

Background: Tudor-SN has been observed in lipid droplets, but its role in lipid homeostasis remains unclear.
Results: Tudor-SN and PPARγ are both regulated by C/EBPβ during adipogenesis and significantly influence the regulation of PPARγ target genes.
Conclusion: Tudor-SN functions as a co-activator of PPARγ in adipogenesis.
Significance: The study has elucidated a new functional mechanism for the regulation of adipogenesis.
Keywords: Adipogenesis, C/EBP Transcription Factor, Histone Deacetylase, Histone Modification, Peroxisome Proliferator-activated Receptor (PPAR), C/EBPβ, HDAC, PPARγ, Tudor-SN
Abstract
Adipogenesis, in which mesenchymal precursor cells differentiate into mature adipocytes, is a well orchestrated process. In the present study we identified Tudor-SN as a novel co-activator of the transcription factor peroxisome proliferator-activated receptor γ (PPARγ). We provide the first evidence that Tudor-SN and PPARγ exist in the same complex. Both are up-regulated by the early factor C/EBPβ during adipogenesis and significantly influence the regulation of PPARγ target genes in both 3T3-L1 pre-adipocyte and mouse embryonic fibroblasts (MEF) upon exposure to a mixture of hormonal mixture. Moreover, aP2-PPARγ response element (PPRE) interacts with both PPARγ and Tudor-SN, and the gene transcriptional activation of PPRE-luc is enhanced by ectopic expression of Tudor-SN. Deletion of Tudor-SN protein (MEF-KO) affects but does not completely abolish the association of PPARγ and aP2-PPRE. Loss-of-function studies further verified that Tudor-SN is required for adipogenesis, as deletion of Tudor-SN (MEF-KO) impairs dexamethasone, 3-isobutyl-1-methylxanthine, and insulin (DMI)-induced adipocyte differentiation and the expression of PPARγ target genes, such as aP2 and adipsin. Furthermore, H3 acetylation levels were lower in MEF-KO than MEF-WT. Both HDAC1 and HDAC3 are stably associated with PPARγ in MEF-KO, whereas only a small amount of association was observed in MEF-WT after 5 days of treatment during adipogenesis. PPARγ requires various co-activators or co-repressors, which may dynamically associate with and regulate the higher order chromatin remodeling of the promoter region of PPARγ-bound target genes; Tudor-SN is likely one of these co-activators.
Introduction
Adipose tissue is crucial for whole-body insulin sensitivity and energy homeostasis (1). It also plays essential roles in regulating inflammatory responses and angiogenesis via the secretion of adipokine (2). In mammals, brown adipose tissue dissipates energy through thermogenesis, whereas white adipose tissue, which is the predominant type of fat in adults, serves as a storage depot for excess energy. The number of adipocytes is primarily determined in early adulthood, whereas the subsequent increase in fat mass is mainly due to the accumulation and storage of triglycerides in adipocytes (3). However, a small amount of adipocytes is renewable via the coordination of cell death and neo-adipogenesis from mesenchymal precursor cells. Obesity, which has become a major health problem, is considered a hypertrophic disease that results from an increased number of adipocytes and the enlargement of individual adipocytes. Thus, understanding the molecular mechanisms of adipocyte differentiation is crucial for improved treatment of obesity and its associated diseases.
Adipogenesis is a well orchestrated process in which mesenchymal precursor cells differentiate into mature adipocytes. This process is regulated by a cascade of well characterized transcription factors. In 3T3-L1 pre-adipocytes, the hormonal adipogenic stimulus triggers the immediate expression of CCAAT/enhancer-binding proteins (C/EBPδ and C/EBPβ),4 which further induces the expression of C/EBPα and peroxisome proliferator activated receptor γ (PPARγ) (4, 5). PPARγ is a member of the nuclear hormone receptor superfamily and is required for adipocyte differentiation (6). The PPARγ gene is transcribed from alternative promoters that give rise to two major protein isoforms, PPARγ1 and PPARγ2 (7, 8). Transcriptional activation of PPARγ and other nuclear hormone receptors regulate the participation of co-activators or co-repressors that bridge the association between nuclear receptors and the basal transcription machinery (9, 10). However, the mechanisms by which these cofactors mediate transcriptional regulation in the process of adipogenesis or adipocyte differentiation have not been fully elucidated.
Tudor staphylococcal nuclease (Tudor-SN), also known as SND1 (staphylococcal nuclease domain containing 1) or p100, is a highly conserved and ubiquitously expressed multifunctional protein. Tudor-SN was first identified as a transcriptional co-activator of Epstein-Barr virus nuclear antigen 2 (EBNA2) (11), c-Myb (12), Pim-1 (13), polycystin-1 (PC1) (14), STAT6 (15), and STAT5 (16). Tudor-SN was recently found in the cytosolic lipid droplets of lactating mammary glands from cows and mice (17). Interestingly, the abundant expression of Tudor-SN is closely associated with milk production in response to lactogenic stimuli in cultured mammary cells (18), and Tudor-SN likely plays an important role in lipid homeostasis (19). These data support a potential function of Tudor-SN in regulating adipogenesis and the formation of lipid droplets. Thus, the aim of this study was to investigate the underlying molecular mechanisms by which the Tudor-SN protein regulates adipogenesis. Accordingly, we identified Tudor-SN as a novel and essential co-activator of PPARγ in adipogenesis.
EXPERIMENTAL PROCEDURES
Cell Culture, Differentiation, and Red Oil O Staining
3T3-L1 cells were grown in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum (FBS). For 3T3-L1 differentiation, 0.5 mm 3 isobutyl-1-methylxanthine (Sigma), 10 μg/ml insulin (Sigma), and 1 μm dexamethasone (Sigma) were added for 2 days. On day 3, 10 μg/ml insulin was added. After 48 h, the medium was changed to DMEM with 10% FBS. The Tudor-SN knock-out mouse was generated in the C57BL/6N background.5 Tudor-SN knock-out mouse embryonic fibroblasts (KO-MEFs) were generated after at least six generations of backcrossing to Tudor-SN knock-out C57BL/6N at the Turku Center for Disease Modeling according to a standard procedure and generously sent to us as a gift. MEF cells were grown in DMEM with 15% FBS, and isobutyl-1-methylxanthine, dexamethasone, and insulin complex were added for differentiation. 3T3-L1 cells were transfected using X-tremeGENE DNA transfection reagents (Roche Applied Science) according to the manufacturer's instructions. siRNA was transfected into 3T3-L1 cells using Lipofectamine RNAi MAX (Invitrogen), and Tudor-SN siRNA was generated as reported previously (15). For red oil O staining, cells were fixed for 2 h at room temperature with 10% formalin in phosphate-buffered saline and washed 3 times with distilled water and then stained for 1 h with filtered 0.5% Oil Red O in 60% isopropyl alcohol. After washing three times with distilled water, the cells were examined under a microscope.
