

Background: AMP and fructose 2,6-bisphosphate (Fru-2,6-P2) synergistically inhibit eukaryotic fructose-1,6-bisphosphatase (FBPase). How did this property evolve?
Results: Directed mutations, kinetics, and structure determinations link the central cavity of FBPase to AMP/Fru-2,6-P2 synergism.
Conclusion: A central cavity in any FBPase implies AMP/Fru-2,6-P2 and AMP/Fru-6-P synergism.
Significance: AMP/Fru-2,6-P2 synergism of eukaryotic FBPases probably evolved from AMP/Fru-6-P synergism of an ancestral FBPase.
Keywords: Allosteric Regulation, Crystal Structure, Enzyme Inhibitors, Enzyme Kinetics, Evolution, Gluconeogenesis, Metabolism
Abstract
The effects of AMP and fructose 2,6-bisphosphate (Fru-2,6-P2) on porcine fructose-1,6-bisphosphatase (pFBPase) and Escherichia coli FBPase (eFBPase) differ in three respects. AMP/Fru-2,6-P2 synergism in pFBPase is absent in eFBPase. Fru-2,6-P2 induces a 13° subunit pair rotation in pFBPase but no rotation in eFBPase. Hydrophilic side chains in eFBPase occupy what otherwise would be a central aqueous cavity observed in pFBPase. Explored here is the linkage of AMP/Fru-2,6-P2 synergism to the central cavity and the evolution of synergism in FBPases. The single mutation Ser45 → His substantially fills the central cavity of pFBPase, and the triple mutation Ser45 → His, Thr46 → Arg, and Leu186 → Tyr replaces porcine with E. coli type side chains. Both single and triple mutations significantly reduce synergism while retaining other wild-type kinetic properties. Similar to the effect of Fru-2,6-P2 on eFBPase, the triple mutant of pFBPase with bound Fru-2,6-P2 exhibits only a 2° subunit pair rotation as opposed to the 13° rotation exhibited by the Fru-2,6-P2 complex of wild-type pFBPase. The side chain at position 45 is small in all available eukaryotic FBPases but large and hydrophilic in bacterial FBPases, similar to eFBPase. Sequence information indicates the likelihood of synergism in the FBPase from Leptospira interrogans (lFBPase), and indeed recombinant lFBPase exhibits AMP/Fru-2,6-P2 synergism. Unexpectedly, however, AMP also enhances Fru-6-P binding to lFBPase. Taken together, these observations suggest the evolution of AMP/Fru-2,6-P2 synergism in eukaryotic FBPases from an ancestral FBPase having a central aqueous cavity and exhibiting synergistic feedback inhibition by AMP and Fru-6-P.
Introduction
Fructose-1,6-bisphosphatase (d-fructose-1,6-bisphosphate 1-phosphohydrolase; EC 3.1.3; FBPase)2 catalyzes the penultimate step in gluconeogenesis, hydrolyzing fructose 1,6-bisphosphate (Fru-1,6-P2) to fructose 6-phosphate (Fru-6-P) and phosphate (Pi) (1, 2). Fructose-6-phosphate-1-kinase (PFK-1) in glycolysis opposes the action of FBPase by the ATP-dependent phosphorylation of Fru-6-P (3). The coordinated regulation of FBPase and PFK-1 limits futile cycling in vivo. In mammals and presumably other eukaryotes, fructose 2,6-bisphosphate (Fru-2,6-P2) and AMP inhibit FBPase while activating PFK-1 (3–10). In vivo AMP concentrations are nearly constant due to the action of adenylate kinase (11), whereas the level of Fru-2,6-P2 varies in response to changing nutritional states (10). AMP/Fru-2,6-P2 synergism in the inhibition of eukaryotic FBPases (5, 6, 12, 13), however, lowers the apparent Ki of AMP as concentrations of Fru-2,6-P2 increase, making both AMP and Fru-2,6-P2 dynamic physiological regulators.
Porcine liver FBPase (pFBPase) is a model for eukaryotic FBPases (14). Four identical subunits (molecular mass of 37 kDa) form an approximate square-planar arrangement with D2 symmetry. AMP, which binds 28 Å away from the active site (15, 16), is a non-competitive inhibitor with respect to Fru-1,6-P2 but a competitive inhibitor with respect to catalytically essential divalent cations (Mg2+, Mn2+, or Zn2+) (17, 18). Binding of AMP induces a conformational transition from active R-state to inactive T-state (16, 19). The R- and T-states differ principally by a 15° rotation of the upper relative to the lower subunit pairs of the tetramer and by the displacement of loop 50–72 from an active site-engaged conformation to a disengaged conformation (16). At least two AMP molecules are required for the R- to T-state transition (20). The dimer-dimer interface is the origin of AMP cooperativity (20). Mutations that disrupt interactions between subunit pairs eliminate the cooperative binding of AMP (21, 22). Two AMP-bound intermediate states (IR-state and IT-state) have been identified, both of which have engaged loop 50–72 and subunit pair rotations of 3 and 12°, respectively (23, 24). The IR- and IT-states exist in AMP complexes because mutations Ala54 → Leu and Ile10 → Asp, respectively, destabilize the disengaged conformation of loop 50–72.
Fru-2,6-P2 is a competitive inhibitor with respect to Fru-1,6-P2 (5, 6). Chemical modification, NMR, mutagenesis, and crystallographic studies consistently indicate Fru-2,6-P2 binding to the active site (14, 25–28). In the presence of Fru-2,6-P2, the Ki for AMP falls 10-fold (AMP/Fru-2,6-P2 synergism) (5, 6, 29). Hines et al. (14) suggest that AMP/Fru-2,6-P2 synergism arises from a similar end state induced independently by AMP and Fru-2,6-P2, and indeed, Fru-2,6-P2 and AMP cause comparable subunit pair rotations in pFBPase. Moreover, the crystallographic work is in harmony with similar UV spectra of FBPase in complexes with Fru-2,6-P2 and AMP (14, 29).
Escherichia coli FBPase (eFBPase) has 41% sequence identity with pFBPase but a fundamentally different mechanism of regulation (30–32). eFBPase is subject to feed-forward activation by phosphoenolpyruvate, the binding of which favors an active tetramer over an inactive or less active dimer (30–31). AMP and glucose 6-phosphate (Glc-6-P) are synergistic inhibitors that together drive the enzyme to a T-like state (32). Fru-2,6-P2, although not present in E. coli, is a potent inhibitor of eFBPase (14). Unlike pFBPase, Fru-2,6-P2/AMP synergism is absent in the kinetics of eFBPase, and, consistent with the hypothesis of Hines et al. (14), eFBPase remains in the R-state in the presence of Fru-2,6-P2.
The foregoing begs the following question. If subunit pair rotation in response to Fru-2,6-P2 is necessary for synergism, then why do subunit pairs rotate in pFBPase and not in eFBPase? A large water-filled cavity is present at the center of the pFBPase tetramer. In eFBPase, however, hydrophilic side chains fill the corresponding region. Shown here by directed mutations, kinetics, and structure determinations, the central cavity of pFBPase enables the subunit pair rotation of the tetramer in response to Fru-2,6-P2. Moreover, sequence information implies the existence of bacterial FBPases that probably exhibit AMP/Fru-2,6-P2 synergism, even in organisms unlikely to produce Fru-2,6-P2. The FBPase from one such organism, Leptospira interrogans, exhibits not only AMP/Fru-2,6-P2 synergism but also the AMP-enhanced binding of Fru-6-P. Fru-2,6-P2 as a dynamic regulator of FBPase in eukaryotic systems probably evolved then from a primordial FBPase subject to synergistic feedback inhibition by AMP and Fru-6-P.
EXPERIMENTAL PROCEDURES
Materials
Fru-1,6-P2, NADP+ and AMP came from Sigma. Fru-2,6-P2 was prepared by published protocols (33). Glucose-6-phosphate dehydrogenase and phosphoglucose isomerase were purchased from Roche Applied Sciences. The FBPase-deficient E. coli strain DF 657 was from the Genetic Stock Center at Yale University. Other chemicals were of reagent grade or equivalent.
Directed Mutations of pFBPase
Mutations were accomplished by specific base changes utilizing the TransformerTM site-directed mutagenesis kit (Clontech). The mutagenic primers were as follows: Ser45 → His, 5′-CAAGGCCATCCACACCGCAGTC-3′; Thr46 → Arg, 5′-CAAGGCCATCTCCCGCGCAGTCCGCAAG-3′; Leu186 → Tyr, TGCTTCATGTATGACCCGGCC ATC; Ser45 → His/Thr46 → Arg, 5′-CAAGGCCATCCACCGCGCAGTCCGCAAG-3′ (codons for point mutations are underlined and in boldface type). Ser45 → His/Thr46 → Arg/Leu186 → Tyr pFBPase (Cav− pFBPase) came from sequential rounds of mutation using the latter pair of mutagenic primers. Constructs were confirmed by sequencing the promoter region and the entire open reading frame. The Iowa State University sequencing facility provided DNA sequences using the fluorescent dye-dideoxy terminator method.
