

Background: NPHP and BBS are closely related syndromes, but the underlying mechanisms are unclear.
Results: BBS11 promotes accumulation of NPHP7, changing the properties of NPHP7.
Conclusion: NPHP and BBS gene products may be involved in similar signaling pathways.
Significance: These findings may help to explain the clinical overlap between certain ciliopathies.
Keywords: Genetic Diseases, Protein Degradation, Protein Stability, Transcription Regulation, Ubiquitylation, Bardet-Biedl Syndrome, Ciliopathies, Nephronophthisis
Abstract
Although the two ciliopathies Bardet-Biedl syndrome and nephronophthisis share multiple clinical manifestations, the molecular basis for this overlap remains largely unknown. Both BBS11 and NPHP7 are unusual members of their respective gene families. Although BBS11/TRIM32 represents a RING finger E3 ubiquitin ligase also involved in hereditary forms of muscular dystrophy, NPHP7/Glis2 is a Gli-like transcriptional repressor that localizes to the nucleus, deviating from the ciliary localization of most other ciliopathy-associated gene products. We found that BBS11/TRIM32 and NPHP7/Glis2 can physically interact with each other, suggesting that both proteins form a functionally relevant protein complex in vivo. This hypothesis was further supported by the genetic interaction and synergist cyst formation in the zebrafish pronephros model. However, contrary to our expectation, the E3 ubiquitin ligase BBS11/TRIM32 was not responsible for the short half-life of NPHP7/Glis2 but instead promoted the accumulation of mixed Lys48/Lys63-polyubiquitylated NPHP7/Glis2 species. This modification not only prolonged the half-life of NPHP7/Glis2, but also altered the subnuclear localization and the transcriptional activity of NPHP7/Glis2. Thus, physical and functional interactions between NPHP and Bardet-Biedl syndrome gene products, demonstrated for Glis2 and TRIM32, may help to explain the phenotypic similarities between these two syndromes.
Introduction
Nephronopthisis (NPH)3 is a rare autosomal recessive cystic kidney disease with distinct extra-renal manifestations. The underlying gene defects are heterogeneous, and more than 20 genes have been identified that can cause NPH or NPH-like disease. Although the gene products share no sequence similarities, most of them localize to the primary (or motile) cilium, a microtubular organelle attached to most body cells. Hence, NPH is considered a member of the growing family of ciliopathies, i.e., a group of genetic diseases that affect multiple organs and tissue through developmental abnormalities caused by defective cilia (1). NPH typically involves severe kidney abnormalities that cause kidney failure within the first two decades of life (2). In most cases, cysts and fibrotic tissue replace the normal kidney parenchyma. One prominent manifestation characteristic for NPH is a degeneration of the retina (Senior-Loken syndrome, retinitis pigmentosa), often leading to blindness. NPH shares cerebellar defects including an aplasia of the cerebellar vermis, associated with ataxia and oculomotoric abnormalities with the closely related Joubert syndrome (JBTS). Although many perturbations have been observed in cultured cells and animal model systems associated with the loss of NPH proteins (NPHPs), the molecular functions of NPHPs have remained largely elusive. In the cilium, established primarily in Caenorhabditis elegans, NPH proteins seem to play an essential role in establishing the transition zone and act in modules to contribute to gate-keeping function of the proximal ciliary axoneme (3–5). Genetic analysis in combination with high resolution confocal imaging revealed that NPHPs act in concert with proteins mutated in Meckel-Gruber syndrome (MKS) (5). MKS is like NPH a rare autosomal recessive multisystem disease that typically results in more severe manifestations than NPH, often associated with prenatal death. MKS proteins together with NPHPs are essential for forming the transition zone, a proximal region of the cilium that corresponds to the connecting cilium of the photoreceptor and is highly enriched for NPHPs. A proteomics screen, using NPHPs as baits, recently supported the idea that NPHPs and MKS proteins act in large protein complexes (6). Surprisingly, this screen did not reveal a direct link to a fourth closely related ciliopathy syndrome, the Bardet-Biedl syndrome (BBS), which is characterized by renal cysts, polydactyly, retinal degeneration, hepatic fibrosis, cerebellar abnormalities, situs inversus, obesity, and mental retardation (7). In addition to 12 designated BBS genes (BBS1–12), MKS1 and NPHP6 (CEP290) have been identified in patients with BBS manifestations, underlining the close link between these four ciliopathies.
NPHP7 is an unusual NPH family member. It is the only known transcriptional regulator linked to the NPH/JBTS/MKS/BBS ciliopathies. Currently, only two families have been identified with recessive NPHP7 mutations (8, 9). NPHP7 was originally described as a Gli-related Krüppel-like zinc finger protein (Glis2) (10, 11) that represses transcription through interaction with CtBP1 (C-terminal binding protein 1) (12). NPHP7/Glis2 is one of three family members (GLIS1–3) that are closely related to the Gli family of transcription factors. Glis2 contains five C2H2-type zinc fingers, which are required for DNA recognition. NPHP7/Glis2 antagonizes canonical Wnt signaling through interaction with β-catenin (13) and suppresses Hedgehog signaling and opposes Gli1 activity by binding to the cis-regulatory sequences of Snai1 and Wnt4 (14). Recently, NPHP7/Glis2 was identified as a fusion partner of CBFA2T3 in acute myeloid leukemia, implicating NPHP7/Glis2 in the control of bone morphogenetic protein (15, 16). NPHP7/Glis2 is required to maintain a normal renal structure and function; deletion of NPHP7/Glis2 in mice shortens the life span of these animals because of the development of renal fibrosis; gene expression profiling suggests that NPHP7/Glis2 suppresses genes involved in inflammation, fibrosis, and tissue remodeling (8, 17). In the absence of NPHP7/Glis2, gene products implicated in epithelial to mesenchymal transition are up-regulated, including TGFβ, Vimentin, Snail, and Slug. NPHP7/Glis2 localizes primarily to the nucleus, requiring a region within zinc finger 3 (18), and can recruit other interacting proteins such as p120 catenin to the nucleus (19). However, both NPHP7/Glis2 and Glis3 have also been identified in the primary cilium (8, 20). Based on their relationship to Gli family members, it has been speculated that accumulation of NPHP7/Glis2 and Glis3 within the ciliary compartment is controlled by a signaling pathway similar to the Hedgehog pathway, known to trigger processing and activation of Gli family members within the cilium (reviewed in Ref. 21).
BBS11/TRIM32, similar to Glis2, is an outsider within the BBS family. Only one family with typical BBS manifestations (obesity, retinopathy, polydactyly, hypogonadism, and renal and cardiac abnormalities) has been described so far, harboring a mutation (P130S) in the B-box of TRIM32/BBS11 (22). BBS11/TRIM32 is a member of the TRIM family, characterized by a tripartite TRIM/RBCC motif (RING, B-box, coiled-coil) (23). TRIM proteins were originally identified as regulators of innate immunity that are produced in response to interferons (24) but are now implicated in a broad range of functions and abnormalities, including transcriptional regulation, muscle homeostasis, and cancer (25–27). BBS11/TRIM32 contains six C-terminal NHL repeats, and a mutation located within the third NHL repeat (D487N) is associated with limb girdle muscular dystrophy type 2H, an autosomal recessive muscular disorder (28). Other mutations within the C-terminal domain (29), as well as corresponding mouse and Drosophila models (26, 30, 31), confirm the role of BBS11/TRIM32 in muscle homeostasis. Mediated by the RING domain, BBS11/TRIM32 acts as an E3 ubiquitin ligase and promotes degradation of several targets, including actin (32), PIASγ (33), Abl interactor 2 (34), dysbindin (35), X-linked inhibitor of apoptosis (XIAP) (36), p73 transcription factor (37), and thin filaments and Z-bands during fasting (38).
We observed that NPHP7/Glis2 is a labile protein with a short half-life. Because BBS11/TRIM32 physically and genetically interacted with NPHP7/Glis2, we speculated that NPHP7/Glis2 is a substrate of the E3 ubiquitin ligase BBS11/TRIM32. However, BBS11/TRIM32 facilitated the accumulation of ubiquitylated NPHP7/Glis2, partially co-localized with NPHP7/Glis2 in the nucleus, facilitated the accumulation of Glis/NPHP7 in nuclear subcompartments, and altered the transcriptional profile of NPHP7/Glis2. Collectively, our data demonstrate a functional and genetic link between members of the NPH and BBS gene family, suggesting that simultaneous mutations in both family members might occur and determine the clinical manifestations.