RNA Isolation and Quantitative Real-time PCR
Total RNA was isolated from cells at different differentiation time points using TRIzol reagent (Invitrogen). Reverse transcription PCR was performed using the Revert Aid First Strand cDNA synthesis kit (Fermentas) according to the manufacturer's protocol. Quantitative real-time PCR was performed using Step-One plus (ABI) and the DNA double-strand-specific reagent SYBR Green I for detection (Roche Applied Science). -Fold changes were calculated using the ΔΔCt method. The results were normalized to GAPDH levels.
The primer sequences were as follows: Mus GAPDH sense (5′-CCTGGAGAAACCTGCCAAGT-3′), antisense, (5′-TGGGAGTTGCTGTTGAAGTC-3′); Mus Tudor-SN sense (5′-TCCGGGACCTCAAGTACACCA-3′), antisense (5′-GACCACACTGCCGTCTCGAA-3′); Mus PPARγ sense (5′-TGTCGGTTTCAGAAGTGCCTTG-3′), antisense (5′-TTCAGCTGGTCGATATCACTGGAG-3′); Mus adipsin sense (5′-GCAACCGCAGGGACACTT-3′), antisense (5′-TTGCCATTGCCACAGACG-3′); Mus aP2 sense (5′-AGCATCATAACCCTAGATGGCG-3′), antisense (5′-CATAACACATTCCACCACCAGC-3′).
Co-immunoprecipitation
3T3-L1 cells or MEF-WT and MEF-KO cells were harvested at different time points during differentiation as indicated in the figures. Total cell lysates (TCLs) were prepared as previously described (20). 3T3-L1 TCLs were incubated with anti-Tudor-SN antibody or rabbit polyclonal IgG antibody (Santa Cruz Biotechnology) as a control followed by incubation with protein G-Sepharose (Millipore). The bound proteins were separated by SDS-PAGE and blotted with anti-Tudor-SN antibody or anti-PPARγ antibody (Santa Cruz Biotechnology). The mouse monoclonal anti-Tudor-SN antibody was generated against the SN4 domain (amino acids 507–674) of Tudor-SN by the Institute of Medical Technology, University of Tampere, Finland. TCLs of MEF-WT and MEF-KO were incubated with an anti-PPARγ or anti-IgG antibody as a negative control and immunoblotted with anti-PPARγ, anti-PTB-associated splicing factor (PSF; Santa Cruz), anti-HDAC1 (Abcam), anti-HDAC3 (Abcam), or anti-Tudor-SN antibody; 10% of the TCL was used as input. Anti-H3 antibody (Abcam) and anti-Ace-H3 antibody (Millipore) were used to detect the presence of H3 and Ace-H3.
GST Pulldown Assays
GST pulldown experiments were performed as previously described (21). GST, GST-SN, or GST-TSN fusion proteins were produced in BL21 Escherichia coli and purified using glutathione-Sepharose 4B beads (Amersham Biosciences) according to the manufacturer's instructions. The bead-bound GST fusion proteins were incubated with TCL overnight at 4 °C with head-over-tail rotation and then washed 5 times with binding buffer containing 75 mm NaCl. The bound proteins were separated by SDS-PAGE and blotted with an anti-PPARγ antibody.
Immunofluorescence
Cells were grown and differentiated for 8 days in 35-mm dishes, then fixed for 10 min in 4% paraformaldehyde. After washing with PBS, the cells were permeabilized in 0.5% Triton X-100 for 10 min. PBS containing 5% BSA was used for blocking. Anti-Tudor-SN (rabbit 1:100, Abcam) and anti-PPARγ (Mus 1:50, Santa Cruz) antibodies were incubated with the cells at 4 °C overnight. The cells were then washed with PBST (PBS with 0.1% Tween 20) and incubated with Texas Red-conjugated goat anti-rabbit and fluorescein isothiocyanate (FITC)-labeled goat anti-mouse secondary antibodies (1:500, Invitrogen) for 1 h at room temperature. After three washes in PBS, the cells were stained with 4′,6′-diamidino-2-phenylindole (DAPI) to visualize the nuclei. Images were acquired using a OLYMPUS IX 71 microscope.
Luciferase Assay
3T3-L1 cells were plated in a 12-well plate at a density of 3 × 104 cells per well, grown to 60–80% confluence, and transfected with appropriate plasmids using X-tremeGENE DNA Transfection Reagents (Roche Applied Science). After 48 h, the cells were lysed with reporter lysis buffer (Promega), and luciferase activity was measured as described previously (21). The luciferase values were normalized to β-galactosidase activity and are presented as the mean relative luciferase activity of three independent experiments.
The plasmid containing the full-length cDNA of PPARγ (pcDNA3.1-PPARγ) was kindly provided by Prof. Han (Nankai University, Tianjin, China). PPRE-luc, which contains a PPARγ-responsive element, was kindly provided by Dr. Lluis Fajas (Institut National de la Santé Recherche Médicale, Montpellier, France). The plasmid containing the Tudor-SN promoter-luciferase reporter gene (pGL3-PL) was constructed by Jinsai, Beijing, China. The plasmid containing the full-length cDNA of C/EBPβ was kindly provided by Prof. Zhu (Nankai University, Tianjin, China). pSG5-p100-FLAG and β-gal were constructed as previously described (21). For all experiments, empty pSG5 vector DNA was used to balance the different amounts of DNA used in the various transfections.
Chromatin Immunoprecipitation
Formaldehyde was added to the differentiated 3T3-L1, MEF-WT, and MEF-KO cells at a final concentration of 1%. Cross-linking was stopped by the addition of glycine to a final concentration of 100 mm. The TCLs were harvested with SDS lysis buffer. Soluble chromatin was prepared via sonication with a Vibra Cell 500 watt sonicator (Sonics and Materials). After centrifugation, the samples were precleared with protein G beads and immunoprecipitated with anti-C/EBPβ, anti-PPARγ, anti-Tudor-SN, or rabbit polyclonal IgG antibodies. The immunocomplexes were then incubated with protein G beads. The beads were washed 3 times with buffer containing 10 mm Tris-HCl (pH 8), 140 mm NaCl, 1 mm EDTA, and 0.1% Triton X-100 and once with TE buffer (10 mm Tris-HCl (pH 8), 1 mm EDTA). The chromatin fragments were eluted from the beads with elution buffer containing 62.5 mm Tris-HCl (pH 6.8), 200 mm NaCl, 2% SDS, and 10 mm dithiothreitol, and cross-links were reverted by heating at 65 °C overnight. DNA was extracted with phenol/chloroform, ethanol-precipitated, and analyzed for the presence of the Tudor-SN or PPARγ promoter and ap2 PPARγ response element (PPRE) region by PCR. The extract aliquoted before the immunoprecipitation step (total input chromatin) was also used for PCR analysis.
