

Background: At the redox potential of NADH/NAD+, at least four mitochondrial sites produce superoxide/H2O2.
Results: We compare their capacities in situ in isolated mitochondria.
Conclusion: Maximum capacities of complexes were 2-oxoglutarate dehydrogenase > pyruvate dehydrogenase > branched-chain 2-oxoacid dehydrogenase > complex I.
Significance: H2O2 production from 2-oxoacid dehydrogenases can be considerable but may previously have been misattributed to complex I.
Keywords: Mitochondria, Pyruvate Dehydrogenase Complex, Rat, Reactive Oxygen Species (ROS), Skeletal Muscle, 2-Oxoglutarate Dehydrogenase Complex, NADH Autofluorescence, α-Ketoglutarate Dehydrogenase, Branched-chain 2-Oxoacid Dehydrogenase Complex, Branched-chain Ketoacid Dehydrogenase
Abstract
Several flavin-dependent enzymes of the mitochondrial matrix utilize NAD+ or NADH at about the same operating redox potential as the NADH/NAD+ pool and comprise the NADH/NAD+ isopotential enzyme group. Complex I (specifically the flavin, site IF) is often regarded as the major source of matrix superoxide/H2O2 production at this redox potential. However, the 2-oxoglutarate dehydrogenase (OGDH), branched-chain 2-oxoacid dehydrogenase (BCKDH), and pyruvate dehydrogenase (PDH) complexes are also capable of considerable superoxide/H2O2 production. To differentiate the superoxide/H2O2-producing capacities of these different mitochondrial sites in situ, we compared the observed rates of H2O2 production over a range of different NAD(P)H reduction levels in isolated skeletal muscle mitochondria under conditions that favored superoxide/H2O2 production from complex I, the OGDH complex, the BCKDH complex, or the PDH complex. The rates from all four complexes increased at higher NAD(P)H/NAD(P)+ ratios, although the 2-oxoacid dehydrogenase complexes produced superoxide/H2O2 at high rates only when oxidizing their specific 2-oxoacid substrates and not in the reverse reaction from NADH. At optimal conditions for each system, superoxide/H2O2 was produced by the OGDH complex at about twice the rate from the PDH complex, four times the rate from the BCKDH complex, and eight times the rate from site IF of complex I. Depending on the substrates present, the dominant sites of superoxide/H2O2 production at the level of NADH may be the OGDH and PDH complexes, but these activities may often be misattributed to complex I.
Introduction
Mitochondria may generate superoxide anion radical (“superoxide”) or H2O2 from at least 10 distinct sites in the electron transport chain and associated pathways (such as the Krebs cycle and β-oxidation). Respiratory complexes I and III are usually described as the principal producers (1–9), but many other mitochondrial enzymes can also reduce oxygen prematurely, most notably complex II (10).
In complex I there are two sites of superoxide production: the flavin in the NADH-oxidizing site (site IF)4 and the ubiquinone-reducing site (site IQ) (11). In complex III, superoxide arises from the quinol-oxidizing site (site IIIQo) (12–14). In complex II, the flavin site of complex II (site IIF) generates superoxide and/or H2O2 (10). Other sites include mitochondrial glycerol-3-phosphate dehydrogenase (15), the electron transferring flavoprotein/ETF:ubiquinone oxidoreductase system of fatty acid β-oxidation (16), dihydroorotate dehydrogenase (15, 17), and the dihydrolipoamide dehydrogenase of 2-oxoacid dehydrogenase complexes (18): 2-oxoglutarate dehydrogenase (OGDH) complex (19–21), branched-chain 2-oxoacid dehydrogenase (BCKDH) complex (22), and pyruvate dehydrogenase (PDH) complex (20, 23). Proline dehydrogenase has also been implicated (24). Most studies on mitochondrial production of superoxide and H2O2 have concentrated on the maximum capacities of one or other of these sites when electron flow is blocked by electron transport chain inhibitors, but techniques to assay the rates from several different sites during normal electron flow are now becoming available (15, 16, 25–27).
Complex I is often credited as the primary site of matrix superoxide production when electron flow from Krebs cycle intermediates into the rest of the respiratory chain is inhibited by the addition of rotenone to block site IQ (1–9). However, other sites within the matrix can also be important producers of superoxide/H2O2 under these conditions (19, 20, 23). Together with site IF, these matrix sites (including the OGDH, BCKDH, and PDH complexes) comprise the NADH/NAD+ isopotential group, i.e. they operate at the relatively low redox potential of the NADH/NAD+ pool. The matrix sites are flavoenzymes. Reduced flavins are good electron donors to oxygen (28), as shown for several isolated flavoenzymes (19, 20, 29, 30). The relative contribution of each site in the NADH/NAD+ isopotential group to superoxide/H2O2 production by isolated mitochondria has not been measured. This may be because they probably all respond to the reduction state of the NADH pool, making it hard to assign individual activities.
The objective of the present study was to measure the relative maximum capacities for superoxide/H2O2 production of the matrix NADH/NAD+-linked enzyme complexes OGDH, BCKDH, PDH, and complex I under optimal conditions in situ in isolated muscle mitochondria. We show that complex I is not the highest capacity matrix superoxide/H2O2-producing enzyme in the presence of rotenone. Instead, the OGDH complex has the greatest capacity followed by the PDH complex and the BCKDH complex. Site IF of complex I has the lowest capacity in this isopotential group.
EXPERIMENTAL PROCEDURES
Animals, Mitochondria, and Reagents
Female Wistar rats (Harlan Laboratories), age 5–8 weeks, were fed chow ad libitum and given free access to water. Mitochondria were isolated from hind limb skeletal muscle at 4 °C in Chappell-Perry buffer (CP1; 100 mm KCl, 50 mm Tris, 2 mm EGTA (pH 7.4 at 4 °C) by standard procedures (31). The animal protocol was approved by the Buck Institute Animal Care and Use Committee (IACUC) in accordance with IACUC standards. All reagents were from Sigma except Amplex UltraRed (Invitrogen) and atpenin A5 (Santa Cruz).
Complex I-deficient mutant (Ndufa1S55A/Y) and wild-type control (Ndufa1+/Y) male mice were used at 24 weeks of age. Mutant mice were developed by Ndufa1S55A allele knock-in at the native locus by homologous recombination on the X chromosome on the 129/Sv genetic background.5 Animals were fed ad libitum and given free access to water. Mitochondria were isolated from hind limb skeletal muscle as above (31), except tissue was disrupted with no more than five strokes in a glass-Teflon homogenizer. Mitochondria were resuspended in CP1, and protein was measured by the biuret method.
Superoxide/H2O2 Production
Rates of superoxide/H2O2 production were measured collectively as rates of H2O2 production, as two superoxide molecules are dismutated by endogenous or exogenous superoxide dismutase to yield one H2O2. H2O2 was detected using horseradish peroxidase and Amplex UltraRed (9). Mitochondria (0.3 mg of protein·ml−1) were suspended at 37 °C in non-phosphorylating medium containing 120 mm KCl, 5 mm Hepes, 5 mm KH2PO4, 2.5 mm MgCl2, 1 mm EGTA, 0.3% (w/v) bovine serum albumin (pH 7.0 at 37 °C), 5 units·ml−1 horseradish peroxidase, 25 units·ml−1 superoxide dismutase, 50 μm Amplex UltraRed, and 1 μg·ml−1 oligomycin. For measurement of H2O2 production from the PDH complex, the medium also contained 1 mm dichloroacetic acid and 450 nm free Ca2+ (achieved by addition of 575 μm total Ca2+, calculated using the program MaxChelator). Reactions were monitored fluorometrically in a Varian Cary Eclipse spectrofluorometer (λexcitation = 560 nm, λemission = 590 nm) with constant stirring and calibrated with known amounts of H2O2 in the presence of all relevant substrates, as some substrates quenched the fluorescence (9).
H2O2 production rates in Fig. 11 were corrected for losses of H2O2 caused by peroxidase activity in the matrix to give a better estimate of actual superoxide/H2O2 production rates. Rates were mathematically corrected to those that would have been observed in these mitochondria after pretreatment with 1-chloro-2,4-dinitrobenzene to deplete glutathione and decrease glutathione peroxidase and peroxiredoxin activity as described (27, 32) using an empirical equation,
where rates are in nmol H2O2·min−1·mg of protein−1.
FIGURE 11.
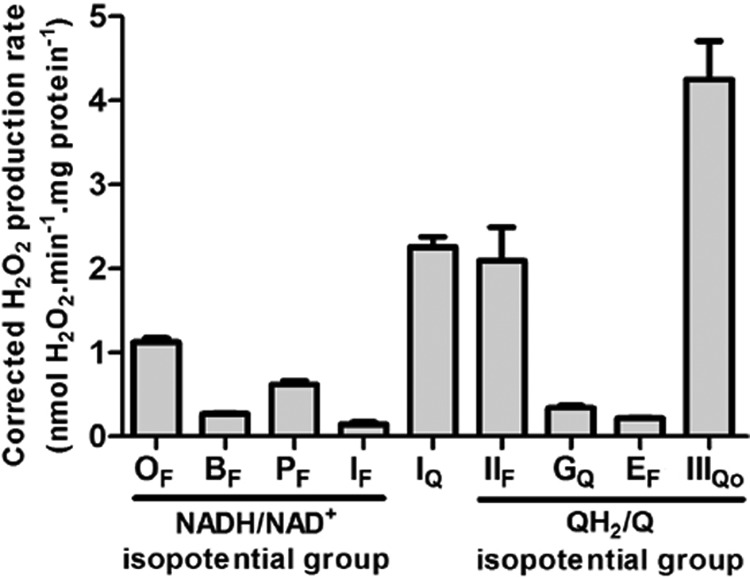
Maximum rates of superoxide/H2O2 production from characterized sites of the mitochondrial respiratory chain. The components of the NADH/NAD+ isopotential group are: OF, flavin site of 2-oxoglutarate dehydrogenase; BF, flavin site of branched-chain 2-oxoacid dehydrogenase; PF, flavin site of pyruvate dehydrogenase; IF, flavin site of complex I. The ubiquinone binding site of complex I (site IQ) is between isopotential groups. The components of the QH2/Q isopotential group are: IIF, flavin site of complex II; GQ, quinone site of mitochondrial glycerol-3-phosphate dehydrogenase; EF, flavin site of the electron transferring flavoprotein/ETF:ubiquinone oxidoreductase system; IIIQo, Qo binding site of complex III. All data were mathematically corrected for matrix peroxidase activity using the 1-chloro-2,4-dinitrobenzene correction described under “Experimental Procedures.” Data for OF, BF, PF, and IF are from Figs. 5C, 7C, 9C, and 2D (plus aspartate/ATP); other sites are replotted from (16). The maximum rates from OF, BF, and PF were corrected by subtracting the rate from site IF at the same NAD(P)H reduction level. Data are the means ± S.E. (n ≥ 3).