EXPERIMENTAL PROCEDURES
Reagents and Plasmids
Full-length human NPHP7/Glis2 (NM_032575) and BBS11/TRIM32 (NM_012210) were synthesized by GeneArt (Invitrogen). Full-length and truncated versions with different N-terminal tags (FLAG, V5, and YFP) were generated in expression vectors (PCDNA6; Invitrogen) as previously described (39, 40). Full-length PML in PCDNA6 with V5 and FLAG tag was generated from a clone containing the full-length PML cDNA (ImaGenes). The luciferase reporter containing the mIns2 promoter was kindly provided by Dr. K. Fererri, and the Gli1.eGFP and luciferase reporter GBS12-Luc were kindly provided by Dr. R. Toftgard. TRIM32.P130S, TRIM32.D487N, and TRIM32ΔRING constructs were generated by quick change PCR with overlapping forward and reverse primers, using wild-type TRIM32 as a template. Ubiquitin was cloned into a HA-tagged pMT123 plasmid, using NotI and EcoRI restriction sites. K48R and K63R ubiquitin mutants were kindly provided by Dr. I. Dikic. The GST.TRIM32 fusion protein was generated by cloning full-length TRIM32 into GST-pGEX4T1, using Mlu and NotI sites. FLAG.TRIM28 was generated, using a full-length cDNA of TRIM28 (ImaGenes). MG132 (Calbiochem) was used at a concentration of 10 μm. Antibodies used in this study included mouse anti-FLAG M2 (Sigma), anti-V5 (Serotec), anti-GFP (MBL), anti-β-actin (Sigma-Aldrich), anti-HA 12CA5 (Roche Applied Science), anti-Lamin A (Sigma-Aldrich), anti-α-tubulin (sigma), anti-PML (PG-M3) (Santa-Cruz), anti-ubiquitin-K63 specific (Millipore), and anti-FK1 (Biomol) antibodies. Secondary HRP-coupled antibodies against rabbit and mouse IgG were from Dako and GE Healthcare. Cy3- and Cy5-conjugated antibodies were from Jackson ImmunoResearch.
Cell Culture and Transfections
DMEM supplemented with 10% FBS (Biochrome) was used to culture HEK 293T cells. The cells were transfected, using calcium phosphate or the TransPEI transfection method (Eurogentec, Cologne, Germany).
Co-immunoprecipitation and Western Blotting
After 24 h of transfection, cells were washed with PBS and lysed, using lysis buffer (20 mm Tris, pH 7.5, 1% Triton X-100, 50 mm NaCl, 50 mm NaF, 15 mm Na4P2O7, 0.1 mm EDTA) supplemented with protease inhibitor mixture complete (Roche Applied Science). Centrifugation (15,000 × g for 15 min at 4 °C) was followed by ultracentrifugation (100,000 × g for 30 min at 4 °C); the lysates were then incubated with 30 μl of FLAG M2 Sepharose beads (Sigma) for ∼2 h at 4 °C in an overhead shaker. After incubation, the beads were washed with lysis buffer, and the proteins were eluted in 2× Laemmli buffer supplemented with β-mercaptoethanol or DTT. The bound proteins were further resolved by SDS-PAGE, and the interaction was detected by Western blotting.
Ubiquitylation Assay
HEK 293T cells were transfected with the plasmids as indicated along with HA-tagged ubiquitin. After 24 h of transfection, the cells were washed with PBS and subjected to lysis using radioimmune precipitation assay buffer (1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 150 mm NaCl, 50 mm NaF, 2 mm EDTA, 13.7 mm Na2HPO4, 6.3 mm NaH2PO4). The lysates were clarified by ultracentrifugation and incubated with FLAG-M2 beads at 4 °C for 2 h. After washing four times with lysis buffer, bound proteins were further resolved by SDS-PAGE gel and stained with HA antibody (Roche Applied Science) to detect ubiquitylated proteins.
Immunofluorescence
HEK 293T cells were cultured on 6-well plates containing coverslips pretreated with polylysine to ensure the attachment of the cells. After 24 h of transfection, cells were fixed with 4% paraformaldehyde and treated with 0.25–0.5% Triton X-100 containing PBS. The cells were then blocked with (2% horse serum, 0.5% Tween 20) and incubated with primary antibody for 1 h. After removing the primary antibody, secondary antibody was added, followed by an additional incubation period of 30–45 min. After removing excess secondary antibody, nuclear staining was performed using DAPI. After several washing steps, coverslips were mounted on a clear microscopic slide, using prolong gold anti-fade reagent (Invitrogen). Images were captured with LSM 510 confocal or Apotome microscope (Zeiss).
Luciferase Assay
HEK 293T cells were split into 12-well plates and transiently transfected with the plasmids as indicated in triplicate along with the luciferase reporter construct and a β-galactosidase expression vector. After 24 h of transfection, the cells were lysed with Tropix lysis buffer (Applied Biosystems) for 15 min at room temperature. Lysates were centrifuged at 15,000 × g for 5 min. Supernatant was transferred to a 96-well plate, and luciferase activity was measured and normalized to β-galactosidase activity to correct for transfection efficiency.
Zebrafish Embryo Manipulation
zGlis2 and zTRIM32 sequences were described elsewhere (22, 41). The transgenic zebrafish line Wt1b::GFP was described recently (42). Antisense morpholino oligonucleotides (MO) were designed by Gene Tools to target either the translation start or an exon splice donor site causing splicing defects of the mRNA. The efficacy of the zTRIM32 MO was verified in zebrafish embryos, using GFP fused to the MO target sequence. The MOs were diluted in 200 mm KCl, 10 mm Hepes, and 0.1% phenol red (Sigma). The injection amounts varied between 0.1 and 0.5 mm (as indicated); the injection volume was 4.6 nl/embryo. Toxic side effects of injection reagent were compensated by co-injecting p53 morpholino (0.1 mm). In combined knockdown experiments, the total morpholino concentration was balanced to ∼0.3 mm using control morpholino.
RESULTS
Co-immunoprecipitation between Glis2 and TRIM32
Co-expression and immunoprecipitation of tagged proteins in HEK 293T cells revealed that Glis2 interacts with components of the BBSome (41) and TRIM32 (Figs. 1 and 2A). Recombinant GST-tagged TRIM32, incubated with the lysates of HEK 293T cells transiently transfected with FLAG-tagged Glis2, precipitated Glis2 (Fig. 2B). Truncational analysis mapped the interaction with TRIM32 to amino acids 141–359 of Glis2 (Fig. 2C), a region that includes the five zinc fingers between amino acids 168 and 317 (NCBI NP_115964). Reciprocal mapping revealed that the first 256 amino acids of TRIM32, encompassing the N-terminal Ring and B-box domain, were sufficient to immobilize Glis2 (Fig. 2D).
FIGURE 1.
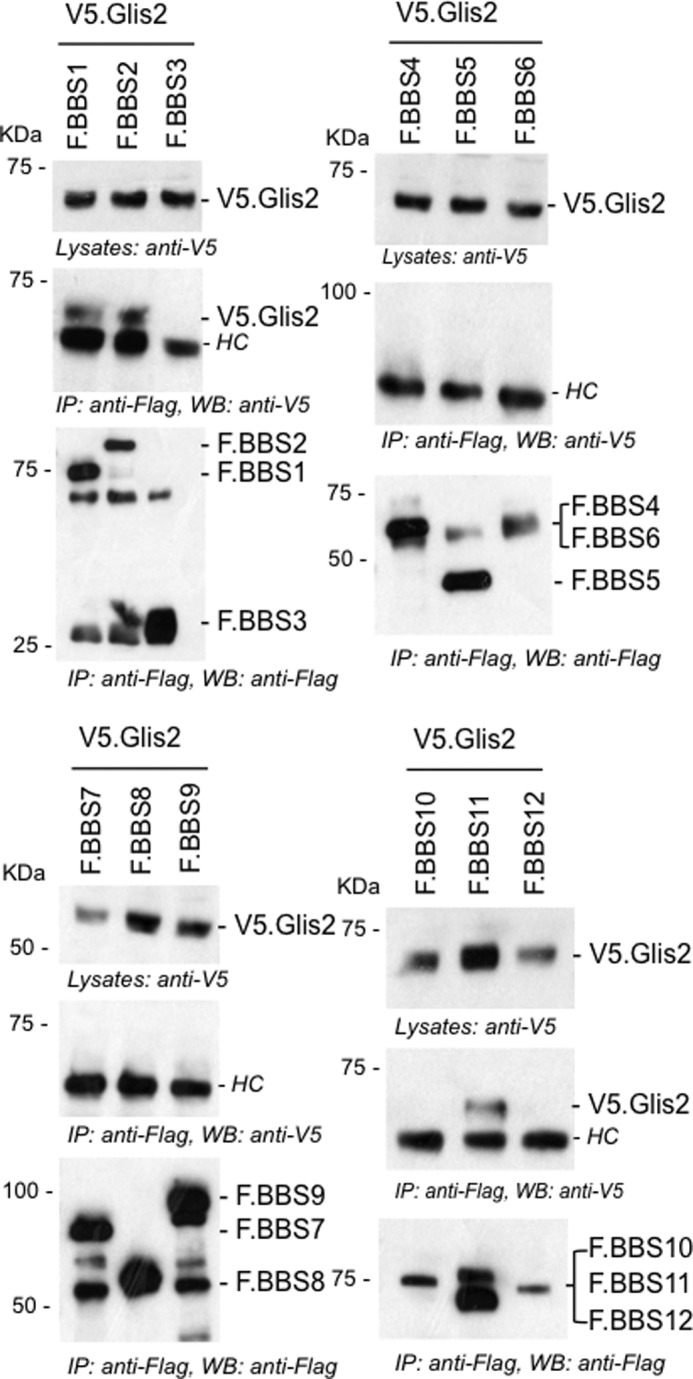
Glis2 interacts with BBS1, BBS2, and BBS11/TRIM32. FLAG-tagged BBS proteins (BBS1–12) were transiently co-transfected with V5.Glis2 into HEK 293T cells. After 24 h of transfection, cells were lysed, and FLAG-tagged BBS proteins were immunoprecipitated with FLAG-M2 beads. The precipitates were loaded onto 10% SDS gel and analyzed by Western blotting using anti-V5 and anti-FLAG antibodies. V5.Glis2 interacted with F.BBS1, F.BBS2, and F. BBS11/TRIM32, but not with the other BBS proteins. Antibody heavy chains were marked with HC. IP, immunoprecipitation; WB, Western blotting.