Expression and Purification of Wild-type and Mutant pFBPases
E. coli strain DF 657 was used for expression of pFBPases. The cell culture of E. coli DF657, transformed with plasmid, grew to A600 of 1.0, at which time transcription was induced by the addition of isopropyl-β-d-thiogalactopyranoside (final concentration 1 mm). The culture was maintained (with shaking, 37 °C) for an additional 16 h before harvesting. The supernatant solution of a cell-free extract was loaded onto a Cibracon Blue-Sepharose column previously equilibrated with 20 mm Tris-HCl, pH 7.5, and 5 mm MgCl2. The column was washed with 20 mm Tris-HCl, pH 7.5. Enzyme was eluted with 5 mm AMP and 20 mm Tris-HCl, pH 7.5. The pH of eluted protein solution was adjusted to 8.5 before loading onto a DEAE-Sepharose column equilibrated with 20 mm Tris-HCl, pH 8.3. Purified enzyme was eluted with a NaCl gradient (0–0.5 m) in 10 mm Tris-HCl, pH 8.3, and then dialyzed extensively against 50 mm Hepes, pH 7.4, for kinetic investigations and for crystallization experiments. Purity and protein concentrations of FBPase preparations were confirmed by SDS-PAGE (34) and by the Bradford assay (35), respectively.
Cloning, Expression, and Purification of lFBPase
The gene for lFBPase (UniProt ID Q8F421) was synthesized and cloned into a pET24b plasmid from Genscript. The integrity of the construct was confirmed by sequencing the promoter region and the entire open reading frame as above for mutant constructs of pFBPase. Expression of lFBPase followed the procedure outlined above for pFBPase. The supernatant component of the cell-free extract was subject to ammonium sulfate fractionation. The 30–50% ammonium sulfate fraction was collected and dialyzed in 20 mm Tris, pH 8.0, before loading onto a DEAE-Sepharose column. Purified enzyme was eluted with a NaCl gradient (0–0.5 m) in 20 mm Tris-HCl, pH 8.0. Enzyme was dialyzed extensively against 50 mm Hepes, pH 7.4, for kinetics studies. Purity and protein concentrations of lFBPase preparations were confirmed by SDS-PAGE (34) and by Bradford assay (35), respectively.
Kinetics
Phosphoglucose isomerase and glucose-6-phosphate dehydrogenase were used as coupling enzymes in assays for FBPase (1). For specific activity measurements, reduction of NADP to NADPH was monitored by absorbance at 340 nm. Other assays used the same coupling enzymes but monitored the formation of NADPH by its fluorescence emission at 470 nm using an excitation wavelength of 340 nm. Assays were performed at 22 °C in 50 mm Hepes, pH 7.5. Assay solutions for wild-type and mutant pFBPases contained EDTA and KCl at concentrations of 10 μm and 150 mm, respectively, whereas KCl was absent in the assay solution for lFBPase. Data for AMP inhibition with respect to Mg2+ and Fru-2,6-P2 inhibition with respect to Fru-1,6-P2 were fit to several models using Grafit (36), with best fits of data to models reported here.
Tryptophan Fluorescence
Fluorescence measurements were made at room temperature using a 1-cm2 quartz cuvette on an SLM Amico 8000 fluorometer. The excitation wavelength was 295 nm, and emission scans were integrated from 310 to 370 nm. Fluorescence scans (repeat of three scans for each data point) were performed after the additions of small volumes of ligand to 2 ml of a 2 μm lFBPase solution. The total volume of added titrant did not exceed 1% of the initial volume. Data fitting was carried out using protocols in the literature (37).
Crystallization of Cav− pFBPase
The method of hanging drops combined 2 μl of protein solution with 2 μl of precipitant solution. Wells contained 500 μl of the precipitant solution. Crystals of the Fru-2,6-P2 complex grew from a protein solution (10 mg/ml Cav− FBPase, 25 mm Hepes, pH 7.4, 5 mm MgCl2, and 5 mm Fru-2,6-P2) combined with a precipitant solution (100 mm Hepes, pH 7.4, 8% (w/v) polyethylene glycol 3350, 27% (v/v) glycerol, and 5% (v/v) t-butyl alcohol). Crystals of the AMP complex grew from a protein solution (10 mg/ml Cav− FBPase, 25 mm Hepes, pH 7.4, 5 mm MgCl2, and 5 mm AMP) combined with a precipitant solution (100 mm Hepes, pH 7.4, 12% (w/v) polyethylene glycol 3350, 23% (v/v) glycerol, and 5% (v/v) t-butyl alcohol). Crystals were of equal dimensions (0.2–0.4 mm), growing in ∼3 days at 20 °C, and were transferred directly from droplet to liquid nitrogen as cryoprotectants were included in the conditions of crystallization.
X-ray Data Collection, Structure Determination, and Refinement
Data were collected at Iowa State University from single crystals on a Rigaku R-AXIS IV++ rotating anode/image plate system using CuKα radiation from an Osmic confocal optics system and a temperature of 110 K. The program d*trek (38) was used in reducing data.
Cav− FBPase with Fru-2,6-P2 and AMP are isomorphous to R- and T-state FBPase complexes, respectively. R-state FBPase (PDB code 1CNQ) and T-state FBPase (PDB code 1EYJ) were used as initial models for molecular replacement. The resulting models underwent refinement using CNS (39) with force constants and parameters of stereochemistry from Engh and Huber (40). A cycle of refinement consisted of slow cooling from 1000 to 300 K in steps of 25 K followed by 100 cycles of conjugate gradient minimization and concluded by the refinement of individual thermal parameters. Restraints of 1.5 Å2 on nearest neighbor and next-to-nearest neighbor main chain atoms, 2.0 Å2 on nearest neighbor side chain atoms, and 2.5 Å2 on next-to-nearest neighbor side chain atoms were employed in thermal parameter refinement. Water molecules were added to difference electron density of 2.5σ or better until no significant decrease was evident in the Rfree value. Water molecules in the final models were within acceptable donor-acceptor distances to each other and/or a polar group of the protein. Stereochemistry of the models was analyzed by use of MolProbity (41).
RESULTS
Rationale for Directed Mutations
Directed mutations have identified residues essential for AMP binding (20, 42), regulation (43–46), cooperativity (21–22, 43), and Fru-2,6-P2 binding (47); however, no mutation eliminates AMP/Fru-2,6-P2 synergism while retaining potent AMP and Fru-2,6-P2 inhibition. Because eFBPase shares 41% identity with pFBPase but lacks AMP/Fru-2,6-P2 synergism, some subset of the remaining 59% of residues must be responsible.
An overview of FBPase showing the relationship of ligand binding sites and loop 50–72 to the central cavity appears in Fig. 1. The number of water molecules in the central cavities of pFBPases, varying in quaternary state and loop conformation (PDB codes 1CNQ, 1NUY, 1YYZ, 2F3D, 2QVV, 2QVU, 1EYJ, and 1FRP), averages to 31, and that of a similar ensemble of eFBPases (PDB codes 2OX3, 2QVR, 2GQ1, and 2Q8M) averages to 14 (supplemental Fig. S1). The difference in water structures of the central cavities of pFBPase and eFBPase is not solely due to the resolution of crystal structures because cavities with the highest numbers of discrete sites for water molecules in eFBPase (2GQ1; 22 water molecules, 1.5 Å resolution) and pFBPase (1YYZ; 48 water molecules, 1.9 Å resolution) follow the trend established by the ensemble averages (Fig. 1). Order/disorder transitions of water molecules in the cavity could be a factor in AMP/inhibitor synergism. Only three positions in or near the central cavity differ in residue type between eFBPase and pFBPase. Ser45, Thr46, and Leu186 in the porcine enzyme are His37, Arg38, and Tyr180, respectively, in eFBPase (Fig. 1). Side chains of Ser45/His37 point into the central cavity, whereas side chains of Thr46/Arg38 and Leu186/Tyr180 are at the edge. If the central cavity is essential to synergism, then mutations that replace residues of pFBPase with those of eFBPase should significantly reduce AMP/Fru-2,6-P2 synergism.
FIGURE 1.

Overview of FBPases. A, alignment of R-state (cyan, product complex, PDB code 1CNQ) and T-state (magenta, AMP complex, PDB code 1EYJ) pFBPases map the locations of AMP binding, active sites, and the central cavity. B, an expanded view of A, showing subunit C2 with loop 50–72 in engaged (cyan) and disengaged (magenta) conformations. C, the central cavity of Leu54 pFBPase (left, IR-state, AMP complex, 1.9 Å, PDB code 1YYZ) and the central region of eFBPase (right, ammonium sulfate complex, 1.5 Å, PDB code 2GQ1). Comparable regions exhibit significant differences in hydration levels. The surface renderings are of subunits C1 and C4 of the tetramer, with ball-and-stick models representing selected residues from subunits C2 and C3 and water molecules (red spheres). The icon with associated arrow indicates the region viewed and viewing direction, respectively. Image generated with PyMOL (57).
Kinetics of Wild-type and Mutant pFBPases
His45, Arg46, and Cav− FBPase have substantial activity relative to wild-type enzyme (Table 1). The Km for substrate for Tyr186 pFBPase is 2-fold greater than that for wild-type enzyme. Mutations cause only minor changes in the Ka for Mg2+. AMP is a competitive inhibitor with respect to Mg2+ for all mutants. Hill coefficients of ∼2 indicate positive cooperativity in AMP binding. The Ki of AMP for Arg46 pFBPase decreases 6-fold, whereas for other mutants, the Ki for AMP inhibition is comparable with that of wild-type enzyme. His45, Arg46, and Cav− pFBPases exhibit residual activity (10–20% of Vmax) at saturating levels of AMP. This phenomenon has been attributed to active AMP complexes in intermediate quaternary states of the tetramer (24, 45). Fru-2,6-P2 is a potent inhibitor, competitive with respect to Fru-1,6-P2 for all FBPases here. In general, cavity mutations have relatively minor effects on activity and ligand binding affinities.