NAD(P)H Redox State
Experiments used 0.3 mg of mitochondrial protein·ml−1 at 37 °C in parallel with measurements of H2O2 production in the same non-phosphorylating medium with the same additions. The reduction state of endogenous NAD(P)H was determined by autofluorescence (most of the signal is from NADH bound in the matrix, and NADPH hardly changes in the present experiments, but for full disclosure we call it “NAD(P)H”) using a Shimadzu RF5301-PC at λexcitation = 365 nm, λemission = 450 nm (11, 27). NAD(P)H was assumed to be 0% reduced after 5 min without added substrate and 100% reduced with saturating substrate (e.g. 5 mm malate plus 5 mm glutamate) and 4 μm rotenone. Intermediate values were determined as % reduced NAD(P)H relative to the 0 and 100% values. 3-Methyl-2-oxopentanoate (α-keto-methylvalerate (KMV)) decreased the NAD(P)H signal. To correct for this, 3-methyl-2-oxopentanoate was titrated from 0–20 mm, and the dependence of fluorescence on 3-methyl-2-oxopentanoate concentration was determined. The corrected fluorescence l0 was calculated as l0 = 10(C×slope)×l, where l = observed fluorescence, and C = 3-methyl-2-oxopentanoate concentration.
Permeabilized Mitochondria
Mitochondria were permeabilized with alamethicin according to Grivennikova et al. (33). Intact mitochondria (25–35 mg of protein·ml−1) were diluted 20-fold with medium at 25 °C comprising 0.25 m sucrose, 10 mm Hepes/KOH (pH 7.4), 0.2 mm EDTA, bovine serum albumin (1 mg·ml−1), 2.5 mm MgCl2, and alamethicin (40 μg·ml−1). The suspension was incubated for 5 min, diluted 2.5-fold with the same buffer but ice-cold and lacking MgCl2 and alamethicin, then centrifuged at 30,000 × g for 15 min. The permeabilized mitochondria were suspended in 0.25 m sucrose, 10 mm HEPES (pH 7.4), 0.2 mm EDTA, and 10 mg · ml−1 bovine serum albumin and stored on ice.
Complex I Activity
NADH:quinone oxidoreductase activity was assayed at 30 °C as a decrease of A340 with 100 μm NADH as substrate and 100 μm ubiquinone-1 (Q1) as acceptor in the presence of 1 mm KCN (33) in medium comprising 0.25 m sucrose, 10 mm HEPES (pH 7.4), 0.2 mm potassium EDTA, and permeabilized mitochondria (0.125 mg of protein·ml−1). A340 was measured in an Olis DW2 dual-beam spectrophotometer in split beam mode. The rotenone-sensitive linear rates of NADH oxidation over 60 s were converted to molar units with ϵ = 6.22 mm·cm−1.
Western Blot
To determine the amount of complex I, 0.4 μg of mitochondrial protein was boiled in NU-PAGE loading buffer. Proteins were separated by 4–12% NU-PAGE gradient gel using 1× MES buffer (Invitrogen) and transferred to a nitrocellulose membrane. Anti-complex I 75-kDa subunit (NDUFS1) (Santa Cruz: sc-271510) was used at a 1:1000 dilution. Secondary antibody was horseradish peroxidase-conjugated goat anti-mouse (Bio-Rad) at 1:50,000 dilution. Chemiluminescence was generated with SuperSignal West Pico (Thermo Scientific) and quantified with Image J software (National Institutes of Health).
RESULTS AND DISCUSSION
The aim of this study was to determine the relative maximum capacities of different matrix sites of superoxide/H2O2 production in the NADH/NAD+ isopotential group in intact skeletal muscle mitochondria. Electrons leak from the respiratory chain to generate superoxide or H2O2 at two different redox potentials (Eh): at the isopotential group of redox carriers around the NADH/NAD+ pool at Eh ∼ −280 mV and at the isopotential group of redox carriers around the QH2/Q pool at Eh ∼+20 mV (26, 27, 34) (Fig. 1). At each isopotential group, an important determinant of the rate of superoxide or H2O2 production is the redox state; the more reduced species will generally leak electrons to oxygen at a faster rate.
FIGURE 1.
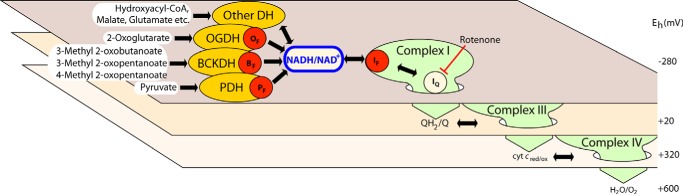
The NADH/NAD+ isopotential group and its component superoxide/H2O2-producing enzymes. The three planes represent different isopotential groups of redox centers in mitochondria. Each group contains multiple redox centers operating at about the same redox potential (Eh): centers around NADH/NAD+ at Eh ∼ −280 mV, around QH2/Q at Eh ∼ +20 mV, and around cytochrome c at Eh ∼ +320 mV (34). The normal flow of electrons from substrate dehydrogenases through NADH and the respiratory complexes of the electron transport chain to oxygen are indicated by the large green arrows dropping down through the isopotential planes. Electrons from NAD-linked substrates enter the NADH/NAD+ pool at Eh ∼ −280 mV through NAD-linked dehydrogenases (DH) including the OGDH, BCKDH, and PDH complexes and flow into complex I (site IF). In the absence of rotenone they then drop down via site IQ to QH2/Q in the next isopotential pool through complex III to cytochrome c and ultimately to their final acceptor, oxygen. In the presence of rotenone (red blunted arrow) other sites of superoxide and H2O2 production are fully oxidized and, therefore, do not leak electrons to O2, and only the sites in the NADH/NAD+ isopotential pool are active. These sites (red dots) are site IF, the flavin of the OGDH complex (OF), the flavin of the BCKDH complex (BF), and the flavin of the PDH complex (PF) (see Ref. 26).
The NADH/NAD+ isopotential group contains several enzymes, but of interest here are the superoxide/H2O2 producing enzymes, which are typically flavoenzymes. Reduced flavins and flavoproteins generate radicals (19, 20, 28). In particular, the flavin sites of complex I (site IF) and the OGDH complex may be important sources of matrix superoxide/H2O2 (19, 20, 30). There is no evidence that the BCKDH complex produces superoxide or H2O2, but it shares with the other 2-oxoacid dehydrogenase complexes a dihydrolipoamide dehydrogenase subunit that is likely a source of superoxide/H2O2 production. Isolated PDH complex generates superoxide/H2O2 (20, 23), but the physiological relevance is uncertain. Other members of the NADH/NAD+ isopotential group, such as malate dehydrogenase and isocitrate dehydrogenase, have not been shown to produce superoxide or H2O2 (35). Here we describe conditions under which complex I and the OGDH, BCKDH, and PDH complexes are largely distinct from each other, analyze the conditions required for maximum rates in skeletal muscle mitochondria, and compare the maximum rates of superoxide/H2O2 production by each complex.
Complex I Flavin (Site IF)
Complex I (NADH-ubiquinone oxidoreductase) oxidizes NADH to NAD+ and reduces ubiquinone (Q) to ubiquinol (QH2). During this process two electrons are transferred through multiple redox centers, and four protons are pumped from the matrix to the intermembrane space. The enzyme is fully reversible, either oxidizing NADH and reducing Q (and pumping protons) in the forward reaction or oxidizing QH2 and reducing NAD+ in the reverse reaction driven by protonmotive force (34).
Complex I produces superoxide at two different internal sites: the NADH-oxidizing flavin site, IF, and the ubiquinone-reducing site, IQ (11, 36–40), although site IQ activity has yet to be measured in the reconstituted complex (41). The production of superoxide from site IF occurs when electrons leak to O2 from the fully reduced flavin, FMNH2 (30). As electrons can only be transferred to oxygen when the flavin is reduced, it is likely that the steady-state concentration of reduced FMNH2 is an important determinant of the rate of superoxide production from this site. Similarly, the steady-state reduction level of FMN depends on the steady-state redox state of its reductant, NADH (i.e. the NADH/NAD+ ratio). Therefore, the steady-state reduction level of NADH/NAD+ in situ predicts the rate of superoxide production from site IF (27). Indeed, as the NAD(P)H/NAD(P)+ ratio increases, the rate of superoxide production attributed to site IF in complex I increases steeply (26, 27, 32).
The relationship between superoxide/H2O2 production from site IF and the reduction state of NAD(P)H can be revealed by titrating mitochondria with a substrate that reduces NAD+ in the presence of rotenone to prevent its rapid reoxidation (squares, Fig. 2D). Malate was chosen here because malate dehydrogenase is thought to reduce NAD+ directly without generating superoxide or H2O2 (35) or providing sufficient Krebs cycle intermediates to support superoxide/H2O2 production from other sites. To test the robustness of the relationship between superoxide produced from site IF and NAD(P)H reduction state, we added physiologically relevant components such as amino acids and nucleotides to the medium. In general, this did not change the established relationship between the observed rate of H2O2 production and the NAD(P)H reduction level (squares, Fig. 2D). However, the addition of two biologically relevant compounds, ATP and aspartate, decreased the observed rate of mitochondrial H2O2 production (Fig. 2A) without changing the reduction state of NAD(P)H (Fig. 2D) or the NADH:Q oxidoreductase activity of complex I (Fig. 2B).
FIGURE 2.

