

Background: G-quadruplex forming DNA of gene promoter associated with cell death and growth arrest.
Results: G-quadruplex forming DNA at c-Myc promoter Pu27 destabilizes proteins at telomere and inhibits DNA repair molecules.
Conclusion: Pu27 shows extensive DNA damage primarily at telomere that contributes to cell death.
Significance: Learning how Pu27 destabilizes at telomeric region is crucial to understanding G-quadruplex-mediated cancer biology.
Keywords: Cell Death, DNA Damage, Myc, Survivin, Telomeres, ATM, Alt Cell, G-quadruplex, Phospho-H2AX, Shelterin
Abstract
Quadruplex-forming DNA sequences are present throughout the eukaryotic genome, including in telomeric DNA. We have shown that the c-Myc promoter quadruplex-forming sequence Pu-27 selectively kills transformed cells (Sedoris, K. C., Thomas, S. D., Clarkson, C. R., Muench, D., Islam, A., Singh, R., and Miller, D. M. (2012) Genomic c-Myc quadruplex DNA selectively kills leukemia. Mol. Cancer Ther. 11, 66–76). In this study, we show that Pu-27 induces profound DNA damage, resulting in striking chromosomal abnormalities in the form of chromatid or chromosomal breaks, radial formation, and telomeric DNA loss, which induces γ-H2AX in U937 cells. Pu-27 down-regulates telomeric shelterin proteins, DNA damage response mediators (RAD17 and RAD50), double-stranded break repair molecule 53BP1, G2 checkpoint regulators (CHK1 and CHK2), and anti-apoptosis gene survivin. Interestingly, there are no changes of DNA repair molecules H2AX, BRCA1, and the telomere maintenance gene, hTERT. ΔB-U937, where U937 cells stably transfected with deleted basic domain of TRF2 is partially sensitive to Pu-27 but exhibits no changes in expression of shelterin proteins. However, there is an up-regulation of CHK1, CHK2, H2AX, BRCA1, and survivin. Telomere dysfunction-induced foci assay revealed co-association of TRF1with γ-H2AX in ATM deficient cells, which are differentially sensitive to Pu-27 than ATM proficient cells. Alt (alternating lengthening of telomere) cells are relatively resistant to Pu-27, but there are no significant changes of telomerase activity in both Alt and non-Alt cells. Lastly, we show that this Pu-27-mediated sensitivity is p53-independent. The data therefore support two conclusions. First, Pu-27 induces DNA damage within both telomeric and nontelomeric regions of the genome. Second, Pu-27-mediated telomeric damage is due, at least in part, to compromise of the telomeric shelterin protein complex.
Introduction
G-quadruplex-forming2 sequences are present in the genomes of all species that have been studied (1–3). These sequences occur naturally in the telomeres as well as promoter regions (4–6). The Pu-27 sequence is a part of the nuclease hypersensitivity element III1 element of the c-Myc promoter that controls 85–90% of c-Myc transcription. Pu-27 is a Gly/Cys-rich sequence that forms an intramolecular DNA quadruplex structure, which later results in c-Myc silencing. A substantial proportion (∼80%) of c-Myc expression is regulated by the nuclease hypersensitivity element III1 element, of which Pu-27 is a critical part. An oligonucleotide encoding this sequence, Pu-27, has been shown to inhibit the proliferation of tumor cells derived from different tissues including breast, lung, and blood. The Pu-27 oligonucleotide forms a stable G-quadruplex structure, as demonstrated by circular dichroism. We believe that this characteristic contributes to its non-antisense growth inhibitory effects.
In addition to G-quadruplexes scattered throughout the genome, putative quadruplex-forming sequences are found in the telomeres and are believed to be crucial for the preservation of chromosome stability (7). The structural characteristics of the telomere, including protein binding and DNA secondary structure, prevent their recognition as DNA double-stranded breaks (DSBs) (8). The telomeric repeat binding factor 2 (TRF2) plays a crucial role in protecting chromosome ends against instability (9–11). TRF2 binds directly to telomeric DNA and co-associates with five other proteins (TRF1, TIN2, TPP1, POT1, and Rap1), forming a nucleoprotein complex called “shelterin” (12). Multiple lines of evidence suggest that TRF2 protects the human telomere from inappropriate end to end fusions (11, 13). The TRF2 N-terminal domain, which includes the basic domain of TRF2, condenses the telomeric end by inserting a negative torsion, stimulates the invasion of telomeric single strand DNA inside a homologous duplex (1), and favors telomeric loop formation (11).
Eukaryotic cells have evolved a complex, multidimensional response to DNA damage (14). When cells sense DNA damage or replication arrest, cell cycle checkpoints are activated that lead to cell cycle arrest. This, in turn, buys time for repair before the DNA damage can be passed on to daughter cells. In addition to checkpoint activation, the DNA damage response induces transcriptional machinery, improves DNA repair pathways and, if the level of damage is severe, initiates cell death (15). All of these processes are tightly regulated so that the genetic material can be accurately maintained and replicated within the cell.
An important component of this complex response to DNA damage is the ATM (ataxia-telangiectasia mutated) gene, which belongs to the phosphoinositol 3-kinase family (16–18). ATM is primarily activated following DNA DSB by autophosphorylation of its serine residue 1981 (19, 20). This, in turn, leads to the phosphorylation of multiple downstream proteins which are involved in DNA damage recognition, cell cycle arrest, and apoptosis such as p53, H2AX, CHK2, and Smc1 (21). ATM is considered to be one of the main transducers of the telomere damage signal arising from telomeric erosion during senescence (22). TRF2 was reported to be a direct inhibitor of ATM at the telomere (23, 24). Mutations in ATM lead to faulty telomere maintenance in mammalian cells (16). ATM influences the interaction between telomeres and the nuclear matrix. Therefore, alteration in telomere chromatin could be at least partly responsible for the pleiotropic phenotype of the ATM gene defect (25). There is also evidence that ATM is recruited to the telomere during the G2 phase of cell cycle. It has been suggested that telomere ends need to be recognized as DNA damage to complete telomere replication and to acquire a structure that is essential for proper function (26–28). However, the exact role of ATM at telomeres remains largely unclear because of its multiple functions. It is particularly challenging to distinguish a possible role in telomere replication from its role at dysfunctional telomeres and in induction of cell checkpoints, repair, and apoptosis. It has been proposed that the replication fork is buried in telomeres leading to a transient ATM and ATR/ATM DNA damage response, which is not enough to stop cell propagation but probably is required for proper telomere processing (26).
In this study, we demonstrate that Pu-27 somehow causes extensive DNA damage, inducing a brisk DNA damage response. We explore the important role of the various molecules involved in DNA damage response in Pu-27-induced cell death.