Isolation of Pre-adipocytes and Mature Adipocytes
Adipocytes were isolated from C57BL/6 mice. Briefly, epididymal fat pads were weighed and rinsed in DMEM/F-12 medium, and the tissue was then minced to a very fine consistency in a new Petri dish. The minced tissue was treated with collagenase H (1 mg/ml) (Roche Applied Science) in HEPES solution (29.8 g of HEPES, 8.78 g of NaCl, 4.66 g of KCl, 1.12 g of d-glucose, 18.8 g of BSA, 0.132 g of CaCl2, double distilled H2O up to 1 liter) in a shaking water bath at 37 °C and 115 rpm, then filtered through a 100-μm filter. After centrifugation at 500 × g for 5 min, the supernatant and the pellet were collected separately. To obtain mature adipocytes, the supernatant fraction was filtered through a 70-μm filter, washed with culture medium 3 times, and planted in a culture vessel at 37 °C. To obtain pre-adipocytes, the pellet fraction was incubated with red blood cell lysis buffer for 5 min, then resuspended in an equal volume of culture medium (DMEM/F-12 + 10% FBS). The cell suspension was filtered through a 40-μm filter and collected in a new 50-ml tube. After centrifugation at 500 × g for 5 min, the supernatant was removed, and the pellet was resuspended in culture medium, then planted in a culture vessel at 37 °C.
RESULTS
Tudor-SN Is Required for Hormone-induced Adipocyte Differentiation
3T3-L1 is a well characterized pre-adipocyte cell line that can differentiate into mature adipocytes upon exposure to a hormonal mixture of dexamethasone, 3-isobutyl-1-methylxanthine, and insulin (DMI). We thus utilized 3T3-L1 cells to investigate the potential role of Tudor-SN in adipogenesis. The endogenous Tudor-SN protein was knocked down by transfection with Tudor-SN siRNA or ectopically expressed by transfection of a pSG5-Tudor-SN-FLAG plasmid. As shown in Fig. 1A and in the statistical analysis in Fig. 1B, the endogenous Tudor-SN protein was efficiently knocked down (second lane) (p < 0.01) or overexpressed (fourth lane) (p < 0.05) in 3T3-L1 pre-adipocytes. Scramble siRNA (first lane) or empty plasmid pSG5 (third lane) were used as negative controls. The different transfected 3T3-L1 pre-adipocytes were then cultured to confluence and treated with the DMI hormone mixture. After 8 days of treatment, the cells were stained with Oil Red O (ORO). The stained cells were calculated and statistically analyzed. As shown in Fig. 1, C and D, less droplets were observed in the cells in which endogenous Tudor-SN was knocked down (siRNA) compared with the control group (Scr) (p < 0.01). Conversely, ectopic expression of Tudor-SN resulted in the increased accumulation of lipid droplets and in a larger fraction of cells compared with the empty vector control group (pSG5) (p < 0.05).
FIGURE 1.

The effect of Tudor-SN on the differentiation of 3T3-L1 and MEF cells. A, 3T3-L1 cells were transfected with pSG5-Tudor-SN plasmid, pSG5 vector, Tudor-SN siRNA, or scramble (Scr) siRNA, and the transfection efficiency was detected by Western blot. B, statistical analysis of the transfection efficiency. The graph represents densitometric units normalized to β-actin for each protein and is representative of the S.D. Statistical analysis was performed using the independent-samples Student's t test; *, p < 0.05(n = 3); **, p < 0.01(n = 3). C, after transfection with siRNA or plasmids, 3T3-L1 cells were treated with DMI for 8 days. The cells were stained with Oil Red O to observe lipid droplet formation. D, statistical analysis of lipid droplet formation. Oil Red O was washed away with 60% isopropyl alcohol and measured at an optical density (OD) of 500. Statistical analysis was performed using the independent-samples Student's t test; *, p < 0.05(n = 3); **, p < 0.01(n = 3). E, expression levels of Tudor-SN in MEF-WT and MEF-KO cells. F, knock out of Tudor-SN severely affects adipogenesis. MEF-WT and MEF-KO cells were treated with DMI for 8 days. The cells were stained with Oil Red O to observe lipid droplet formation.
To further investigate the role of Tudor-SN in adipogenesis, we also performed an ORO staining experiment using embryonic fibroblasts prepared from wild-type mouse (MEF-WT) or Tudor-SN knock-out mouse (MEF-KO) embryos. Fig. 1E demonstrates that the Tudor-SN protein was completely abolished in MEF-KO cells. After 8 days of treatment with DMI, the cells were stained with ORO. MEF-WT cells efficiently differentiated into adipocytes with a large amount of lipid droplets (Fig. 1F, left panel). By contrast, no obvious lipid droplets were observed in MEF-KO cells (Fig. 1F, right panel) under the same conditions. These results demonstrate that Tudor-SN is essential for hormone-induced adipogenesis.
Tudor-SN Associates with PPARγ and Is Up-regulated during Adipogenesis
PPARγ is a direct and essential regulator of adipogenesis. We, therefore, hypothesized that Tudor-SN might function as a co-activator of PPARγ. We first investigated the expression profiles of Tudor-SN and PPARγ during adipocyte differentiation. As expected, the expression pattern of Tudor-SN (Fig. 2A, upper panel) was the same as that of PPARγ (middle panel) during adipogenesis. As shown in Fig. 2A, equal amounts of endogenous β-actin were present in different samples (lower panel), and the expression kinetics of the PPARγ (middle panel) and Tudor-SN (upper panel) proteins were the same. During adipogenesis, the expression level of both proteins gradually increased starting on day 2 and reached a peak at day 5 or 6. The time-dependent expression pattern is further illustrated in Fig. 2B. In line with the level of protein expression, real-time PCR analyses further confirmed the parallel expression of Tudor-SN (Fig. 2C) and PPARγ (Fig. 2D), and the mRNA level also gradually increased during adipogenesis.
FIGURE 2.

The expression of Tudor-SN and PPARγ are both enhanced in 3T3-L1 cells during adipogenesis. A, TCLs were harvested at different time points during the differentiation process as indicated and incubated with anti-Tudor-SN antibody, anti-PPARγ antibody, and anti-β-actin-antibody as a control. B, the band intensity was quantified using densitometry and normalized to the band intensity of β-actin. Shown are mRNA levels of Tudor-SN (C) and PPARγ (D) during adipogenesis. The expression levels were normalized to GAPDH levels.