Superoxide/H2O2 production with malate as substrate; complex I and the OGDH complex. A, Amplex UltraRed traces illustrate that H2O2 production rates with 5 mm malate as substrate were decreased by the addition of 2.5 mm ATP and 1.5 mm aspartate. Mitochondria were suspended in non-phosphorylating medium, and 4 μm rotenone was added where indicated. A representative trace is shown; numbers indicate mean rates in pmol of H2O2·min−1·mg of protein−1 from four replicates. a.u., arbitrary units. B, complex I activity measured in alamethicin-permeabilized mitochondria as the rotenone-sensitive NADH:Q-oxidoreductase activity at 30 °C was unaffected by addition of ATP plus aspartate. C, schematic to illustrate that in isolated mitochondria ATP and aspartate may indirectly inhibit the OGDH complex by decreasing its substrates 2-oxoglutarate (through aspartate aminotransferase (AAT)) and CoA (through inhibition of succinate thiokinase), increasing its inhibitory product succinyl CoA (through inhibition of succinate thiokinase) and through direct inhibitory effects of ATP on the enzyme. D, dependence of superoxide/H2O2 production on the redox state of NAD(P)H (measured by autofluorescence). Malate was titrated from 20 μm–5 mm in the presence of 4 μm rotenone either in the presence or absence of 1.5 mm aspartate and 2.5 mm ATP. Data in A are representative traces. Data in B and D are the means ± S.E. (n = 3).
ATP and aspartate may affect other matrix enzymes, for example the OGDH complex (Fig. 2C). ATP is a negative regulator of many Krebs cycle enzymes, including OGDH (42, 43). It may also product-inhibit succinate thiokinase, which generates succinate and ATP from succinyl-CoA (44); thus, ATP will lower free CoA, a substrate for the OGDH complex, and raise succinyl-CoA, a potent product inhibitor of the complex (45). Aspartate will drive the aspartate aminotransferase reaction, aspartate + 2-oxoglutarate ↔ oxaloacetate + glutamate, to remove 2-oxoglutarate. To check this, we inhibited aminotransferases using aminooxyacetate, which eliminated the effect of aspartate addition (not shown). Thus, the effects of aspartate and ATP on mitochondrial H2O2 production point to 2-oxoglutarate and CoA-dependent production of superoxide by the OGDH complex in the physiological (forward) reaction (19). The non-additivity of the effects of aspartate and ATP (Fig. 2A) supports this conclusion, as they each affect the same process (Fig. 2C).
We found no other agents that further decreased the rate of H2O2 production driven by oxidation of malate in the presence of rotenone, so we assume that the residual rate in the presence of ATP and aspartate (Fig. 2D) was solely attributable to site IF (any NADH-dependent contribution from dihydrolipoamide dehydrogenase (18) under these conditions would also be included in our measurements of H2O2 production by site IF). Therefore, the lower set of points in Fig. 2D can be used to predict the rate from site IF at any measured reduction state of NAD(P)H in situ.
The maximum observed rates of H2O2 production from site IF in muscle mitochondria when NAD+ is highly reduced occur when the size of the NADH+NAD+ pool and the concentrations of other effectors are set by the system. These rates may be very different from the maximum rates achievable with the isolated enzyme (30) or in submitochondrial particles (46, 47), where the concentrations of effectors can be independently optimized. The same argument applies to the other sites analyzed below. When comparing the capacities of different sites in situ, the maximum rates in intact mitochondria are more relevant.
Based on the logic outlined above, we propose that the effects of ATP and aspartate on the rate of H2O2 production with malate as substrate were mediated by effects on the OGDH complex, not direct effects on complex I. Furthermore, the OGDH complex may often be the predominant source of superoxide/H2O2 when classical complex I substrate mixes are used.
2-Oxoacid Dehydrogenase Complexes
The 2-oxoacid dehydrogenase complexes catalyze the oxidation of 2-oxoacids (2-oxoglutarate, branched-chain 2-oxoacids, or pyruvate) to yield the corresponding acyl-CoAs and NADH. Multiple copies of three enzymatic components are organized into complexes (48). The first reaction is the irreversible decarboxylation of the 2-oxoacid, catalyzed by a specific 2-oxoacid dehydrogenase (E1) that requires the cofactor thiamine pyrophosphate. The second reaction is catalyzed by dihydrolipoamide acyltransferase (E2) and requires CoA to generate an acyl-CoA. The third reaction is catalyzed by dihydrolipoamide dehydrogenase (E3) and requires FAD+ and NAD+ to reoxidize the dihydrolipoamide. E1 and E2 are unique to the individual complexes, but E3 is the same gene product in each complex and catalyzes an identical reaction (49); it is also present in the glycine cleavage system. The covalently attached lipoate group in E2 connects the three active sites of E1, E2, and E3 and channels substrates through the complexes. E2 forms the catalytic core and establishes the structural foundation for the complex, as both E1 and E3 bind to it.
2-Oxoglutarate Dehydrogenase Complex
The OGDH complex catalyzes the conversion of 2-oxoglutarate to succinyl-CoA in the Krebs cycle. The mammalian enzyme is regulated by ATP, ADP, calcium, and substrate availability. It is inhibited by its products, succinyl-CoA and NADH (42, 43).
The OGDH complex is an important source of reactive oxygen species in the matrix (19–21, 50, 51). It is a major source of H2O2 in isolated brain mitochondria at high NADH/NAD+ (20, 21, 52) and of superoxide in neurons during glutamate excitotoxicity (51). The E3 component of the purified enzyme contains the redox-active flavin that reduces oxygen (53, 54) and generates substantial superoxide/H2O2 when NAD+ is limiting (19–21). E3 is abundant in skeletal muscle mitochondria (55), and the midpoint potential of the flavin is sufficiently negative (∼−280 mV) to make it a good electron donor to oxygen (56).
When E3 functions within the OGDH or PDH complexes, the reduced lipoyl residue may equilibrate with the FADH* semiquinone formed by 1e− oxidation of FADH2 by O2. Because of this, formation of superoxide by E3 is associated with generation of lipoate thiyl radicals (19, 50) (Fig. 3). This side reaction may occur when FAD is reduced in either the forward reaction (from 2-oxoglutarate oxidation in the presence of CoA leading to reduction of the dihydrolipoyl residue) or the reverse reaction (from NADH oxidation). Efficient addition of O2 to thiyl radicals has not been observed (57), but thiyl radicals are very reactive species, which in the case of the OGDH and PDH complexes, are more damaging than superoxide/H2O2 (19). Intrinsic thiyl radical formation underlies an important regulatory mechanism in the OGDH and PDH complexes, irreversibly inactivating the corresponding E1s when NAD+ is lacking (19, 50). Inactivation is favored by particular matrix environments, e.g. the reduced state of NADH, and depends on thioredoxin and endogenous thiol-disulfide pools. The isolated E1 components of the OGDH and PDH complexes provide still other intermediates to react with oxygen, supposedly generating either H2O2 or peracids (58), but occurrence of this side reaction in the native complexes has not been shown.
FIGURE 3.
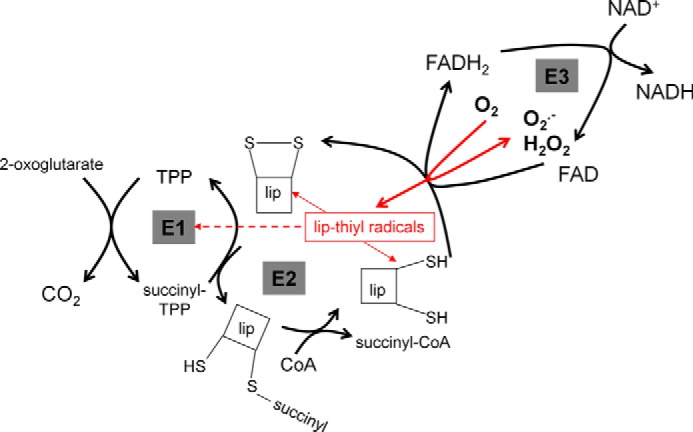
Generation of superoxide and H2O2 by the 2-oxoglutarate dehydrogenase complex. The complex contains multiple copies of the three major components: thiamin pyrophosphate (TPP)-dependent E1, E2, which contains the lipoyl group (lip), and E3, which contains FAD. Side reactions involving the E3-bound FAD and the E2-bound lipoyl residues when NAD is limiting are shown in red. Oxidation of FADH2 by O2 can generate superoxide or H2O2 (19–21). When it generates superoxide, the resulting FADH* can reduce lipoyl residues to thiyl radicals (19, 50), which may inactivate E1 (dashed line) or dismutate (arrows to the reduced and oxidized lipoyl species).
In our experiments (Fig. 4, A and B), the observed H2O2 production with 2-oxoglutarate in skeletal muscle mitochondria was strongly stimulated by ADP. This probably occurred by two mechanisms, (a) lowering the Kd of OGDH for its substrate and (b) providing substrate for succinate thiokinase, which regenerates CoA, the substrate of the OGDH complex, and simultaneously removes the potent inhibitor succinyl-CoA (42, 43, 45).
FIGURE 4.

The properties of superoxide/H2O2 production by the OGDH complex. A, typical Amplex UltraRed traces indicate that mitochondria suspended in non-phosphorylating medium with 2.5 mm 2-oxoglutarate (2-OG) as substrate generated H2O2 at relatively low rates in the absence of the positive regulator ADP. Rates were greatly enhanced by the addition of 4 μm rotenone and were dependent on the ADP concentration. a.u., arbitrary units. B, mean rates of H2O2 production with 2.5 mm 2-oxoglutarate and 4 μm rotenone in the presence of either 2.5 mm ATP or 2.5 mm ADP. Data are the mean ± S.E. (n = 3). C, Effect of 1 mm malonate on the rates of H2O2 production with 2.5 mm 2-oxoglutarate as substrate in the presence of 2.5 mm ADP. D, effect of 2 mm succinyl phosphonate on H2O2 production rates in intact and alamethicin-permeabilized mitochondria. In the permeabilized mitochondria, 50 μm 2-oxoglutarate was oxidized in the presence of 100 μm CoA. In the intact mitochondria, 50 μm 2-oxoglutarate was oxidized in the presence of 2.5 mm ADP and 4 μm rotenone. E, Amplex UltraRed traces show the effect of different concentrations of KMV in the presence of 2.5 mm 2-oxoglutarate and 2.5 mm ADP. F, mean rates of H2O2 production when OGDH was inhibited by increasing concentrations of 2-methyl-3-oxopentanoate in the presence of 2.5 mm 2-oxoglutarate and 2.5 mm ADP. Data are the means ± S.E. (n = 3). Panels A, C, D, and E show representative traces; additions are indicated by arrows; numbers indicate mean rates in pmol of H2O2·min−1·mg of protein−1 (n ≥ 3).