EXPERIMENTAL PROCEDURES
Cells and Cell Culture
Human histiocytic lymphoma U937 cells (ATCC) and human colon carcinoma HCT 116 p53+/+, HCT116 p53+/−, and HCT p53−/− cells were bought from the Core Cell Center of Johns Hopkins University, ATM mouse wild type, and null (334 ATM+/+, 4a ATM+/+, and 695 ± 743−/−) cells (gift from MS Turker, Oregon Health Science University, Portland, OR), and lung adenocarcinoma cell lines A549 and Sk-Lu-1 (obtained from ATCC) were maintained in RPMI or DMEM supplemented with 10% FCS. 5 × 104 cells were plated in 6-well plates and treated with 10 μm of Pu-27 or control cytosine-rich oligonucleotide (CRO) next day. The cells were harvested for respective assay on different days.
Western Blots
After treatments, the cells were lysed in modified radioimmune precipitation assay buffer (9 mm urea, 75 mm Tris, pH 7.5, 150 mm NaCl, 0.5% sodium deoxicolate, 1 mm EDTA) placed on ice for 30 min. During this time, cells were mixed by inverting the tube several times every 10 min. Extracts were centrifuged at 12,000 rpm with a table top centrifuge for 10 min to sediment insoluble material. Lysates were quantitated using Bio-Rad BSA protein assay, and 10–20-μg quantities of proteins were separated on 10% or 15% bis-acrylamide gels and then transferred onto PVDF membranes. Proteins were visualized using TRF2 (Novus Biological), γH2AX, and β-actin (Sigma) antibodies. After treatment with Pu-27 and mutated Pu-27, the cells were lysed at the indicated time with radioimmune precipitation assay buffer.
Transfection
Transfection of U937 human histiocytic lymphoma cancer cell was conducted by electroporation (29, 30) in nonsupplemented medium at 280 V and 960 microfarads by means of a Gene Pulser system and a 0.4-cm electrode gap in Gene Pulser cuvettes (Bio-Rad). Cells in the initial log phase of growth were harvested by centrifugation, washed once with medium, and resuspended at room temperature to a concentration of 4 × 107 cells/0.4 ml. Aliquots of 0.4 ml of cells were transfected with 2 μg of the plasmid pLPC TRF2 ΔB (Addgene), which contains the TRF2 gene in which the basic domain of amino acids 1–44 is deleted. Electroporated cells were incubated at 37 °C overnight, and stably transfected cells were selected and maintained with puromycin (1 μg/ml). Expression was checked by Western blotting.
Immunofluorescence Staining
Cells were fixed in 4% formaldehyde and permeabilized in 0.1% Triton X-100 in 0.02% BSA in PBS for 2 min at room temperature. For immunolabeling experiments, cells were blocked in 20% goat serum with 2% bovine serum albumin in PBS for 15 min at room temperature, incubated with primary antibody, washed in 0.2% in PBS and incubated with the following secondary antibodies: goat anti-rabbit Alexa-546 (Fig. 3B, red) and goat anti-mouse Alexa-488 (Fig. 3B, green). Nuclei were visualized using DAPI by confocal analysis obtained with a Zeiss LSM 510 META Laser Scanning Microscope (Zeiss, Oberkochen, Germany).
FIGURE 3.

A, ATM deficient cells are more sensitive to Pu-27. MTT assay was done in 4a ATM+/+, 695 ATM+/−, and 525 ATM−/− cells derived from mice treated with Pu-27 and showed that both hetero- and homozygous cells are sensitive, but not wild type cells. *, p values for 695 ATM+/− and 525 ATM−/− are < 0.001, respectively, and that for 334 ATM+/+ is 0.145 at 10 μm of Pu-27 treatment. B, Pu-27-induced H2AX phosphorylation (γH2AX) at dysfunctional telomere by uncapping TRF1, a member of shelterin complex. 695 ATM+/− and 334 ATM+/+ cells treated with Pu-27 for 2 days and immunostained with TRF1 (green) and γ-H2AX (red). Co-localization of TRF1 with γ-H2AX in ATM deficient cells represents the presence of telomere dysfunction-induced foci, which suggests that Pu-27 destabilizes telomere-shelterin homeostasis.
MTT Cell Proliferation Assay
The cells were seeded into 96-well plates (Corning, Lowell, MA) at 1500 cells/well. Twenty-four hours after cells were seeded, different concentrations of Pu27 and CRO dissolved in water were added to the medium directly for an additional 96 h. MTT (5 mg/ml; Sigma) was added to each well. After a 4-h incubation, reduced MTT was solubilized in 10% SDS, 0.1 n HCl) plate reader. Background absorbance of the medium in the absence of cells was subtracted. All samples were assayed in triplicate, and the mean for each experiment was calculated.
Measurement of Telomerase Activity
Telomerase activity in U937, ΔB-U937, A549, and SL-Lu-1 was detected using a quantitative telomerase detection kit (Allied Biotech, Inc., Germantown, MD) according to the manufacturer's protocol. The assay is based on PCR amplification of the telomeric DNA. Briefly, 1 mg of cell extract from 1 × 105 cells of different cell line was added telomeric repeats (TTAGGG) onto the 3′ end of the substrate oligonucleotide and quantitative telomerase detection premix and was amplified with a 7500 fast real time machine (Allied Biotech, Inc.). The PCR products were detected by measuring the increase in fluorescence caused by the binding of SYBR Green to double-stranded DNA. A heat-inactivated cell extract served as a negative control. The real time PCR conditions were as follows: telomerase reaction for 20 min at 25 °C, PCR initial activation step for 10 min at 95 °C, followed by 35 cycles of denaturation for 30 s at 95 °C, annealing for 30 s at 60 °C, and extension for 30 s at 72 °C.
Annexin V/PI Analyses for Cell Death/Apoptosis
Apoptotic cells were measured using an annexin V-FITC/propidium iodide (PI) kit (BioVision, Palo Alto, CA) and detected by flow cytometry according to the manufacturer's protocol. Briefly, after 4 and 5 days of treatment with Pu-27, the cells were harvested, collected, resuspended in binding buffer (pH 7.5, 10 mm HEPES, 2.5 mm CaCl2, and 140 mm NaCl), incubated with annexin V-FITC/PI for 10 min in the dark, and then analyzed by flow cytometry. Early stage apoptotic cells stain positive for annexin V-FITC, whereas those in the late stages of apoptosis or necrosis stain positive for both annexin V-FITC and PI. The data were analyzed using the Modfit and Cell Quest software programs (Becton, Dickinson and Company).
γH2AX Detection by Flow Cytometry
Measurement of H2AX phosphorylation was performed by anti-phospho-histone H2AX (Ser139), clone JBW301, FITC conjugate (Millipore, Temecula, CA). U937 and ΔB-U937 suspension cells were treated with 10 μm Pu-27, mutated Pu-27 (as a control), and CRO (as a control) for 3 days. Cells were harvested at the indicated time points and washed to remove residual media. The cells were then resuspended, fixed with 2% paraformaldehyde in PBS at 37 °C for 10 min, and washed in 0.5% BSA-PBS and permeabilized cells with 0.125% Triton X-100 in 0.5% BSA-PBS for 1 min; the cells were washed with 0.5% BSA-PBS and incubated with 3 μg/150 ml of anti-phospho-H2AX Ser139 antibodies conjugated with the FITC fluorochrome for 30 min in the dark at room temperature. The cells were then washed to remove excess antibody, and FITC was measured on a Becton Dickinson FACSCalibur flow cytometer.