A co-immunoprecipitation assay was performed to determine whether Tudor-SN and PPARγ form a complex in cells. Cell lysates of confluent 3T3-L1 cells (day 0) and DMI-induced differentiated cells at day 2 and day 4 were immunoprecipitated with an anti-Tudor-SN antibody, and the precipitated PPARγ protein was then detected by anti-PPARγ immunoblotting. As shown in Fig. 3A, PPARγ co-precipitated with Tudor-SN (lower panel, lane 2) but not IgG antibody, which was used as a negative control (lane 3). Interestingly, the association increased as the duration of DMI treatment increased. On days 0 and 2, the Tudor-SN protein precipitated only a small amount of PPARγ (lower panel, lanes 2 and 5), whereas on day 4 the co-precipitated PPARγ was remarkably increased (lower panel, lane 8). These data demonstrate that endogenous Tudor-SN interacts with PPARγ during adipogenesis. To map the interaction domain of Tudor-SN with PPARγ, a GST pulldown assay was performed. Different GST fusion proteins were bound to glutathione-conjugated beads and incubated with TCLs of 3T3-L1 cells. As shown in Fig. 3B (top panel), the SN domain of Tudor-SN readily associated with PPARγ but not the TSN domain or GST negative control.
FIGURE 3.

Tudor-SN interacts with PPARγ. A, endogenous Tudor-SN associates with PPARγ in vivo. The TCLs of 3T3-L1 cells at days 0, 2, and 4 of DMI treatment were immunoprecipitated (IP) with anti-Tudor-SN or anti-IgG as a negative control. The bound proteins were separated by SDS-PAGE and blotted with an anti-Tudor-SN antibody (upper panel) or anti-PPARγ antibody (lower panel); 10% of the TCL was loaded as the input. B, Tudor-SN interacts with PPARγ via its SN domain. The TCLs of 3T3-L1 cells were incubated with GST-SN or GST-TSN fusion proteins, respectively, and GST was used as a negative control. Bound proteins were separated by SDS-PAGE and immunoblotted with an anti-PPARγ antibody (top panel); 10% of the TCL was used as the input. The expression levels of the GST fusion proteins were visualized by Coomassie Blue staining (bottom panel). C, 3T3-L1 cells after adipogenesis were fixed and incubated with rabbit polyclonal anti-Tudor-SN and mouse monoclonal anti-PPARγ antibodies followed by Texas Red-conjugated goat anti-rabbit and FITC-labeled goat anti-mouse secondary antibodies. Images were acquired with an OLYMPUS IX 71 microscope with a ×20 objective. D, expression of Tudor-SN and PPARγ in primary pre-adipocytes and mature adipocytes. Primary pre-adipocytes and mature adipocytes were isolated as described. The TCLs of these two cell types were incubated with an anti-Tudor-SN antibody or anti-PPARγ antibody. E, endogenous Tudor-SN associates with PPARγ in pre-adipocytes. TCLs were immunoprecipitated (IP) with an anti-PPARγ antibody or anti-IgG as a negative control and then blotted with an anti-Tudor-SN antibody.
In addition, an immunofluorescence assay was performed to investigate the in vivo localization of Tudor-SN and PPARγ. As shown in Fig. 3C, in adipocytes differentiated from 3T3-L1 cells (DMI treatment for 8 days), both PPARγ (a, green) and Tudor-SN (b, red) were mainly distributed in the nucleus, with only a small portion in the cytosolic fraction. The merged picture illustrates the co-localization of these two proteins (c, yellow). To further investigate the physiological relevance of these novel findings, we detected the expression of Tudor-SN in primary adipocytes isolated from C57BL/6 mice. Because the primary adipose tissue is a mixture of mature adipocytes and a stromal vascular cells (SVCs) fraction, which contains SVCs and pre-adipocytes, we first separated these cells and then detected the expression of the Tudor-SN and PPARγ proteins. As shown in Fig. 3D, PPARγ protein was detected in both pre-adipocytes and mature adipocytes (lower panel), whereas Tudor-SN was present in the pre-adipocytes but not the mature adipocytes (upper panel). Fig. 3E demonstrates that Tudor-SN (lower panel) was co-precipitated with PPARγ (upper panel) in the pre-adipocytes. These data further demonstrate that endogenous Tudor-SN and PPARγ proteins form a physical complex in vivo and suggest that Tudor-SN and PPARγ may function cooperatively in the differentiation of pre-adipocytes into mature adipocytes.
C/EBPβ Induces Tudor-SN Expression during Adipogenesis
The transcription factor CCAAT/enhancer binding protein (C/EBP) is responsible for transcriptional regulation of PPARγ expression (21, 22). Because Tudor-SN associates with PPARγ and both proteins exhibit parallel expression levels after hormonal mixture treatment, we predicted that the regulation of Tudor-SN expression might be the same as that of PPARγ. By BLAST comparison, we identified a C/EBP binding motif in the promoter region of both the mouse and human Tudor-SN genes (Fig. 4A), in agreement with an earlier report of a well conserved C/EBP binding motif in the rat Tudor-SN promoter region (23). Thus, a luciferase assay was performed to examine whether C/EBPβ is involved in regulating Tudor-SN gene transcription. 3T3-L1 cells were co-transfected with plasmids containing the Tudor-SN promoter-luciferase reporter gene (pGL3-PL) and C/EBPβ expression plasmids. As shown in Fig. 4B, the ectopic expression of C/EBPβ enhanced the gene transcriptional activation of Tudor-SN in a dose-dependent manner. We next analyzed the binding of C/EBPβ to the DNA promoter region in the Tudor-SN gene using chromatin immunoprecipitation (ChIP) assays. Chromatin samples were prepared from confluent 3T3-L1 cells on day 0 and differentiated cells on days 2 and 4, and the chromatin was then immunoprecipitated with specific antibodies against C/EBPβ; normal mouse lgG was used as a negative control. As shown in Fig. 4C, no obvious binding of C/EBPβ to the Tudor-SN promoter was observed on day 0 (upper panel, lane 2), whereas more C/EBPβ was recruited to the Tudor-SN promoter region at day 2 and day 4 (upper panel, lanes 5 and 8). There was no binding in the −2000-bp region of the Tudor-SN promoter (lower panel, negative control), which did not contain a C/EBPβ binding motif. The C/EBPβ binding motif in the promoter region of PPARγ was used as a positive control (lower panel). These results demonstrate that Tudor-SN is also regulated by the transcriptional factor C/EBPβ in adipogenesis.
FIGURE 4.

C/EBPβ activates Tudor-SN gene expression. A, the Tudor-SN promoter region contains a C/EBPβ binding site as indicated (highlighted box) in rat, mouse, and human. B, C/EBPβ enhances the gene transcriptional activity of Tudor-SN. 3T3-L1 cells were co-transfected with pGL3-PL-Luc reporter gene constructs carrying the Tudor-SN promoter region (0.5 μg) and β-galactosidase vector (0.5 μg) with different amounts of the C/EBPβ expression plasmid. Normalized luciferase activity is presented. The mean normalized luciferase values of three independent experiments with S.D. are shown. C, chromatin immunoprecipitation (IP) assays of the association of C/EBPβ with the Tudor-SN promoter in 3T3-L1 cells. Cross-linked chromatin from 3T3-L1 cells was incubated with antibodies against C/EBPβ or lgG antibody as a negative control. The immunoprecipitates were analyzed by PCR with primers specific for the mouse Tudor-SN promoter at different time points and primers amplifying a region outside the C/EBPβ binding sites (lower panel) as a negative control. The Input represents the PCR product of total chromatin DNA detection.