TABLE 1.
Parameters from the kinetics of wild-type and mutant forms of pFBPases and wild typelFBPase
Values in parenthesis are standard deviations in the last significant digit.
| Specific activitya | Km Fru-1,6-P2b | Hill coefficient Mg2+c | Ka Mg2+d | Hill coefficient AMPe | Ki AMPf | Ki Fru-2,6-P2g | |
|---|---|---|---|---|---|---|---|
| μmol s−1 mg−1 | μm | mm | μm2 | μm | |||
| WT pFBPase | 25 (1) | 1.47 (7) | 1.7 (1) | 0.70 (5) | 2.2 (1) | 0.76 (8) | 0.14 (1) |
| S45H pFBPase | 11.9 (7) | 1.07 (7) | 1.6 (1) | 1.0 (1) | 1.8 (1) | 0.35 (5) | 0.053 (9) |
| T46R pFBPase | 12.3 (9) | 0.68 (8) | 1.8 (2) | 0.93 (9) | 2.4 (2) | 4.8 (2) | 0.27 (5) |
| L186Y pFBPase | 21.0 (7) | 1.1 (1) | 1.6 (1) | 0.68 (4) | 1.7 (2) | 0.58 (9) | 0.16 (1) |
| Cav− pFBPase | 15.2 (9) | 1.1 (2) | 1.8 (1) | 0.82 (6) | 2.2 (2) | 0.56 (4) | 0.14 (1) |
| lFBPase | 15 (1) | 1.0 (1) | 0.77 (8) | 1.3 (1) | 2.2 (2) | 0.58 (5) | 0.05 (1) |
a Determined at saturating Fru-1,6-P2 (20 μm) and saturating Mg2+ (5 mm for wild-type pFBPase and lFBPase and 10 mm for mutant pFBPases). Reported values come from five repetitions.
b Determined at saturating Mg2+ (5 mm for wild-type pFBPase and lFBPase and 10 mm for mutant pFBPases). The concentrations of Fru-1,6-P2 vary from 0.3 to 20 μm.
c Hill coefficient for Mg2+ employed 0.2–20 mm Mg2+ and saturating Fru-1,6-P2 (20 μm).
d Determined by using concentrations in Mg2+ from 0.5 to 5 mm and Fru-1,6-P2 from 0.5 to 10 μm.
e Determined by using concentrations in AMP from 0.1 to 100 μm and saturating Fru-1,6-P2 (20 μm) and Mg2+ (10 mm).
f Determined by using concentrations in Mg2+ from 0.5 to 5 mm with saturating Fru-1,6-P2 (20 μm). AMP concentrations range from 0 to 5 μm for wild-type pFBPase and lFBPase, from 0 to 3 μm for His45 pFBPase, from 0 to 20 μm for Arg46 pFBPase, and from 0 to 5 μm for Tyr186 and Cav− pFBPases.
g Determined by using Mg2+ (5 mm for wild-type pFBPase and lFBPase and 10 mm for mutant pFBPases) and Fru-1,6-P2 concentrations from 0.5 to 10 μm. Concentrations of Fru-2,6-P2 range from 0 to 1 μm for wild-type pFBPase, from 0 to 0.5 μm for His45 pFBPase and lFBPase, from 0 to 1 μm for Arg46 pFBPase, and from 0 to 1 μm for Tyr186 pFBPase and Cav− pFBPases.
Data in Fig. 2 represent the linear increase in fluorescence with time due to the production of NADPH. In previous work (14), each curve was fit by an empirical relationship with adjustable parameters for maximal and minimum velocities and a Hill coefficient and then replotted as relative velocity versus [AMP]. An alternative model-based definition of synergism (Scheme 1) provides an analytical expression valid at all concentrations of Fru-2,6-P2, including an infinite concentration.
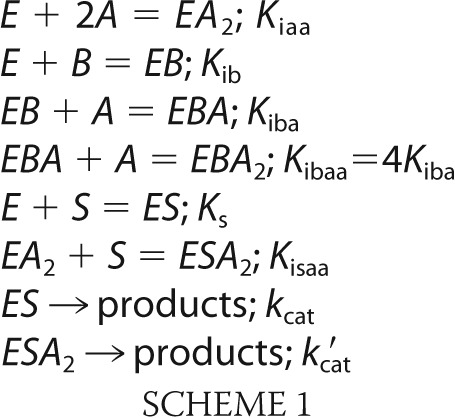 |
In Scheme I, substrate S binds to FBPase subunits independently (48), resulting in an ES complex that contributes to activity (kcat). Two molecules of AMP (represented by A) bind with a Hill coefficient of 2 and affect an R- to T-state transition (20). The ESA2 complexes for some mutant pFBPases retain residual activity (kcat′ is nonzero). Fru-2,6-P2 (represented by B) and substrate are mutually exclusive in binding to the enzyme (5, 6); however, the binding of AMP and substrate are not mutually exclusive (43). AMP and Fru-2,6-P2 bind to distinct sites (EBA and EBA2). The Hill coefficient for AMP becomes unity in the presence of Fru-2,6-P2. Hence, the equilibrium constant (Kibaa) for the binding of A to EBA equals 4Kiba, as required for the independent binding of ligands to two identical sites.
FIGURE 2.

AMP/Fru-2,6-P2 synergism for wild-type and mutant FBPases. A–E, from assays using wild-type, His45, Arg46, Tyr186, and Cav− pFBPases, respectively. Reaction velocities as a function of AMP concentration are taken at 0 (○), 0.1 (□), 0.3 (▵), 1 (●), 3 (■), and 10 μm (▴) Fru-2,6-P2. F, from AMP inhibition assays with wild-type lFBPase at 0 (○) and 0.33 μm (+) Fru-2,6-P2. In all assays, the concentration of Fru-1,6-P2 is 20 μm, and that of Mg2+ is 10 mm.
Equation 1 gives the velocity v based on Scheme 1.
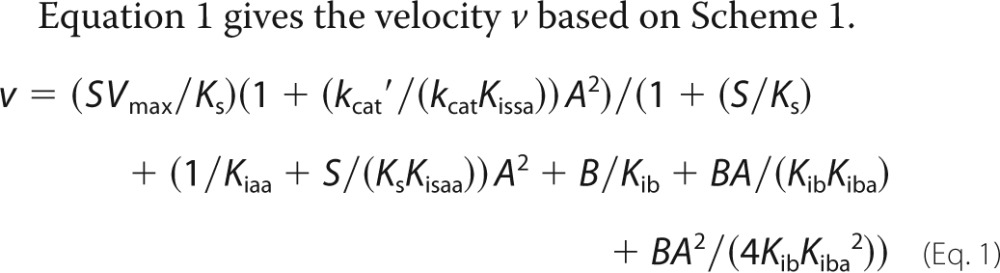 |
In fitting Equation 1, Vmax, kcat′/(kcatKissa), (1/Kiaa + S/(KsKisaa)), and Kiba are adjustable parameters. Ks and Kib are determined by separate experiments for each enzyme (Km and Ki Fru-2,6-P2 of Table 1, respectively), and S is 20 μm for all determinations. Values for parameters for each FBPase are shown in Table 2 with fits of Equation 1 in Fig. 2.
TABLE 2.
Fru-2,6-P2/AMP synergism of FBPases
Values in parentheses are uncertainties in the last significant digit.
| WT pFBPase | S45H pFBPase | T46R pFBPase | L186Y pFBPase | Cav− pFBPase | WT lFBPase | |
|---|---|---|---|---|---|---|
| Vmax (arbitrary units min−1) | 0.194 (4) | 0.88 (3) | 0.434 (8) | 0.495 (9) | 0.47 (1) | 0.48 (1) |
| (k′cat/(kcatKissa) (μm−2) | 0a | 0.0018 (4) | 0.0006 (1) | 0.0017 (2) | 0.0024 (3) | 0a |
| Kiba (μm) | 0.51 (4) | 0.30 (2) | 4.1 (5) | 0.28 (3) | 0.20 (2) | 0.21 (1) |
| (1/Kiaa + S/(KsKisaa)) (μm−2) | 0.78 (6) | 0.85 (6) | 1.7 (1) | 0.35 (3) | 0.72 (4) | 0.17 (2) |
| A0.5B=∞ (μm) | 0.42 (4) | 0.71 (6) | 1.4 (1) | 0.29 (3) | 0.60 (4) | 0.14 (2) |
| A0.5B=0 (μm) | 3.9 (3) | 2.4 (2) | 11 (1) | 2.3 (2) | 2.0 (2) | 2.1 (2) |
| A0.5B=0/A0.5B=∞ | 9 (1) | 3.4 (9) | 8 (1) | 8 (2) | 3.3 (6) | 15 (4) |
a Value fixed to zero, reflecting full inhibition at saturating concentrations of AMP.
Rewriting Equation 1 with A as the independent variable leads to Equation 2,
where a = (v0.5)(1/Kiaa + S/KSKisaa + B/4KibKiba2) − SVmaxk′cat/KSKissakcat; b = (v0.5)(B/(KibKiba)); c = (v0.5)(1 + S/KS + B/Kib) − SVmax/KS; and v0.5 is one-half of the sum of the maximum (at A = 0) and minimum (at A = ∞) velocities for a given concentration of B. The quadratic formula provides A0.5 at any value of B including B = ∞ (Table 2). Synergism is defined here as A0.5B=0/A0.5B=∞. For wild-type, Arg46, and Tyr186 pFBPases, A0.5 decreases significantly as Fru-2,6-P2 concentration increases. On the other hand, His45 and Cav− pFBPases show a two-thirds loss of AMP/Fru-2,6-P2 synergism (Table 2).
