

Background: The H2S-generating human enzyme cystathionine β-synthase (CBS) is inhibited by NO• and CO.
Results: NO• binds to the ferrous heme in human CBS much more quickly than CO and much more tightly than currently thought.
Conclusion: Results support the physiological role of NO• in CBS regulation.
Significance: CBS may integrate the cross-talk among NO•, CO, and H2S, major modulators in human (patho)physiology.
Keywords: Cell Signaling, Enzyme Kinetics, Heme, Hydrogen Sulfide, Redox Regulation
Abstract
The hexa-coordinate heme in the H2S-generating human enzyme cystathionine β-synthase (CBS) acts as a redox-sensitive regulator that impairs CBS activity upon binding of NO• or CO at the reduced iron. Despite the proposed physiological relevance of this inhibitory mechanism, unlike CO, NO• was reported to bind at the CBS heme with very low affinity (Kd = 30–281 μm). This discrepancy was herein reconciled by investigating the NO• reactivity of recombinant human CBS by static and stopped-flow UV-visible absorption spectroscopy. We found that NO• binds tightly to the ferrous CBS heme, with an apparent Kd ≤0.23 μm. In line with this result, at 25 °C, NO• binds quickly to CBS (kon ∼ 8 × 103 m−1 s−1) and dissociates slowly from the enzyme (koff ∼ 0.003 s−1). The observed rate constants for NO• binding were found to be linearly dependent on [NO•] up to ∼ 800 μm NO•, and >100-fold higher than those measured for CO, indicating that the reaction is not limited by the slow dissociation of Cys-52 from the heme iron, as reported for CO. For the first time the heme of human CBS is reported to bind NO• quickly and tightly, providing a mechanistic basis for the in vivo regulation of the enzyme by NO•. The novel findings reported here shed new light on CBS regulation by NO• and its possible (patho)physiological relevance, enforcing the growing evidence for an interplay among the gasotransmitters NO•, CO, and H2S in cell signaling.
Introduction
The past three decades have witnessed the emergence of “biologic gases” as key modulators of several (patho)physiological functions in human health. Merely considered as toxic gases in the past, CO, NO•, and H2S have been more recently shown to be enzymatically produced to control various physiologic processes such as blood flow, immune defense against invading pathogens, cellular stress response, apoptosis, inflammation, and energy metabolism (1–3). Moreover, these signaling gaseous molecules (also known as “gasotransmitters”) show a remarkable and intricate interplay in terms of regulating each other's synthesis and breakdown (for review see Refs. 2, 4).
Hydrogen sulfide (H2S) is enzymatically synthesized in humans by three enzymes: cystathionine β-synthase (CBS)4 and cystathionine γ-lyase in the transsulfuration pathway, and 3-mercaptopyruvate sulfurtransferase. CBS and cystathionine γ-lyase are considered the prominent enzymatic sources of H2S, their distribution, abundance, and contribution to H2S synthesis appearing to be tissue-specific (1, 5, 6). Recent studies on 3-mercaptopyruvate sulfurtransferase contributed to reevaluate its physiological relevance in H2S generation (7).
Human CBS is a 63-kDa, 551-amino acid long cytoplasmic protein, assembled as a homotetramer. Each monomer is composed of three structural domains: a central catalytic core domain containing a pyridoxal 5′-phosphate cofactor in the active site, flanked by a C-terminal regulatory domain with two binding sites for the allosteric activator S-adenosylmethionine (8) and an N-terminal domain with a B-type heme (9–11).
The heme moiety in CBS has been the focus of great research efforts aimed at understanding its regulatory role, mainly because this cofactor appears to be an evolutionary hallmark, being absent in CBS enzymes from unicellular eukaryotes like Saccharomyces cerevisiae and Trypanosoma cruzi (12). The heme iron of human CBS both in the oxidized and reduced state is low spin, hexa-coordinate, being axially bound by the endogenous ligands His-65 and Cys-52. This heme displays a remarkably low reduction potential (−350 mV versus standard hydrogen electrode) (13) and acts as a redox-dependent gas sensor because of its ability to react with O2 and to bind CO and NO• in the reduced form. Despite the low reduction potential and the fact that most studies on the CBS heme reactivity were carried out in the presence of a large excess of the potent chemical reductant sodium dithionite, it has been recently shown that the diflavin enzyme methionine synthase reductase is able to reduce the CBS heme with NADPH as the electron donor (14, 15), conferring greater physiologic relevance to ferrous CBS and its adducts with CO and NO•.