Tudor-SN Enhances PPARγ-mediated Transcription
Because Tudor-SN and PPARγ form a physical complex in vivo, we hypothesized that these two proteins may act as co-factors to regulate gene transcription activation in adipogenesis. The adipocyte-selective fatty acid-binding protein aP2 is a direct target gene of PPARγ, which has a PPRE. To determine if Tudor-SN acts as a co-activator to participate in PPARγ-mediated target gene transcription, the primers targeting aP2-PPRE were used in chromatin immunoprecipitation assays. As expected, aP2-PPRE was observed in the PPARγ complex precipitated by either the anti-PPARγ antibody (Fig. 5A, upper panel, lane 2) or anti-Tudor-SN antibody (lane 3), whereas no aP2-PPRE product was obtained in the IgG negative control. To exclude nonspecific binding in the ChIP assay, the promoter region located outside the PPRE was used as a negative control (Fig. 5A, lower panel). To further demonstrate the functional binding of Tudor-SN with the PPRE, a luciferase assay was performed in 3T3-L1 cells by transfecting luciferase-based reporter constructs containing a PPAR-responsive element (PPRE-TK-luc), plasmids containing the full-length cDNA of PPARγ or Tudor-SN, and β-gal expression plasmids. As shown in Fig. 5B, ectopic expression of either Tudor-SN or PPARγ alone or together or in combination enhanced the luciferase activity containing of PPRE-transfected cells, particularly when the proteins were overexpressed together. However, no effect was observed when the control reporter vector TK-Luc, which does not contain a PPRE (data not shown), was used. These data indicate that Tudor-SN can be recruited to and recognize the PPRE binding site together with PPARγ, which strongly suggests that Tudor-SN is a co-activator of the transcription factor PPARγ in regulating adipogenesis.
FIGURE 5.

Tudor-SN combines with the PPRE region and activates downstream gene expression. A, association of Tudor-SN with the PPRE of the aP2 promoter in differentiated 3T3-L1 cells. Cross-linked chromatin from 4-day-differentiated 3T3-L1 cells was incubated with antibodies against PPARγ and Tudor-SN or lgG antibody as a negative control. Primers to the aP2 PPRE region were used for PCR, and primers amplifying a region outside the PPARγ and Tudor-SN binding site were used as a negative control. IP, immunoprecipitate. B, Tudor-SN enhances the transcriptional activity of genes containing a PPRE. 3T3-L1 cells were cotransfected with constructs carrying the PPRE promoter region linked with the Luc reporter gene (0.5 μg) and β-galactosidase vector (0.5 μg) together with Tudor-SN or PPARγ expression plasmids. Normalized luciferase activity is presented. The mean normalized luciferase values of three independent experiments with the S.D. are shown. C, chromatin immunoprecipitation assays demonstrating that knock out of Tudor-SN affects the association of PPARγ with its downstream gene aP2. Cross-linked chromatin from 0-, 5-, and 8-day differentiated MEF-WT or MEF-KO cells were incubated with antibodies against PPARγ or IgG as a negative control, and the primers for the aP2 PPRE region were used for PCR. D, statistical analysis of the ChIP-qPCR results was performed using the independent-samples Student's t test. The results are the means± S.D. *, p < 0.05 (n = 3); ***, p < 0.001(n = 3). E, mRNA expression of aP2 and adipsin in MEF-WT or MEF-KO cells during adipogenesis. Total RNA was harvested from cells treated with DMI at the indicated time points. Real-time PCR analysis was performed to detect the mRNA levels of aP2 and adipsin. Expression levels were normalized to GAPDH mRNA expression.
To further confirm that Tudor-SN is likely a co-activator in PPARγ-mediated gene transcription, a ChIP assay was performed to detect the association of PPARγ with the PPRE in the presence or absence of Tudor-SN. As shown in Fig. 5C, more PPRE was associated with PPARγ during adipogenesis in MEF-WT cells (lower panel), but no obvious changes were observed in MEF-KO cells (upper panel). Statistical analysis (Fig. 5D) further indicated that during adipogenesis the amount of PPRE that was recognized by PPARγ was significantly higher in MEF-WT cells (p < 0.001) than in MEF-KO cells.
To investigate whether Tudor-SN plays physiological roles in adipogenesis in vivo, we detected the expression levels of the adipocyte markers aP2 and adipsin in MEF-WT or MEF-KO cells treated with DMI. As shown in Fig. 5E, although the mRNA levels of aP2 and adipsin gradually increased in MEF-WT cells, there was no obvious change in MEF-KO cells. This result is consistent with the data demonstrating that DMI treatment did not induce adipogenesis in MEF-KO cells (Fig. 1F). These data indicate that Tudor-SN is a novel co-activator of PPARγ.
Appropriate regulation of gene expression requires the coordination of activating and repressing signals modulating DNA accessibility. We have previously reported that Tudor-SN is capable of enhancing histone acetyltransferase activity via recruitment of CREB-binding protein (CBP) to STAT6 (15). Very recently we also identified PSF as a repressor of STAT6-mediated transcription that functions through recruitment of histone deacetylase (HDAC) to the STAT6 transcriptional complex (24). We thus investigated whether a lack of Tudor-SN in MEF cells affects histone acetylation. As shown in Fig. 6A, endogenous histone3 (H3) of MEF-WT (upper panel, lanes 1 and 2) or MEF-KO (upper panel, lanes 3 and 4) cells was immunoprecipitated with an anti-H3 antibody to equal extents and then blotted with an anti-Ace-H3 antibody to detect the acetylation level of H3. Surprisingly, less H3 was acetylated in MEF-KO cells (lower pane, lanes 3 and 4) than in MEF-WT cells (lower panel, lanes 1 and 2). The down-regulation of histone de-acetylation and the up-regulation of histone acetylation have been reported to be essential for adipogenesis, and HDACs might inhibit the adipogenic program by directly repressing the transcriptional activity of pro-adipogenic transcription factors (25). Therefore, we performed a co-immunoprecipitation assay to determine whether HDAC is recruited to the PPARγ complex in MEF-KO cells. TCLs of confluent (day 0) or differentiated cells at days 5 and 8 were immunoprecipitated with an anti-PPARγ antibody, and the precipitated complex was detected by blotting with anti-PSF, anti-HDAC1, anti-HDAC3, or anti-Tudor-SN. During adipogenesis, association of PPARγ and Tudor-SN was observed in a time-dependent manner in MEF-WT cells (Fig. 6B, lower panel). At day 0, only a small amount of Tudor-SN was detected in the PPARγ complex, whereas at days 5 and 8, much more Tudor-SN was recruited to PPARγ. No obvious PSF (second panel) was detected in the complex, whereas a small amount of HDAC1 (third panel) or HDAC3 (fourth panel) was detected at days 5 and 8. However, in MEF-KO cells (Fig. 6C), in which Tudor-SN was deleted, PSF associated with PPARγ in a time-dependent manner (second panel), and HDAC1 (third panel) or HDAC3 (lower panel) was stably associated with PPARγ. These results support our previous results demonstrating that Tudor-SN can regulate the histone modification (15) and affect chromatin structure, resulting in the enhanced association of transcription factors with their target genes.
