, not applicable.
FIGURE 8.

Structural homologs of the C-terminal domain of VesB. A, stereo view of superposition of the C-terminal domain of VesB (green and purple) and the N-terminal domain of S pilus chaperone (blue) (PDB code 1L4I) (65). The N and C termini of the C-terminal domain of VesB are labeled. Note that the C-terminal domain of VesB has two additional β-strands and two additional short helices (purple). B, stereo view of the superposition of the C-terminal domains of VesB (green and purple) and cucumisin (orange) (PDB 3VTA) (57). Pro-VesB contains two additional short helices (purple).
Interface between the Protease and C-terminal Domains
In the crystals, the two domains of pro-VesB interact with each other with a buried surface area of 1220 Å2 (Fig. 5B). This interface is classified by the PISA server as insignificant (46), although physiologically relevant interfaces with the same extent of buried surface area have been reported (47). Hence, it is uncertain whether the two domains of pro-VesB have a different or flexible relative orientation with respect to each other in solution or during secretion via the T2S apparatus.
DISCUSSION
In this study, we show that the protease domain of VesB is related to trypsin in sequence, structure, and substrate specificity. Upon sequence alignment of trypsin and VesB, the catalytic triad and the activation site of VesB became apparent (Fig. 6B). Mutating these motifs rendered VesB inactive, suggesting that the catalytic triad and the activation site are essential for activity, but have no impact on extracellular secretion of VesB (Fig. 4). Our enzymatic studies suggested that VesB may preferentially cleave peptides that have an Arg at the P1 position and showed that VesB is inhibited by the serine protease inhibitors, leupeptin and benzamidine (Figs. 2 and 3, Table 1). Last, the crystal structure revealed that the N-terminal protease domain of pro-VesB is structurally homologous to mammalian trypsin of the S1A subfamily and the C-terminal domain contains an Ig-fold (Figs. 5–8).
Bacterial serine proteases belonging to the S1A subfamily were once considered rare and only thought to exist in certain Streptomyces species (48, 49). With the rapidly growing number of sequenced genomes, however, it is becoming increasingly clear that additional bacterial species also carry genes for trypsin-like enzymes. Besides the V. cholerae vesB, vesA, and vesC, genes encoding proteases of the chymotrypsin family are also found in other marine Gram-negative bacteria, many belonging to the Vibrionaceae and Shewanellaceae families. Previously, a study was carried out to determine the nature of acquiring trypsin-encoding genes in bacterial genomes. A phylogenetic tree based on the sequence and structure of trypsins from various mammalian species and Streptomyces griseus revealed that these proteases originate from a common ancestor (50). A later study performed a similar analysis looking at a larger selection of trypsin-like proteases from a variety of species, but only including a single Streptomyces protease as bacterial representative (48). Based on the data presented in the latter study it is difficult to determine the evolutionary relationship between the trypsin-like proteases. It is not clear whether trypsin encoding genes originated in bacterial cells, and were maintained during subsequent evolutionary steps in eukaryotic species but lost in many bacterial species, or if the genes have been transferred horizontally between species.
A way to distinguish and further analyze the relationship between the serine proteases of the chymotrypsin family is by determining how the activity of these proteases is regulated. For several trypsin-like proteases, which function in blood coagulation or complement formation, activation by Na+ plays a well established role (41, 51). It has been shown that in these proteins the residue equivalent to Tyr225 in thrombin plays a key role in the proper formation of the Na+-binding site (41). In VesB, Tyr250 is equivalent in position to Tyr225 of thrombin (Figs. 6B and 7C) and hence it would be possible that VesB is activated by sodium ions. However, addition of NaCl does not appear to increase purified VesB activity. In the crystal structure of pro-VesB no density representing a sodium ion at the expected position is evident, although it should be noted that the zymogen form of thrombin also lacks sodium (Fig. 7D) (43). These results are not too surprising for two reasons.
First, elegant protein engineering studies on S. griseus trypsin, which contains a Pro at the equivalent position of Tyr225 in thrombin and is not activated by Na+, have shown that up to 19 residues need to be altered, in addition to the Pro to Tyr change near the Na+ activation site, to obtain a S. griseus trypsin variant that is fully Na+-sensitive (52). Hence, the presence of Tyr at the position equivalent to Tyr225 in thrombin is a necessary but not sufficient requirement for a member of the trypsin-like family to become activated by Na+, which is also supported by studies on complement mannan-binding lectin-associated serine protease 2 (53).