FIGURE 2.
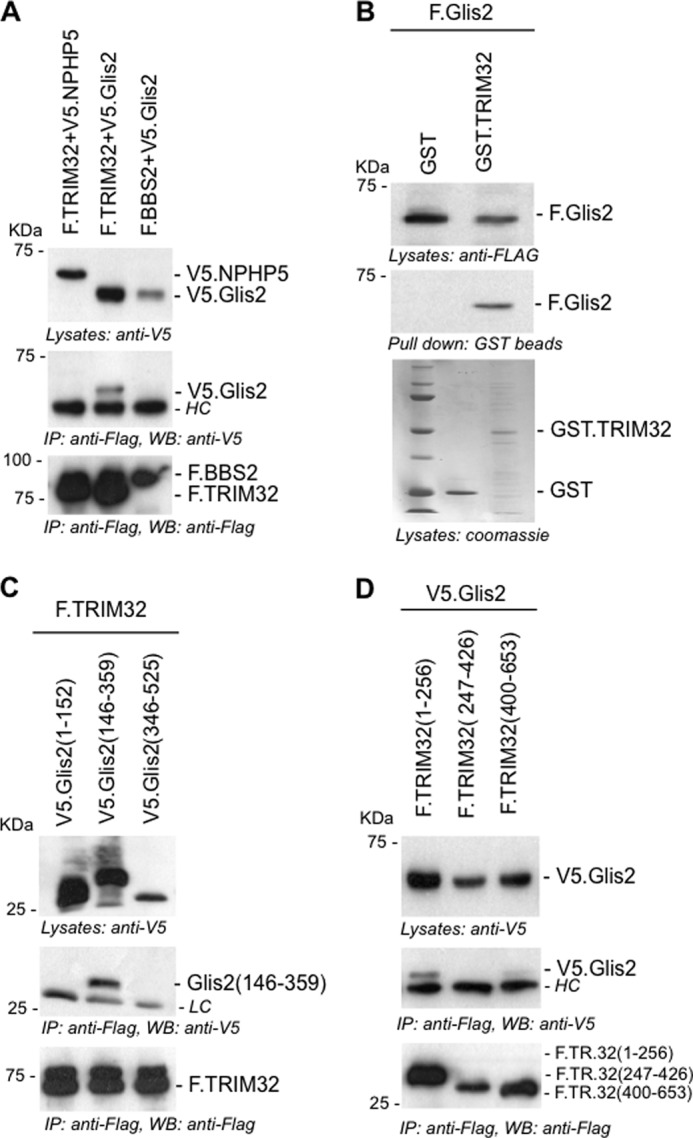
TRIM32 interacts with the zinc finger domains of Glis2. A, FLAG-tagged TRIM32 (F.TRIM32) or F.BBS2 were transiently co-expressed in HEK 293T cells with V5-tagged Glis2 (V5.Glis2) or V5.NPHP5. Precipitation of F.TRIM32 immobilized V5.Glis2, but not V5.NPHP5. B, lysates of HEK 293T cells, transiently transfected with F.Glis2, were incubated with recombinant GST.TRIM32 or GST. F.Glis2 was pulled down by GST.TRIM32 but not by GST. Expression levels of GST.TRIM32 and GST were checked by Coomassie staining. C, truncations of Glis2, V5.Glis2 (amino acids 1–152), V5.Glis2 (amino acids 346–525), and V5.Glis2 (amino acids 146–359) were generated to map the interaction with TRIM32. F.TRIM32 interacted with the zinc finger-containing fragment of V5.Glis2, spanning amino acids 146–359. D, FLAG-tagged fragments of TRIM32 were co-expressed with V5.Glis2. Only the N-terminal fragment of TRIM32, spanning amino acids 1–256, interacted with Glis2. HC, antibody heavy chains; LC, antibody light chains; IP, immunoprecipitation; WB, Western blotting.
Genetic Interaction between TRIM32 and Glis2
MO-mediated knockdown of Glis2 in zebrafish causes pronephric cyst formation and other abnormalities including cilia motility (41). Depletion of TRIM32 in zebrafish reportedly reduces the size of the Kupffer's vesicle and inhibits melanosome transport (33). To establish a genetic interaction between TRIM32 and Glis2, we performed knockdown of zTRIM32 targeting the translational initiation site (ATG MO). Knockdown of zTRIM32 resulted in pronephric cyst formation in a dose-dependent manner (Fig. 3A), supporting the hypothesis that TRIM32 mutations can be associated with a renal phenotype. Individual depletion of zTRIM32 and zGlis2 resulted in 25 and 15% of cyst formation, approximately. However, in combined zGlis2/zTRIM32 knockdown, we observed that pronephric cyst formation was enhanced to ∼60% (Fig. 3B), indicating a genetic interaction between these two gene products.
FIGURE 3.
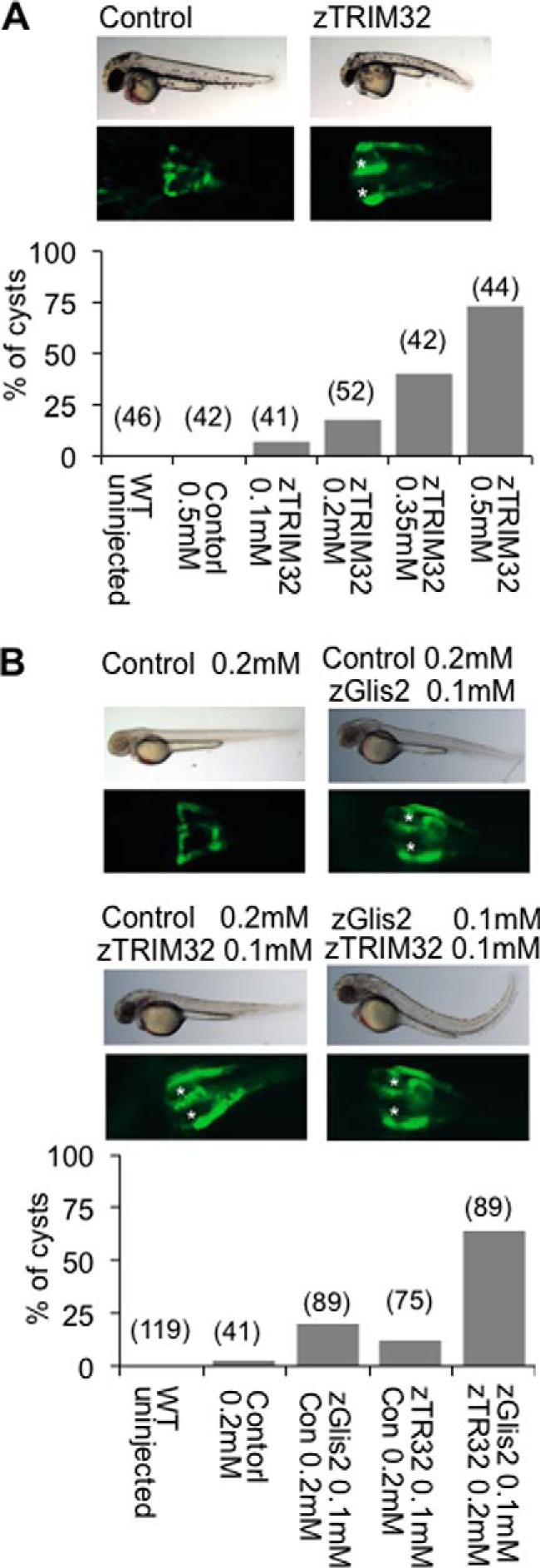
Combined depletion of zGlis2 and zTRIM32 enhances cyst formation of the zebrafish pronephros. A, WT1-GFP transgenic zebrafish embryos, highlighting the proximal pronephros, were injected with control or zTRIM32-specific MO and scored for cyst formation at 55 h postfertilization. Knockdown of zTRIM32 resulted in pronephric cyst formation in a dose-dependent manner. The pronephric cysts of zTRIM32-deficient zebrafish embryos are marked with asterisks in the GFP-labeled pronephric ducts. No cyst formation was observed in zebrafish embryos injected with control MO. B, zebrafish embryos were injected with different MO combinations as indicated. Low doses of zTRIM32 MO enhanced the cyst formation in zebrafish embryos injected with zGlis2 MO.