FIGURE 6.
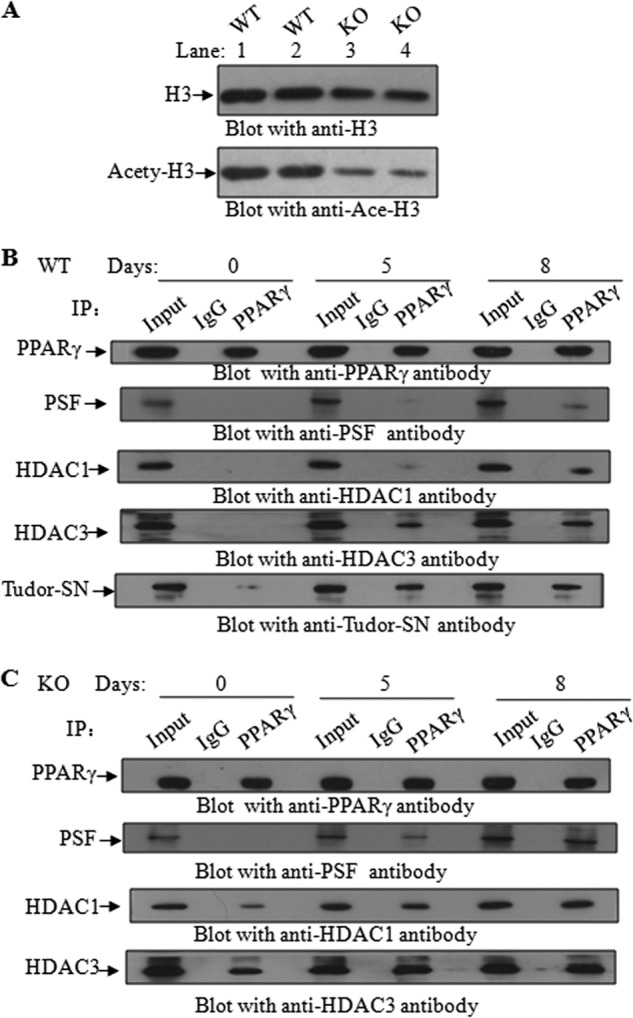
Tudor-SN affects H3 acetylation during adipogenesis. A, knock out of Tudor-SN reduces the acetylation level of histone H3. TCLs of MEF-WT and MEF-KO cells were immunoprecipitated (IP) with an anti-H3 antibody and then blotted with an anti-Ace-H3 antibody. B and C, TCLs of MEF-WT or MEF-KO cells at 0, 5, and 8 days of DMI treatment were immunoprecipitated with anti-PPARγ or anti-IgG as a negative control. The bound proteins were separated by SDS-PAGE and immunoblotted with anti-PPARγ, anti-PSF, anti-HDAC1, anti-HDAC3, and anti-Tudor-SN antibodies; 10% of the TCL was used as input.
DISCUSSION
Adipocyte differentiation or adipogenesis is a complex process that is regulated by a cascade of sequentially acting transcription factors and chromatin modifying coregulators. Palacios et al. (19) first reported that the rat homologue of human Tudor-SN, SND p102, was present in newly formed lipid droplets in liver parenchyma and cultured hepatocytes. They further demonstrated a specific, positive association of SND p102 and phospholipid in lipoprotein particles in hepatocytes. Therefore, Tudor-SN might be involved in adipogenesis, although the mechanism is not clear. The nuclear receptor PPAR, which is a ligand-activated transcription factor, is considered the master regulator of adipogenesis. Moreover, PPARγ is the most extensively characterized member of the PPAR family (23, 26). The results presented in this study indicate that Tudor-SN is likely a novel key component of PPARγ coactivators in adipogenesis.
During adipocyte differentiation, treatment with a hormonal mixture (DMI) induces the expression of transcription factor C/EBPs, which in turn recognize the promoter regions of the target genes, thereby inducing and enhancing gene transcription and protein expression. In the present study we demonstrated that the promoter region of Tudor-SN gene also contains a C/EBP binding sequence, although recognition and association are dependent on DMI treatment. Both the mRNA and protein levels of Tudor-SN increased in a time-dependent manner with DMI treatment, which is consistent with the expression pattern of PPARγ during adipogenesis. Accordingly, the physiological association of Tudor-SN and PPARγ was also enhanced in both 3T3 L1 cells and MEF cells.
The interaction between Tudor-SN and PPARγ suggests that these two proteins may function in the same complex and that Tudor-SN might have potential functions in regulating the expression of adipose factors. Furthermore, the association of PPARγ and Tudor-SN in primary pre-adipocytes supports the physiological relevance of Tudor-SN in adipogenesis. Subcellular fractionation assays indicated that Tudor-SN and PPARγ are mainly present in the nucleus at day 8 of adipogenesis, which further suggests that the predominant expression pattern of Tudor-SN in adipogenesis is similar to that of PPARγ. In general, Tudor-SN is mainly localized in the cytoplasm in 3T3-L1 pre-adipocytes, and the translocation of Tudor-SN from the cytoplasm to the nucleus may be due to phospho-modification of Tudor-SN caused by DMI treatment. For instance, we have identified several threonine or serine phosphorylation sites in the Tudor-SN protein under different stress stimulations (data not shown), although the underlying mechanism remains to be established.