To test whether the observed H2O2 production with 2-oxoglutarate was downstream of the OGDH complex, we added malonate, a competitive inhibitor of succinate dehydrogenase (59). However, malonate also inhibits OGDH by acting at the regulatory site responsible for activation by 2-oxoglutarate (60, 61) and may affect 2-oxoglutarate distribution across the mitochondrial inner membrane. Malonate completely inhibits superoxide production by succinate dehydrogenase when succinate concentration is low; inhibition is effectively instantaneous (10). However, with 2-oxoglutarate as substrate, malonate caused a time-dependent inhibition of H2O2 production. As shown in Fig. 4C, the initial rate (first 10–20 s after rotenone addition) was minimally affected by malonate, but strong inhibition developed over the next few minutes. The time dependence of malonate inhibition, not observed in succinate-dependent H2O2 production, suggests that the malonate effect was on the catalytically slow conformational transition involved in substrate-dependent activation of OGDH rather than by competitive inhibition of succinate dehydrogenase.
A good way to test the hypothesis that the OGDH complex is responsible for the majority of the H2O2 production observed when 2-oxoglutarate or malate is oxidized would be to inhibit OGDH. Substrate analogs such as succinyl phosphonate are tightly bound and highly specific competitive inhibitors of OGDH (62–65). Unfortunately, succinyl phosphonate was a poor inhibitor of the OGDH complex in mitochondria from rat skeletal muscle compared with those from liver, brain, and kidney (65) (Fig. 4D). In alamethicin-permeabilized skeletal muscle mitochondria, succinyl phosphonate strongly inhibited 2-oxoglutarate-dependent production of superoxide/H2O2 (Fig. 4D), showing that poor transport of succinyl phosphonate explained its weak action in intact rat skeletal muscle mitochondria. Branched-chain 2-oxoacids at millimolar concentrations are known inhibitors of OGDH (66, 67). Fig. 4, E and F, show that 20 mm KMV inhibited OGDH-dependent H2O2 production >80%. However, even at high concentrations 3-methyl-2-oxopentanoate did not fully inhibit all H2O2 generation because it was also a substrate for H2O2 production by BCKDH (see below).
The production of superoxide/H2O2 by NAD+-linked oxidoreductases is expected to depend on the reduction state of the NADH/NAD+ pool (11, 19, 20, 26, 27). As discussed above, the reversibility of the complex I-catalyzed reaction implies that NADH/NAD+ should be close to equilibrium with the superoxide-producing flavin center of complex I. However, the increased matrix NADH/NAD+ ratio should also increase the rate of superoxide/H2O2 production from the OGDH complex because this side reaction is stimulated by the lack of the terminal substrate NAD+ (19, 50).
Fig. 5, A and B, shows the dependence of the rate of H2O2 production and level of NAD(P)H reduction on 2-oxoglutarate concentration. Combining these data revealed a steep relationship between the H2O2 production rate and the reduction state of NAD(P)H as 2-oxoglutarate concentration was varied (squares, Fig. 5C). Importantly, with malate as substrate (and ATP and aspartate present to inhibit superoxide/H2O2 production by the OGDH complex and reveal that from site IF), there was a different relationship between H2O2 production and NAD(P)H reduction (circles, Fig. 5C). This indicated that (a) as NAD(P) became >∼80% reduced, the rate of superoxide/H2O2 production increased steeply from both the OGDH complex and site IF, but with very different slopes, and (b) the OGDH complex produced superoxide/H2O2 much more slowly using electrons from NADH than in the forward reaction from 2-oxoglutarate. As mentioned above, the isolated E3 component of the OGDH complex readily oxidizes NADH and reduces the flavin to generate superoxide/H2O2 at relatively high rates (20, 21). However, within the native complex, the 2-oxoglutarate-dependent rate of superoxide/H2O2 production is faster than the NADH-dependent rate (68). In our experiments when the NADH pool was reduced in the absence of 2-oxoglutarate (circles, Fig. 5C), superoxide/H2O2 was produced at a much lower rate than with 2-oxoglutarate (squares, Fig. 5C), indicating that superoxide/H2O2 production by the OGDH complex in isolated mitochondria occurred at high rates only in the forward reaction in the presence of 2-oxoglutarate.
FIGURE 5.
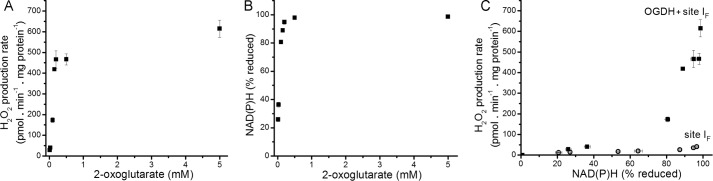
Relationship between the observed rate of H2O2 production by the OGDH complex and the reduction state of NAD(P)H. A, dependence of the rate of H2O2 production on 2-oxoglutarate concentration in non-phosphorylating medium in the presence of 2.5 mm ADP after the addition of 4 μm rotenone. B, dependence of %NAD(P)H reduction on 2-oxoglutarate concentration in non-phosphorylating medium in the presence of 2.5 mm ADP after the addition of rotenone (100% reduction was subsequently established by addition of 5 mm malate). C, relationship between the observed rate of H2O2 production from the OGDH complex (plus site IF) and NAD(P)H reduction state (filled squares), obtained by combining panels A and B; open symbols are the replotted data for site IF alone from Fig. 2D. Where not visible, error bars are contained within the points. Data are the means ± S.E. (n = 4).
Branched-chain 2-Oxoacid Dehydrogenase Complex
The BCKDH complex has strong control over oxidation of the branched-chain amino acids valine, leucine, and isoleucine. These amino acids are converted by specific branched-chain amino acid aminotransferases to their respective branched-chain 2-oxoacids, 3-methyl-2-oxobutanoate (KIV), 4-methyl-2-oxopentanoate (KIC), and KMV, then oxidized by the BCKDH complex to an acyl-CoA, CO2, and NADH. The complex is phosphorylated and inactivated by BCKDH kinase and dephosphorylated and activated by BCKDH phosphatase, both bound to the E2 core. The kinase belongs to the same family as pyruvate dehydrogenase kinase (69) and is regulated allosterically by inhibitors such as 3-methyl-2-oxobutanoate (KIV) (70) and at the expression level (71).
Fig. 6 shows H2O2 production during oxidation of branched-chain 2-oxoacids. The addition of 3-methyl-2-oxopentanoate in the presence of antimycin A stimulated rapid H2O2 production (Fig. 6A). Under these conditions, several potential sites of superoxide/H2O2 production may be active, including complexes I, II, and III. The rate was decreased by myxothiazol (inhibits site Qo of complex III, green bar), indicating that site IIIQo was recruited in this condition. It was further decreased by atpenin A5 (red bar), an inhibitor of the quinone-binding site of complex II, indicating that 3-methyl-2-oxopentanoate induces superoxide/H2O2 production from complex II in the reverse reaction. Both of these effects are driven by reduction of the ubiquinone (Q) pool. A surrogate measure of this is the steady-state reduction level of cytochrome b566 (14, 27). Fig. 6B shows that the addition of 3-methyl-2-oxopentanoate in the presence of antimycin reduced cytochrome b and the Q pool. The addition of rotenone prevented the flow of electrons from 3-methyl-2-oxopentanoate into the Q pool (Fig. 6B), eliminating the contributions from site IIIQo and site IIF and revealing the maximum rate of superoxide/H2O2 production caused by oxidation of 3-methyl-2-oxopentanoate at the level of the NADH isopotential group (Fig. 6A, white and blue bars), i.e. by the BCKDH complex plus site IF.
FIGURE 6.

The branched-chain 2-oxoacid dehydrogenase complex produces superoxide/H2O2. A, mean rates of H2O2 production with 20 mm KMV in non-phosphorylating medium in the presence of 2 μm antimycin A (beige bar), 2 μm antimycin A plus 2 μm myxothiazol (green bar), 2 μm antimycin A, 2 μm myxothiazol and 2 μm atpenin A5 (red bar), 2 μm antimycin A plus 4 μm rotenone (white bar), and 4 μm rotenone (blue bar). Data are the means ± S.E. (n = 3). B, cytochrome b566 reduction state in non-phosphorylating medium in the presence of 20 mm 3-methyl-2-oxopentanoate plus 4 μm rotenone or plus 2 μm antimycin A. C, typical Amplex UltraRed traces indicate that mitochondria suspended in non-phosphorylating medium with different concentrations of 3-methyl-2-oxopentanoate as substrate generated H2O2. Rates were greatly enhanced by the addition of 4 μm rotenone. a.u., arbitrary units. Arrows indicate additions; numbers indicate mean rates in pmol of H2O2·min−1·mg of protein−1 (n ≥ 3). D, mean rates of H2O2 production in non-phosphorylating medium in the presence of the branched-chain 2-oxoacids KMV or KIC at different concentrations and with 20 mm 3-methyl-2-oxopentanoate in the presence of 4 μm rotenone and 450 nm free Ca2+ plus 1 mm dichloroacetate (DCA), 1 mm isoleucine, 1 mm leucine, 1 mm carnitine, or 2.5 mm ADP. Data are the means ± S.E. (n = 3).
The rate of superoxide/H2O2 production from the BCKDH complex plus site IF was titrated with KMV (Fig. 6C) and 4-methyl-2-oxopentanoate (KIC). The highest rates were obtained with 20 mm 3-methyl-2-oxopentanoate (Fig. 6D). We tested whether they were further stimulated by putative inhibitors of BCKDH kinase. However, the addition of leucine or isoleucine (72) or dichloroacetate plus CaCl2 did not increase H2O2 production nor did carnitine, which should recycle inhibitory acyl-CoA products (73) and regenerate CoA, a required substrate of BCKDH (Fig. 6D).
In the presence of oligomycin, ADP inhibited the rate of superoxide/H2O2 production with 3-methyl-2-oxopentanoate as substrate (Fig. 6D). This is the opposite of its effect with 2-oxoglutarate as substrate (Fig. 4), supporting the hypothesis that the observed H2O2 production with 3-methyl-2-oxopentanoate was largely from the BCKDH complex and not a residual activity of the OGDH complex using 3-methyl-2-oxopentanoate as a weak substrate.