Chromosome and Telomere Fluorescence in Situ Hybridization (FISH) Analysis
U937 cells were treated with Pu-27 and CRO oligonucleotides for 2 and 5 days. Metaphases were prepared by standard protocols. Briefly, cells were treated with 0.1 μg/ml colcemid (Invitrogen) for 6 h, exposed to hypotonic 75 mm KCl solution for 20 min at 37 °C, and fixed in methanol:acetic acid (3:1) three times. Slides were stained with Giemsa, and chromosome aberrations were analyzed under a BX51 microscope (Olympus, Tokyo, Japan). A minimum of 150 metaphases were analyzed between two separate experiments to identify chromatid- and chromosome-type aberrations. To see the effects of Pu-27 on wild type karyotype, we extracted peripheral blood lymphocyte from healthy donor using HISTOPAQUE (Sigma Diagnostics, Inc.) and treated with PHA (Invitrogen) for stimulation. For telomere FISH, the slides with metaphase preparations were treated with 100 μg/ml RNase A for 10 min at 37 °C, fixed in 4% formaldehyde, and rinsed in PBS. The slides were denatured using 70% formamide/2× saline sodium citrate buffer (3 m NaCl, 0. 3 m sodium citrate, pH 7.0) at 75 °C for 2 min, followed by dehydration in ethanol series. Peptide-nucleic acid telomere probes (DAKO, Carpinteria, CA) were denatured at 75 °C for 5 min. The denatured probes were added to the fixed cells on slides and kept in a hot humidified dark chamber at 37 °C for 3 h. Slides were then washed in 70% formamide/2× saline sodium citrate buffer at 32 °C for 15 min and in sodium phosphate buffer (0.1 m NaH2PO4, 0.1 m Na2HPO4, pH 8.0, and 0.1% Nonidet P-40) for 5 min. Slides were counterstained with DAPI, photographed using a BX61 microscope and a cooled CCD Exi Aqua camera (Q-imaging, BC, Canada), and scored for telomere signal loss in 75–100 metaphases.
RESULTS
Pu-27 Induces Extensive Chromosome Aberrations in U937 Cells
Karyotype analysis of U937 cells exposed to 5 μm Pu-27 for up to 5 days demonstrated frequent metaphase abnormalities and extensive chromosomal damage. This damage included chromatid breaks, chromosome breaks, dicentics, and radials, as well as extensive breaks and simple breaks (Fig. 1 and Table 1) throughout the genome including at the ends of chromosomes. The number of abnormal metaphases and chromosomal breaks were more numerous on day 2 (48.1–63.2%) than day 5 (31.8–33.3%). The frequency of metaphases with simple chromosomal breaks was also greater on day 2 (30–39%) than day 5 (23%) after treatment with Pu-27. Similarly, the metaphases exhibiting multi-radial chromosomes and extensive chromosomal breakage was also higher at day 2 (26–39.7%) compared with 5-day exposure (11.4–17.5%). Although significant numbers of cells with chromosomal abnormalities were still present at day 5 compared with control treated cells (Table 1). The loss of telomere-specific FISH signals was also frequently identified (Fig. 1D), which suggests the occurrence of large telomeric loss. We investigated the effects of Pu-27 on peripheral blood lymphocyte (as nontransformed WT cells) from a healthy donor (Table 2). The effect of Pu-27 on WT karyotypes is significantly less than it is on cancerous human histiocytic lymphoma U937 cells. The “abnormal metaphase” and “cells with radials with extensive breaks” in malignant cells are 48–63 and 24–40%, whereas in WT cells they are 14–18 and 0–3.5%, respectively. Thus, this pronounced Pu-27-induced chromosomal hypersensitivity of U937 cells suggests that DNA repair pathways may be targeted by Pu-27 in the genome.
FIGURE 1.
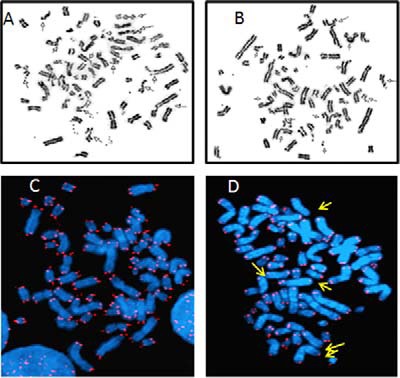
Pu-27 induces chromosomal aberrations in U937 cells. U937 cells were treated with Pu-27 for 5 days and were subjected to karyotype analysis and telomere-specific FISH. A and B, cells treated with Pu-27 showed extensive DNA damage throughout the chromosome. C and D, cells left untreated (C) and cells treated with Pu-27 (D) showed numerous chromosomal end telomeric signals. The arrow indicates several of the abnormalities.
TABLE 1.
Chromosome aberrations of Pu-27-treated U937 cells
# represents the number of the experiment.
| Sample | Total metaphase studied | Abnormal metaphase | Chromatid breaks | Chromosome breaks | Cells with radials and extensive breaks | Frequency of metaphases with simple break |
|---|---|---|---|---|---|---|
| U937 control #1 | 103 | 3 (2.9%) | 1 (1.0%) | 0 | 1 (1.0%) | 0.02 |
| U937 control #2 | 93 | 3 (3.2%) | 0 | 0 | 3 (3.2%) | 0 |
| U937 Pu-27#1 (day 2) | 77 | 37 (48.1%) | 14 (18.2%) | 3 (3.9%) | 20 (26.0%) | 0.30 |
| U937 Pu-27#2 (day 2) | 68 | 43 (63.2%) | 11 (16.2%) | 3 (4.4%) | 27 (39.7%) | 0.39 |
| U937 Pu-27#1 (day 5) | 120 | 40 (33.3%) | 7 (5.8%) | 12 (10%) | 21 (17.5%) | 0.23 |
| U937 Pu-27#2 (day 5) | 88 | 28 (31.8%) | 12 (13.6%) | 3 (3.4%) | 10 (11.4%) | 0.23 |
TABLE 2.