In addition, we further demonstrated that the Tudor-SN-PPARγ complexes could recognize and interact with the PPRE of the PPARγ target genes and subsequently activate gene transcription. We observed the interaction of aP2-PPRE with both PPARγ and Tudor-SN as well as the enhancement of gene transcriptional activation of PPRE-luc upon ectopic expression of Tudor-SN. By contrast, deletion of the Tudor-SN protein (in MEF-KO) affected but did not completely abolish the association of PPARγ with aP2-PPRE. Consistent with this concept, loss-of-function studies demonstrated that Tudor-SN is required for DMI-induced adipogenesis in vivo: 1) in MEF-KO cells deletion of Tudor-SN impairs DMI-induced adipocyte differentiation; 2) in MEF-KO cells DMI treatment did not induce the enhanced expression of PPARγ target genes, such as aP2 and adipsin; 3) the expression of PPARγ and its upstream transcriptional factors were significantly increased in MEF-KO cells (data not shown), which indicates that the low expression of adipokines caused by Tudor-SN deletion cannot be restored despite compensatory up-regulation of PPARγ and other related transcription factors. These observations demonstrate that Tudor-SN is required for adipocyte differentiation and is vital for the successful transcriptional activity of several genes involved in adipogenesis. Interestingly, Tudor-SN is not detectable in the primary mature adipocytes of C57BL/6N mice, consistent with our recent findings that Tudor-SN is absent in the terminally differentiated cells known as end cells, which no longer have the ability to divide, such as polymorphonuclear leukocytes (data not shown). Tudor-SN might play important roles in adipogenesis in vivo and then gradually degrade in mature adipocytes. The molecular mechanism is now under investigation.
The three-dimensional structure of Tudor-SN indicates that it is composed of five repeats of SN-like (staphylococcal nuclease) domains followed by a Tudor domain. The SN-like domains of Tudor-SN lack catalytic activity and have been proposed to function as interaction domains. For example, Tudor-SN acts as a coactivator by interacting with STAT6 and STAT5 through the SN-like domains (21). Accordingly, we clarified that the SN domain but not the Tudor domain of the Tudor-SN protein associates with PPARγ. We previously identified Tudor-SN as a coactivator that increases gene transcription by enhancing histone acetylation and through the recruitment and stabilization of transcriptional complexes (15). We previously reported that Tudor-SN brings CBP to STAT6 response elements, resulting in enhanced histone acetylation activity and nucleosome unfolding and facilitating the access of the STAT6-Tudor-SN protein complex to the basal transcription machinery (15, 21). Consistent with this concept, as a novel co-activator of PPARγ, the absence of Tudor-SN might negatively affect protein-protein interactions, causing inadequate docking or linkage between CBP/p300 and the polymerase II basal transcription machinery and resulting in failed adipogenesis. In support of this mechanism 1) we observed a lower level of histone acetylation in MEF-KO cells, and 2) PSF has been reported to inhibit transcription by recruiting the mSin3A-HDAC complex to deacetylate histones and modulate chromatin structure, whereas we observed increased HDAC1 or HDAC3 in the PPARγ-associated transcription complex in the absence of Tudor-SN in MEF-KO cells, even with DMI treatment. The effect of Tudor-SN on PPARγ-mediated adipogenesis is similar to that of PPAR-interacting protein (PRIP) and PPARγ-binding protein (PBP), which also play important roles in mediating adipocyte differentiation. PBP has emerged as a critical component in the large TRAP-DRIP-ARC-Mediator complex, which plays an important role in connecting CBP/p300-bound coactivators with the polymerase II-containing preinitation complex (27), whereas PPAR-interacting protein (PRIP) and the PRIP-binding protein PIMT were recently shown to serve as linkers between CBP- and PBP-anchored cofactor complexes (28). Our data further demonstrate a functional link between histone acetylation/deacetylation in the regulation of PPARγ-mediated adipogenesis.
In summary, the present study identified Tudor-SN as a novel co-activator of PPARγ in adipogenesis (Fig. 7). The results presented here provide the first evidence that Tudor-SN and the nuclear transcription factor PPARγ exist in the same complex and that both are regulated by the early factor C/EBPβ during adipogenesis and exert significant influence on the regulation of PPARγ target genes. PPARγ requires various co-activators or co-repressors, which may dynamically associate with and regulate the higher-order chromatin remodeling of the promoter region of PPARγ-bound genes to regulate the transcription of its target genes. For instance, PPARγ is likely to associate with either the histone acetyltransferase (Tudor-SN-CBP-PBP) complex as a coactivator to enhance histone acetylation or with the HDAC (PSF-HDAC1) complex as a co-repressor to inhibit transcriptional activity through deacetylation. This novel finding may have implications for the development of new strategies for the prevention and treatment of obesity. We are currently working on Tudor-SN transgenic and knock-out mice to confirm the role of Tudor-SN in adipogenesis under physiological and pathological conditions and related mechanisms.
FIGURE 7.

Tudor-SN is a novel coactivator of PPARγ in adipogenesis.
This work was supported by grants from the National Science Foundation for Distinguished Young Scholars of China (31125012) and the National Science Foundation of China (31370749, 31100967, 31170830).
J. Saarikettu, T. Fashe, E. Arretxe, M. Pesu, I. Junttila, J. Partanen, P. Sipilä, M. Poutanen, J. Yang, and O. Silvennoinen, submitted for publication.
- C/EBP
- CCAAT/enhancer-binding protein
- MEF
- mouse embryonic fibroblast
- PPARγ
- peroxisome proliferator-activated receptor γ
- SN
- staphylococcal nuclease
- TCL
- total cell lysate
- PSF
- PTB-associated splicing factor
- PPRE
- PPARγ response element
- DMI
- dexamethasone, 3-isobutyl-1-methylxanthine, and insulin
- ORO
- Oil Red O
- CBP
- cAMP-response element-binding protein (CREB)-binding protein
- H3
- histone3
- HDAC
- histone deacetylase
- SND
- staphylococcal nuclease domain
- PBP
- PPARγ-binding protein.