TRIM32 Facilitates Accumulation of Glis2
Several attempts failed to detect endogenous Glis2 by Western blot analysis (Ref. 14), suggesting that Glis2 is a labile transcriptional regulator present only at very low levels. Because TRIM32 is an E3 ubiquitin ligase that targets several proteins for proteasomal degradation (33–38), we hypothesized that TRIM32 interacts with Glis2 to curtail Glis2 protein levels. Surprisingly, co-expression of TRIM32 increased the protein levels of Glis2 (Fig. 4A). Consistent with the proteasomal degradation of Glis2, the proteasome inhibitor MG132 increased the levels of Glis2 in transfected HEK 293T cells; however, the increase in protein Glis2 protein levels was even more pronounced by TRIM32. MG132 had no further effect on Glis2 protein levels in the presence of TRIM32 (Fig. 4B), suggesting that TRIM32 acts in the same pathway, preventing ubiquitylation-dependent protein degradation. Accumulation of ubiquitylated Glis2 in the presence of MG132 further confirmed that Glis2 protein levels are controlled by ubiquitylation-mediated proteasomal degradation (Fig. 4C). Inhibition of protein synthesis by cycloheximide revealed that co-expression of TRIM32 prolonged the half-life of Glis2 (Fig. 4, D and E), supporting the hypothesis that TRIM32 interferes with the proteasomal degradation of Glis2. In contrast, TRIM32 did not alter the protein levels of Glis3, another Glis family member (Fig. 4F). BBS1, which also interacted with Glis2, did not affect Glis2 protein levels (Fig. 4G), whereas noninteracting truncations of TRIM32 did not alter Glis2 protein stability (Fig. 4H). These findings suggest that TRIM32 specifically targets Glis2 but not the other Glis proteins and that an interaction with TRIM32 is required to promote the accumulation of Glis2 protein.
FIGURE 4.

TRIM32 stabilizes Glis2. A, HEK 293T cells were transiently transfected with V5.Glis2 together with a control vector, F.TRIM32, or F.CD2AP. After 24 h of transfection, cells were lysed; proteins were resolved on SDS-PAGE gel and detected by Western blotting, using anti-V5 and anti-FLAG antibodies. Protein levels of Glis2 were quantified using LabImage 1D software and normalized to β-actin protein levels. The bars (right panel) represent the relative protein levels from three independent experiments. B, HEK 293T cells, transfected with V5.Glis2, were treated with DMSO or the proteasomal inhibitor MG132 (12 μm) for 2 h and co-transfected with F.TRIM32 as indicated. MG132 treatment increased Glis2 protein levels but had no apparent effect in the presence of TRIM32. The bars (right panel) represent the quantification of Glis2 protein levels normalized to β-actin levels from three independent experiments. C, HEK 293T cells were transfected with F.Glis2 and HA-tagged ubiquitin. After 24 h of transfection, cells were treated with MG132 or DMSO (control). F.Glis2 was precipitated using FLAG-M2 beads. Ubiquitylated Glis2 (Ubi-Glis2) was detected using an anti-HA antibody, demonstrating an increase of ubiquitylated Glis2 species in the presence of MG132. D, HEK 293T cells, transfected with Glis2 alone or together with TRIM32, were treated with cycloheximide (30 μg) to inhibit protein synthesis. Glis2 protein levels were followed over a time course of 9 h by Western blot analysis. Glis2 protein levels remained stable in the presence of TRIM32. Anti-β-actin staining was used to control for equal loading. E, the graph demonstrates the decline of Glis2 protein levels after inhibition of protein synthesis with cycloheximide in the absence (purple line) or presence (blue line) of TRIM32. The results represent the average of three experiments; β-actin was used as a loading control (see Fig. 3D). F, protein levels of V5.Glis3 remained unaffected in the presence of different levels of F.TRIM32. G, protein levels of Glis2 were not increased by the presence of BBS1. H, the N-terminal truncation of F.TRIM32 (1–256), interacting with Glis2, was sufficient to increase Glis2 protein levels, whereas noninteraction F.TRIM32 truncations failed to affect Glis2 protein levels. IP, immunoprecipitation; WB, Western blotting.
TRIM32 Promotes the Accumulation of K63-linked Polyubiquitylated Glis2
Because Glis2 levels are controlled by ubiquitylation and proteasomal degradation, whereas TRIM32 interferes with Glis2 degradation and prolongs its half-life, we investigated whether TRIM32 blocks ubiquitylation. Surprisingly, TRIM32 enhanced the ubiquitylation of Glis2 (Fig. 5A). The two patient mutations did not alter the ubiquitylation pattern of Glis2, and a TRIM32 fragment, encompassing the RING finger and the B-box, was sufficient to enhance ubiquitylation of Glis2 (Fig. 5B). We replaced amino acids Cys20, Cys39, and His41 by alanine to destroy the ubiquitin ligase activity of TRIM32 (35). The in vivo ubiquitin assays did not reveal any difference between the ubiquitylation of Glis2 in the presence of wild-type and mutant TRIM32. Only some of the higher molecular weight species were reduced in the presence of mutant TRIM32 (Fig. 5C), likely because the mutant TRIM32 no longer performed self-ubiquitylation. Polyubiquitin specific antibodies revealed that Glis2 is largely polyubiquitylated, which was increased by TRIM32 (Fig. 5D). Glis2 harbors one group II WW PPLP motif as well as several group IV WW (ST) P motifs recognized by HECT ubiquitin ligases (43). We tested several HECT ubiquitin ligases, including Nedd4-2, WWP1, WWP2, Smurf1, Smurf2, and AIP4; however, none of these E3 ligases had a significant effect on Glis2 ubiquitylation (Fig. 6A). The length of the ubiquitin chain, the targeted lysine, and the type of ubiquitin molecule attached to the target protein determine the downstream consequences of ubiquitylation (44). Polyubiquitin chains linked through K48 typically target proteins for proteasomal degradation, whereas K63-linked polyubiquitin chains control trafficking, activity, and interactions with other proteins (45). Ubiquitylation of Glis2 was reduced in the presence of the ubiquitin K48R mutant and slightly less by the ubiquitin K63R mutant (Fig. 6B), suggesting that the polyubiquitylated Glis2 contained more K48- than K63-linked ubiquitin chains. In the presence of the TRIM32 RING mutant, ubiquitylation of Glis2 was reduced both by both K48R and K63R of ubiquitin (Fig. 6C). To support this observation, we analyzed the ubiquitylated Glis2 in the presence of MG132 versus TRIM32, using ubiquitin K63 specific antibodies (Fig. 7). Although no K63-containing ubiquitin chains were detectable in the presence of MG132 (Fig. 7A), K63 ubiquitin-containing Glis2 became detectable in the presence of TRIM32 (Fig. 7B). Our findings suggest that Glis2 typically contains K48 polyubiquitin chains that target Glis2 for rapid degradation. TRIM32, although not directly involved in Glis2 ubiquitylation, appears to shift the ratio between K48 and K63 polyubiquitylation, facilitating the accumulation of K63-polyubiquitylated Glis2.
FIGURE 5.

TRIM32 promotes the accumulation of ubiquitylated Glis2. A, HEK 293T cells were transfected with HA-tagged ubiquitin in combination with the plasmids as indicated. After 24 h of transfection, F.Glis2 was precipitated using FLAG-M2 beads; ubiquitylated Glis2 (Ubi-Glis2, top panel) was detected using an anti-HA antibody. Autoubiquitylation of TRIM32 was detectable except for the TRIM32.D487N mutation (lane 5, bottom panel). B, the Glis2-interacting, N-terminal domain of TRIM32 (aminio acid 1–256) was sufficient to enhance ubiquitylation of Glis2. C, the accumulation of ubiquitylated Glis2 was preserved in the presence of E3 ligase-deficient TRIM32 ΔRING mutant. F.Glis2 was co-transfected with V5.TRIM32 lacking E3 ligase activity (TRIM32 ΔRING) and V5.TRIM32 WT. Consistent with the elimination of the E3 ligase activity, no autoubiquitylation was detectable for the TRIM32 ΔRING mutant (lane 4, bottom panel). D, HEK 293T cells were transfected with the plasmids as indicated. Polyubiquitylation of F.Glis2 was detected in the presence of the E3 ligase-deficient TRIM32 ΔRING mutant, using anti-Fk1 antibody. IP, immunoprecipitation; WB, Western blotting.
FIGURE 6.

TRIM32 promotes K63-linked ubiquitylation of Glis2. A, HECT E3 ubiquitin ligases did not increase Glis2 ubiquitylation. Ubiquitylation of F.Glis2 was monitored in the presence of different HECT E3 ubiquitin ligases, and ubiquitylated Glis2 was detected by an anti-HA antibody. B, HEK 293T cells were transfected with constructs as indicated, and ubiquitylation of F.Glis2 was compared in the presence of the ubiquitin K48R and K63R mutants. K48R ubiquitin reduced the ubiquitylation more efficiently, suggesting that in the absence of TRIM32, Glis2 is predominantly K48-ubiquitylated. C, in vivo ubiquitylation assay was performed to assess the ubiquitylation of Glis2 in the presence of wild-type HA.ubiquitin (Ubi-WT), HA.ubiquitin K48R (Ubi-K48R), and HA.ubiquitin K63R (Ubi-K63R). The TRIM32 ΔRING was used to prevent autoubiquitylation of TRIM32. Both K48R and K63R ubiquitin reduced Glis2 ubiquitylation to a similar degree, suggesting that TRIM32 shifts Glis2 ubiquitylation toward K63 ubiquitin chain species. IP, immunoprecipitation; WB, Western blotting.
FIGURE 7.