AMP Complex of Cav− pFBPase (Protein Data Bank Code 4GWS)
Crystals of the AMP complex of Cav− pFBPase (space group P21212, a = 60.24 Å, b = 164.29 Å, and c = 79.14 Å) belong to the same space group and lattice as crystals of canonical T-state pFBPase. A C1-C2 dimer exists in the asymmetric unit. Both monomers in the model start from residue 9. Electron density for residues 60–72 in the dynamic loop is weak and is associated with high B-parameters. Statistics for data collection and refinement are shown in Table 3.
TABLE 3.
Statistics of data collection and refinement for Cav− FBPase
| Fru-2,6-P2 complex | AMP complex | |
|---|---|---|
| Space group | I222 | P21212 |
| Unit cell lengths (Å) | 54.00, 81.22, 165.06 | 60.24, 164.29, 79.14 |
| Unit cell angles (degrees) | 90, 90, 90 | 90, 90, 90 |
| Resolution limit (Å) | 3.00 | 2.75 |
| No. of measurements | 26,045 | 50,125 |
| No. of unique reflections | 7478 | 17,334 |
| Completeness of data (%) | ||
| Overall | 98.4 | 82.0 |
| Last shell/resolution range (Å) | 99.9/3.11–3.00 | 85.5/2.85–2.75 |
| Rsyma | ||
| Overall | 0.163 | 0.100 |
| Last shell/resolution range (Å) | 0.516/3.11–3.00 | 0.272/2.85–2.75 |
| No. of reflections in refinement | 7104 | 16,412 |
| Number of atoms | 2489 | 5399 |
| Number of solvent sites | 88 | 281 |
| Rfactorb | 0.229 | 0.203 |
| Rfreec | 0.284 | 0.262 |
| Mean B (Å2) overall/protein | 54/54 | 22/22 |
| Mean B (Å2) for Fru-2,6-P2 | 47 | |
| Mean B (Å2) for AMP | 29 | |
| Root mean square deviations | ||
| Bond lengths (Å) | 0.026 | 0.023 |
| Bond angles (degrees) | 1.8 | 1.8 |
| Ramachandran statistics | ||
| Preferred (%) | 98.6 | 98.7 |
| Outlier (%) | 0 | 0 |
a Rsym = ΣjΣi|Iij − 〈Ij〉|/ΣiΣjIij, where i runs over multiple observations of the same intensity, and j runs over all crystallographically unique intensities.
b Rfactor = Σ ‖Fobs| − |Fcalc‖/Σ|Fobs|, where |Fobs| > 0.
c Rfree based upon 10% of the data randomly culled and not used in the refinement.
The AMP complex of Cav− pFBPase is in the T-state, with a 15° subunit pair rotation and a disengaged loop 50–72. One molecule each of Fru-6-P, Mg2+, Pi, and AMP binds to each subunit. The root mean square deviation in Cα carbons between AMP complexes of Cav− and wild-type pFBPase (both in the T-state) is 0.3 Å. Further details regarding the structure of the AMP complex of pFBPase can be found in the literature (16).
Fru-2,6-P2 Complex of Cav− pFBPase (PDB Code 4GWU)
Crystals of the Fru-2,6-P2 complex of Cav− pFBPase (space group I222, a = 54.00 Å, b = 81.22 Å, and c = 165.06 Å) belong to the same space group and lattice as crystals of wild-type pFBPase in its R-state (PDB code 1CNQ). The asymmetric unit is a monomer. Electron density for residues 1–9 and 52–70 is absent. Electron density for residues 22–27 at the AMP binding site is weak, consistent with the previously reported R-state structure (49). Side chains of His45 and Tyr186 are clear in electron density maps, whereas the side chain of Arg46 is partially disordered (Fig. 3). Electron density confirms the presence of bound Fru-2,6-P2. Mg2+ at metal site-1 is in contact with the 1-hydroxyl group and 2-phosphoryl group of Fru-2,6-P2.
FIGURE 3.

Conformational changes of Cav−pFBPase induced by Fru-2,6-P2. A, subunit pair rotations induced by Fru-2,6-P2 in wild-type (blue, 13°, PDB code 2QVV) and Cav− pFBPase (magenta, 2°) relative to R-state pFBPase (green, PDB code 1CNQ). C1–C2 subunit pairs are aligned, and residues 10–100 in C3–C4 subunit pairs are drawn as Cα traces. B, central cavity in Fru-2,6-P2 complexes of Cav− pFBPase, showing electron density (contour level, 1σ) for mutated residues His45, Arg46, and Tyr186. The image was generated with PyMOL (57). C, conformational differences at the active site for the Fru-2,6-P2 complex of Cav− pPBPase (magenta), the product complex of wild-type pFBPase (green; PDB code 1CNQ), and the Fru-2,6-P2 complex of wild-type pFBPase (blue). Individual subunits are aligned to eliminate differences due to subunit pair rotations. Electron density (contour level, 1σ) is provided for Fru-2,6-P2 and Mg2+. D, conformational differences at the AMP site for the Fru-2,6-P2 complex of Cav− pFBPase (magenta), the AMP complex of Leu54 pFBPase (cyan; PDB code 1YYZ), and the R-state product complex of wild-type pFBPase (green; PDB code 1CNQ). Dotted lines indicate hydrogen bonds. The relative positions of helices H1 and H2 are similar in the Fru-2,6-P2 complex of Cav− pFBPase and AMP complex of Leu54 pFBPase, which have subunit pair rotations of 2 and 3°, respectively, relative to the R-state product complex of wild-type pFBPase. Icons represent the region viewed and viewing direction.
Superposition of the entire tetramer of the Fru-2,6-P2 complex of Cav− pFBPase onto the wild-type R-state, wild-type Fru-2,6-P2 complex, and wild-type T-state yields root mean square deviations in Cα coordinates of 0.75, 2.3, and 2.5 Å, respectively. The Fru-2,6-P2 complex of Cav− pFBPase is near the canonical R-state, exhibiting a 2° subunit pair rotation as opposed to the 13° rotation of the Fru-2,6-P2 complex of wild-type pFBPase (14) (Fig. 3). The 2° rotation in Fru-2,6-P2 complex of Cav− pFBPase is similar to the 3° rotation of the IR-state induced by AMP in Leu54 pFBPase (23). The shearing of helices H1 and H2 observed in the AMP complex of Leu54 pFBPase is evident in the Fru-2,6-P2 complex of Cav− pFBPase, as is the loss of the hydrogen bond between Asn35 and Thr14 and the movement of Ile10 from its hydrophobic pocket (Fig. 3); however, unlike the AMP complex of Leu54 FBPase, disruption of the Glu192-Thr39 hydrogen bond across the C1-C4 subunit interface does not occur.
Fru-2,6-P2 induces local conformational change in Cav− pFBPase similar to that induced in wild-type pFBPase (Fig. 3) (14). Loop 121–126 moves toward Fru-2,6-P2 probably in response to hydrogen bonds formed between backbone amide groups of Ser123 and Ser124 and the 2-phosphoryl group of Fru-2,6-P2. α-Carbons of Glu97 and Glu98 move toward the Mg2+ at site-1, most likely a consequence of altered metal coordination due to the absence of metal at site-2. Movements in residues 97–98 and 121–126 may block Asp74 from forming a functionally essential hydrogen bond with the C-terminal end of loop 50–72. Loop 50–72 itself is disordered, and metal cations are absent from sites 2 and 3. The conformation of loop 264–274 in the Fru-2,6-P2 complex of Cav− pFBPase is nearly identical to that of the product complex of wild-type pFBPase (R-state), having not undergone the conformational change observed in the R- to T-state transition.
Sequence Analysis of Type I FBPases
The alignment and hierarchical clustering of 307 Type I FBPase sequences from the Uniprot database (50) employed the ClustalW Web server on the GenomeNet database. Comparisons of sequence positions occupied by signature residues responsible for AMP inhibition, tetramer stability, Glc-6-P inhibition, phosphoenolpyruvate activation, and cavity formation segregate FBPases into groups with mutually exclusive regulatory mechanisms. Position 45, for instance, is a small side-chain residue in eukaryotic FBPases but large and hydrophilic in E. coli-like FBPases (Fig. 4). Residue 112 is critical for AMP inhibition (42) and is lysine in the vast majority of eukaryotic and E. coli-like FBPases. The salt link between Lys42 and Glu192 is essential to tetramer stability, and position 192 is glutamate in eukaryotic FBPase but a non-charged residue type in E. coli-like FBPases (30, 31). Positive charges at positions 228 and 38 are important for Glu-6-P inhibition and anion activation in eFBPase, respectively, both of which are absent in eukaryotic FBPases (30–32).
FIGURE 4.