Fig. 7, A and B, show the dependence of the rate of H2O2 production and level of NAD(P)H reduction on 3-methyl-2-oxopentanoate concentration. Fig. 7C shows that there was a steep relationship between superoxide/H2O2 production by the BCKDH complex (plus site IF), and NAD(P)H level as 3-methyl-2-oxopentanoate was varied. NAD(P) was maximally only 40% reduced, perhaps because of kinetic limitation by the activity of the BCKDH complex, which is inhibited by NADH (73). However, the important observation is that there was a different relationship between H2O2 production and NAD(P)H in the presence of 3-methyl-2-oxopentanoate (squares, Fig. 7C) and in the presence of malate plus aspartate and ATP (circles, Fig. 7C), indicating that when 3-methyl-2-oxopentanoate is oxidized a different site of superoxide/H2O2 production is recruited, most likely the BCKDH complex. Fig. 7C shows that the contribution of site IF to the total observed H2O2 production with 20 mm 3-methyl-2-oxopentanoate in these conditions was <20%. It also indicates that the enzyme only produces superoxide/H2O2 at high rates in the forward reaction (during 3-methyl-2-oxopentanoate oxidation), as the observed rates of H2O2 production at the same reduction level of NAD(P)H were always higher in the presence of 3-methyl-2-oxopentanoate.
FIGURE 7.
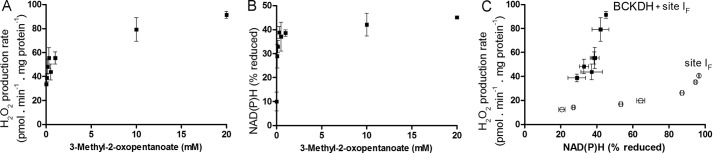
Relationship between the observed rate of H2O2 production by the BCKDH complex and the reduction state of NAD(P)H. A, dependence of the rate of H2O2 production on KMV concentration in non-phosphorylating medium after the addition of 4 μm rotenone. B, dependence of %NAD(P)H reduction on 3-methyl-2-oxopentanoate concentration in non-phosphorylating medium after the addition of rotenone (100% reduction was subsequently established by the addition of 5 mm malate and glutamate). C, relationship between the observed rate of H2O2 production from the BCKDH complex (plus site IF) and NAD(P)H reduction state (filled squares) obtained by combining panels A and B; the open symbols are the replotted data for site IF alone from Fig. 2D. Where not visible, error bars are contained within the points. Data are the means ± S.E. (n = 4).
Pyruvate Dehydrogenase Complex
Mammalian PDH complex is mechanistically similar to the other 2-oxoacid dehydrogenases. Its regulation is similar to that of the BCKDH complex. It is rapidly and reversibly controlled by two mechanisms: end-product inhibition and enzyme phosphorylation/dephosphorylation by specific kinases and phosphatases.
In experiments to assess superoxide/H2O2 production by the PDH complex, a primary concern was removal of inhibitory acetyl-CoA, which competes potently with CoA (Ki = 5–10 μm) (74). In isolated mitochondria, acetyl-CoA can be removed by (a) the addition of malate to generate oxaloacetate, which will condense with acetyl-CoA to generate citrate through citrate synthase, and (b) the addition of carnitine to convert acetyl-CoA to acetylcarnitine, catalyzed by carnitine acetyltransferase (Fig. 8A). Removal of acetyl-CoA by either pathway should promote flux through the PDH complex, but the downstream carbon flows will be different. The use of malate as co-substrate with pyruvate promotes Krebs cycle carbon flux, as evident from the decrease in H2O2 production when ATP and aspartate were added to suppress that flux (Fig. 8B). However, when carnitine was co-substrate with pyruvate, H2O2 production was not appreciably affected by ATP and aspartate (Fig. 8B). In this system Krebs cycle carbon flows were likely limited as acetylcarnitine was exported from the matrix in exchange for carnitine on the carnitine-acylcarnitine translocase (75, 76).
FIGURE 8.

Experimental design for measuring superoxide/H2O2 production from the pyruvate dehydrogenase complex. A, carbon flows with pyruvate plus carnitine or pyruvate plus malate as substrates. MDH, malate dehydrogenase. B, H2O2 production in non-phosphorylating medium with 2.5 mm pyruvate plus 5 mm malate or 2.5 mm pyruvate plus 5 mm carnitine as substrate. The contribution from other sites during oxidation of these substrate pairs was assessed by the sensitivity of the rates to 2.5 mm ATP and 1.5 mm aspartate (ASP). 4 μm rotenone was added where indicated. a.u., arbitrary units. C, Amplex UltraRed traces in non-phosphorylating medium show that at a low pyruvate concentration (25 μm) the rates of H2O2 production were low. The presence of 450 nm free Ca2+ and 1 mm dichloroacetate (DCA) increased the observed rate at low pyruvate concentration. D, rates of H2O2 production in non-phosphorylating medium at low (25 μm) and high (2.5 mm) pyruvate concentrations in the presence and absence of 450 nm free Ca2+ and 1 mm dichloroacetate. Data are the means ± S.E. (n = 3). Panels B and C show representative traces; numbers indicate mean rates in pmol of H2O2·min−1·mg of protein−1 (n = 3).
A second concern was the phosphorylation state of the enzyme (77). Three serines on the α-subunits of E1 may be phosphorylated by four pyruvate dehydrogenase kinases (PDK1–4) (78), inactivating PDH (79). Dephosphorylation is accomplished by two pyruvate dehydrogenase phosphate phosphatases (PDP1 and PDP2). The activities of pyruvate dehydrogenase kinase and dehydrogenase phosphate phosphatase are highly regulated. Pyruvate dehydrogenase kinase is inhibited by high concentrations of the E1 substrate pyruvate and activated by acetyl-CoA and NADH, products of both the PDH complex and β-oxidation (80). The non-metabolizable pyruvate analog dichloroacetate inhibits the kinase and thereby activates PDH (81). PDP1, the dominant isoform in muscle, requires Mg2+ and is stimulated by Ca2+ (82) because PDP1 binding to E2 requires Ca2+ (83), and Ca2+ decreases the Km of PDP1 for Mg2+ (84).
In skeletal muscle mitochondria, H2O2 production after the addition of a low concentration of pyruvate (25 μm) was increased >3-fold by dichloroacetate and CaCl2 (Fig. 8, C and D). However, at high pyruvate concentrations (2.5 mm), dichloroacetate and CaCl2 did not affect the rate (Fig. 8D). These data indicate that at low pyruvate concentrations pyruvate dehydrogenase kinase phosphorylated and inactivated PDH.
Based on these observations, the experimental conditions chosen for assay of superoxide/H2O2 production by the PDH complex were a high concentration of pyruvate in the presence of dichloroacetate and CaCl2. The H2O2 production rate (Fig. 9A) and the steady-state reduction level of NAD(P)H (Fig. 9B) were titrated in parallel with carnitine to achieve different steady-state concentrations of inhibitory acetyl-CoA. The data were combined to reveal the relationship between the rate of H2O2 production from the PDH complex (plus site IF) and NAD(P)H reduction state as PDH activity was altered (squares, Fig. 9C). Similar to the other 2-oxoacid dehydrogenase complexes described above, the rate of superoxide/H2O2 production from the PDH complex increased as the NAD(P)H/NAD(P)+ pool was reduced, and PDH was activated by removal of acetyl CoA. As with the OGDH complex, the increase in PDH complex-dependent superoxide/H2O2 production at higher NADH/NAD+ was ascribed to the lack of the terminal substrate NAD+, as this lack stimulates superoxide production by both PDH and OGDH complexes (19). In contrast, increasing the reduction state of NAD(P) in the absence of pyruvate did not cause rapid superoxide/H2O2 production (circles, Fig. 9C). These observations indicate that superoxide/H2O2 production upon oxidation of NADH by the E3 component of the PDH complex was not favored in these intact mitochondria. Thus, superoxide/H2O2 production by the PDH complex in isolated mitochondria occurs at high rates only in the forward reaction (during pyruvate oxidation). The E3 subunit is also present in the glycine cleavage system. However, there was no additional H2O2 production when glycine was added to rotenone-inhibited mitochondria (<5 pmol of H2O2·min−1·mg of protein−1) (not shown).
FIGURE 9.
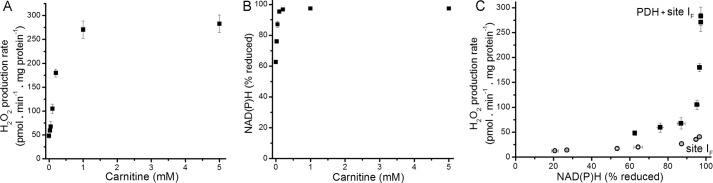
Relationship between the observed rate of H2O2 production by the PDH complex and the reduction state of NAD(P)H. A, dependence of the rate of H2O2 production on carnitine concentration in non-phosphorylating medium in the presence of 2.5 mm pyruvate and 4 μm rotenone. B, dependence of %NAD(P)H reduction on carnitine concentration in non-phosphorylating medium after the addition of rotenone (100% reduction was subsequently established by the addition of 5 mm malate). C, relationship between the observed rate of H2O2 production from the PDH complex (plus site IF) and the NAD(P)H reduction state (filled squares), obtained by combining panels A and B; the open symbols are the data for site IF alone from Fig. 2D. Where not visible, error bars are contained within the points. Data are the means ± S.E. (n = 3).
Effect of Decreasing the Amount of Complex I on Superoxide/H2O2 Production
The MWFE subunit of complex I encoded by the X-linked Ndufa1 gene is essential for complex I assembly (85). Conversion of the evolutionarily conserved serine 55 to alanine (S55A) decreases complex I assembly/activity in mammalian cells (86). Fig. 10A confirms that mitochondria from mutant mice carrying the S55A substitution in the MWFE protein had lower levels of complex I.
FIGURE 10.