Chromosome aberrations of Pu27-treated normal peripheral lymphocytes
| Sample | Total metaphases studied | Abnormal metaphases | Chromatid breaks | Chromosome breaks | Cells with radials and extensive break | No. breaks/metaphase (excluding radials) |
|---|---|---|---|---|---|---|
| UNT | 119 | 4 (3.4%) | 5 (4.2%) | 2 (1.7%) | 0 | 0.06 |
| Pu27 day 1 | 129 | 12 (9.3%) | 11 (8.5%) | 2 (1.6%) | 0 | 0.1 |
| Pu27 day 3 | 134 | 19 (14.2%) | 12 (9%) | 14 (10.4%) | 0 | 0.19 |
| Pu27 day 5 | 141 | 26 (18.4%) | 27 (19.1%) | 18 (12.8%) | 5 (3.5%) | 0.32 |
Pu-27 Induces Phosphorylation of H2AX
To further characterize the DNA (chromosomal) damage induced by Pu-27, we measured the induction of double-stranded breaks using phosphorylated H2AX (termed γ-H2AX) as a marker. Although it was not present in untreated cells, γ-H2AX began to appear after 6 h of exposure to Pu-27 (data not shown) and was present throughout the treatment period. On days 1, 2, and 3, mean γ-H2AX expressions in Pu-27-treated U937 cells were 22, 32, and 28%, respectively, whereas in untreated cells mean γ-H2AX expression is only 5% (Fig. 2, A and B). TRF2 is a telomere-binding protein and is essential for t-loop formation. Overexpression of a dominant-negative variant of TRF2 such as (ΔB-TRF2) results in telomere uncapping as well as loss of the G-rich strand (11). U937 cells transfected with ΔB-TRF2 (ΔB-U937) became resistant to Pu-27 treatment (Fig. 2A). In ΔB-U937 cells very minimal changes (5, 5, and 2% on days 1, 2, and 3, respectively, compared with untreated only 2%) in mean γ-H2AX were observed during first 3 days of Pu-27 treatment. These results suggest that there was an enormous induction of γ-H2AX in U937 cells compared with ΔB-U937 cells when treated with Pu-27.
FIGURE 2.
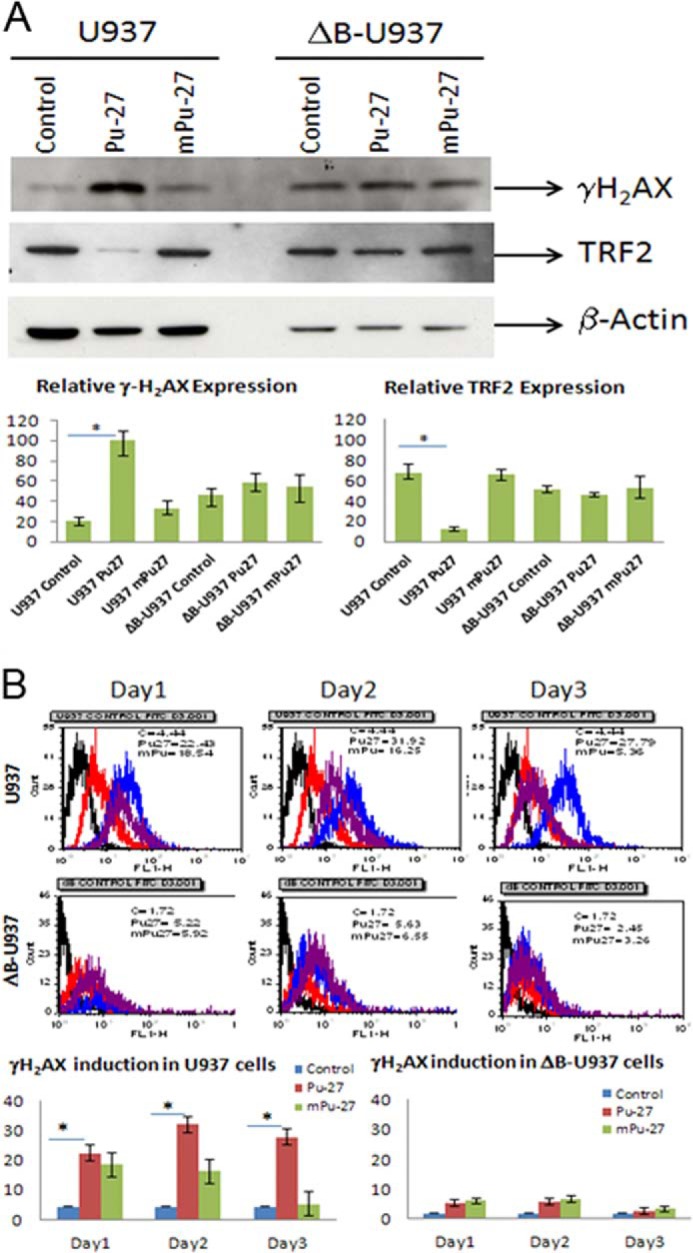
Pu-27 induces phosphorylation of H2AX (γH2AX). U937 cells and ΔB-U937 cells treated with Pu-27 and were subjected to Western blot and FACS analysis. A, 3-day treatment of Pu-27 showed profound up-regulation of γH2AX expression in U937 cells but almost no changes in dB-U937 cells. Relative expressions of TRF2 in U937 and ΔB-U937 cells were also shown. TRF2 expression is down-regulated in U937 cells and no changes in ΔB-U937 cells when treated with Pu-27 (consistent with RT-PCR data in Fig. 6). Bottom panel, relative expression presented in bar graphs. *, p < 0.05. B, by FACS analysis γH2AX has shown continued to be highly expressed from days 1–3, and again there was no significant change in ΔB-U937 cells. Bottom panels, relative expression presented in bar graphs. *, p < 0.05.
ATM Deficient Cells Are More Sensitive than ATM Proficient Cells to Pu-27
ATM is an important upstream regulator in DNA damage response pathway (19). TRF2 has been reported as a direct modulator of ATM on telomeres (22–24). We sought to investigate the sensitivity of Pu-27 in ATM proficient and deficient mouse fibroblast cells: 4a ATM+/+, 695 ATM+/−, and ATM 525−/−. Interestingly, both heterozygous and homozygous ATM deficient cells (+/− and −/−) cells were sensitive to Pu-27, whereas ATM proficient wild type cells (ATM+/+) cells were relatively resistant to Pu-27. This indicated that, in wild type ATM+/+ cells, DNA damaged by Pu-27 responded well, and cells were repaired in an ATM-dependent manner. In the heterozygous ATM+/− or homozygous ATM−/− cells, repair of Pu-27 mediated DNA damage was inefficient, leading to chromosomal instability and cell death (Fig. 3A). 334 ATM+/+ cells were also resistant to Pu-27 (data not shown).