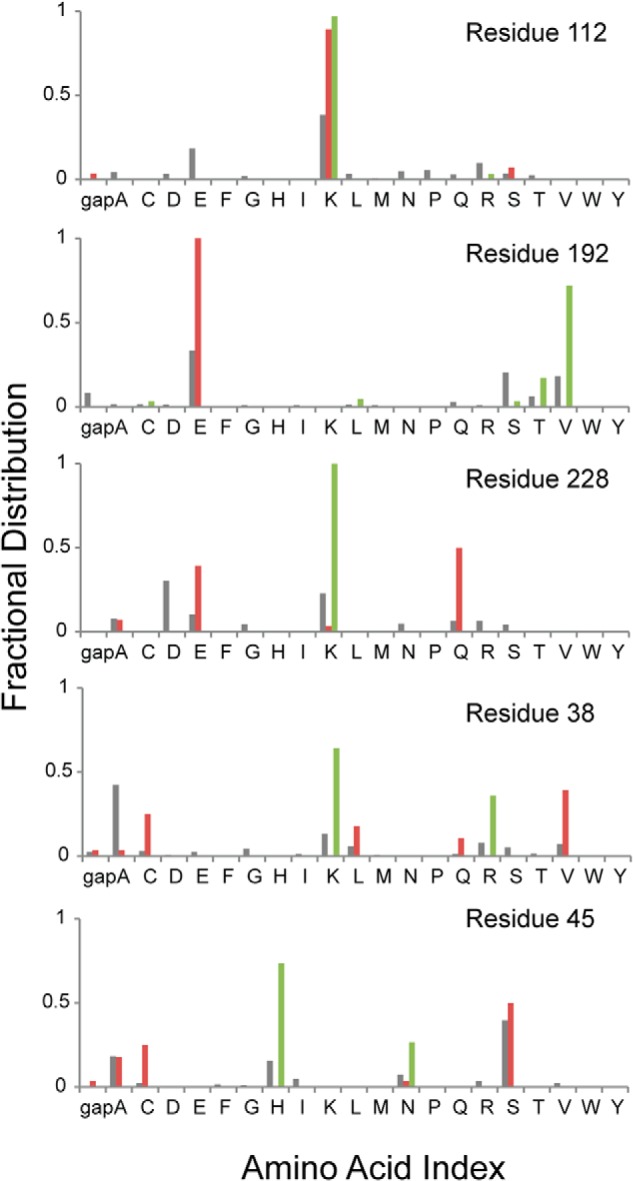
Conservation of signature residues associated with regulatory mechanisms of FBPase. Amino acid types associated with sequence positions essential for AMP inhibition (residue 112), tetramer formation (residue 192), Glc-6-P inhibition (residue 228), anion activation (residue 38), and central cavity formation (residue 45) are presented from top to bottom. Gray, red, and green columns represent the amino acid type for all FBPases, eukaryotic FBPases, and E. coli-like FBPases, respectively.
The association of residue type with regulatory mechanism facilitates the prediction of regulatory properties of FBPases that have yet to be studied experimentally. Sequences of eukaryotic FBPases fall into blocks of relatively high similarity corresponding to fungi, mammalian muscle, mammalian liver, and plants (Fig. 5). The eukaryotic FBPases should be stable tetramers subject to synergistic inhibition by AMP and Fru-2,6-P2 (signature residues are present for the AMP site, tetramer stability, and central cavity). A block with 64 sequences in the middle of the similarity matrix includes eFBPase. E. coli-like FBPases have residue types associated with phosphoenolpyruvate activation, a dynamic equilibrium between dimer and tetramer forms, AMP inhibition, no central cavity, and, hence, no AMP/Fru-2,6-P2 synergism. More than half of all similarity blocks have scant or no experimental data. FBPases in the large block at the top right of the similarity matrix, for instance, should lack all known mechanisms of FBPase inhibition (AMP and/or Glc-6-P inhibition or reversible disulfide bond formation) and yet be stable tetramers.
FIGURE 5.

Similarity matrix of Type I FBPase sequences and the prediction of structural and regulatory properties. The fraction of identical residues in sequence pairs are represented by color according to the scale to the right of the similarity matrix. The positions of eFBPase, pFBPase, and lFBPase are marked by vertical and horizontal dotted lines. FBPases from eukaryotic systems and E. coli-like FBPases are indicated by brackets. Regulatory properties are predicted (gray bar) according to residue types associated with key positions in the amino acid sequence. AMP inhibition requires serine or threonine for residue 31, lysine or arginine for residue 112, and tyrosine for residue 113. Tetramer stability requires lysine or arginine for residue 42 and glutamate for residue 192. Glc-6-P inhibition requires tyrosine for residue 216, lysine or arginine for residue 228, and glutamine for residue 231. Anion activation requires glycine for residue 14, lysine or arginine for residue 38, lysine or arginine for residue 88, and not glutamate for residue 192. The presence of a central cavity (AMP/Fru-2,6-P2 synergism) requires small side chains (glycine, alanine, cysteine, serine, or gap) for residue 45.
Kinetics of lFBPase
FBPases from Leptospira are between eukaryotic and E. coli-like FBPases in the similarity matrix but resemble eukaryotic FBPases, having signature residues for AMP inhibition, tetramer formation, and AMP/Fru-2,6-P2 synergism. A bacterial FBPase with properties of a eukaryotic FBPase would suggest a pathway for the evolution of regulatory mechanisms, but the predicted properties of lFBPase first need confirmation by direct measurement.
Potassium activates pFBPase at millimolar concentrations (1, 2, 51) but inhibits lFBPase. Hence, assays here for lFBPase omit KCl. lFBPase catalyzes the hydrolysis of Fru-1,6-P2 with specific activity and Km values comparable with those of pFBPase. Mg2+ is an essential cofactor, but unlike pFBPase, Mg2+ activation is not cooperative in lFBPase catalysis (Table 1). Loss of Mg2+ cooperativity may be due to the change in residue type (aspartate to glutamate) at position 68 (pFBPase numerology). The mutation Asp68 → Glu in pFBPase reduces Mg2+ cooperativity (46), and eFBPase has glutamate at position 68 with no Mg2+ cooperativity (52). All other Mg2+-binding residues are identical among the three wild-type FBPases under consideration here.
AMP inhibition of lFBPase is comparable with that of pFBPase. The Hill coefficient for AMP inhibition is close to 2. AMP is a competitive inhibitor with respect to Mg2+ (Ki = 0.58 ± 0.05 μm) and a non-competitive inhibitor with respect to Fru-1,6-P2 (Ki = Kis = 0.83 ± 0.05 μm).
Although the genome does not reveal an obvious candidate for a fructose-6-phosphate-2-kinase in L. interrogans, Fru-2,6-P2 is a potent inhibitor of lFBPase (Table 1), competitive with substrate. In the presence of saturating substrate (20 μm) and Mg2+ (5 mm), the concentration of Fru-2,6-P2 causing 50% inhibition is 0.33 ± 0.02 μm, with a Hill coefficient 1.05 ± 0.08.
Fru-2,6-P2 enhances AMP inhibition of lFBPase by 15-fold (Table 2), confirming the prediction from sequence analysis. The Hill coefficient for AMP inhibition drops from 2.2 to unity upon the addition of Fru-2,6-P2. AMP at a concentration that causes 50% inhibition (1.8 μm) enhances Fru-2,6-P2 inhibition by 2-fold (Fig. 6).
FIGURE 6.

Synergism between active site ligands and AMP in lFBPase. A, Fru-2,6-P2 inhibition in the absence (○) and presence (●) of AMP ([AMP] = I0.5-AMP = 1.8 μm). All assays are carried out with saturating Fru-1,6-P2 (20 μm) and Mg2+ (10 mm). B–D, titrations for Fru-1,6-P2 (B), Fru-2,6-P2 (C), and Fru-6-P (D) performed in the absence (open symbols) and presence (filled symbols) of AMP ([AMP] = I0.5-AMP = 1.8 μm) reveal binding synergism between AMP and only Fru-6-P and Fru-2,6-P2. Each titration data point represents integration of the Trp219 emission spectrum from 310 to 370 nm.
Anion activators, such as PEP or citrate, activate eFBPase by promoting tetramer formation (31). To test the effect of PEP on lFBPase, Mg2+-initialized assays were used (31). PEP activation is not present for lFBPase. Instead, millimolar levels of PEP cause moderate inhibition. Assays of Glc-6-P inhibition followed the published protocol for eFBPase (32). Glc-6-P inhibition becomes evident only at millimolar levels, ∼50-fold weaker than Glc-6-P inhibition of eFBPase. Glc-6-P inhibits pFBPase and lFBPase at comparable concentrations.
Sedimentation Equilibrium
lFBPase and pFBPase at three protein concentrations (A280 = 0.3, 0.5, and 0.7) were sedimented to equilibrium at angular speeds of 15,000, 18,000 and 21,000 rpm. Sedimentation equilibrium data were fit to single component and dimer-tetramer equilibrium models with UltraScan (53). Regardless of model, pFBPase and lFBPase give similar apparent molecular masses, 111.9 and 112.1 kDa, respectively. The determined molecular masses are less than the calculated mass of the tetramer (140 kDa).
Tryptophan Fluorescence
lFBPase has but one tryptophan (Trp219), which on the basis of structural similarity is near the common binding sites for the 6-phosphoryl groups of substrate, product, and Fru-2,6-P2. Steady-state fluorescence emission from Trp219 increases as levels of substrate, product, or Fru-2,6-P2 increase, whereas increasing concentrations of AMP cause little change. Substrate, product, Fru-2,6-P2, and AMP have no effect on fluorescence emission from 100 μm tryptophan in the absence of lFBPase. The fluorescence change caused by Fru-6-P, Fru-1,6-P2, or Fru-2,6-P2 is arguably due to a change in the local environment of Trp219. Indeed, at saturating concentrations, the three active site ligands induce nearly identical limiting changes in fluorescence emission.