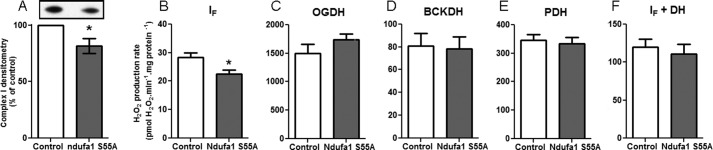
Superoxide/H2O2 production by mitochondria from complex I-deficient Ndufa1S55A/Y mice. A, densitometry of Western blots of mitochondria from wild type and mutant mice probed for the 75-kDa NDUFS1 subunit of complex I (inset, representative Western blot. Left, control; right, NdufaS55A/Y). B–F, superoxide/H2O2 generation in non-phosphorylating medium after the addition of 4 μm rotenone by mitochondria from control (white bars) or complex I deficient (NdufaS55A/Y) mice (gray bars) in the presence of 5 mm malate, 2.5 mm ATP, and 1.5 mm aspartate (B), 2.5 mm 2-oxoglutarate and 2.5 mm ADP (C), 20 mm 3-methyl-2-oxopentanoate (D), 2.5 mm pyruvate and 5 mm carnitine (E), and 5 mm malate (F). DH, NADH dehydrogenases. Data in A are expressed as % of paired control from five independent paired skeletal muscle mitochondrial preparations; error bars indicate 95% confidence limits; *, p < 0.05 by 95% confidence interval. Data in B–F are the means ± S.E. (n = 5); *, p < 0.05 by Student's t test.
If the assignment of superoxide/H2O2 production in the presence of malate, aspartate, and ATP (circles, Fig. 2D) to site IF of complex I is correct, this production should decrease proportionally to the decrease in complex I in mitochondria from the mutant mice; Fig. 10B shows that this was indeed the case. Conversely, if we have correctly assigned superoxide/H2O2 production with 2-oxoacid substrates (Figs. 5C, 7C, and 9C) to the appropriate 2-oxoacid dehydrogenase complex, this production should not be affected by a decrease in complex I; Fig. 10, C, D, and E, shows that no decrease in H2O2 production was observed with these substrates in the mutant (from Fig. 10B the contribution of site IF would be only 0.5, 9, and 2% respectively, too small to register). Similarly, we assign superoxide/H2O2 production with malate as substrate (squares, Fig. 2D) to the OGDH complex plus site IF; Fig. 10F shows that no decrease was observed in the mutant (the contribution of site IF would be only 6%). These observations strongly support our assignments of superoxide/H2O2 production to the specific sites mentioned.
Superoxide production from site IQ of complex I should also decrease proportionally to the decrease in complex I. We did not observe the predicted decrease when site IQ was driven by succinate oxidation, but this can be explained by measured compensatory increases in the amount of complex II and the magnitude of the proton-motive force during succinate oxidation (not shown).
Mice lacking apoptosis-inducing factor also have compromised complex I assembly, and superoxide/H2O2 generation from site IQ was decreased in brain mitochondria from these mice (87). However, H2O2 production driven by glutamate plus malate in the presence of rotenone was not, suggesting that “the major source of superoxide/H2O2 in this model may lie outside complex I” (see Ref. 87). Our results (Fig. 10) confirm these observations in a different model and support the conclusion that the OGHD complex is the major site of superoxide/H2O2 generation with glutamate plus malate as substrate in the presence of rotenone.
Maximum Capacities of Specific Sites of Superoxide/H2O2 Production in Intact Skeletal Muscle Mitochondria
Fig. 11 puts the in situ capacities of the superoxide/H2O2 producing sites of the NADH/NAD+ isopotential group in context. It shows the maximum rates from the OGDH, BCKDH, and PDH complexes, site IF, site IQ, site IIF, mitochondrial glycerol-3-phosphate dehydrogenase, electron transferring flavoprotein/ETF:ubiquinone oxidoreductase, and site IIIQo (all after correction for matrix peroxidase activity using the correction described under “Experimental Procedures”). For this correction, we assumed that 100% of the superoxide/H2O2 from site IF and the 2-oxoacid dehydrogenase complexes was superoxide or H2O2 produced to the matrix, as the rates were insensitive to the addition of exogenous superoxide dismutase (not shown). Of the nine sites examined, site IF had the lowest maximum capacity in skeletal muscle mitochondria. The maximum rate of superoxide/H2O2 production by the OGDH complex was ∼8 times higher than the maximum rate of superoxide production from site IF; the maximum rate from the PDH complex was more than four times higher, and the maximum rate from the BCKDH complex was almost twice the rate from site IF. Although not as high as the maximal rates from the producers with the greatest capacities, IQ, IIF, and IIIQo, the rates from the OGDH, PDH, and BCKDH complexes were a much higher proportion of the rate from the NADH/NAD+ isopotential group in the presence of rotenone than we and many others originally envisaged (1–9, 25–27).
Conclusions
When electron transport through complex I is inhibited by rotenone, the NADH/NAD+ pool becomes reduced, and the enzymes in the isopotential group around this pool, specifically the OGDH, BCKDH, and PDH complexes and site IF, generate more superoxide and/or H2O2. The contributions of the different sites depend on the conditions. Because site IF is close to equilibrium with the matrix NADH/NAD+ pool, its rate of superoxide production is a simple function of the redox state of the NADH/NAD+, described by the lower set of points in Fig. 2D.
Although they tend to become more reduced and generate superoxide/H2O2 at higher rates in the presence of their specific substrates when NADH is high, the redox centers that produce superoxide/H2O2 in the OGDH, BCKDH, and PDH complexes are not in equilibrium with the NADH/NAD+ pool. This is clear from Figs. 5C, 7C, and 9C, which show that the OGDH, BCKDH, and PDH complexes do not generate superoxide/H2O2 at high rates in the absence of 2-oxoglutarate, 3-methyl-2-oxopentanoate, or pyruvate, respectively (lower sets of points) even when NADH/NAD+ is high. Therefore, superoxide/H2O2 production at the OGDH (or BCKDH or PDH) complex does not have a unique relationship to NADH/NAD+; instead it also depends on the concentration of 2-oxoglutarate (or branched chain 2-oxoacid or pyruvate) and on the activation state of the enzyme.
Under optimal conditions for each system in mitochondria isolated from rat skeletal muscle, the OGDH complex can produce superoxide/H2O2 at about eight times the rate from site IF (Fig. 5C), the PDH complex can produce superoxide/H2O2 at about four times the rate from site IF (Fig. 9C), and the BCKDH complex can produce superoxide/H2O2 at almost twice the rate from site IF (Fig. 7C). Depending on the substrates present and the conditions, the dominant sites of superoxide/H2O2 production at the level of the NADH/NAD+ isopotential pool may be the OGDH and PDH complexes, but in the past their superoxide/H2O2 production may often have been misattributed to complex I.
This work was supported, in whole or in part, by National Institutes of Health Grant TL1 AG032116 (to C. L. Q.). This work was also supported by The Ellison Medical Foundation Grant AG-SS-2288-09 (to M. D. B.), the Brazilian Government through the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) e ao Conselho de Nacional de Desenvolvimento Científico e Tecnológico programa Ciências Sem Fronteiras (CNPq-CSF) (to R. L. S. G.), the Carlsberg Foundation (to M. H.-M.), the Center for Excellence in Apoptosis Research funds from Massachusetts Technology Collaborative Grant A00000000004448 (to N. Y.), and Russian Foundation of Basic Research (RFBR) Grant 12-04-01541 (to V. I. B.).
C. Kim and N. Yadava, unpublished data.
- site IF
- flavin in the NADH-oxidizing site of complex I
- site IQ
- ubiquinone-reducing site of complex I
- site IIF
- flavin site of complex II
- site IIIQo
- quinol-oxidizing site of complex III
- OGDH
- 2-oxoglutarate dehydrogenase
- PDH
- pyruvate dehydrogenase
- BCKDH
- branched-chain 2-oxoacid (or α-ketoacid) dehydrogenase
- KMV
- 3-methyl-2-oxopentanoate (α-keto-methylvalerate)
- KIV
- 3-methyl-2-oxobutanoate (α-keto-isovalerate)
- KIC
- 4-methyl-2-oxopentanoate (α-ketoisocaproate)
- Eh
- operating redox potential
- Q
- ubiquinone
- QH2
- ubiquinol
- E1
- 2-oxoacid dehydrogenase
- E2
- dihydrolipoamide acyltransferase
- E3
- dihydrolipoamide dehydrogenase
- PDP
- pyruvate dehydrogenase phosphatase
- ETF
- electron transferring flavoprotein.