REFERENCES
- 1. Rosen E. D., Spiegelman B. M. (2006) Adipocytes as regulators of energy balance and glucose homeostasis. Nature 444, 847–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Flier J. S. (2004) Obesity wars. Molecular progress confronts an expanding epidemic. Cell 116, 337–350 [DOI] [PubMed] [Google Scholar]
- 3. Spalding K. L., Arner E., Westermark P. O., Bernard S., Buchholz B. A., Bergmann O., Blomqvist L., Hoffstedt J., Näslund E., Britton T., Concha H., Hassan M., Rydén M., Frisén J., Arner P. (2008) Dynamics of fat cell turnover in humans. Nature 453, 783–787 [DOI] [PubMed] [Google Scholar]
- 4. Morrison R. F., Farmer S. R. (2000) Hormonal signaling and transcriptional control of adipocyte differentiation. J. Nutr. 130, 3116S–3121S [DOI] [PubMed] [Google Scholar]
- 5. Rosen E. D., Walkey C. J., Puigserver P., Spiegelman B. M. (2000) Transcriptional regulation of adipogenesis. Genes Dev. 14, 1293–1307 [PubMed] [Google Scholar]
- 6. Tontonoz P., Spiegelman B. M. (2008) Fat and beyond. The diverse biology of PPARγ. Annu. Rev. Biochem. 77, 289–312 [DOI] [PubMed] [Google Scholar]
- 7. Zhu Y., Qi C., Korenberg J. R., Chen X. N., Noya D., Rao M. S., Reddy J. K. (1995) Structural organization of mouse peroxisome proliferator-activated receptor γ (mPPAR γ) gene. Alternative promoter use and different splicing yield two mPPAR γ isoforms. Proc. Natl. Acad. Sci. U.S.A. 92, 7921–7925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Vidal-Puig A., Jimenez-Liñan M., Lowell B. B., Hamann A., Hu E., Spiegelman B., Flier J. S., Moller D. E. (1996) Regulation of PPAR γ gene expression by nutrition and obesity in rodents. J. Clin. Invest. 97, 2553–2561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Janknecht R., Hunter T. (1996) Transcription. A growing coactivator network. Nature 383, 22–23 [DOI] [PubMed] [Google Scholar]
- 10. Kamei Y., Xu L., Heinzel T., Torchia J., Kurokawa R., Gloss B., Lin S. C., Heyman R. A., Rose D. W., Glass C. K., Rosenfeld M. G. (1996) A CBP integrator complex mediates transcriptional activation and AP-1 inhibition by nuclear receptors. Cell 85, 403–414 [DOI] [PubMed] [Google Scholar]
- 11. Tong X., Drapkin R., Yalamanchili R., Mosialos G., Kieff E. (1995) The Epstein-Barr virus nuclear protein 2 acidic domain forms a complex with a novel cellular coactivator that can interact with TFIIE. Mol. Cell. Biol. 15, 4735–4744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dash A. B., Orrico F. C., Ness S. A. (1996) The EVES motif mediates both intermolecular and intramolecular regulation of c-Myb. Genes Dev. 10, 1858–1869 [DOI] [PubMed] [Google Scholar]
- 13. Leverson J. D., Koskinen P. J., Orrico F. C., Rainio E. M., Jalkanen K. J., Dash A. B., Eisenman R. N., Ness S. A. (1998) Pim-1 kinase and p100 cooperate to enhance c-Myb activity. Mol. Cell 2, 417–425 [DOI] [PubMed] [Google Scholar]
- 14. Low S. H., Vasanth S., Larson C. H., Mukherjee S., Sharma N., Kinter M. T., Kane M. E., Obara T., Weimbs T. (2006) Polycystin-1, STAT6, and P100 function in a pathway that transduces ciliary mechanosensation and is activated in polycystic kidney disease. Dev. Cell 10, 57–69 [DOI] [PubMed] [Google Scholar]
- 15. Välineva T., Yang J., Palovuori R., Silvennoinen O. (2005) The transcriptional co-activator protein p100 recruits histone acetyltransferase activity to STAT6 and mediates interaction between the CREB-binding protein and STAT6. J. Biol. Chem. 280, 14989–14996 [DOI] [PubMed] [Google Scholar]
- 16. Paukku K., Yang J., Silvennoinen O. (2003) Tudor and nuclease-like domains containing protein p100 function as coactivators for signal transducer and activator of transcription 5. Mol. Endocrinol. 17, 1805–1814 [DOI] [PubMed] [Google Scholar]
- 17. Keenan T. W., Winter S., Rackwitz H. R., Heid H. W. (2000) Nuclear coactivator protein p100 is present in endoplasmic reticulum and lipid droplets of milk secreting cells. Biochim. Biophys. Acta 1523, 84–90 [DOI] [PubMed] [Google Scholar]
- 18. Broadhurst M. K., Wheeler T. T. (2001) The p100 coactivator is present in the nuclei of mammary epithelial cells, and its abundance is increased in response to prolactin in culture and in mammary tissue during lactation. J. Endocrinol. 171, 329–337 [DOI] [PubMed] [Google Scholar]
- 19. Palacios L., Ochoa B., Gómez-Lechón M. J., Castell J. V., Fresnedo O. (2006) Overexpression of SND p102, a rat homologue of p100 coactivator, promotes the secretion of lipoprotein phospholipids in primary hepatocytes. Biochim. Biophys. Acta 1761, 698–708 [DOI] [PubMed] [Google Scholar]
- 20. Gao X., Zhao X., Zhu Y., He J., Shao J., Su C., Zhang Y., Zhang W., Saarikettu J., Silvennoinen O., Yao Z., Yang J. (2012) Tudor staphylococcal nuclease (Tudor-SN) participates in small ribonucleoprotein (snRNP) assembly via interacting with symmetrically dimethylated Sm proteins. J. Biol. Chem. 287, 18130–18141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yang J., Aittomäki S., Pesu M., Carter K., Saarinen J., Kalkkinen N., Kieff E., Silvennoinen O. (2002) Identification of p100 as a coactivator for STAT6 that bridges STAT6 with RNA polymerase II. EMBO J. 21, 4950–4958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ntambi J. M., Buhrow S. A., Kaestner K. H., Christy R. J., Sibley E., Kelly T. J., Jr., Lane M. D. (1988) Differentiation-induced gene expression in 3T3-L1 pre-adipocytes. Characterization of a differentially expressed gene encoding stearoyl-CoA desaturase. J. Biol. Chem. 263, 17291–17300 [PubMed] [Google Scholar]
- 23. Rodríguez L., Ochoa B., Martínez M. J. (2007) NF-Y and Sp1 are involved in transcriptional regulation of rat SND p102 gene. Biochem. Biophys. Res. Commun. 356, 226–232 [DOI] [PubMed] [Google Scholar]
- 24. Dong L., Zhang X., Fu X., Zhang X., Gao X., Zhu M., Wang X., Yang Z., Jensen O. N., Saarikettu J., Yao Z., Silvennoinen O., Yang J. (2011) PTB-associated splicing factor (PSF) functions as a repressor of STAT6-mediated Ig ϵ gene transcription by recruitment of HDAC1. J. Biol. Chem. 286, 3451–3459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Haberland M., Carrer M., Mokalled M. H., Montgomery R. L., Olson E. N. (2010) Redundant control of adipogenesis by histone deacetylases 1 and 2. J. Biol. Chem. 285, 14663–14670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Farmer S. R. (2006) Transcriptional control of adipocyte formation. Cell Metab. 4, 263–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Qi C., Surapureddi S., Zhu Y. J., Yu S., Kashireddy P., Rao M. S., Reddy J. K. (2003) Transcriptional coactivator PRIP, the peroxisome proliferator-activated receptor γ (PPARγ)-interacting protein, is required for PPARγ-mediated adipogenesis. J. Biol. Chem. 278, 25281–25284 [DOI] [PubMed] [Google Scholar]
- 28. Misra P., Qi C., Yu S., Shah S. H., Cao W. Q., Rao M. S., Thimmapaya B., Zhu Y., Reddy J. K. (2002) Interaction of PIMT with transcriptional coactivators CBP, p300, and PBP differential role in transcriptional regulation. J. Biol. Chem. 277, 20011–20019 [DOI] [PubMed] [Google Scholar]