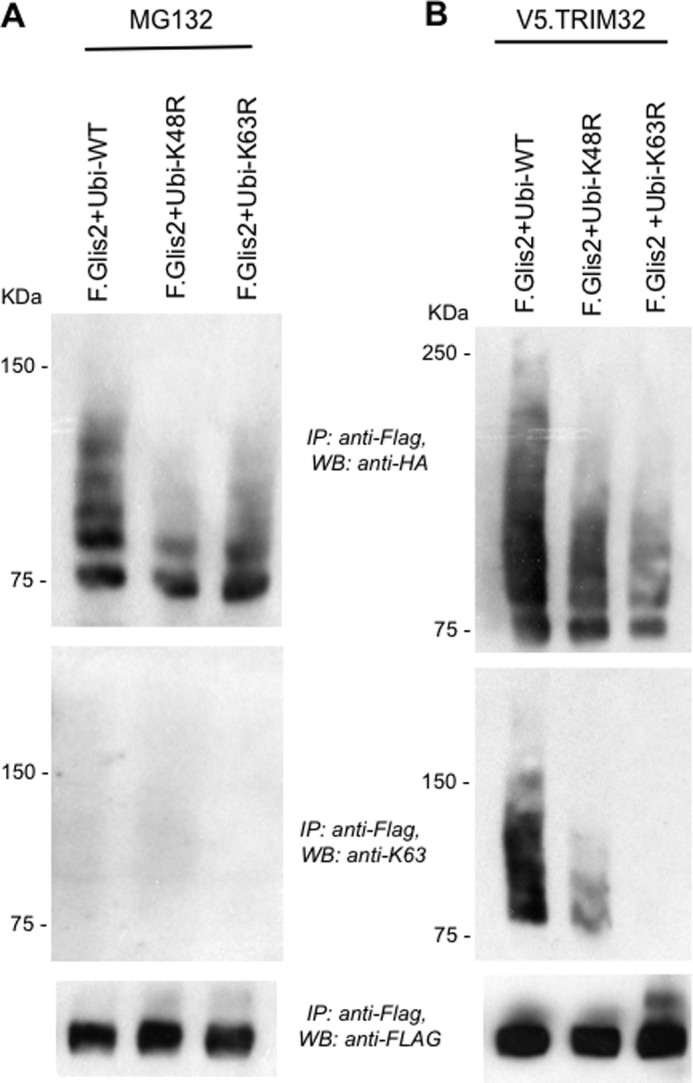
TRIM32 facilitates the accumulation of K63 ubiquitin chain-containing Glis2. Glis2 was co-expressed with HA-tagged, wild-type ubiquitin (Ubi-WT), HA-tagged ubiquitin K48R (Ubi-K48R), or HA-tagged ubiquitin K63R (Ubi-K63R) in HEK 293T cells. In A, cells were incubated for the last 2 h with 12 μm MG132. In B, cells were co-transfected with V5-tagged TRIM32 (V5.TRIM32). K63-linked ubiquitin chain accumulated in the presence of V5.TRIM32, but not after incubation with MG132, using a K63 specific antibody. IP, immunoprecipitation; WB, Western blotting.
TRIM32 Alters the Subcellular Localization of Glis2
TRIM32 has recently been reported to mediate K63-linked ubiquitylation of MITA; K63 polyubiquitylation is required for MITA to interact with TBK1 in response to viral infections (46). We speculated that the TRIM32-mediated shift in Glis2 K48/K63 polyubiquitylation may also alter the interaction partners of Glis2, affecting its subcellular localization. Immunofluorescence experiments in HEK 293T cells using YFP-tagged Glis2 (YFP.Glis2) revealed a diffuse nuclear localization of Glis2 consistent with previous observations (18) (Fig. 8A). Co-expression of FLAG-tagged TRIM32 changed the localization of Glis2 and recruited the transcriptional repressor into specific subnuclear compartments (Fig. 8A). The localization of Glis2 remained unchanged in the presence of TRIM28, another TRIM family member (Fig. 8B). The N-terminal truncation of TRIM32, mediating the interaction with Glis2, was sufficient to drive the recruitment of Glis2 (Fig. 8C). TRIM32, although predominantly present in the cytoplasm, partially co-localized with Glis2 in these subnuclear compartments (Fig. 8D). Glis2 interacted with PML in the presence of TRIM32, but not in the presence of CD2AP (Fig. 9). However, the TRIM32-mediated nuclear aggregates of Glis2 were larger than the antibody-labeled PML bodies and only in some cases appeared to encompass PML bodies (Fig. 8E). The striking changes of Glis2 in the presence of TRIM32 support the hypothesis that TRIM32 alters the Glis2 ubiquitylation pattern, which affects its interactions and subcellular localization.
FIGURE 8.
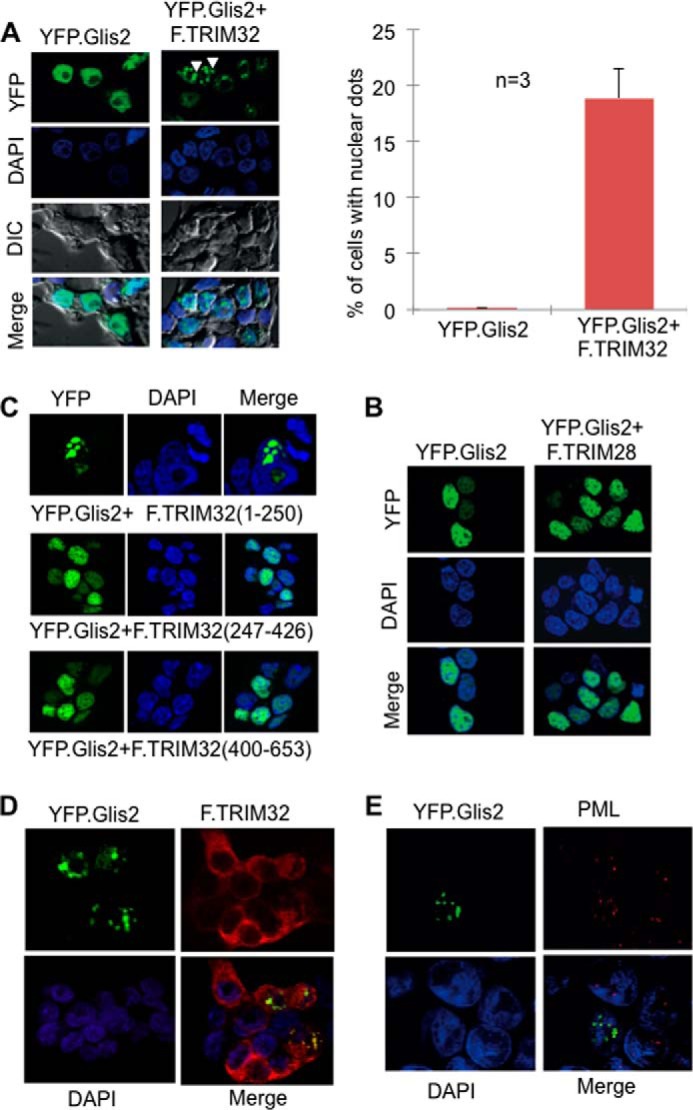
TRIM32 recruits Glis2 into subnuclear domains. A, immunofluorescence analysis of HEK 293T cells expressing YFP.Glis2 (green) alone or in the presence of TRIM32. Nuclei were stained with Hoechst blue. YFP.Glis2 alone showed a diffuse nuclear localization (left panel). The arrowheads point highlight the recruitment of YFP.Glis2 into specific subnuclear domains in the presence of TRIM32. Cells with Glis2 localizing nuclear dots were counted and quantified (right panel). DIC, differential interference contrast. B, the N-terminal domain of F.TRIM32, spanning the Glis2-interacting amino acids 1–256, is sufficient to drive the recruitment to nuclear aggregates (top panel), whereas the two noninteracting fragments (F.TRIM32 (amino acids 247–426) and F.TRIM32 (amino acids 400–653)) failed to alter the subcellular localization of Glis2. C, no recruitment of Glis2 was detected in the presence of TRIM28, another TRIM family member. D, YFP.Glis2 and F.TRIM32 partially co-localized within the nucleus, suggesting that Glis2 facilitates the nuclear import of TRIM32. YFP.Glis2 and FLAG-tagged TRIM32 were transiently co-expressed in HEK 293T cells. F.TRIM32 was detected using a polyclonal FLAG rabbit antibody followed by Cy3-conjugated donkey anti-rabbit IgG (red). E, only a subset of PML bodies (red) co-localized with the YFP.Glis2-positive nuclear aggregates in HEK 293T cells. Endogenous PML was detected using anti-PML mouse antibody followed by Cy3-conjugated donkey anti-mouse IgG.
FIGURE 9.

Glis2 interacts with PML only in the presence of TRIM32. HEK 293T cells were transfected with the plasmids as indicated. Precipitation of F.PML did not immobilize V5.Glis2 (left panel). However, V5.Glis2 was detectable in the immunoprecipitates of F.PML in the presence of V5.TRIM32 (middle panel), but not in the presence of a control protein (V5.CD2AP) (right panel). IP, immunoprecipitation; WB, Western blotting.