REFERENCES
- 1. Hansford R. G., Hogue B. A., Mildaziene V. (1997) Dependence of H2O2 formation by rat heart mitochondria on substrate availability and donor age. J. Bioenerg. Biomembr. 29, 89–95 [DOI] [PubMed] [Google Scholar]
- 2. Barja G., Herrero A. (1998) Localization at complex I and mechanism of the higher free radical production of brain nonsynaptic mitochondria in the short-lived rat than in the longevous pigeon. J. Bioenerg. Biomembr. 30, 235–243 [DOI] [PubMed] [Google Scholar]
- 3. Kushnareva Y., Murphy A. N., Andreyev A. (2002) Complex I-mediated reactive oxygen species generation. Modulation by cytochrome c and NAD(P)+ oxidation-reduction state. Biochem. J. 368, 545–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. St-Pierre J., Buckingham J. A., Roebuck S. J., Brand M. D. (2002) Topology of superoxide production from different sites in the mitochondrial electron transport chain. J. Biol. Chem. 277, 44784–44790 [DOI] [PubMed] [Google Scholar]
- 5. Andreyev A. Y., Kushnareva Y. E., Starkov A. A. (2005) Mitochondrial metabolism of reactive oxygen species. Biochemistry 70, 200–214 [DOI] [PubMed] [Google Scholar]
- 6. Murphy M. P. (2009) How mitochondria produce reactive oxygen species. Biochem. J. 417, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brand M. D. (2010) The sites and topology of mitochondrial superoxide production. Exp. Gerontol. 45, 466–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Quinlan C. L., Treberg J. R., Brand M. D. (2011) Mechanisms of mitochondrial free radical production and their relationship to the aging process. In Handbook of the Biology of Aging (Masoro E. J., Austad S. N., eds) 7th Ed., pp. 47–61, Elsevier, Amsterdam [Google Scholar]
- 9. Quinlan C. L., Perevoschikova I. V., Goncalves R. L., Hey-Mogensen M., Brand M. D. (2013) The determination and analysis of site-specific rates of mitochondrial reactive oxygen species production. Methods Enzymol. 526, 189–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Quinlan C. L., Orr A. L., Perevoshchikova I. V., Treberg J. R., Ackrell B. A., Brand M. D. (2012) Mitochondrial complex II can generate reactive oxygen species at high rates in both the forward and reverse reactions. J. Biol. Chem. 287, 27255–27264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Treberg J. R., Quinlan C. L., Brand M. D. (2011) Evidence for two sites of superoxide production by mitochondrial NADH-ubiquinone oxidoreductase (complex I). J. Biol. Chem. 286, 27103–27110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kramer D. M., Roberts A. G., Muller F., Cape J., Bowman M. K. (2004) Q-cycle bypass reactions at the Qo site of the cytochrome bc1 (and related) complexes. Methods Enzymol. 382, 21–45 [DOI] [PubMed] [Google Scholar]
- 13. Muller F. L., Roberts A. G., Bowman M. K., Kramer D. M. (2003) Architecture of the Qo site of the cytochrome bc1 complex probed by superoxide production. Biochemistry 42, 6493–6499 [DOI] [PubMed] [Google Scholar]
- 14. Quinlan C. L., Gerencser A. A., Treberg J. R., Brand M. D. (2011) The mechanism of superoxide production by the antimycin-inhibited mitochondrial Q-cycle. J. Biol. Chem. 286, 31361–31372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Orr A. L., Quinlan C. L., Perevoshchikova I. V., Brand M. D. (2012) A refined analysis of superoxide production by mitochondrial sn-glycerol-3-phosphate dehydrogenase. J. Biol. Chem. 287, 42921–42935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Perevoshchikova I. V., Quinlan C. L., Orr A. L., Gerencser A. A., Brand M. D. (2013) Sites of superoxide and hydrogen peroxide production during fatty acid oxidation in rat skeletal muscle mitochondria. Free Radic. Biol. Med. 61, 298–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Forman H. J., Kennedy J. (1975) Superoxide production and electron transport in mitochondrial oxidation of dihydroorotic acid. J. Biol. Chem. 250, 4322–4326 [PubMed] [Google Scholar]
- 18. Kareyeva A. V., Grivennikova V. G., Cecchini G., Vinogradov A. D. (2011) Molecular identification of the enzyme responsible for the mitochondrial NADH-supported ammonium-dependent hydrogen peroxide production. FEBS Lett. 585, 385–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bunik V. I., Sievers C. (2002) Inactivation of the 2-oxo acid dehydrogenase complexes upon generation of intrinsic radical species. Eur. J. Biochem. 269, 5004–5015 [DOI] [PubMed] [Google Scholar]
- 20. Starkov A. A., Fiskum G., Chinopoulos C., Lorenzo B. J., Browne S. E., Patel M. S., Beal M. F. (2004) Mitochondrial α-ketoglutarate dehydrogenase complex generates reactive oxygen species. J. Neurosci. 24, 7779–7788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tretter L., Adam-Vizi V. (2004) Generation of reactive oxygen species in the reaction catalyzed by α-ketoglutarate dehydrogenase. J. Neurosci. 24, 7771–7778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ambrus A., Adam-Vizi V. (2013) Molecular dynamics study of the structural basis of dysfunction and the modulation of reactive oxygen species generation by pathogenic mutants of human dihydrolipoamide dehydrogenase. Arch. Biochem. Biophys. 538, 145–155 [DOI] [PubMed] [Google Scholar]
- 23. Fisher-Wellman K. H., Gilliam L. A., Lin C. T., Cathey B. L., Lark D. S., Darrell Neufer P. (2013) Mitochondrial glutathione depletion reveals a novel role for the pyruvate dehydrogenase complex as a key HO-emitting source under conditions of nutrient overload. Free Radic. Biol. Med. 65, 1201–1208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. White T. A., Krishnan N., Becker D. F., Tanner J. J. (2007) Structure and kinetics of monofunctional proline dehydrogenase from Thermus thermophilus. J. Biol. Chem. 282, 14316–14327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Brand M. D., Orr A. L., Perevoschikova I. V., Quinlan C. L. (2013) The role of mitochondrial function and cellular bioenergetics in aging and disease. Br. J. Dermatol. 169, 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Quinlan C. L., Perevoshchikova I. V., Hey-Mogensen M., Orr A. L., Brand M. D. (2013) Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol. 1, 304–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Quinlan C. L., Treberg J. R., Perevoshchikova I. V., Orr A. L., Brand M. D. (2012) Native rates of superoxide production from multiple sites in isolated mitochondria measured using endogenous reporters. Free Radic. Biol. Med. 53, 1807–1817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Massey V. (1994) Activation of molecular oxygen by flavins and flavoproteins. J. Biol. Chem. 269, 22459–22462 [PubMed] [Google Scholar]
- 29. Zhang L., Yu L., Yu C. A. (1998) Generation of superoxide anion by succinate-cytochrome c reductase from bovine heart mitochondria. J. Biol. Chem. 273, 33972–33976 [DOI] [PubMed] [Google Scholar]
- 30. Kussmaul L., Hirst J. (2006) The mechanism of superoxide production by NADH:ubiquinone oxidoreductase (complex I) from bovine heart mitochondria. Proc. Natl. Acad. Sci. U.S.A. 103, 7607–7612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Affourtit C., Quinlan C. L., Brand M. D. (2012) Measurement of proton leak and electron leak in isolated mitochondria. Methods Mol. Biol. 810, 165–182 [DOI] [PubMed] [Google Scholar]
- 32. Treberg J. R., Quinlan C. L., Brand M. D. (2010) Hydrogen peroxide efflux from muscle mitochondria underestimates matrix superoxide production. A correction using glutathione depletion. FEBS J. 277, 2766–2778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Grivennikova V. G., Kapustin A. N., Vinogradov A. D. (2001) Catalytic activity of NADH-ubiquinone oxidoreductase (complex I) in intact mitochondria. Evidence for the slow active/inactive transition. J. Biol. Chem. 276, 9038–9044 [DOI] [PubMed] [Google Scholar]
- 34. Nicholls D. G., Ferguson S. J. (2013) Bioenergetics 4, p. 107, Academic Press Inc., London [Google Scholar]
- 35. Chan P. C., Bielski B. H. (1974) Enzyme-catalyzed free radical reactions with nicotinamide adenine nucleotides. II. Lactate dehydrogenase-catalyzed oxidation of reduced nicotinamide adenine dinucleotide by superoxide radicals generated by xanthine oxidase. J. Biol. Chem. 249, 1317–1319 [PubMed] [Google Scholar]
- 36. Lambert A. J., Brand M. D. (2004) Inhibitors of the quinone-binding site allow rapid superoxide production from mitochondrial NADH:ubiquinone oxidoreductase (complex I). J. Biol. Chem. 279, 39414–39420 [DOI] [PubMed] [Google Scholar]
- 37. Lambert A. J., Brand M. D. (2004) Superoxide production by NADH:ubiquinone oxidoreductase (complex I) depends on the pH gradient across the mitochondrial inner membrane. Biochem. J. 382, 511–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lambert A. J., Buckingham J. A., Boysen H. M., Brand M. D. (2008) Diphenyleneiodonium acutely inhibits reactive oxygen species production by mitochondrial complex I during reverse, but not forward electron transport. Biochim. Biophys. Acta 1777, 397–403 [DOI] [PubMed] [Google Scholar]
- 39. Lambert A. J., Buckingham J. A., Brand M. D. (2008) Dissociation of superoxide production by mitochondrial complex I from NAD(P)H redox state. FEBS Lett. 582, 1711–1714 [DOI] [PubMed] [Google Scholar]
- 40. Orr A. L., Ashok D., Sarantos M. R., Shi T., Hughes R. E., Brand M. D. (2013) Inhibitors of ROS production by the ubiquinone-binding site of mitochondrial complex I identified by chemical screening. Free Radic. Biol. Med. 65, 1047–1059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pryde K. R., Hirst J. (2011) Superoxide is produced by the reduced flavin in mitochondrial complex I. A single, unified mechanism that applies during both forward and reverse electron transfer. J. Biol. Chem. 286, 18056–18065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Qi F., Pradhan R. K., Dash R. K., Beard D. A. (2011) Detailed kinetics and regulation of mammalian 2-oxoglutarate dehydrogenase. BMC Biochem. 12, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bunik V., Raddatz G., Strumilo S. (2013) Translating enzymology into metabolic regulation. The case of the 2-oxoglutarate dehydrogenase multienzyme complex. Curr. Chem. Biol. 7, 74–93 [Google Scholar]
- 44. Lambeth D. O., Tews K. N., Adkins S., Frohlich D., Milavetz B. I. (2004) Expression of two succinyl-CoA synthetases with different nucleotide specificities in mammalian tissues. J. Biol. Chem. 279, 36621–36624 [DOI] [PubMed] [Google Scholar]