CO was shown to bind ferrous CBS with high affinity (Kd1 = 1.5 μm and Kd2 = 68 μm), leading to enzyme inhibition (Ki = 5.6 μm) (16). An electrostatic interaction between the Cys-52 thiolate and the guanidinium group of Arg-266 has been identified as a key element in communicating the heme CO binding to the pyridoxal 5′-phosphate active site (13, 17, 18). The reaction with CO proceeds at very low rates showing hyperbolic dependence on CO concentration (14–16, 19). CO binding has been proposed to be limited by the slow dissociation rate (0.0166 s−1 at 24.5 °C) of Cys-52 from the CBS heme (19).
In contrast with the high affinity for CO, NO• was reported to bind ferrous CBS very weakly (Kd = 30–281 μm), also resulting in enzyme inhibition (Ki = 320 μm), based on studies with a NO• releaser (DEA-NONOate) (20, 21). Whereas the CO-bound ferrous adduct is hexa-coordinate keeping the His-65 ligand in place, NO• binding to CBS results in a penta-coordinate iron-nitrosyl species where both endogenous axial ligands are lost, as shown by visible absorption and EPR spectroscopy (20). Notably, both the NO•- and CO-bound adducts are promptly oxidized by O2 back to the ferric state, resulting in a recovery of the enzymatic activity (22).
Involvement of CO and NO• in CBS regulation is supported by a number of (patho)physiological observations. Elevated NO• levels resulting from renal ischemia-reperfusion in mice have been shown to inhibit CBS and cause an increase in homocysteine that proved to be detrimental for the kidney (23). In a mouse model of brain hypoxia, cerebral vasodilation has been reported to be associated with CBS regulation by CO. The CO-generating oxygen sensor heme oxygenase HO-2 is down-regulated in hypoxia, and the lower than basal CO levels result in a de-repression of CBS-catalyzed H2S generation, which mediates vasodilation of precapillary arterioles (24).
Despite the several observations pointing to NO• and CO as physiological regulators of CBS, only the parameters determined for CO binding to the enzyme, particularly its low dissociation constant, appear consistent with such a role (14, 19, 20). The reported very weak binding of NO• to ferrous CBS is indeed hardly compatible with the nanomolar to few micromolar physiological concentrations of NO•.
Herein we investigated by static and stopped-flow absorption spectroscopy the NO• binding properties of ferrous human CBS using solutions of authentic NO• gas and observed that NO• binds much more tightly (approximately 1000-fold) than previously reported (20) and >100-fold faster than CO under similar experimental conditions, without being limited by Cys-52 dissociation. Overall, the data presented here provide a mechanistic basis for in vivo regulation of CBS by NO•.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification
Human CBS cDNA was cloned into a pET28b vector with restriction sites for NdeI and XhoI, yielding an N-terminally His-tagged protein, as described in Ref. 25. The resulting vector was used to transform Escherichia coli BL21(DE3) Rosetta cells. Cells were grown at 37 °C in Luria-Bertani broth containing 25 mg/liter kanamycin (Nzytech) and 34 mg/liter chloramphenicol (Nzytech) until A600 nm reached 0.4–0.5. Protein overexpression was induced by the addition of 0.5 mm isopropyl-β-d-thiogalactopyranoside (Nzytech). δ-Aminolevulinic acid (Sigma) was added to the culture medium to a final concentration of 75 mg/liter. Following a 4-h incubation at 22 °C and 120 rpm, cells were harvested by centrifugation and the pellet resuspended in buffer A (50 mm potassium phosphate, 300 mm KCl, pH 7.0, 10% glycerol; 10 ml of buffer per liter of culture) containing 1 mg/ml lysozyme (AppliChem), 1 mm phenylmethylsulfonyl fluoride (Merck), and a few grains of deoxyribonuclease I (AppliChem). After a 30-min incubation on ice, cells were disrupted by sonication, centrifuged at 8200 × g (5 min, 4 °C), and imidazole (10 mm) was added to the cleared supernatant. Purification of the His-tagged protein was performed by affinity chromatography, using a HisTrap FF crude 1-ml column (GE Healthcare) previously equilibrated with buffer A containing 10 mm imidazole (buffer B). The column was washed with 15 volumes of buffer B at 1 ml/min, and the protein was eluted in 20 volumes of a linear imidazole gradient to a final concentration of 500 mm. Following imidazole removal in a PD10 desalting column (GE Healthcare), the purified protein was concentrated in a Vivaspin 15R 30-kDa tube (Sartorius). Protein purity was assessed by SDS-PAGE and concentration determined by the Bradford method. Concentration of the ferric heme in the isolated protein was determined using ϵ428 nm = 92,700 m−1 cm−1 (22).