TRIM32 Modulates the Transcriptional Activities of Glis2
Glis2 interacts with β-catenin and has been shown to function as a repressor for TCF/LEF-dependent gene activation (13). TRIM32, although itself not displaying any inhibitory activity on β-catenin-mediated TOPflash activation, completely reversed the Glis2-mediated inhibition of β-catenin-dependent TOPflash activation (Fig. 10A). Although Glis2 interacts with CtBP1 to function as a transcriptional repressor in most instances (12), Glis2 activates the mIns2 (mouse insulin-2) promoter by binding to the Glis binding sites of the mIns2 promoter (18). Here, TRIM32 had a modest repressor function, inhibiting the Glis2-mediated mIns2 activation by ∼50% (Fig. 10B).
FIGURE 10.
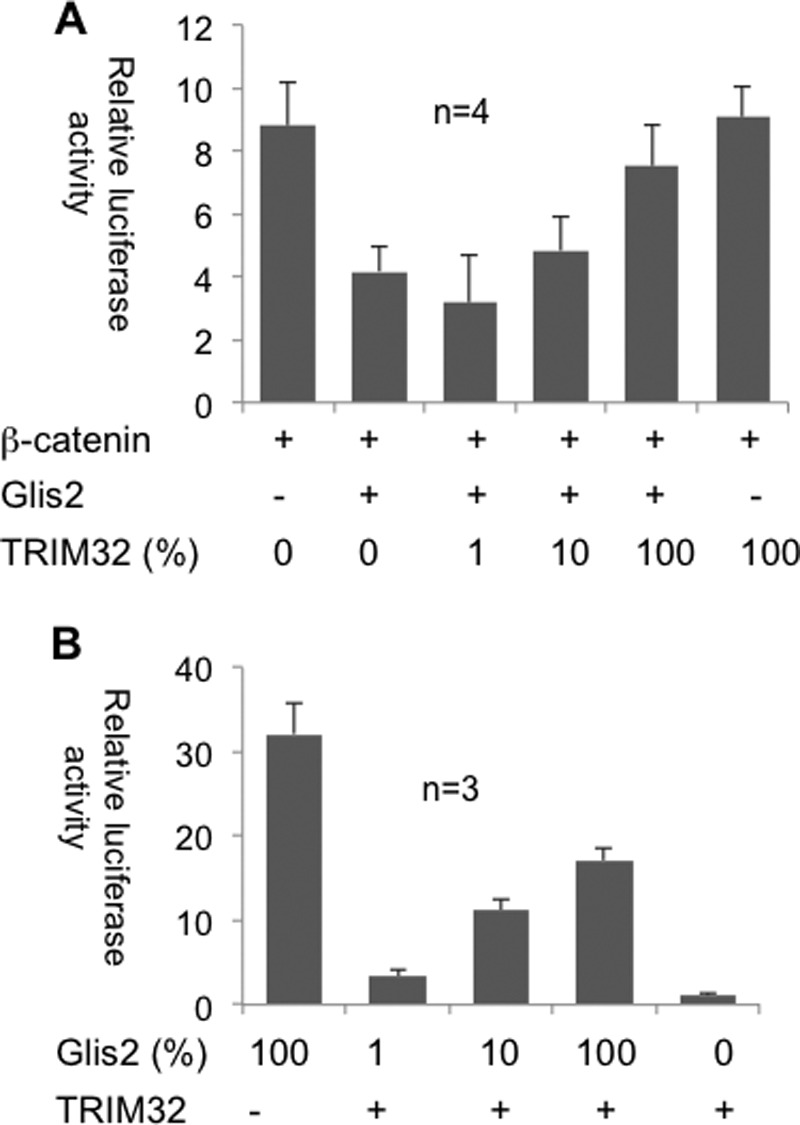
TRIM32 modulates the transcriptional activity of Glis2. A, the repressive effect of Glis2 on β-catenin-dependent transcriptional activity (TOPflash) was reversed by the co-transfection of TRIM32. Four independent experiments were performed in triplicate, and the values were normalized to β-galactosidase. B, TRIM32 inhibits the transcriptional activation of mIns2 (mouse insulin-2 promoter) by Glis2. HEK 293T cells were transfected with the depicted constructs to quantify the effect of TRIM32 on the transcriptional activation of the mIns2 promoter by Glis2.
DISCUSSION
Structural or functional defects of cilia cause a broad spectrum of human disease manifestations now collectively termed ciliopathies (1). Based on the combination of predominant phenotypes, ciliopathies have been grouped into distinct autosomal recessive syndromes. MKS, JBTS, BBS, and NPH are closely related and share several clinical features, including renal dysfunction, retinal degeneration, mental retardation, and cerebellar and hepatic abnormalities (7). The molecular basis for this clinical overlap is currently evolving. There is extensive genetic overlap, and MKS mutations have recently been shown to cause BBS and vice versa, whereas NPHP6/CEP290 has been identified in patients with MKS and JBTS (reviewed in Ref. 7). Seven BBS proteins (BBS1, 2, 4, 5, 7, 8, and 9) form the BBSome, a coat-like protein complex implicated in the trafficking of cargo molecules to the cilium (reviewed in Ref. 47). Although ciliary molecules have been linked to a BBSome-dependent transport, it is currently unknown whether MKS or NPHP gene products utilize BBSome-dependent trafficking to reach the cilium. We observed that NPHP7/Glis2 interacts with BBS1 and BSS2, but attempts to detect endogenous NPHP7/Glis2 by antibodies in native tissues have failed so far (Ref. 14 and data not shown),4 limiting the possibility to determine whether BBS11 and NPHP7 share overlapping trafficking pathways. One explanation for the difficulties to detect endogenous NPHP7/Glis2 appears to be its short half-life, caused by ubiquitylation-dependent proteasomal degradation. Because our candidate screen identified BBS11/TRIM32 as NPHP7/Glis2-interacting protein, we speculated that the RING finger E3 ubiqutin ligase BBS11/TRIM32 targets NPHP7/Glis2 for K48-linked polyubiquitylation and degradation. Surprisingly, BBS11/TRIM32 prevented the degradation of NPHP7/Glis2 and promoted its accumulation. We noted that in the presence of BBS11/TRIM32, a higher proportion of ubiquitylated NPHP7/Glis2 species contained K63-linked polyubiquitin chains, implicated in signaling or trafficking functions rather than degradation. Consistent with the proposed function of K63 chains, we found that the presence of BBS11/TRIM32 altered the intranuclear localization of NPHP7/Glis2, promoting the accumulation in large nuclear domains. BBS11/TRIM32 also facilitated the interaction with PML; however, the NPHP7/Glis2-positive intranuclear domains were substantially larger than PML bodies and only partially overlapped with PML bodies. TRIM32 facilitated the accumulation of ubiquitylated, K63-containing Glis2 species independent of its E3 ligase activity. Because this property required the interaction between TRIM32 and Glis2, TRIM32 could act as an adaptor protein that recruits ubiquitin E3 ligases to attach K63 chains to Glis2 or block interaction with the E3 ligase responsible for K48 ubiquitylation, thereby allowing other E3 ligases to attach K63 chains to Glis2.
Closely related to the Gli transcription factors, the Glis family members (Glis1–3) of transcriptional modulators have been established as important developmental regulators in Hedgehog (14) and Wnt (13) signaling and in the inhibition of pathways that promote epithelial to mesenchymal transition and fibrosis (8). Belonging to the subfamily of Krüppel-like zinc finger proteins, the Glis transcriptional regulators contain a highly conserved DNA-binding domain with five C2H2-type zinc finger motifs that recognize the consensus Glis DNA binding sequence (G/C)TGGGGGGT(A/C) (10, 11, 18). To determine whether BBS11/TRIM32 modifies the transcriptional profile of NPHP7/Glis2, we tested some of the recently established target genes. Surprisingly, BBS11/TRIM32 reversed the inhibitory effect of Glis2 on β-catenin-mediated TOPflash activation. This finding suggests that interaction with NPHP7/Glis2 may affect the nuclear import of BBS11/TRIM32, which then not only alters the intranuclear localization of NPHP7/Glis2 but also affects its transcriptional activation, for example by sequestering NPHP7/Glis2 away from LEF/TCF binding sites. Indeed, we observed increased nuclear BBS11/TRIM32 and co-localization with NPHP7/Glis2. The mouse insulin-2 promoter was recently discovered as a transcriptional target of NPHP7/Glis2. In contrast to most transcriptional repressor functions, NPHP7/Glis2 activates this promoter (18). Although BBS11/TRIM32 by itself had no affect, it reduced the NPHP7/Glis2-mediated activation by ∼50%, providing another example that BBS1/TRIM32-mediated changes of ubiquitin composition and intranuclear localization affect the function of NPHP7/Glis.