Data from titrations of 6-phosphoryl ligands in the presence and absence of AMP are fit to Equation 3,
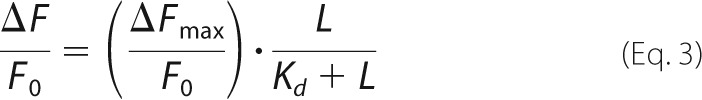 |
where ΔF is the change in fluorescence upon the addition of ligand L, Fo is the fluorescence in the absence of ligand, and Kd is the dissociation constant. AMP does not affect the binding of Fru-1,6-P2 (Fig. 6 and Table 4); however, AMP at a concentration that causes 50% inhibition (1.8 μm) enhances the binding of Fru-2,6-P2 by ∼2-fold, consistent with data from kinetics. AMP enhancement of Fru-6-P binding to lFBPase is also 2-fold, suggesting that the reverse effect (Fru-6-P enhancement of AMP binding) is comparable with Fru-2,6-P2 enhancement (15-fold; Table 2). Fru-6-P enhancement of AMP binding cannot be measured because of assay design (coupling of Fru-6-P production to NADPH formation).
TABLE 4.
Effect of AMP on the binding of ligands to the active site
S.D. in the last significant digit is given parenthetically.
| Ligand | Kd ([AMP] = 0) | Kd ([AMP] = 1.8 μm) |
|---|---|---|
| μm | μm | |
| Fru-1,6-P2 | 0.77 (9) | 0.73 (5) |
| Fru-2,6-P2 | 0.32 (7) | 0.17 (2) |
| Fru-6-P | 105 (9) | 50 (7) |
DISCUSSION
Ser45 → His and Cav− pFBPases represent first instances of modified pFBPases that retain potent AMP and Fru-2,6-P2 inhibition with greatly reduced AMP/Fru-2,6-P2 synergism. Kinetic parameters for catalysis, Mg2+ activation, AMP inhibition, and Fru-2,6-P2 inhibition of mutant enzymes investigated here are similar to those of wild-type pFBPase. Reduced Fru-2,6-P2/AMP synergism in Cav− pFBPase is not due to the disruption of the active or allosteric binding sites but rather to an impaired quaternary response; Cav− pFBPase exhibits only a 2° subunit pair rotation in contrast to the 13° subunit pair rotation for the wild-type enzyme. Conceivably, Cav− mutations could stabilize the R-state (or destabilize the T-state), making the quaternary transition unfavorable; however, previously reported mutations that destabilize quaternary states (Ala54 → Leu and Ile10 → Asp, for instance) broadly impact the kinetics of pFBPase. Moreover, impairment of the R- to T-state transition is specific to Fru-2,6-P2. AMP drives Cav− pFBPase to a canonical T-state, which is structurally indistinguishable from the T-state of the wild-type enzyme.
The effect of Fru-2,6-P2 must be more than the displacement of loop 50–72 from its engaged conformation. Loop 50–72 is disordered in the Fru-2,6-P2 complex of Cav− pFBPase, but the enzyme remains in an R-like state. In contrast, the loop 50–72 acquires its disengaged conformation in the Fru-2,6-P2 complex of wild-type pFBPase, and the enzyme is in a near-T quaternary state. Structural evidence here indicates the subunit pair rotation must go beyond 2° to allow the disengaged conformer.
The solvated central cavity, absent in Cav− pFBPase, evidently enables the requisite subunit pair rotation in response to the binding of Fru-2,6-P2. The binding of AMP to Leu54 pFBPase causes a 3° subunit pair rotation, retaining the engaged conformation of loop 50–72 (23) and an organized water structure in the central cavity (Fig. 1). In contrast, in the T-state of Leu54 pFBPase, loop 50–72 and the water structure in the cavity are disordered. Arguably, an order-to-disorder transition of water molecules within the central cavity, caused by the movement of loop 50–72 from its engaged conformation, provides the requisite motive force for subunit pair rotation. In Cav− pFBPase, the order-to-disorder transition of water structure is inoperative, and consequently movement of loop 50–72 from its engaged conformation results in virtually no subunit pair rotation. The mutation of Ser45 → His alone is largely responsible for this effect, suggesting a critical role for Ser45 in maintaining an organized water structure when loop 50–72 is engaged. The volume of the histidyl side chain (137 Å3) relative to that of serine (89 Å3) is probably a factor; however, the side chain of serine in this specific context may offer the capacity, not shared by other amino acid types, to “seed” a network of hydrogen-bonded water molecules.
Fru-2,6-P2/AMP synergism, although reduced, is still present in His45 and Cav− pFBPase. The 2° subunit pair rotation caused by Fru-2,6-P2 in Cav− pFBPase is similar to changes induced by AMP in Leu54 pFBPase (IR-state, 3° subunit pair rotation). Structural changes at the C1-C4 interface due to small subunit pair rotations could be the structural basis for the weak synergism that remains in Cav− pFBPase. Indeed, wild-type and mutant pFBPases here exhibit reduced AMP cooperativity in the presence of Fru-2,6-P2, which may come from conformational changes at the C1-C4 interface (such as the weakening of the hydrogen bond between Glu192 and Thr39) within the first few degrees of subunit pair rotation. The model above suggests that an engaged dynamic loop in the presence of bound Fru-2,6-P2 would eliminate Fru-2,6-P2/AMP synergism altogether, and in fact, such is the case for eFBPase; there is no Fru-2,6-P2/AMP synergism, and the Fru-2,6-P2 complex of wild-type eFBPase reveals a dynamic loop in its engaged conformation (14).
The failure of the loop 50–72 to achieve a disengaged conformation in its Fru-2,6-P2 complex of Cav− pFBPase is also consistent with the results of targeted molecular dynamics simulations, which indicate that the transition of loop 50–72 from an engaged to disengaged conformation comes late in the R- to T-state transition (24). Moreover, previous work ties the conformational change in loop 264–274 to subunit pair rotation from the IR- to IT-state (from 3 to 12°, respectively) (23, 24). The Fru-2,6-P2 complexes of Cav− and wild-type pFBPases reveal a conformational change in loop 264–274 only for the latter complex.
The foregoing analysis supports the following hypothesis. Any tetrameric FBPase with signature residues of a central cavity should exhibit AMP/Fru-2,6-P2 synergism. Hence, all eukaryotic organisms for which sequences of FBPases are known should exhibit AMP/Fru-2,6-P2 synergism.
Fru-2,6-P2 is not present in bacteria (54), so the evolution of AMP/Fru-2,6-P2 synergism from a bacterial ancestor is problematic. E. coli FBPase employs Glc-6-P as a synergistic inhibitor with respect to AMP (32). Unlike Fru-2,6-P2, Glc-6-P is an allosteric non-competitive inhibitor with respect to substrate. Vastly different kinetic properties and a distinct allosteric binding site for Glc-6-P in eFBPase make Glc-6-P an unlikely precursor of Fru-2,6-P2 through evolution. The analysis of Type I FBPase sequences, however, indicates a central cavity in lFBPase. Moreover, AMP/Fru-6-P binding synergism in lFBPase suggests an evolutionary pathway from AMP/Fru-6-P synergism in bacteria to AMP/Fru-2,6-P2 synergism in eukaryotic organisms.
At some point in evolution, a primordial FBPase may have existed as an equilibrium mixture of dimers and tetramers (Fig. 7). This primordial FBPase may have been subject to AMP inhibition and feedback inhibition by Fru-6-P. One pathway of evolution stabilizes the tetramer through the creation of an anion activation site. Over time, this form of FBPase acquires a site for Glc-6-P inhibition that acts synergistically with AMP, becoming the present day eFBPase.
FIGURE 7.
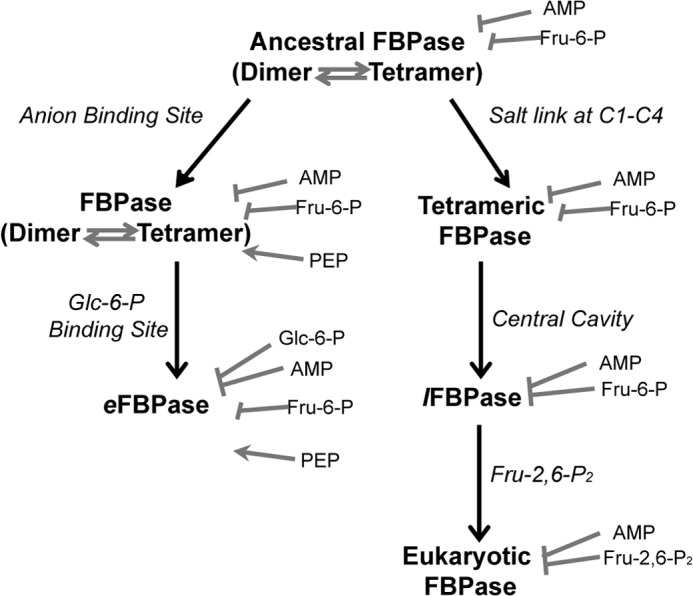
Proposed evolution of the regulatory properties of FBPases. Different pathways to a stable tetramer (anion binding site versus salt link formation) led to E. coli-like and eukaryotic mechanisms of regulation.