Preparation of NO•- and CO-equilibrated Solutions
Higher N oxides in the NO• gas were preliminarily removed by flushing the gas through a 5 m NaOH trap and a second water trap. Stock solutions of NO• were then prepared by equilibrating thoroughly degassed water with the pure gas at 1 atm. The concentration of NO• in solution was determined by titration of reduced bovine cytochrome c oxidase monitored by visible absorption spectroscopy (26). Stock solutions of CO were prepared by equilibrating thoroughly degassed buffer (50 mm potassium phosphate, 300 mm KCl, 10% glycerol, pH 7.0) with the pure gas at 1 atm, yielding 1 mm CO at 20 °C.
Static UV-visible Absorption Spectroscopy
Static UV-visible absorption spectra were recorded in an Agilent Cary-60 spectrophotometer, in a rubber cap sealed quartz cuvette, made anaerobic by nitrogen flushing. CBS (1.3–2.2 μm in heme) was flushed with nitrogen and transferred anaerobically into the cuvette. Glucose oxidase (4 units/ml), catalase (13 μg/ml), superoxide dismutase (SOD, 60 units/ml), and glucose (2 mm) were added to scavenge contaminant oxygen, hydrogen peroxide, and superoxide anion. After recording the spectrum of as isolated oxidized CBS, the protein was reduced with sodium dithionite (ranging from 11.3 to 45 μm) diluted from a freshly prepared 45 mm stock solution. Dithionite concentration was determined using ϵ314 nm = 8043 m−1 cm−1 (27). Aliquots of NO• or CO were added to reduced CBS using gas-tight Hamilton syringes. After each addition, spectral changes were monitored in real time, and when no more changes were observed, a new addition was immediately made.
Apparent Kd for NO• was determined by fitting the data to the equation:
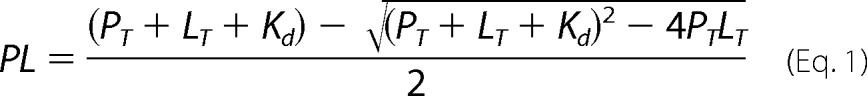 |
where PL is the concentration of NO•-bound CBS, and PT and LT are the total concentration of CBS and NO•, respectively. In the case of CO, two apparent Kd had to be assumed to satisfactorily fit the data, in agreement with Refs. 16, 19.
Time-resolved Absorption Spectroscopy
Stopped-flow experiments were performed in a thermostated instrument (DX.17MV, Applied Photophysics, Leatherhead, UK), equipped with a photodiode array (light path, 1 cm). For reactions studied in the multiwavelength mode, to avoid light artifacts, we reduced the intensity of the incident white light beam and employed a filter cutting UV light at λ <360 nm. Time-resolved absorption spectra were recorded with an acquisition time of 10 ms/spectrum according to a logarithmic or a linear time scale.
All reactions were investigated at 25 °C, in 50 mm potassium phosphate, 300 mm KCl, 10% glycerol, pH 7.0. Recombinant human CBS was diluted to a final concentration of 1.4–8.0 μm (in heme content) and flushed with nitrogen. Glucose oxidase (4 units/ml), catalase (13 μg/ml), SOD (6–90 units/ml), and glucose (2 mm) were then added, and the protein was placed on ice and protected from light (to prevent possible damaging photoreactions). CBS reduction was achieved by addition of 90 μm sodium dithionite, diluted from a freshly prepared 45 mm stock solution. Reduced CBS (as such or after addition of CO or NO•) was mixed in the stopped-flow apparatus with solutions containing CO, NO•, O2, or sulfite, and the spectra were recorded over time. Solutions of NO• at different concentrations were prepared by diluting the NO• stock solution into anaerobic reaction buffer containing glucose, glucose oxidase, catalase, and SOD at the concentrations mentioned above. In control experiments, the NO• concentration was shown not to be affected by this enzymatic scavenging system in the course of the experiments (data not shown). O2 solutions at different concentrations were prepared by diluting O2-equilibrated buffer into degassed buffer containing catalase (13 μg/ml) and SOD (90 units/ml).
Data Analysis
Data were analyzed with MATLAB software (Mathworks, South Natick, MA). Global fit analysis of the spectral data was performed by singular value decomposition analysis combined with curve fitting (28).