Pu-27 Induces H2AX Phosphorylation at Dysfunctional Telomere by Uncapping TRF1, a Member of Shelterin Complex
Pu-27 induced extensive DNA damage throughout the chromosomes, including telomeres. Therefore we next investigated whether Pu-27 promotes the uncapping of telomeres. ATM deficient mouse fibroblasts (695ATM) were more sensitive to Pu-27 compared with ATM proficient cells (4a ATM) (Fig. 3A). ATM deficient and proficient cells were treated with 10 μm Pu-27 for 2 days and were subjected to examine by confocal microscopy. We found profound phosphorylation of γ-H2AX (red) probably reflecting the presence of DSB only in ATM depleted cells. In the same experiment, we also asked whether this phosphorylation was due to dysfunctional telomeres. Double immunofluorescent staining of ATM cells was performed with TRF1 (green), a good hallmark of interphase chromosomes. Interestingly, there was abundant telomere dysfunction-induced foci co-localized with γ-H2AX (yellow) in Pu-27 sensitive cells. We did not see any significant telomere dysfunction-induced foci in ATM proficient cells (334 ATM) treated with Pu-27 (Fig. 3B).
U937 Cells Lacking the Shelterin Complex Relatively Resistant to Pu-27-induced Cytotoxicity
To further assess possible interactions between Pu-27 and the shelterin complex on telomeres, we employed a stable cell line transfected with ΔB-TRF2 wherein the basic domain of TRF2, a component of shelterin complex has been deleted (11). This ΔB-U937 cell line is negatively affected by the disruption of shelterin; they grow more slowly but can nonetheless be propagated. U937 and ΔB-U937 cells were treated with different doses of Pu-27 and a control oligonucleotide, mutated Pu-27, and viability was assessed by MTT assay on day 6. U937 cells were sensitive, as we have reported before, whereas ΔB-U937 cells were partially (∼20%) sensitive to cytotoxic effects of Pu-27 (Fig. 4A). Annexin-PI staining of U937 cells revealed (Fig. 4B) that almost 11% cells was positive for annexin-V, PI, or both when treated with Pu-27 for 5 days in U937 cells, and in ΔB-U937, it was only 3%. Overall, this is an important result because the effect of ΔB-U937 is on the telomere shelterin complex, as well as perhaps on the other quadruplex forming sequences throughout the rest of the genome. This implies that the toxic effects of Pu-27 are mediated primarily through inhibitions of telomeric and nontelomeric function.
FIGURE 4.

Pu-27 induces significant cell death in U937 cells but not in dB-U937 cells. A, MTT assay of U937 and ΔB-U937 cells shows U937 cells are sensitive, and ΔB-U937 cells are relatively resistant to Pu-27. CRO is used as a control. *, p < 0.002 at 10 μm of Pu-27 treatment. B, U937 and ΔB-U937 cells were treated with Pu-27 for 4 and 5 days, and dead cells were measured with annexin/PI staining. Almost 10% of cells are positive for annexin, PI, and annexin-PI in U937 cells, but few cells were positive in ΔΒ-U937 cells, at least after 5 days of treatment. *, p < 0.001 in both days 4 and 5.
Pu-27-mediated Optimum Sensitivity Requires Intact Shelterin Complex and Not by Inhibiting Quantitative Telomerase
Previously, we showed that human histiocytic lymphoma U937 cells were sensitive to G-quadruplex Pu-27 (22). Chromosomal telomeric DNA consists of tandem G-rich repeats (23) and is protected by a complex of proteins called shelterin (33, 34). We hypothesized that the cytotoxic effects of Pu-27 to U937 cells might be due, at least in part, to destabilization of the shelterin protein complex. Indeed, we found that U937 cells treated with Pu-27 showed down-regulation of key components of the shelterin complex TRF2, TRF1, and TIN2 (Fig. 2A; see also Fig. 6A).
FIGURE 6.

Pu-27 inhibits the molecules related to DNA damage repair machinery in U937. A, RT-PCR of U937 and ΔB-U937 cells treated with Pu-27 for 3 days. Pu-27 inhibits ATM, RAD17, RAD50, CHK14, and CHK2 but not H2AX, BRCA1, and telomerase reverse transcriptase in U937 cells. B, ΔB-U937 cells showed no changes of TRF2, TRF1, POT1, TIN2, RAD17, RAD50, and 53BP1; down-regulation of ATM; up-regulation of CHK1 and CHK2; and H2AX and BRCA1.
Next, we tested whether the cytotoxic effects of Pu-27 might be due, in part, to effects on telomerase. RT-PCR analysis revealed no changes of telomerase reverse transcriptase mRNA levels in Pu-27-treated U937 cells (see Fig. 6A). Several cell lines do not require telomerase reverse transcriptase for maintenance of telomeres. Alt (alternating lengthening of telomere) cells have the ability to replicate indefinitely and maintain the length of telomere in the absence of telomerase (35, 36). A549 cells require telomerase for telomere maintenance and growth and are sensitive to Pu-27. In contrast, Sk-Lu1, an Alt cell line that does not depend on telomerase for replication is relatively resistant to Pu-27 (Fig. 5A).
FIGURE 5.

Pu-27 exerted its sensitivity through intact shelterin complex and not by inhibiting telomerase qualitatively. A, Sk-Lu-1, an Alt lung cancer cell line, is relatively resistant to Pu-27, whereas A549 non-Alt lung cancer cell line is sensitive to Pu-27. *, p < 0.003 at 10 μm of Pu-27 treatment. B, telomerase activity in Alt (Sk-Lu-1), non-Alt (A549), U937, and ΔB-U937 cells after treatment of Pu-27 (PU) and CRO (as a control). Statistical analysis on the relative telomerase activities among various treatments has been performed with the χ2 test for independence. Among control and Pu-27-treated A549, Sk-Lu-1 (Alt cells) cells, H0 cannot be rejected, p = 0.97; among control and Pu-27-treated U937 and dB cells, H0 cannot be rejected, p = 0.925; these results signify no difference among the various groups.
The effect of Pu-27 on telomerase activity was further investigated by quantitative telomerase assay in U937, ΔB-U937, A549, and Sk-Lu-1 cells. There was no difference of telomerase activity among Pu-27-treated and untreated cells (Fig. 5B). These data indicate that Pu-27 perhaps exerted its sensitivity to U937 not by reducing telomerase qualitatively but rather by compromising the telomerase-binding site by destabilizing the shelterin complex.
DNA Damage Response Repair Machinery Is Compromised in Pu-27-treated U937
Pu-27 seems to disrupt the telomeric shelterin complex (TRF2, TRF1, POT1, and TIN2) (Fig. 6A) and may also cause DNA damage in nontelomeric regions (Fig. 1). Next, U937 and ΔB-U937 cells treated with Pu27 for 3 days were analyzed for transcript levels of the molecules that regulate the DNA damage repair by real time RT-PCR. Down-regulation of upstream kinase ATM, DNA damage response mediators (RAD17, RAD50), and DSB repair factor 53BP1 and G2 checkpoint molecules (CHK1 and CHK2) was observed. Interestingly, there were no changes of DNA repair molecules (H2AX and BRCA1) and telomere maintenance gene telomerase reverse transcriptase (Fig. 6A) in U937 cells. ΔB-U937 cells, comparing to U937 cells presented no changes of telomeric shelterin proteins (TRF2, TRF1, POT1, and TIN2), DNA damage response mediators (RAD17 and RAD50) and DSB repair factor 53BP1; down-regulation of upstream kinase ATM; up-regulation of G2 checkpoint molecules (CHK1 and CHK2); and DNA repair molecules (H2AX and BRCA1) (Fig. 6B). These data indicate that Pu-27 resulting in DNA damage and leaving the G2 checkpoint without repair might contribute to cellular death.