Although NPHP7/Glis2 has been characterized in great detail (8, 10–13, 15–19, 48), the endogenous protein has evaded its detection, suggesting that this protein is extremely labile and/or expressed at low levels. Most studies have therefore relied on heterologous expression of NPHP7/Glis2 in cultured cells, limiting the significance of the observed perturbations for human physiology and disease. We found that NPHP7/Glis2 and BBS11/TRIM32 expressed in the human embryonic kidney cell line 293T form robust protein complexes. Given the technical limitations, we validated this result with recombinant proteins and used the zebrafish pronephros model to provide further evidence for a genetic interaction between these two proteins. Only one Bardet-Biedl typical BBS11 mutation has been identified so far (22, 49), replacing the proline at position 130 with a serine. BBS11/TRIM32-deficient mice only display phenotypic abnormalities consistent with the limb girdle muscular dystrophy type 2H observed in humans with BBS11/TRIM32 mutations, questioning the relevance of BBS11/TRIM32 mutations as an underlying cause for the Bardet-Biedl syndrome. However, the phenotypes observed after knockdown of BBS11/TRIM32 in zebrafish are clearly consistent with a ciliopathy. Resolving this apparent controversy, our findings provide an alternative explanation as to why patients and animal models with BBS11/TRIM32 mutations rarely display BBS-like symptoms. We identified potential interactions between BBS proteins and NPHP7/Glis2 and propose that additional mutations in NPHP7/Glis2 or other ciliopathy genes are required to elicit a BBS11/TRIM32-associated Bardet-Biedl syndrome. The concept of compound heterozygote mutations appears to be particularly relevant for the Bardet-Biedl syndrome (7, 50, 51), but the overall mutational load likely applies to the manifestations of other ciliopathies.
Acknowledgments
We thank Christina Engel and Barbara Müller for excellent technical support and the members of the Renal Division for helpful discussions and feedback.
This work was supported by Deutsche Forschungsgemeinschaft Grant KFO 201 (to S. S. L. and A. K.-Z.), Sonderforschungsbereich 746 (to T. S. and G. W.), and the European Community's Seventh Framework Program Grant Agreement 241955 (SYSCILIA; to G. W.).
- NPH
- nephronopthisis
- BSS
- Bardet-Biedl syndrome
- NPHP
- NPH protein
- JBTS
- Joubert syndrome
- MKS
- Meckel-Gruber syndrome
- K48
- Lys48
- K63
- Lys63
- MO
- morpholino oligonucleotide.
REFERENCES
- 1. Hildebrandt F., Benzing T., Katsanis N. (2011) Ciliopathies. N. Engl. J. Med. 364, 1533–1543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wolf M. T., Hildebrandt F. (2011) Nephronophthisis. Pediatr. Nephrol. 26, 181–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jauregui A. R., Nguyen K. C., Hall D. H., Barr M. M. (2008) The Caenorhabditis elegans nephrocystins act as global modifiers of cilium structure. J. Cell Biol. 180, 973–988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Williams C. L., Masyukova S. V., Yoder B. K. (2010) Normal ciliogenesis requires synergy between the cystic kidney disease genes MKS-3 and NPHP-4. J. Am. Soc. Nephrol. 21, 782–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Williams C. L., Li C., Kida K., Inglis P. N., Mohan S., Semenec L., Bialas N. J., Stupay R. M., Chen N., Blacque O. E., Yoder B. K., Leroux M. R. (2011) MKS and NPHP modules cooperate to establish basal body/transition zone membrane associations and ciliary gate function during ciliogenesis. J. Cell Biol. 192, 1023–1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sang L., Miller J. J., Corbit K. C., Giles R. H., Brauer M. J., Otto E. A., Baye L. M., Wen X., Scales S. J., Kwong M., Huntzicker E. G., Sfakianos M. K., Sandoval W., Bazan J. F., Kulkarni P., Garcia-Gonzalo F. R., Seol A. D., O'Toole J. F., Held S., Reutter H. M., Lane W. S., Rafiq M. A., Noor A., Ansar M., Devi A. R., Sheffield V. C., Slusarski D. C., Vincent J. B., Doherty D. A., Hildebrandt F., Reiter J. F., Jackson P. K. (2011) Mapping the NPHP-JBTS-MKS protein network reveals ciliopathy disease genes and pathways. Cell 145, 513–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zaghloul N. A., Katsanis N. (2009) Mechanistic insights into Bardet-Biedl syndrome, a model ciliopathy. J. Clin. Invest. 119, 428–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Attanasio M., Uhlenhaut N. H., Sousa V. H., O'Toole J. F., Otto E., Anlag K., Klugmann C., Treier A. C., Helou J., Sayer J. A., Seelow D., Nürnberg G., Becker C., Chudley A. E., Nürnberg P., Hildebrandt F., Treier M. (2007) Loss of GLIS2 causes nephronophthisis in humans and mice by increased apoptosis and fibrosis. Nat. Genet. 39, 1018–1024 [DOI] [PubMed] [Google Scholar]
- 9. Halbritter J., Porath J. D., Diaz K. A., Braun D. A., Kohl S., Chaki M., Allen S. J., Soliman N. A., Hildebrandt F., Otto E. A. (2013) Identification of 99 novel mutations in a worldwide cohort of 1,056 patients with a nephronophthisis-related ciliopathy. Hum. Genet. 132, 865–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang F., Jetten A. M. (2001) Genomic structure of the gene encoding the human GLI-related, Kruppel-like zinc finger protein GLIS2. Gene 280, 49–57 [DOI] [PubMed] [Google Scholar]
- 11. Zhang F., Nakanishi G., Kurebayashi S., Yoshino K., Perantoni A., Kim Y. S., Jetten A. M. (2002) Characterization of Glis2, a novel gene encoding a Gli-related, Kruppel-like transcription factor with transactivation and repressor functions. Roles in kidney development and neurogenesis. J. Biol. Chem. 277, 10139–10149 [DOI] [PubMed] [Google Scholar]
- 12. Kim S. C., Kim Y. S., Jetten A. M. (2005) Kruppel-like zinc finger protein Gli-similar 2 (Glis2) represses transcription through interaction with C-terminal binding protein 1 (CtBP1). Nucleic Acids Res. 33, 6805–6815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kim Y. S., Kang H. S., Jetten A. M. (2007) The Kruppel-like zinc finger protein Glis2 functions as a negative modulator of the Wnt/beta-catenin signaling pathway. FEBS Lett. 581, 858–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li B., Rauhauser A. A., Dai J., Sakthivel R., Igarashi P., Jetten A. M., Attanasio M. (2011) Increased hedgehog signaling in postnatal kidney results in aberrant activation of nephron developmental programs. Hum. Mol. Genet 20, 4155–4166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gruber T. A., Larson Gedman A., Zhang J., Koss C. S., Marada S., Ta H. Q., Chen S. C., Su X., Ogden S. K., Dang J., Wu G., Gupta V., Andersson A. K., Pounds S., Shi L., Easton J., Barbato M. I., Mulder H. L., Manne J., Wang J., Rusch M., Ranade S., Ganti R., Parker M., Ma J., Radtke I., Ding L., Cazzaniga G., Biondi A., Kornblau S. M., Ravandi F., Kantarjian H., Nimer S. D., Döhner K., Döhner H., Ley T. J., Ballerini P., Shurtleff S., Tomizawa D., Adachi S., Hayashi Y., Tawa A., Shih L. Y., Liang D. C., Rubnitz J. E., Pui C. H., Mardis E. R., Wilson R. K., Downing J. R. (2012) An Inv(16)(p13.3q24.3)-encoded CBFA2T3-GLIS2 fusion protein defines an aggressive subtype of pediatric acute megakaryoblastic leukemia. Cancer Cell 22, 683–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Masetti R., Pigazzi M., Togni M., Astolfi A., Indio V., Manara E., Casadio R., Pession A., Basso G., Locatelli F. (2013) CBFA2T3-GLIS2 fusion transcript is a novel common feature in pediatric, cytogenetically normal AML, not restricted to FAB M7 subtype. Blood 121, 3469–3472 [DOI] [PubMed] [Google Scholar]
- 17. Kim Y. S., Kang H. S., Herbert R., Beak J. Y., Collins J. B., Grissom S. F., Jetten A. M. (2008) Kruppel-like zinc finger protein Glis2 is essential for the maintenance of normal renal functions. Mol. Cell Biol. 28, 2358–2367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vasanth S., ZeRuth G., Kang H. S., Jetten A. M. (2011) Identification of nuclear localization, DNA binding, and transactivating mechanisms of Kruppel-like zinc finger protein Gli-similar 2 (Glis2). J. Biol. Chem. 286, 4749–4759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hosking C. R., Ulloa F., Hogan C., Ferber E. C., Figueroa A., Gevaert K., Birchmeier W., Briscoe J., Fujita Y. (2007) The transcriptional repressor Glis2 is a novel binding partner for p120 catenin. Mol. Biol. Cell 18, 1918–1927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kang H. S., Beak J. Y., Kim Y. S., Herbert R., Jetten A. M. (2009) Glis3 is associated with primary cilia and Wwtr1/TAZ and implicated in polycystic kidney disease. Mol. Cell Biol. 29, 2556–2569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kang H. S., ZeRuth G., Lichti-Kaiser K., Vasanth S., Yin Z., Kim Y. S., Jetten A. M. (2010) Gli-similar (Glis) Kruppel-like zinc finger proteins. Insights into their physiological functions and critical roles in neonatal diabetes and cystic renal disease. Histol. Histopathol. 25, 1481–1496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chiang A. P., Beck J. S., Yen H. J., Tayeh M. K., Scheetz T. E., Swiderski R. E., Nishimura D. Y., Braun T. A., Kim K. Y., Huang J., Elbedour K., Carmi R., Slusarski D. C., Casavant T. L., Stone E. M., Sheffield V. C. (2006) Homozygosity mapping with SNP arrays identifies TRIM32, an E3 ubiquitin ligase, as a Bardet-Biedl syndrome gene (BBS11). Proc. Natl. Acad. Sci. U.S.A. 103, 6287–6292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Micale L., Chaignat E., Fusco C., Reymond A., Merla G. (2012) The tripartite motif. Structure and function. Adv. Exp. Med. Biol. 770, 11–25 [PubMed] [Google Scholar]