An alternative pathway of evolution also leads to a stable FBPase tetramer through the formation of Glu192–Lys42 salt links. Because this form of the tetramer is at all times catalytically competent, a robust mechanism of inhibition would presumably co-evolve. Mutation of position 45 from histidine to serine would create a nascent central cavity in the primordial enzyme with no effect on activity. The introduction of the Glu192–Lys42 salt link in a Ser45 ancestral FBPase would create a stable tetramer with a central cavity. The central cavity would enable AMP/Fru-6-P synergism as a counterpoint to the enhanced activity of a stable tetramer. Over time, Fru-2,6-P2 replaces Fru-6-P as a dynamic regulator. Crystal structures of Fru-2,6-P2 bound to pFBPase clearly show that all atoms, excepting the 2-phosphoryl group, occupy positions corresponding to those of Fru-6-P (14).
L. interrogans is a pathogenic bacterium with a metabolism strikingly different from that of mammals (55). L. interrogans lacks hexokinase (as well as other enzymes of glycolysis) and cannot capture and directly use glucose from exogenous sources (55, 56). Instead, lipid is the principal energy source for L. interrogans.
The apparent role of gluconeogenesis in L. interrogans is the generation of Fru-6-P as a precursor for sugar-nucleotide biosynthesis (56). Given that a gene coding for glucose 6-phosphate dehydrogenase is absent in L. interrogans (56), Fru-6-P must enter the non-oxidative pentose phosphate pathway in order to produce ribose 5-phosphate. The conversion of Fru-1,6-P2 into Fru-6-P in L. interrogans then represents the first committed step in ribose 5-phosphate biosynthesis and de novo nucleotide biosynthesis. In this scheme, Fru-6-P is a direct feedback inhibitor, and AMP is an allosteric feedback inhibitor. High levels of Fru-6-P should affect only lFBPase, due to the absence of a glycolytic pathway. When nucleotides are in demand, the concentrations of AMP and Fru-6-P (the latter being in equilibrium with the pool of ribose 5-phosphate) should be low; however, rising concentrations of AMP and Fru-6-P may synergistically support each other in the feedback inhibition of lFBPase.
Leptospira is the causative agent of leptospirosis, a disease of global concern, causing renal and hepatic damage and, in some instances, sudden and fatal hemoptysis (55). Infections in livestock and domestic animals can expose humans to Leptospira infections through sanitary waste or contaminated drinking water. Little information is known about the metabolism of L. interrogans by direct experimentation. Given the current status of our knowledge, however, one could regard FBPase from Leptospira as a potential target in the development of agents of anti-microbial growth.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grant NS 10546.
The atomic coordinates and structure factors (codes 4GWS and 4GWU) have been deposited in the Protein Data Bank (http://wwpdb.org/).

This article contains supplemental Fig. S1.
- FBPase
- fructose-1,6-bisphosphatase
- pFBPase
- porcine liver FBPase
- eFBPase
- Escherichia coli FBPase
- lFBPase
- Leptospira interrogans FBPase
- Fru-1,6-P2
- fructose 1,6-bisphosphate
- Fru-2,6-P2
- fructose 2,6-bisphosphate
- Fru-6-P
- fructose 6-phosphate
- PFK-1
- fructose-6-phosphate-1-kinase
- Glc-6-P
- glucose 6-phosphate
- PDB
- Protein Data Bank.
REFERENCES
- 1. Benkovic S. J., deMaine M. M. (1982) Mechanism of action of fructose 1,6-bisphosphatase. Adv. Enzymol. Relat. Areas Mol. Biol. 53, 45–82 [DOI] [PubMed] [Google Scholar]
- 2. Tejwani G. A. (1983) Regulation of fructose-bisphosphatase activity. Adv. Enzymol. Relat. Areas Mol. Biol. 54, 121–194 [DOI] [PubMed] [Google Scholar]
- 3. Blangy D., Buc H., Monod J. (1968) Kinetics of the allosteric interactions of phosphofructokinase from Escherichia coli. J. Mol. Biol. 31, 13–35 [DOI] [PubMed] [Google Scholar]
- 4. Taketa K., Pogell B. M. (1965) Allosteric inhibition of rat liver fructose 1,6-bisphosphatase by adenosine 5′-monophosphate. J. Biol. Chem. 240, 651–662 [PubMed] [Google Scholar]
- 5. Pilkis S. J., El-Maghrabi M. R., Pilkis J., Claus T. (1981) Inhibition of fructose-1,6-bisphosphatase by fructose 2,6-bisphosphate. J. Biol. Chem. 256, 3619–3622 [PubMed] [Google Scholar]
- 6. Van Schaftingen E., Hers H.-G. (1981) Inhibition of fructose-1,6-bisphosphatase by fructose 2,6-biphosphate. Proc. Natl. Acad. Sci. U.S.A. 78, 2861–2863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pilkis S. J., El-Maghrabi M. R., Pilkis J., Claus T. H., Cumming D. A. (1981) Fructose 2,6-bisphosphate. A new activator of phosphofructokinase. J. Biol. Chem. 256, 3171–3174 [PubMed] [Google Scholar]
- 8. Van Schaftingen E., Jett M.-F., Hue L., Hers H.-G. (1981) Control of liver 6-phosphofructokinase by fructose 2,6-bisphosphate and other effectors. Proc. Natl. Acad. Sci. U.S.A. 78, 3483–3486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Uyeda K., Furuya E., Luby L. J. (1981) The effect of natural and synthetic d-fructose 2,6-bisphosphate on the regulatory kinetic properties of liver and muscle phosphofructokinase. J. Biol. Chem. 256, 8394–8399 [PubMed] [Google Scholar]
- 10. Okar D. A., Lange A. J. (1999) Fructose-2,6-bisphosphate and control of carbohydrate metabolism in eukaryotes. Biofactors 10, 1–14 [DOI] [PubMed] [Google Scholar]
- 11. Derr R. F., Zieve L. (1972) Adenylate energy charge. Relation to guanylate energy charge and the adenylate kinase equilibrium constant. Biochem. Biophys. Res. Commun. 49, 1385–1390 [DOI] [PubMed] [Google Scholar]
- 12. Han P. F., Han G. Y., Hayes R. L., Moore C. L., Johnson J. (1983) Synergistic effect of AMP and fructose 2,6-bisphosphate on the protection of fructose 1,6-bisphosphatase against inactivation by trypsin. Experientia 39, 1305–1307 [DOI] [PubMed] [Google Scholar]
- 13. Kruger N. J., Beevers H. (1984) Effect of fructose 2,6-bisphosphate on the kinetic properties of cytoplasmic fructose 1,6-bisphosphatase from germinating castor bean endosperm. Plant Physiol. 76, 49–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hines J. K., Chen X., Nix J. C., Fromm H. J., Honzatko R. B. (2007) Structures of mammalian and bacterial fructose-1,6-bisphosphatase reveal the basis for synergism in AMP/fructose 2,6-bisphosphate inhibition. J. Biol. Chem. 282, 36121–36131 [DOI] [PubMed] [Google Scholar]
- 15. Ke H. M., Zhang Y. P., Lipscomb W. N. (1990) Crystal structure of fructose-1,6-bisphosphatase complexed with fructose 6-phosphate, AMP and magnesium. Proc. Natl. Acad. Sci. U.S.A. 87, 5243–5247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Choe J. Y., Fromm H. J., Honzatko R. B. (2000) Crystal structures of fructose 1,6-bisphosphatase. Mechanism of catalysis and allosteric inhibition revealed in product complexes. Biochemistry 39, 8565–8574 [DOI] [PubMed] [Google Scholar]
- 17. Scheffler J. E., Fromm H. J. (1986) Regulation of rabbit liver fructose-1,6-bisphosphatase by metals, nucleotides, and fructose 2,6-bisphosphate as determined from fluorescence studies. Biochemistry 25, 6659–6665 [DOI] [PubMed] [Google Scholar]
- 18. Liu F., Fromm H. J. (1988) Purification and characterization of fructose-1,6-bisphosphatase from bovine brain. Arch. Biochem. Biophys. 260, 609–615 [DOI] [PubMed] [Google Scholar]
- 19. Zhang Y., Liang J. Y., Huang S., Lipscomb W. N. (1994) Toward a mechanism for the allosteric transition of pig kidney fructose-1,6-bisphosphatase. J. Mol. Biol. 244, 609–624 [DOI] [PubMed] [Google Scholar]
- 20. Nelson S. W., Honzatko R. B., Fromm H. J. (2002) Hybrid tetramers of porcine liver fructose-1,6-bisphosphatase reveal multiple pathways of allosteric inhibition. J. Biol. Chem. 277, 15539–15545 [DOI] [PubMed] [Google Scholar]
- 21. Shyur L.-F., Aleshin A. E., Honzatko R. B., Fromm H. J. (1996) Site-directed mutagenesis of residues at subunit interfaces of porcine fructose-1,6-bisphosphatase. J. Biol. Chem. 271, 3005–3010 [DOI] [PubMed] [Google Scholar]
- 22. Shyur L.-F., Aleshin A. E., Honzatko R. B., Fromm H. J. (1996) Biochemical properties of mutant and wild-type fructose-1,6-bisphosphatases are consistent with the coupling of intra- and intersubunit conformational changes in the T- and R-state transition. J. Biol. Chem. 271, 33301–33307 [DOI] [PubMed] [Google Scholar]