Second, VesB is secreted and needs to function in the external milieu like the gut and/or aquatic habitats, sites with highly variable Na+ ion concentrations. For example, at aquatic environmental sites where V. cholerae is frequently isolated, the NaCl concentration varies between 0.2 and 3.0% (54). These conditions are quite different from that of blood, where the sodium ion concentration is precisely controlled. Hence, Tyr250 plays most likely a structural role and is not involved in Na+ activation of this V. cholerae protease. Taken together, our findings suggest that a tyrosine in trypsin-like proteases equivalent to thrombin Tyr225 cannot be used as the sole indicator of Na+-mediated activation.
The C-terminal domain of VesB is structurally similar to Ig-folds of PapD-like chaperones, subtilisin-like proteases, and several extracellular proteins from Vibrio. The role of the Ig-fold in VesB is currently unknown; however, understanding the role of Ig-folds in other proteins may shed light on a possible function of the VesB Ig-fold. The PapD chaperones reside in the periplasm of Gram-negative bacteria and bind to type 1 pilin subunits via a donor-strand complementation mechanism to target them to the Usher protein for outer membrane translocation and pilus assembly on the cell surface (55). The chaperone-mediated stabilization of the pilin subunits is evident in a papD-deficient strain, in which DegP degrades the pilin subunits (55, 56). The subtilisin-like serine protease cucumisin contains a C-terminal Ig-fold domain that interacts with its catalytic domain (57) and in the Aeromonas sobria subtilisin-like protein ASP, the Ig-fold domain is located close to the active site; therefore, it has been proposed that this domain is involved in substrate specificity (58). The Ig-fold domain of yet another subtilisin-like protease, KP-43 from Bacillus, is suggested to provide stability, as KP-43 without its Ig-fold domain could not be expressed (59). Although the Ig-fold domain may similarly provide stability to VesB, as we are unable to detect VesB when expressed without its C-terminal domain in V. cholerae, the Ig-fold may alternatively, or in addition, provide substrate specificity although it is located relatively far (∼20 Å) from the active site (Fig. 5B).
Other possible roles of the Ig-fold domain may relate to the extracellular secretion or surface association of VesB. VesB, GbpA, ChiA, PrtV, and RbmA are extracellular Vibrio proteins that contain Ig-fold domains (44, 45, 60, 61). Some of these proteins (VesB, GbpA, and ChiA) have been shown to utilize the T2S system for outer membrane translocation (5, 62, 63). Although the Ig-fold domains may be involved in the transport of VesB and other extracellular proteins via this translocation system, it is also possible that they assist in surface association of these proteins similar to the Ig-fold domain of GbpA, which binds to the bacterial cell surface (44). Surface localization of VesB may optimally allow for the immediate uptake and intracellular delivery of peptides generated by VesB-mediated proteolysis.
In summary, we have shown that VesB has a structure and specificity profile resembling that of eukaryotic trypsin-like proteases. Additionally, the structure of the C-terminal domain of VesB revealed an Ig-fold domain that may be involved in one or more different functions such as stabilizing the protease domain, co-defining substrate specificity, binding to the bacterial surface, and being part of a yet undefined secretion motif of the T2S system (2).
Acknowledgments
We thank the staff of beamline 8.2.2 at ALS for assistance during data collection. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the United States Department of Energy under Contract No. DE-AC02-05CH11231.
This work was supported, in whole or in part, by National Institutes of Health Grants R01AI049294 and R01AI081705 (to M. S.) and R01AI034501 (to W. G. J. H.) from the NIAID.
S. Gadwal, T. Johnson, and M. Sandkvist, manuscript in preparation.
- T2S
- type II secretion
- Boc
- t-butoxycarbonyl
- AMC
- 7-amino-4-methylcoumarin
- CAPS
- 3-(cyclohexylamino)propanesulfonic acid
- Z
- benzyloxycarbonyl.