Pu-27-treated Cellular Sensitivity Is Independent of p53
p53 is considered as one of the most important molecule regulating ATM-mediated DNA damage response and cellular death. Therefore we investigated the role of p53 in Pu-27-regulated cell death. HCT116 cells with heterozygous and homozygous deletion of p53 were treated with Pu27 showing resistance to Pu27, irrespective of the presence or absence of p53 (Fig. 7).
FIGURE 7.
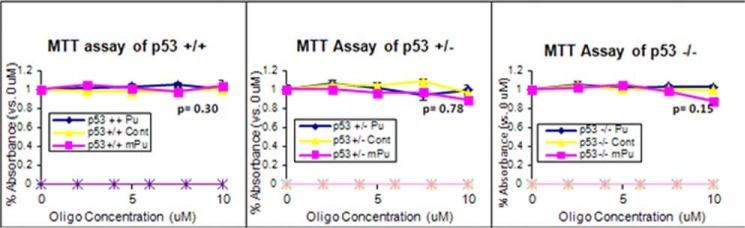
Pu-27-treated cellular sensitivity was independent of p53. HCT116 p53+/+, HCT116 p53+/−, and HCT116 p53−/− cells were treated with Pu27. These cells show resistance to Pu-27 irrespective of their p53 status (p = 0.30, 0.78, and 0.15 in p53+/+, p53+/−, and p53−/− cells, respectively, at 10 μm of Pu-27 treatment).
DISCUSSION
The G-rich quadruplex including Pu-27, enters cells (13) and prompts extensive chromosomal damage. The DNA damage appears to be predominantly in the form of double-stranded breaks as reflected by increases in phosphorylated H2AX. We noticed extensive breaks throughout the chromosome complement following exposure to Pu-27. We also found breaks involved at the telomeric end of the chromosome (Fig. 1D) by FISH as evident by less FISH positive chromosome and more FISH positive chromatid in Pu-27-treated cells. During prolonged incubations, we observed greater disruption of chromosomes and chromatids on day 2 versus day 5, perhaps because cells with extensive damage died early on (Table 1). Cells sense disruption of the shelterin protein complex as double-stranded breaks (33). It has been reported that a small molecule can alter the shelterin integrity and trigger DNA damage response at telomeres (34).
Here, we showed for the first time that a natural G-quadruplex, Pu-27, an important regulator of c-Myc transcription (24), preferentially damages telomeres, partly by disrupting and dismantling the shelterin complex, which is a natural protector of telomeres. One of the mechanisms of cell death caused by Pu-27 is most likely caused by disruption of telomere function/replication. This idea is supported by three main lines of evidences. First, cells that do not depend on telomerase for telomere replication (i.e., alternative lengthening of telomeres) are relatively resistant to the effects of Pu-27. Second, direct quantitation of telomeres in Pu-27-treated cells indicates the loss of several FISH-positive chromosomal ends. Third, introduction of a dominant-negative member of the shelterin complex in U937 cells ΔB-U937 cells, where the N-terminal TRF2 basic domain is deleted, mostly prevents Pu-27-induced cell death. These latter results suggest that an intact shelterin complex is required for the “anti-telomeric” action of Pu-27.
On the other hand, cytogenetic analysis of Pu-27-treated cells shows clear evidence of chromosomal breakage in nontelomeric regions. Whether this damage is a result of interference with the replication of G-rich quadruplex regions of the DNA or mechanical disruption secondary to nondysjunction of telomeres is not clear.
Previously, we argued that Pu-27-mediated cell killing involved interference with c-Myc transcription (24). c-Myc is a proto oncogenic transcription factor related to tumor growth in a number of mouse models (35). Its overexpression causes cell death in fibroblast and myeloid cell lines (36) and induces apoptosis by producing oxidative stress in hepatic cells (37) and inhibition of c-Myc expression sensitizes to TNF-induced apoptosis (38). Therefore, interference with c-Myc expression remains a viable alternative to that of inhibition of telomere replication. Regardless of the precise mechanism(s) involved in Pu-27 cytotoxicity, it clearly does not involve p53-dependent cell death in as much as Pu-27 sensitivity in HCT116 cells is not affected by the presence or absence of p53. The exact mechanism by which Pu-27 causes dysfunction in the c-Myc promoter or the telomere shelterin complex is not known. Indeed, the two effects could be related; multiple levels of evidence suggest that c-Myc has some role in chromosomal rearrangement and remodeling through the telomere (39), and there is direct interaction of c-Myc with catalytic subunit of telomerase (40). The G-quadruplex-interactive telomerase inhibitor, telomestatin (SOT-095), in combination with imatinib has been shown effective against leukemia (41) and the anti-leukemic effect could conceivably involve interference with c-Myc expression, telomerase activity, or both. Quantitative telomerase activity in cell lysates pretreated with Pu-27 exposed that Pu-27 at least does not quantitatively interfere with the telomerase activity of both Alt and non-Alt cells (Fig. 5B). However, whether Pu-27 can interfere with telomerase activity qualitatively and quantitatively in single cellular level remains to be investigated. Cancer therapy directed against telomerase results in cells being mitochondrially adapted and modified with an alternate pathway to lengthening cells (37). Pu-27 could potentially be a good therapeutic strategy for cancer treatment because it appeared to be not directly inhibiting telomerase.
ATM deficient cells are more sensitive to Pu-27 (Fig. 3A) and exhibit abundant telomere dysfunction-induced foci (Fig. 3B). The precise mechanism(s) by which Pu-27 exerts its sensitivity in these ATM deficient cells are not quite explored. ATM is an upstream key regulator of DNA damage response pathways. Therefore, it would be normal to expect that ATM deficient cells are unable to repair the DNA damage properly and are more vulnerable to Pu-27 treatment. The shelterin complex is also not properly protected in ATM deficient cells, making them more susceptible to a DNA-damaging agent like Pu-27. The concept of synthetic lethality (42) in the context of cancer therapy provides a conceptual framework for the cancer-specific cytotoxic agent. G-quadruplex DNA has been shown as a molecular target for induction of synthetic lethality in cancer cells (13). It would be interesting to see whether ATM gene is synthetically lethal to c-Myc or any other genes targeted by Pu-27.
In general, telomerase is more active in cancer cells than normal quiescent cells (31). This may represent an opportunity for the development of agents similar to Pu-27. The apparently greater dependence of cancer cells on telomerase activity may suggest that agents that interfere with this crucial enzymatic activity have real promise for the development of new therapies. Lastly, we have addressed the role of p53 in Pu-27-mediated DNA damage response. It has been postulated that DNA damage response pathway might facilitate tumor suppression by triggering p53 in response to persistent DNA injury and genomic instability that attends tumor progression (32). We have shown that Pu-27 sensitivity in HCT116 cells is not correlated with the presence or absence of p53 (Fig. 7), suggesting that the effect of Pu-27 is p53-independent.