- 23. Iancu C. V., Mukund S., Fromm H. J., Honzatko R. B. (2005) R-state AMP complex reveals initial steps of the quaternary transition of fructose-1,6-bisphosphatase. J. Biol. Chem. 280, 19737–19745 [DOI] [PubMed] [Google Scholar]
- 24. Gao Y., Iancu C. V., Mukind S., Choe J. Y., Honzatko R. B. (2013) Mechanism of displacement of catalytically essential loop from the active site of mammalian fructose-1,6-bisphosphatase. Biochemistry 52, 5206–5216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ganson N. J., Fromm H. J. (1985) Nuclear magnetic resonance studies of fructose 2,6-bisphosphate and adenosine 5′-monophosphate interaction with bovine liver fructose-1,6-bisphosphatase. J. Biol. Chem. 260, 2837–2843 [PubMed] [Google Scholar]
- 26. Liu F., Fromm H. J. (1988) Interaction of fructose 2,6-bisphosphate and AMP with fructose-1,6-bisphosphatase as studied by nuclear magnetic resonance spectroscopy. J. Biol. Chem. 263, 9122–9128 [PubMed] [Google Scholar]
- 27. Liu F., Fromm H. J. (1988) Relationship between thiol group modification and the binding site for fructose 2,6-bisphosphate on rat liver fructose-1,6-bisphosphatase. J. Biol. Chem. 263, 10035–10039 [PubMed] [Google Scholar]
- 28. Xue Y., Huang S., Liang J.-Y., Lipscomb W. N. (1994) Crystal structure of fructose-1,6-bisphosphatase complexed with fructose 2,6-bisphosphate, AMP and Zn2+ at 2.0-Å resolution. Aspects of synergism between inhibitors. Proc. Natl. Acad. Sci. U.S.A. 91, 12482–12486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. McGrane M. M., El-Maghrabi M. R., Pilkis S. J. (1983) The interaction of fructose 2,6-bisphosphate and AMP with rat hepatic fructose 1,6-bisphosphatase. J. Biol. Chem. 258, 10445–10454 [PubMed] [Google Scholar]
- 30. Hines J. K., Fromm H. J., Honzatko R. B. (2006) Novel allosteric activation site in Escherichia coli fructose-1,6-bisphosphatase. J. Biol. Chem. 281, 18386–18393 [DOI] [PubMed] [Google Scholar]
- 31. Hines J. K., Fromm H. J., Honzatko R. B. (2007) Structures of activated fructose-1,6-bisphosphatase from Escherichia coli. Coordinate regulation of bacterial metabolism and the conservation of the R-state. J. Biol. Chem. 282, 11696–11704 [DOI] [PubMed] [Google Scholar]
- 32. Hines J. K., Kruesel C. E., Fromm H. J., Honzatko R. B. (2007) Structures of inhibited fructose-1,6-bisphosphatase from Escherichia coli. Distinct allosteric inhibition sites for AMP and glucose 6-phosphate and the characterization of a gluconeogenic switch. J. Biol. Chem. 282, 24697–24706 [DOI] [PubMed] [Google Scholar]
- 33. Van Schaftingen E., Hers H.-G. (1981) Formation of fructose 2,6-bisphosphate from fructose 1,6-bisphosphate by intramolecular cyclisation followed by alkaline hydrolysis. Eur. J. Biochem. 117, 319–323 [DOI] [PubMed] [Google Scholar]
- 34. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 35. Bradford M. M. (1976) A rapid sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 36. Leatherbarrow R. J. (2009) GraFit, version 7, Erithacus Software Ltd., Horley, UK [Google Scholar]
- 37. Nelson S. W., Choe J.-Y., Iancu C. V., Honzatko R. B., Fromm H. J. (2000) Tryptophan fluorescence reveals the conformational state of a dynamic loop in recombinant porcine fructose-1,6-bisphosphatase. Biochemistry 39, 11100–11106 [DOI] [PubMed] [Google Scholar]
- 38. Pflugrath J. W. (1999) The finer things in X-ray diffraction data collection. Acta Crystallogr. D 55, 1718–1725 [DOI] [PubMed] [Google Scholar]
- 39. Brünger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J.-S., Kuszewski J., Nilges M., Pannu M. S., Read R. J., Rice L. M., Simonson T., Warren G. L. (1998) Crystallography & NMR system. A new software suite for macromolecular structure determination. Acta Crystallogr. D 54, 905–921 [DOI] [PubMed] [Google Scholar]
- 40. Engh R. A., Huber R. (1991) Accurate bond and angle parameters for x-ray protein structure refinement. Acta Crystallogr. A 47, 392–400 [Google Scholar]
- 41. Chen V. B., Arendall W. B., 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. (2010) MolProbity. All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kelley-Loughnane N., Kantrowitz E. R. (2001) AMP inhibition of pig kidney fructose-1,6-bisphosphatase. Biochim. Biophys. Acta 1548, 66–71 [DOI] [PubMed] [Google Scholar]
- 43. Shyur L. F., Poland B. W., Honzatko R. B., Fromm H. J. (1997) Major changes in the kinetic mechanism of AMP inhibition and AMP cooperativity attend the mutation of Arg49 in fructose-1,6-bisphosphatase. J. Biol. Chem. 272, 26295–26299 [DOI] [PubMed] [Google Scholar]
- 44. Nelson S. W., Choe J. Y., Honzatko R. B., Fromm H. J. (2000) Mutations in the hinge of a dynamic loop broadly influence functional properties of fructose-1,6-bisphosphatase. J. Biol. Chem. 275, 29986–29992 [DOI] [PubMed] [Google Scholar]
- 45. Nelson S. W., Kurbanov F. T., Honzatko R. B., Fromm H. J. (2001) The N-terminal segment of recombinant porcine fructose-1,6-bisphosphatase participates in the allosteric regulation of catalysis. J. Biol. Chem. 276, 6119–6124 [DOI] [PubMed] [Google Scholar]
- 46. Kurbanov F. T., Choe J. Y., Honzatko R. B., Fromm H. J. (1998) Directed mutations in the poorly defined region of porcine liver fructose-1,6-bisphosphatase significantly affect catalysis and the mechanism of AMP inhibition. J. Biol. Chem. 273, 17511–17516 [DOI] [PubMed] [Google Scholar]
- 47. el-Maghrabi M. R., Austin L. R., Correia J. J., Pilkis S. J. (1992) Lysine 274 is essential for fructose 2,6-bisphosphate inhibition of fructose-1,6-bisphosphatase. J. Biol. Chem. 267, 6526–6530 [PubMed] [Google Scholar]
- 48. Nelson S. W., Honzatko R. B., Fromm H. J. (2004) Origin of cooperativity in the activation of fructose-1,6-bisphosphatase by Mg2+. J. Biol. Chem. 279, 18481–18487 [DOI] [PubMed] [Google Scholar]
- 49. Choe J. Y., Poland B. W., Fromm H. J., Honzatko R. B. (1998) Role of a dynamic loop in cation activation and allosteric regulation of recombinant porcine fructose-1,6-bisphosphatase. Biochemistry 37, 11441–11450 [DOI] [PubMed] [Google Scholar]
- 50. UniProt Consortium (2012) Reorganizing the protein space at the Universal Protein Resource (UniProt). Nucleic Acids Res. 40, D71–D75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhang R., Villeret V., Lipscomb W. N., Fromm H. J. (1996) Kinetics and mechanisms of activation and inhibition of porcine liver fructose-1,6-bisphosphatase by monovalent cations. Biochemistry 35, 3038–3043 [DOI] [PubMed] [Google Scholar]
- 52. Kelley-Loughnane N., Biolsi S. A., Gibson K. M., Lu G., Hehir M. J., Phelan P., Kantrowitz E. R. (2002) Purification, kinetic studies, and homology model of Escherichia coli fructose-1,6-bisphosphatase. Biochim. Biophys. Acta 1594, 6–16 [DOI] [PubMed] [Google Scholar]
- 53. Demeler B. (2005) UltraScan. A Comprehensive Data Analysis Software Package for Analytical Ultracentrifugation Experiments. in Modern Analytical Ultracentrifugation: Techniques and Methods. (Scott D. J., Harding S. E., Rowe A. J., eds) pp. 210–229, Royal Society of Chemistry, London [Google Scholar]
- 54. Michels P. A., Rigden D. J. (2006) Evolutionary analysis of fructose 2,6-bisphosphate metabolism. IUBMB Life 58, 133–141 [DOI] [PubMed] [Google Scholar]
- 55. Faine S., Adler B., Bolin C., Perolat P. (1999) Leptospira and Leptospirosis, Medisci, Melbourne [Google Scholar]
- 56. Ren S. X., Fu G., Jiang X. G., Zeng R., Miao Y. G., Xu H., Zhang Y. X., Xiong H., Lu G., Lu L. F., Jiang H. Q., Jia J., Tu Y. F., Jiang J. X., Gu W. Y., Zhang Y. Q., Cai Z., Sheng H. H., Yin H. F., Zhang Y., Zhu G. F., Wan M., Huang H. L., Qian Z., Wang S. Y., Ma W., Yao Z. J., Shen Y., Qiang B. Q., Xia Q. C., Guo X. K., Danchin A., Saint Girons I., Somerville R. L., Wen Y. M., Shi M. H., Chen Z., Xu J. G., Zhao G. P. (2003) Unique physiological and pathogenic features of Leptospira interrogans revealed by whole-genome sequencing. Nature 422, 888–893 [DOI] [PubMed] [Google Scholar]
- 57. DeLano W. L. (2012) The PyMOL Molecular Graphics System, version 1.5.0.4 Schrödinger, LLC, New York [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.