REFERENCES
- 1. Douzi B., Filloux A., Voulhoux R. (2012) On the path to uncover the bacterial type II secretion system. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 367, 1059–1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Korotkov K. V., Sandkvist M., Hol W. G. (2012) The type II secretion system. Biogenesis, molecular architecture and mechanism. Nat. Rev. Microbiol. 10, 336–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. McLaughlin L. S., Haft R. J., Forest K. T. (2012) Structural insights into the type II secretion nanomachine. Curr. Opin. Struct. Biol. 22, 208–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sandkvist M., Morales V., Bagdasarian M. (1993) A protein required for secretion of cholera toxin through the outer membrane of Vibrio cholerae. Gene 123, 81–86 [DOI] [PubMed] [Google Scholar]
- 5. Sikora A. E., Zielke R. A., Lawrence D. A., Andrews P. C., Sandkvist M. (2011) Proteomic analysis of the Vibrio cholerae type II secretome reveals new proteins, including three related serine proteases. J. Biol. Chem. 286, 16555–16566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rawlings N. D., Barrett A. J. (1994) Families of serine peptidases. Methods Enzymol. 244, 19–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hedstrom L. (2002) An overview of serine proteases. Current protocols in protein science (Coligan J. E., et al., eds) Chapter 21, Unit 21 10, pp. 1–8, John Wiley & Sons, Inc., Hoboken, NJ: [DOI] [PubMed] [Google Scholar]
- 8. van Zonneveld A. J., Veerman H., MacDonald M. E., van Mourik J. A., Pannekoek H. (1986) Structure and function of human tissue-type plasminogen activator (t-PA). J. Cell. Biochem. 32, 169–178 [DOI] [PubMed] [Google Scholar]
- 9. Forneris F., Wu J., Gros P. (2012) The modular serine proteases of the complement cascade. Curr. Opin. Struct. Biol. 22, 333–341 [DOI] [PubMed] [Google Scholar]
- 10. Szabo R., Bugge T. H. (2011) Membrane-anchored serine proteases in vertebrate cell and developmental biology. Annu. Rev. Cell Dev. Biol. 27, 213–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rawlings N. D., Barrett A. J., Bateman A. (2012) MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 40, D343–D350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Read R. J., James M. N. (1988) Refined crystal structure of Streptomyces griseus trypsin at 1.7-Å resolution. J. Mol. Biol. 200, 523–551 [DOI] [PubMed] [Google Scholar]
- 13. Yamane T., Iwasaki A., Suzuki A., Ashida T., Kawata Y. (1995) Crystal structure of Streptomyces erythraeus trypsin at 1.9-Å resolution. J. Biochem. 118, 882–894 [DOI] [PubMed] [Google Scholar]
- 14. Uesugi Y., Arima J., Usuki H., Iwabuchi M., Hatanaka T. (2008) Two bacterial collagenolytic serine proteases have different topological specificities. Biochim. Biophys. Acta 1784, 716–726 [DOI] [PubMed] [Google Scholar]
- 15. Syngkon A., Elluri S., Koley H., Rompikuntal P. K., Saha D. R., Chakrabarti M. K., Bhadra R. K., Wai S. N., Pal A. (2010) Studies on a novel serine protease of a [Delta]hapA[Delta]prtV Vibrio cholerae O1 strain and its role in hemorrhagic response in the rabbit ileal loop model. PloS One 5(9):e13122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Filloux A. (2004) The underlying mechanisms of type II protein secretion. Biochim. Biophys. Acta 1694, 163–179 [DOI] [PubMed] [Google Scholar]
- 17. Haft D. H., Varghese N. (2011) GlyGly-CTERM and rhombosortase. A C-terminal protein processing signal in a many-to-one pairing with a rhomboid family intramembrane serine protease. PloS One 6, e28886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. LaRocque R. C., Krastins B., Harris J. B., Lebrun L. M., Parker K. C., Chase M., Ryan E. T., Qadri F., Sarracino D., Calderwood S. B. (2008) Proteomic analysis of Vibrio cholerae in human stool. Infect. Immun. 76, 4145–4151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sikora A. E. (2013) Proteins secreted via the type II secretion system. Smart strategies of Vibrio cholerae to maintain fitness in different ecological niches. PLoS Pathog. 9, e1003126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xu Q., Dziejman M., Mekalanos J. J. (2003) Determination of the transcriptome of Vibrio cholerae during intraintestinal growth and midexponential phase in vitro. Proc. Natl. Acad. Sci. U.S.A. 100, 1286–1291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gordon V. M., Leppla S. H. (1994) Proteolytic activation of bacterial toxins. Role of bacterial and host cell proteases. Infect. Immun. 