Our results demonstrate that the G-quadruplex sequence of the c-Myc promoter region, Pu-27, disrupts shelterin complex at the telomere region in addition to causing extensive damage throughout the chromosome. The Pu-27-treated cells sense these telomeric and nontelomeric double-stranded breaks and are unable to repair them because of suppression of DNA repair machinery leading to p53-independent cell death (Fig. 8).
FIGURE 8.
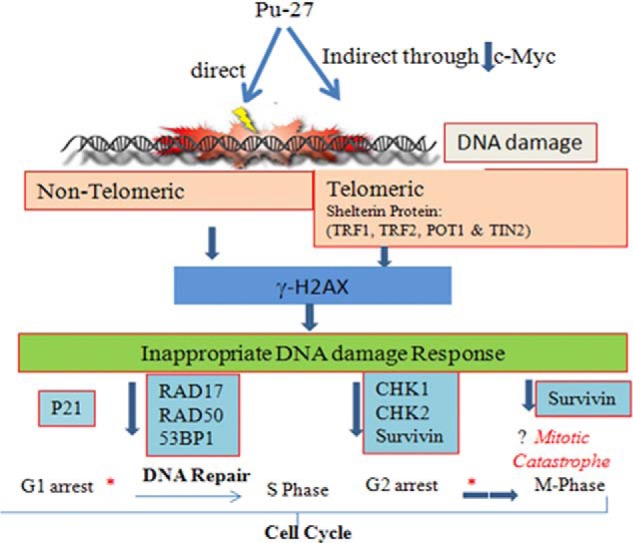
Proposed consequences of Pu-27-induced DNA damage. Pu-27 causes extensive DNA damage telomeric and nontelomeric region directly or indirectly through decreasing expression of c-Myc. This induces γH2AX, activating DNA damage response regulators leading to G1 arrest (43), decreased DNA damage repair, and G2M phase regulatory proteins. Pu-27-induced G1 arrest provides some time to repair. The cell passes through the cell cycle without repair. Then damaged DNA moves quickly through G2 arrest without dividing properly at the M phase (as survivin guards the G2M phase of cell cycle to prevent unwanted apoptosis), leading to cell death perhaps through mitotic catastrophe. Asterisk with thin arrow, cell moves slowly; asterisk with thick arrow, cell moves fast.
Acknowledgments
We express our special thanks to Dr. John Eaton for very helpful scientific revision and input to the manuscript. We also thank Dr. M. S. Turker (Oregon Health Science University, Portland, OR) for the generous gift of ATM cell lines.
This work was supported by Grant COBRE National Institutes of Health: UPHS Grant 1P30GM10639601 (to the James Graham Brown Cancer Center of the University of Louisville).
- G-quadruplex
- guanine-rich quadruplex
- CRO
- cytosine-rich oligonucleotide
- TRF
- telomeric repeat-binding factor
- DSB
- double-stranded break
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- PI
- propidium iodide
- FISH
- fluorescence in situ hybridization.
REFERENCES
- 1. Okazaki S., Tsuchida K., Maekawa H., Ishikawa H., Fujiwara H. (1993) Identification of a pentanucleotide telomeric sequence, (TTAGG)n, in the silkworm Bombyx mori and in other insects. Mol. Cell Biol. 13, 1424–1432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rawal P., Kummarasetti V. B., Ravindran J., Kumar N., Halder K., Sharma R., Mukerji M., Das S. K., Chowdhury S. (2006) Genome-wide prediction of G4 DNA as regulatory motifs. Role in Escherichia coli global regulation. Genome Res. 16, 644–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Robertson H. M., Gordon K. H. (2006) Canonical TTAGG-repeat telomeres and telomerase in the honey bee, Apis mellifera. Genome Res. 16, 1345–1351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Huppert J. L., Balasubramanian S. (2005) Prevalence of quadruplexes in the human genome. Nucleic Acids Res. 33, 2908–2916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Siddiqui-Jain A., Grand C. L., Bearss D. J., Hurley L. H. (2002) Direct evidence for a G-quadruplex in a promoter region and its targeting with a small molecule to repress c-Myc transcription. Proc. Natl. Acad. Sci. U.S.A. 99, 11593–11598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Simonsson T., Pecinka P., Kubista M. (1998) DNA tetraplex formation in the control region of c-Myc. Nucleic Acids Res. 26, 1167–1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Blackburn E. H. (2001) Switching and signaling at the telomere. Cell 106, 661–673 [DOI] [PubMed] [Google Scholar]
- 8. Lundblad V. (2000) DNA ends. Maintenance of chromosome termini versus repair of double strand breaks. Mutat. Res. 451, 227–240 [DOI] [PubMed] [Google Scholar]
- 9. Bilaud T., Brun C., Ancelin K., Koering C. E., Laroche T., Gilson E. (1997) Telomeric localization of TRF2, a novel human telobox protein. Nat. Genet. 17, 236–239 [DOI] [PubMed] [Google Scholar]
- 10. Broccoli D., Smogorzewska A., Chong L., de Lange T. (1997) Human telomeres contain two distinct Myb-related proteins, TRF1 and TRF2. Nat. Genet. 17, 231–235 [DOI] [PubMed] [Google Scholar]
- 11. van Steensel B., Smogorzewska A., de Lange T. (1998) TRF2 protects human telomeres from end-to-end fusions. Cell 92, 401–413 [DOI] [PubMed] [Google Scholar]
- 12. Palm W., de Lange T. (2008) How shelterin protects mammalian telomeres. Annu. Rev. Genet. 42, 301–334 [DOI] [PubMed] [Google Scholar]
- 13. McLuckie K. I., Di Antonio M., Zecchini H., Xian J., Caldas C., Krippendorff B. F., Tannahill D., Lowe C., Balasubramanian S. (2013) G-quadruplex DNA as a molecular target for induced synthetic lethality in cancer cells. J. Am. Chem. Soc. 135, 9640–9643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Friedberg E. C., Bardwell A. J., Bardwell L., Feaver W. J., Kornberg R. D., Svejstrup J. Q., Tomkinson A. E., Wang Z. (1995) Nucleotide excision repair in the yeast Saccharomyces cerevisiae. Its relationship to specialized mitotic recombination and RNA polymerase II basal transcription. Philos. Trans. R. Soc. Lond. B Biol. Sci. 347, 63–68 [DOI] [PubMed] [Google Scholar]
- 15. Zhou B. B., Elledge S. J. (2000) The DNA damage response. Putting checkpoints in perspective. Nature 408, 433–439 [DOI] [PubMed] [Google Scholar]
- 16. Pandita T. K. (2002) ATM function and telomere stability. Oncogene 21, 611–618 [DOI] [PubMed] [Google Scholar]
- 17. Shiloh Y. (2003) ATM and related protein kinases. Safeguarding genome integrity. Nat. Rev. Cancer 3, 155–168 [DOI] [PubMed] [Google Scholar]