- 45. Garland P. B. (1964) Some kinetic properties of pig-heart oxoglutarate dehydrogenase that provide a basis for metabolic control of the enzyme activity and also a stoicheiometric assay for coenzyme A in tissue extracts. Biochem. J. 92, 10C–12C [DOI] [PubMed] [Google Scholar]
- 46. Kareyeva A. V., Grivennikova V. G., Vinogradov A. D. (2012) Mitochondrial hydrogen peroxide production as determined by the pyridine nucleotide pool and its redox state. Biochim. Biophys. Acta 1817, 1879–1885 [DOI] [PubMed] [Google Scholar]
- 47. Grivennikova V. G., Vinogradov A. D. (2013) Partitioning of superoxide and hydrogen peroxide production by mitochondrial respiratory complex I. Biochim. Biophys. Acta 1827, 446–454 [DOI] [PubMed] [Google Scholar]
- 48. Izard T., Aevarsson A., Allen M. D., Westphal A. H., Perham R. N., de Kok A., Hol W. G. (1999) Principles of quasi-equivalence and Euclidean geometry govern the assembly of cubic and dodecahedral cores of pyruvate dehydrogenase complexes. Proc. Natl. Acad. Sci. U.S.A. 96, 1240–1245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Perham R. N. (1991) Domains, motifs, and linkers in 2-oxo acid dehydrogenase multienzyme complexes. A paradigm in the design of a multifunctional protein. Biochemistry 30, 8501–8512 [DOI] [PubMed] [Google Scholar]
- 50. Bunik V. I. (2003) 2-Oxo acid dehydrogenase complexes in redox regulation. Eur. J. Biochem. 270, 1036–1042 [DOI] [PubMed] [Google Scholar]
- 51. Zündorf G., Kahlert S., Bunik V. I., Reiser G. (2009) α-Ketoglutarate dehydrogenase contributes to production of reactive oxygen species in glutamate-stimulated hippocampal neurons in situ. Neuroscience 158, 610–616 [DOI] [PubMed] [Google Scholar]
- 52. Tretter L., Adam-Vizi V. (2005) α-Ketoglutarate dehydrogenase. A target and generator of oxidative stress. Philos. Trans. R. Soc. Lond. B Biol. Sci. 360, 2335–2345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Massey V., Müller F., Feldberg R., Schuman M., Sullivan P. A., Howell L. G., Mayhew S. G., Matthews R. G., Foust G. P. (1969) The reactivity of flavoproteins with sulfite. Possible relevance to the problem of oxygen reactivity. J. Biol. Chem. 244, 3999–4006 [PubMed] [Google Scholar]
- 54. Massey V., Strickland S., Mayhew S. G., Howell L. G., Engel P. C., Matthews R. G., Schuman M., Sullivan P. A. (1969) The production of superoxide anion radicals in the reaction of reduced flavins and flavoproteins with molecular oxygen. Biochem. Biophys. Res. Commun. 36, 891–897 [DOI] [PubMed] [Google Scholar]
- 55. Kunz W. S., Gellerich F. N. (1993) Quantification of the content of fluorescent flavoproteins in mitochondria from liver, kidney cortex, skeletal muscle, and brain. Biochem. Med. Metab. Biol. 50, 103–110 [DOI] [PubMed] [Google Scholar]
- 56. Kunz W. S., Kunz W. (1985) Contribution of different enzymes to flavoprotein fluorescence of isolated rat liver mitochondria. Biochim. Biophys. Acta 841, 237–246 [DOI] [PubMed] [Google Scholar]
- 57. Asmus K. D. (1990) Sulfur-centered free radicals. Methods Enzymol. 186, 168–180 [DOI] [PubMed] [Google Scholar]
- 58. Bunik V. I., Schloss J. V., Pinto J. T., Dudareva N. D., Cooper A. J. L. (2011) A survey of oxidative paracatalytic reactions catalyzed by enzymes that generate carbanionic intermediates. Implications for ROS production, cancer etiology, and neurodegenerative diseases. Adv. Enzymol. Rel. Areas Mol. Biol. 77, 305–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Gutman M., Kearney E. B., Singer T. P. (1971) Control of succinate dehydrogenase in mitochondria. Biochemistry 10, 4763–4770 [DOI] [PubMed] [Google Scholar]
- 60. Bunik V. I., Romash O. G., Gomazkova V. S. (1991) Effect of α-ketoglutarate and its structural analogues on hysteretic properties of α-ketoglutarate dehydrogenase. FEBS Lett. 278, 147–150 [DOI] [PubMed] [Google Scholar]
- 61. Bunik V., Westphal A. H., de Kok A. (2000) Kinetic properties of the 2-oxoglutarate dehydrogenase complex from Azotobacter vinelandii. Evidence for the formation of a precatalytic complex with 2-oxoglutarate. Eur. J. Biochem. 267, 3583–3591 [DOI] [PubMed] [Google Scholar]
- 62. Bunik V. I., Biryukov A. I., Zhukov YuN. (1992) Inhibition of pigeon breast muscle α-ketoglutarate dehydrogenase by phosphonate analogues of α-ketoglutarate. FEBS Lett. 303, 197–201 [DOI] [PubMed] [Google Scholar]
- 63. Biryukov A. I., Bunik V. I., Zhukov Y. N., Khurs E. N., Khomutov R. M. (1996) Succinyl phosphonate inhibits α-ketoglutarate oxidative decarboxylation, catalyzed by α-ketoglutarate dehydrogenase complexes from E. coli and pigeon breast muscle. FEBS Lett. 382, 167–170 [DOI] [PubMed] [Google Scholar]
- 64. Bunik V. I., Denton T. T., Xu H., Thompson C. M., Cooper A. J., Gibson G. E. (2005) Phosphonate analogues of α-ketoglutarate inhibit the activity of the α-ketoglutarate dehydrogenase complex isolated from brain and in cultured cells. Biochemistry 44, 10552–10561 [DOI] [PubMed] [Google Scholar]
- 65. Cheshchevic V., Janssen A. J. M., Dremza I. K., Zavodnik I. B., Bunik V. I. (2010) The OGDHC-exerted control of mitochondrial respiration is increased under energy demand. In Mitochondrial Physiology. The Many Functions of the Organism in Our Cells. (Renner-Sattler K., Gnaiger E., eds.) pp 76–77, Steiger Druck GmbH, Axams, Austria [Google Scholar]
- 66. Patel M. S. (1974) Inhibition by the branched-chain 2-oxo acids of the 2-oxoglutarate dehydrogenase complex in developing rat and human brain. Biochem. J. 144, 91–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Shestopalov A. I., Kristal B. S. (2007) Branched chain keto-acids exert biphasic effects on α-ketoglutarate-stimulated respiration in intact rat liver mitochondria. Neurochem. Res. 32, 947–951 [DOI] [PubMed] [Google Scholar]
- 68. Bunik V. I., Fernie A. R. (2009) Metabolic control exerted by the 2-oxoglutarate dehydrogenase reaction. A cross-kingdom comparison of the crossroad between energy production and nitrogen assimilation. Biochem. J. 422, 405–421 [DOI] [PubMed] [Google Scholar]
- 69. Harris R. A., Popov K. M., Zhao Y., Kedishvili N. Y., Shimomura Y., Crabb D. W. (1995) A new family of protein kinases. The mitochondrial protein kinases. Adv. Enzyme Regul. 35, 147–162 [DOI] [PubMed] [Google Scholar]
- 70. Paxton R., Harris R. A. (1984) Regulation of branched-chain α-ketoacid dehydrogenase kinase. Arch. Biochem. Biophys. 231, 48–57 [DOI] [PubMed] [Google Scholar]
- 71. Harris R. A., Joshi M., Jeoung N. H. (2004) Mechanisms responsible for regulation of branched-chain amino acid catabolism. Biochem. Biophys. Res. Commun. 313, 391–396 [DOI] [PubMed] [Google Scholar]
- 72. Parker P. J., Randle P. J. (1978) Branched chain 2-oxo-acid dehydrogenase complex of rat liver. FEBS Lett. 90, 183–186 [DOI] [PubMed] [Google Scholar]
- 73. Parker P. J., Randle P. J. (1978) Partial purification and properties of branched-chain 2-oxo acid dehydrogenase of ox liver. Biochem. J. 171, 751–757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Wieland O. H. (1983) The mammalian pyruvate dehydrogenase complex. Structure and regulation. Rev. Physiol. Biochem. Pharmacol. 96, 123–170 [DOI] [PubMed] [Google Scholar]
- 75. Ramsay R. R., Tubbs P. K. (1975) The mechanism of fatty acid uptake by heart mitochondria. An acylcarnitine-carnitine exchange. FEBS Lett. 54, 21–25 [DOI] [PubMed] [Google Scholar]
- 76. Pande S. V., Parvin R. (1976) Characterization of carnitine acylcarnitine translocase system of heart mitochondria. J. Biol. Chem. 251, 6683–6691 [PubMed] [Google Scholar]
- 77. Roche T. E., Hiromasa Y., Turkan A., Gong X., Peng T., Yan X., Kasten S. A., Bao H., Dong J. (2003) Essential roles of lipoyl domains in the activated function and control of pyruvate dehydrogenase kinases and phosphatase isoform 1. Eur. J. Biochem. 270, 1050–1056 [DOI] [PubMed] [Google Scholar]
- 78. Yeaman S. J., Hutcheson E. T., Roche T. E., Pettit F. H., Brown J. R., Reed L. J., Watson D. C., Dixon G. H. (1978) Sites of phosphorylation on pyruvate dehydrogenase from bovine kidney and heart. Biochemistry 17, 2364–2370 [DOI] [PubMed] [Google Scholar]
- 79. Linn T. C., Pettit F. H., Reed L. J. (1969) α-Keto acid dehydrogenase complexes. X. Regulation of the activity of the pyruvate dehydrogenase complex from beef kidney mitochondria by phosphorylation and dephosphorylation. Proc. Natl. Acad. Sci. U.S.A. 62, 234–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Kerbey A. L., Randle P. J., Cooper R. H., Whitehouse S., Pask H. T., Denton R. M. (1976) Regulation of pyruvate dehydrogenase in rat heart. Mechanism of regulation of proportions of dephosphorylated and phosphorylated enzyme by oxidation of fatty acids and ketone bodies and of effects of diabetes. Role of coenzyme A, acetyl-coenzyme A, and reduced and oxidized nicotinamide-adenine dinucleotide. Biochem. J. 154, 327–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Stacpoole P. W. (1989) The pharmacology of dichloroacetate. Metabolism 38, 1124–1144 [DOI] [PubMed] [Google Scholar]
- 82. Huang B., Gudi R., Wu P., Harris R. A., Hamilton J., Popov K. M. (1998) Isoenzymes of pyruvate dehydrogenase phosphatase. DNA-derived amino acid sequences, expression, and regulation. J. Biol. Chem. 273, 17680–17688 [DOI] [PubMed] [Google Scholar]
- 83. Pettit F. H., Roche T. E., Reed L. J. (1972) Function of calcium ions in pyruvate dehydrogenase phosphatase activity. Biochem. Biophys. Res. Commun. 49, 563–571 [DOI] [PubMed] [Google Scholar]
- 84. Thomas A. P., Diggle T. A., Denton R. M. (1986) Sensitivity of pyruvate dehydrogenase phosphate phosphatase to magnesium ions. Similar effects of spermine and insulin. Biochem. J. 238, 83–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Yadava N., Houchens T., Potluri P., Scheffler I. E. (2004) Development and characterization of a conditional mitochondrial complex I assembly system. J. Biol. Chem. 279, 12406–12413 [DOI] [PubMed] [Google Scholar]
- 86. Yadava N., Potluri P., Scheffler I. E. (2008) Investigations of the potential effects of phosphorylation of the MWFE and ESSS subunits on complex I activity and assembly. Int. J. Biochem. Cell Biol. 40, 447–460 [DOI] [PubMed] [Google Scholar]
- 87. Chinta S. J., Rane A., Yadava N., Andersen J. K., Nicholls D. G., Polster B. M. (2009) Reactive oxygen species regulation by AIF- and complex I-depleted brain mitochondria. Free Radic. Biol. Med. 46, 939–947 [DOI] [PMC free article] [PubMed] [Google Scholar]