RESULTS
Tight Binding of NO• to Ferrous CBS
The affinity of NO• for the ferrous heme in recombinant human CBS was measured by visible absorption spectroscopy, using solutions of authentic NO• gas (Fig. 1). First, we attempted to reduce CBS with reductants that are unreactive or only slowly reactive with NO•, such as ascorbate plus N,N,N′,N′-tetramethyl-p-phenylenediamine, dithiothreitol, and reduced glutathione. However, none of the tested reductants, even in large molar excess, was able to reduce the CBS heme (data not shown) consistently with the low reduction potential of the cofactor. Therefore, we decided to use dithionite as the reductant. While optimizing the experimental conditions for CBS reduction, we incidentally observed that prolonged (>30 min) incubation of the enzyme with dithionite in strictly anaerobic conditions led to a progressive decay of the dithionite UV band and a blueshift of the 449-nm Soret band of reduced CBS to 427 nm (not shown). Because the protein spectral changes were concomitant with the dithionite decay, we hypothesized that the 427-nm species could result from reaction of the ferrous heme with a product of dithionite decomposition. As sulfite is one of the major dithionite decomposition products, we tested its reactivity toward the ferrous CBS heme by stopped-flow spectroscopy and observed that sulfite reacts slowly with the protein (k = 1.7 × 101 m−1 s−1, at pH 7.0, temperature = 25 °C) with low affinity, apparently resulting in oxidized CBS (Fig. 1A). This incidental observation led us to avoid prolonged incubation of the protein with dithionite during the titrations and to use relatively small amounts of the reductant (≤45 μm), also to minimize possible artifacts because of the reaction with NO•.
FIGURE 1.
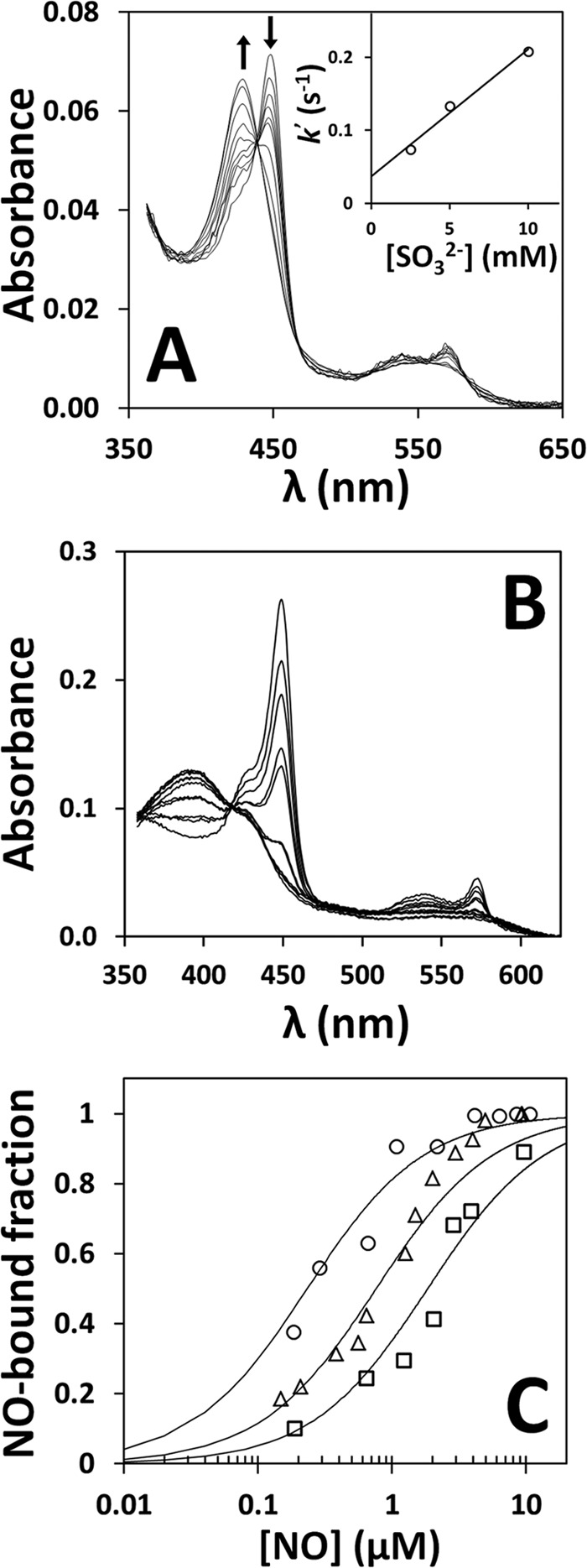
Tight binding of NO• to human CBS. A, reaction of ferrous CBS (1.4 μm in heme content, before mixing) with sulfite (20 mm before mixing). Temperature = 25 °C. Spectra were recorded at 0.01, 0.5, 1.0, 1.5, 2.0, 2.5, 4.1, 7.8, 15.5, and 27.1 s (arrows depict the direction of absorption changes). Inset, dependence of the observed rate constants on sulfite concentration, yielding k = 1.7 × 101 m−1 s−1. B, NO• titration of CBS (2.2 μm in heme), prereduced with 11.3 μm dithionite. Spectra (from two titrations with five data points each) were recorded with ∼2-min intervals, at [NO•] = 0.5, 1.1, 1.6, 2.2, 3.2, 4.3, 6.5, 8.6, 10.8, and 13.0 μm. Buffer was 50 mm potassium phosphate, 300 mm KCl, 10% glycerol, 100 μm EDTA, pH 7.0, containing 2 mm glucose, 4 units/ml glucose oxidase, 13 μg/ml catalase, and 60 units/ml SOD. C, NO• titrations of ferrous CBS (1.3–2.2 μm in heme) measured at 11.3 μm dithionite (Kd = 0.23 μm, circles), 22.5 μm dithionite (Kd = 0.76 μm, triangles), 45 μm dithionite (Kd = 1.84 μm, squares).