- 18. Hurley P. J., Bunz F. (2007) ATM and ATR. Components of an integrated circuit. Cell Cycle 6, 414–417 [DOI] [PubMed] [Google Scholar]
- 19. Pandita T. K., Lieberman H. B., Lim D. S., Dhar S., Zheng W., Taya Y., Kastan M. B. (2000) Ionizing radiation activates the ATM kinase throughout the cell cycle. Oncogene 19, 1386–1391 [DOI] [PubMed] [Google Scholar]
- 20. Bakkenist C. J., Kastan M. B. (2003) DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 421, 499–506 [DOI] [PubMed] [Google Scholar]
- 21. Lavin M. F., Kozlov S. (2007) ATM activation and DNA damage response. Cell Cycle 6, 931–942 [DOI] [PubMed] [Google Scholar]
- 22. Herbig U., Jobling W. A., Chen B. P., Chen D. J., Sedivy J. M. (2004) Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a). Mol. Cell 14, 501–513 [DOI] [PubMed] [Google Scholar]
- 23. Takai H., Smogorzewska A., de Lange T. (2003) DNA damage foci at dysfunctional telomeres. Curr. Biol. 13, 1549–1556 [DOI] [PubMed] [Google Scholar]
- 24. Denchi E. L., de Lange T. (2007) Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1. Nature 448, 1068–1071 [DOI] [PubMed] [Google Scholar]
- 25. Smilenov L. B., Dhar S., Pandita T. K. (1999) Altered telomere nuclear matrix interactions and nucleosomal periodicity in ataxia telangiectasia cells before and after ionizing radiation treatment. Mol. Cell Biol. 19, 6963–6971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Verdun R. E., Crabbe L., Haggblom C., Karlseder J. (2005) Functional human telomeres are recognized as DNA damage in G2 of the cell cycle. Mol. Cell 20, 551–561 [DOI] [PubMed] [Google Scholar]
- 27. Verdun R. E., Karlseder J. (2006) The DNA damage machinery and homologous recombination pathway act consecutively to protect human telomeres. Cell 127, 709–720 [DOI] [PubMed] [Google Scholar]
- 28. Verdun R. E., Karlseder J. (2007) Replication and protection of telomeres. Nature 447, 924–931 [DOI] [PubMed] [Google Scholar]
- 29. Potter H., Weir L., Leder P. (1984) Enhancer-dependent expression of human kappa immunoglobulin genes introduced into mouse pre-B lymphocytes by electroporation. Proc. Natl. Acad. Sci. U.S.A. 81, 7161–7165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Farokhzad O. C., Shelley C. S., Arnaout M. A. (1996) Induction of the CD11b gene during activation of the monocytic cell line U937 requires a novel nuclear factor MS-2. J. Immunol. 157, 5597–5605 [PubMed] [Google Scholar]
- 31. Neidle S. (2010) Human telomeric G-quadruplex. The current status of telomeric G-quadruplexes as therapeutic targets in human cancer. FEBS J. 277, 1118–1125 [DOI] [PubMed] [Google Scholar]
- 32. Meek D. W. (2009) Tumour suppression by p53. A role for the DNA damage response? Nat. Rev. Cancer 9, 714–723 [DOI] [PubMed] [Google Scholar]
- 33. Muraki K., Nyhan K., Han L., Murnane J. P. (2012) Mechanisms of telomere loss and their consequences for chromosome instability. Front. Oncol. 2, 135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hoogstraten D., Bergink S., Ng J. M., Verbiest V. H., Luijsterburg M. S., Geverts B., Raams A., Dinant C., Hoeijmakers J. H., Vermeulen W., Houtsmuller A. B. (2008) Versatile DNA damage detection by the global genome nucleotide excision repair protein XPC. J. Cell Sci. 121, 2850–2859 [DOI] [PubMed] [Google Scholar]
- 35. Miller D. M., Thomas S. D., Islam A., Muench D., Sedoris K. (2012) c-Myc and cancer metabolism. Clin. Cancer Res. 18, 5546–5553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Evan G. I., Wyllie A. H., Gilbert C. S., Littlewood T. D., Land H., Brooks M., Waters C. M., Penn L. Z., Hancock D. C. (1992) Induction of apoptosis in fibroblasts by c-Myc protein. Cell 69, 119–128 [DOI] [PubMed] [Google Scholar]
- 37. Xu Y., Nguyen Q., Lo D. C., Czaja M. J. (1997) c-Myc-dependent hepatoma cell apoptosis results from oxidative stress and not a deficiency of growth factors. J. Cell Physiol. 170, 192–199 [DOI] [PubMed] [Google Scholar]
- 38. Liu H., Lo C. R., Jones B. E., Pradhan Z., Srinivasan A., Valentino K. L., Stockert R. J., Czaja M. J. (2000) Inhibition of c-Myc expression sensitizes hepatocytes to tumor necrosis factor-induced apoptosis and necrosis. J. Biol. Chem. 275, 40155–40162 [DOI] [PubMed] [Google Scholar]
- 39. Louis S. F., Vermolen B. J., Garini Y., Young I. T., Guffei A., Lichtensztejn Z., Kuttler F., Chuang T. C., Moshir S., Mougey V., Chuang A. Y., Kerr P. D., Fest T., Boukamp P., Mai S. (2005) c-Myc induces chromosomal rearrangements through telomere and chromosome remodeling in the interphase nucleus. Proc. Natl. Acad. Sci. U.S.A. 102, 9613–9618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Flores I., Evan G., Blasco M. A. (2006) Genetic analysis of myc and telomerase interactions in vivo. Mol. Cell Biol. 26, 6130–6138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tauchi T., Shin-Ya K., Sashida G., Sumi M., Nakajima A., Shimamoto T., Ohyashiki J. H., Ohyashiki K. (2003) Activity of a novel G-quadruplex-interactive telomerase inhibitor, telomestatin (SOT-095), against human leukemia cells. Involvement of ATM-dependent DNA damage response pathways. Oncogene 22, 5338–5347 [DOI] [PubMed] [Google Scholar]
- 42. Kaelin W. G., Jr. (2005) The concept of synthetic lethality in the context of anticancer therapy. Nat. Rev. Cancer 5, 689–698 [DOI] [PubMed] [Google Scholar]
- 43. Sedoris K. C., Thomas S. D., Clarkson C. R., Muench D., Islam A., Singh R., Miller D. M. (2012) Genomic c-Myc quadruplex DNA selectively kills leukemia. Mol. Cancer Ther. 11, 66–76 [DOI] [PMC free article] [PubMed] [Google Scholar]