- 24. Ozato K., Shin D. M., Chang T. H., Morse H. C., 3rd. (2008) TRIM family proteins and their emerging roles in innate immunity. Nat. Rev. Immunol. 8, 849–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cammas F., Khetchoumian K., Chambon P., Losson R. (2012) TRIM involvement in transcriptional regulation. Adv. Exp. Med. Biol. 770, 59–76 [DOI] [PubMed] [Google Scholar]
- 26. LaBeau-DiMenna E. M., Clark K. A., Bauman K. D., Parker D. S., Cripps R. M., Geisbrecht E. R. (2012) Thin, a Trim32 ortholog, is essential for myofibril stability and is required for the integrity of the costamere in Drosophila. Proc. Natl. Acad. Sci. U.S.A. 109, 17983–17988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hatakeyama S. (2011) TRIM proteins and cancer. Nat. Rev. Cancer 11, 792–804 [DOI] [PubMed] [Google Scholar]
- 28. Frosk P., Weiler T., Nylen E., Sudha T., Greenberg C. R., Morgan K., Fujiwara T. M., Wrogemann K. (2002) Limb-girdle muscular dystrophy type 2H associated with mutation in TRIM32, a putative E3-ubiquitin-ligase gene. Am. J. Hum. Genet. 70, 663–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cossée M., Lagier-Tourenne C., Seguela C., Mohr M., Leturcq F., Gundesli H., Chelly J., Tranchant C., Koenig M., Mandel J. L. (2009) Use of SNP array analysis to identify a novel TRIM32 mutation in limb-girdle muscular dystrophy type 2H. Neuromuscul. Disord. 19, 255–260 [DOI] [PubMed] [Google Scholar]
- 30. Kudryashova E., Wu J., Havton L. A., Spencer M. J. (2009) Deficiency of the E3 ubiquitin ligase TRIM32 in mice leads to a myopathy with a neurogenic component. Hum. Mol. Genet 18, 1353–1367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kudryashova E., Struyk A., Mokhonova E., Cannon S. C., Spencer M. J. (2011) The common missense mutation D489N in TRIM32 causing limb girdle muscular dystrophy 2H leads to loss of the mutated protein in knock-in mice resulting in a Trim32-null phenotype. Hum. Mol. Genet 20, 3925–3932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kudryashova E., Kudryashov D., Kramerova I., Spencer M. J. (2005) Trim32 is a ubiquitin ligase mutated in limb girdle muscular dystrophy type 2H that binds to skeletal muscle myosin and ubiquitinates actin. J. Mol. Biol. 354, 413–424 [DOI] [PubMed] [Google Scholar]
- 33. Albor A., El-Hizawi S., Horn E. J., Laederich M., Frosk P., Wrogemann K., Kulesz-Martin M. (2006) The interaction of Piasy with Trim32, an E3-ubiquitin ligase mutated in limb-girdle muscular dystrophy type 2H, promotes Piasy degradation and regulates UVB-induced keratinocyte apoptosis through NFκB. J. Biol. Chem. 281, 25850–25866 [DOI] [PubMed] [Google Scholar]
- 34. Kano S., Miyajima N., Fukuda S., Hatakeyama S. (2008) Tripartite motif protein 32 facilitates cell growth and migration via degradation of Abl-interactor 2. Cancer Res. 68, 5572–5580 [DOI] [PubMed] [Google Scholar]
- 35. Locke M., Tinsley C. L., Benson M. A., Blake D. J. (2009) TRIM32 is an E3 ubiquitin ligase for dysbindin. Hum. Mol. Genet 18, 2344–2358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ryu Y. S., Lee Y., Lee K. W., Hwang C. Y., Maeng J. S., Kim J. H., Seo Y. S., You K. H., Song B., Kwon K. S. (2011) TRIM32 protein sensitizes cells to tumor necrosis factor (TNFα)-induced apoptosis via its RING domain-dependent E3 ligase activity against X-linked inhibitor of apoptosis (XIAP). J. Biol. Chem. 286, 25729–25738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gonzalez-Cano L., Hillje A. L., Fuertes-Alvarez S., Marques M. M., Blanch A., Ian R. W., Irwin M. S., Schwamborn J. C., Marín M. C. (2013) Regulatory feedback loop between TP73 and TRIM32. Cell Death Dis. 4, e704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cohen S., Zhai B., Gygi S. P., Goldberg A. L. (2012) Ubiquitylation by Trim32 causes coupled loss of desmin, Z-bands, and thin filaments in muscle atrophy. J. Cell Biol. 198, 575–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schäfer T., Pütz M., Lienkamp S., Ganner A., Bergbreiter A., Ramachandran H., Gieloff V., Gerner M., Mattonet C., Czarnecki P. G., Sayer J. A., Otto E. A., Hildebrandt F., Kramer-Zucker A., Walz G. (2008) Genetic and physical interaction between the NPHP5 and NPHP6 gene products. Hum. Mol. Genet 17, 3655–3662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Haribaskar R., Pütz M., Schupp B., Skouloudaki K., Bietenbeck A., Walz G., Schäfer T. (2009) The planar cell polarity (PCP) protein Diversin translocates to the nucleus to interact with the transcription factor AF9. Biochem. Biophys. Res. Commun. 387, 212–217 [DOI] [PubMed] [Google Scholar]
- 41. Kim Y. H., Epting D., Slanchev K., Engel C., Walz G., Kramer-Zucker A. (2013) A complex of BBS1 and NPHP7 is required for cilia motility in zebrafish. PLoS One 8, e72549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Perner B., Englert C., Bollig F. (2007) The Wilms tumor genes wt1a and wt1b control different steps during formation of the zebrafish pronephros. Dev. Biol. 309, 87–96 [DOI] [PubMed] [Google Scholar]
- 43. Mund T., Pelham H. R. (2010) Regulation of PTEN/Akt and MAP kinase signaling pathways by the ubiquitin ligase activators Ndfip1 and Ndfip2. Proc. Natl. Acad. Sci. U.S.A. 107, 11429–11434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Aguilar R. C., Wendland B. (2003) Ubiquitin. Not just for proteasomes anymore. Curr. Opin. Cell Biol. 15, 184–190 [DOI] [PubMed] [Google Scholar]
- 45. Chen Z. J., Sun L. J. (2009) Nonproteolytic functions of ubiquitin in cell signaling. Mol. Cell 33, 275–286 [DOI] [PubMed] [Google Scholar]
- 46. Zhang J., Hu M. M., Wang Y. Y., Shu H. B. (2012) TRIM32 protein modulates type I interferon induction and cellular antiviral response by targeting MITA/STING protein for K63-linked ubiquitination. J. Biol. Chem. 287, 28646–28655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nachury M. V., Seeley E. S., Jin H. (2010) Trafficking to the ciliary membrane. How to get across the periciliary diffusion barrier? Annu. Rev. Cell Dev. Biol. 26, 59–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Thiollier C., Lopez C. K., Gerby B., Ignacimouttou C., Poglio S., Duffourd Y., Guégan J., Rivera-Munoz P., Bluteau O., Mabialah V., Diop M., Wen Q., Petit A., Bauchet A. L., Reinhardt D., Bornhauser B., Gautheret D., Lecluse Y., Landman-Parker J., Radford I., Vainchenker W., Dastugue N., de Botton S., Dessen P., Bourquin J. P., Crispino J. D., Ballerini P., Bernard O. A., Pflumio F., Mercher T. (2012) Characterization of novel genomic alterations and therapeutic approaches using acute megakaryoblastic leukemia xenograft models. J. Exp. Med. 209, 2017–2031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Muller J., Stoetzel C., Vincent M. C., Leitch C. C., Laurier V., Danse J. M., Hellé S., Marion V., Bennouna-Greene V., Vicaire S., Megarbane A., Kaplan J., Drouin-Garraud V., Hamdani M., Sigaudy S., Francannet C., Roume J., Bitoun P., Goldenberg A., Philip N., Odent S., Green J., Cossée M., Davis E. E., Katsanis N., Bonneau D., Verloes A., Poch O., Mandel J. L., Dollfus H. (2010) Identification of 28 novel mutations in the Bardet-Biedl syndrome genes. The burden of private mutations in an extensively heterogeneous disease. Hum. Genet. 127, 583–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Katsanis N., Ansley S. J., Badano J. L., Eichers E. R., Lewis R. A., Hoskins B. E., Scambler P. J., Davidson W. S., Beales P. L., Lupski J. R. (2001) Triallelic inheritance in Bardet-Biedl syndrome, a Mendelian recessive disorder. Science 293, 2256–2259 [DOI] [PubMed] [Google Scholar]
- 51. Badano J. L., Kim J. C., Hoskins B. E., Lewis R. A., Ansley S. J., Cutler D. J., Castellan C., Beales P. L., Leroux M. R., Katsanis N. (2003) Heterozygous mutations in BBS1, BBS2 and BBS6 have a potential epistatic effect on Bardet-Biedl patients with two mutations at a second BBS locus. Hum. Mol. Genet 12, 1651–1659 [DOI] [PubMed] [Google Scholar]