62, 333–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lybarger S. R., Johnson T. L., Gray M. D., Sikora A. E., Sandkvist M. (2009) Docking and assembly of the type II secretion complex of Vibrio cholerae. J. Bacteriol. 191, 3149–3161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sikora A. E., Lybarger S. R., Sandkvist M. (2007) Compromised outer membrane integrity in Vibrio cholerae Type II secretion mutants. J. Bacteriol. 189, 8484–8495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Johnson T. L., Scott M. E., Sandkvist M. (2007) Mapping critical interactive sites within the periplasmic domain of the Vibrio cholerae type II secretion protein EpsM. J. Bacteriol. 189, 9082–9089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Magnusdottir A., Johansson I., Dahlgren L. G., Nordlund P., Berglund H. (2009) Enabling IMAC purification of low abundance recombinant proteins from E. coli lysates. Nat. Methods 6, 477–478 [DOI] [PubMed] [Google Scholar]
- 26. Deng J., Davies D. R., Wisedchaisri G., Wu M., Hol W. G., Mehlin C. (2004) An improved protocol for rapid freezing of protein samples for long-term storage. Acta Crystallogr. D Biol. Crystallogr. 60, 203–204 [DOI] [PubMed] [Google Scholar]
- 27. Kabsch W. (2010) Xds. Acta Crystallogr. Sect. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 29. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Long F., Vagin A. A., Young P., Murshudov G. N. (2008) BALBES. A molecular-replacement pipeline. Acta Crystallogr. D Biol. Crystallogr. 64, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Friedrich R., Fuentes-Prior P., Ong E., Coombs G., Hunter M., Oehler R., Pierson D., Gonzalez R., Huber R., Bode W., Madison E. L. (2002) Catalytic domain structures of MT-SP1/matriptase, a matrix-degrading transmembrane serine proteinase. J. Biol. Chem. 277, 2160–2168 [DOI] [PubMed] [Google Scholar]
- 32. Cowtan K. (2006) The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr. D Biol. Crystallogr. 62, 1002–1011 [DOI] [PubMed] [Google Scholar]
- 33. Cowtan K. (2010) Recent developments in classical density modification. Acta Crystallogr. D Biol. Crystallogr. 66, 470–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Painter J., Merritt E. A. (2006) Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr. D Biol. Crystallogr. 62, 439–450 [DOI] [PubMed] [Google Scholar]
- 37. Chen V. B., Arendall W. B., 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. (2010) MolProbity. All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schechter I., Berger A. (1967) On the size of the active site in proteases. I. Papain. Biochem. Biophys. Res. Commun. 27, 157–162 [DOI] [PubMed] [Google Scholar]
- 39. Fehlhammer H., Bode W., Huber R. (1977) Crystal structure of bovine trypsinogen at 1–8 Å resolution. II. Crystallographic refinement, refined crystal structure and comparison with bovine trypsin. J. Mol. Biol. 111, 415–438 [DOI] [PubMed] [Google Scholar]
- 40. Marquart M., Walter J., Deisenhofer J., Bode W., Huber R. (1983) The geometry of the reactive site and of the peptide groups in trypsin, trypsinogen and its complexes with inhibitors. Acta Crystallogr. Struct. Sci. 39, 480–490 [Google Scholar]
- 41. Dang Q. D., Di Cera E. (1996) Residue 225 determines the Na+-induced allosteric regulation of catalytic activity in serine proteases. Proc. Natl. Acad. Sci. U.S.A. 93, 10653–10656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Di Cera E., Guinto E. R., Vindigni A., Dang Q. D., Ayala Y. M., Wuyi M., Tulinsky A. (1995) The Na+ binding site of thrombin. J. Biol. Chem. 270, 22089–22092 [DOI] [PubMed] [Google Scholar]
- 43. Pozzi N., Chen Z., Gohara D. W., Niu W., Heyduk T., Di Cera E. (2013) Crystal structure of prothrombin reveals conformational flexibility and mechanism of activation. J. Biol. Chem. 288, 22734–22744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wong E., Vaaje-Kolstad G., Ghosh A., Hurtado-Guerrero R., Konarev P. V., Ibrahim A. F., Svergun D. I., Eijsink V. G., Chatterjee N. S., van Aalten D. M. (2012) The Vibrio cholerae colonization factor GbpA possesses a modular structure that governs binding to different host surfaces. PLoS Pathog. 8, e1002373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Songsiriritthigul C., Pantoom S., Aguda A. H., Robinson R. C., Suginta W. (2008) Crystal structures of Vibrio harveyi chitinase A complexed with chitooligosaccharides. Implications for the catalytic mechanism. J. Struct. Biol. 162, 491–499 [DOI] [PubMed] [Google Scholar]
- 46. Krissinel E., Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 47. Korotkov K. V., Johnson T. L., Jobling M. G., Pruneda J., Pardon E., Héroux A., Turley S., Steyaert J., Holmes R. K., Sandkvist M., Hol W. G. (2011) Structural and functional studies on the interaction of GspC and GspD in the type II secretion system. PLoS Pathog. 7, e1002228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rojas A., Doolittle R. F. (2006) The occurrence of type S1A serine proteases in sponge and jelly. J. Mol. Evol. 55, 790–794 [DOI] [PubMed] [Google Scholar]
- 49. Uesugi Y., Usuki H., Iwabuchi M., Hatanaka T. (2009) The role of Tyr-71 in Streptomyces trypsin on the recognition mechanism of structural protein substrates. FEBS J. 276, 5634–5646 [DOI] [PubMed] [Google Scholar]
- 50. Rypniewski W. R., Perrakis A., Vorgias C. E., Wilson K. S. (1994) Evolutionary divergence and conservation of trypsin. Protein Eng. 7, 57–64 [DOI] [PubMed] [Google Scholar]
- 51. Di Cera E., Page M. J., Bah A., Bush-Pelc L. A., Garvey L. C. (2007) Thrombin allostery. Phys. Chem. Chem. Phys. 9, 1291–1306 [DOI] [PubMed] [Google Scholar]
- 52. Page M. J., Bleackley M. R., Wong S., MacGillivray R. T., Di Cera E. (2006) Conversion of trypsin into a Na+-activated enzyme. Biochemistry 45, 2987–2993 [DOI] [PubMed] [Google Scholar]
- 53. Harmat V., Gál P., Kardos J., Szilágyi K., Ambrus G., Végh B., Náray-Szabó G., Závodszky P. (2004) The structure of MBL-associated serine protease-2 reveals that identical substrate specificities of C1s and MASP-2 are realized through different sets of enzyme-substrate interactions. J. Mol. Biol. 342, 1533–1546 [DOI] [PubMed] [Google Scholar]
- 54. Vezzulli L., Pruzzo C., Huq A., Colwell R. R. (2010) Environmental reservoirs of Vibrio cholerae and their role in cholera. Environ. Microbiol. Rep. 2, 27–33 [DOI] [PubMed] [Google Scholar]
- 55. Sauer F. G., Knight S. D., Waksman G. J., Hultgren S. J. (2000) PapD-like chaperones and pilus biogenesis. Semin. Cell Dev. Biol. 11, 27–34 [DOI] [PubMed] [Google Scholar]
- 56. Lindberg F., Tennent J. M., Hultgren S. J., Lund B., Normark S. (1989) PapD, a periplasmic transport protein in P-pilus biogenesis. J. Bacteriol. 171, 6052–6058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Murayama K., Kato-Murayama M., Hosaka T., Sotokawauchi A., Yokoyama S., Arima K., Shirouzu M. (2012) Crystal structure of cucumisin, a subtilisin-like endoprotease from Cucumis melo L. J. Mol. Biol. 423, 386–396 [DOI] [PubMed] [Google Scholar]
- 58. Kobayashi H., Utsunomiya H., Yamanaka H., Sei Y., Katunuma N., Okamoto K., Tsuge H. (2009) Structural basis for the kexin-like serine protease from Aeromonas sobria as sepsis-causing factor. J. Biol. Chem. 284, 27655–27663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Nonaka T., Fujihashi M., Kita A., Saeki K., Ito S., Horikoshi K., Miki K. (2004) The crystal structure of an oxidatively stable subtilisin-like alkaline serine protease, KP-43, with a C-terminal β-barrel domain. J. Biol. Chem. 279, 47344–47351 [DOI] [PubMed] [Google Scholar]
- 60. Edwin A., Rompikuntal P., Björn E., Stier G., Wai S. N., Sauer-Eriksson A. E. (2013) Calcium binding by the PKD1 domain regulates interdomain flexibility in Vibrio cholerae metalloprotease PrtV. FEBS Open Bio 3, 263–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Giglio K. M., Fong J. C., Yildiz F. H., Sondermann H. (2013) Structural basis for biofilm formation via the Vibrio cholerae matrix protein RbmA. J. Bacteriol. 195, 3277–3286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Connell T. D., Metzger D. J., Lynch J., Folster J. P. (1998) Endochitinase is transported to the extracellular milieu by the eps-encoded general secretory pathway of Vibrio cholerae. J. Bacteriol. 180, 5591–5600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kirn T. J., Jude B. A., Taylor R. K. (2005) A colonization factor links Vibrio cholerae environmental survival and human infection. Nature 438, 863–866 [DOI] [PubMed] [Google Scholar]
- 64. Gouet P., Robert X., Courcelle E. (2003) ESPript/ENDscript. Extracting and rendering sequence and 3D information from atomic structures of proteins. Nucleic Acids Res. 31, 3320–3323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Knight S. D., Choudhury D., Hultgren S., Pinkner J., Stojanoff V., Thompson A. (2002) Structure of the S pilus periplasmic chaperone SfaE at 2.2-Å resolution. Acta Crystallogr. D Biol. Crystallogr. 58, 1016–1022 [DOI] [PubMed] [Google Scholar]