CBS (1.3–2.2 μm in heme) was therefore anaerobically reduced with a slight excess of dithionite (11.3–45 μm) and promptly titrated with NO•, monitoring the disappearance of the reduced CBS 449-nm band and the concurrent formation of the broad 396-nm band characteristic of the NO•-bound enzyme (Fig. 1B). As confirmed by global fit analysis, only a single optical transition was detected in the course of the equilibrium titrations, with no evidence for optically detectable intermediates. Notably, as shown in Fig. 1C, we observed a clear dependence of the apparent Kd for NO• on the dithionite concentration employed to keep CBS reduced during the assays. Indeed, at the lowest dithionite concentration tested (11.3 μm), we measured a Kd of 0.23 μm (circles in Fig. 1C), whereas at 22.5 μm and 45 μm dithionite (triangles and squares in Fig. 1C, respectively), we measured higher Kd values (namely, 0.76 μm and 1.84 μm, respectively). This result is probably because of dithionite slowly reacting with NO• over the time of the titration. Based on these observations, we conclude that NO• binds the ferrous heme in CBS with high affinity, with an apparent Kd of 0.23 μm or less.
Despite its high affinity for ferrous CBS, NO• was found to react with ferric CBS only very slowly and with very low affinity, causing reductive nitrosylation of the heme. By anaerobically incubating oxidized CBS at room temperature with 1 mm NO•, and collecting spectra up to 12 h, the reaction was found to proceed with a half-time exceeding 2 h (Fig. 2). Altogether, these results prompted us to measure the on- and off-rate constants of NO• for ferrous CBS.
FIGURE 2.
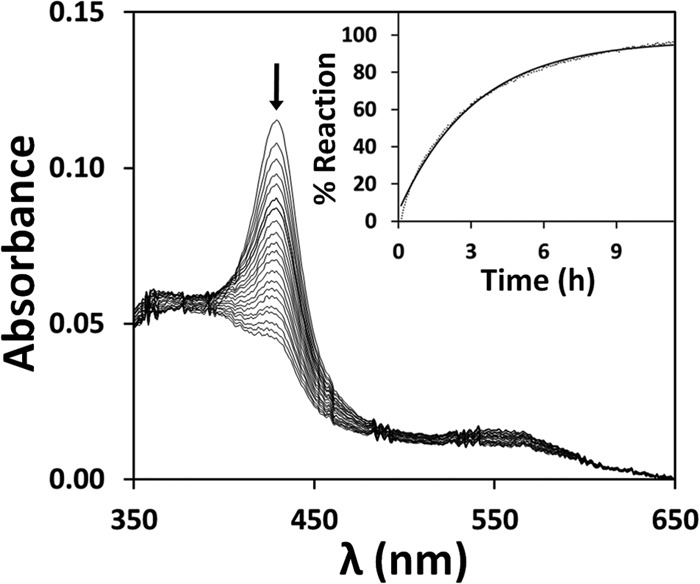
Reductive nitrosylation of ferric CBS. Spectra collected at 25 °C over 12 h, after anaerobic addition of 1.08 mm NO• to 1.2 μm (heme content) ferric CBS (in 50 mm potassium phosphate, 300 mm KCl, 10% glycerol, 100 μm EDTA, pH 7.0, containing 4 units/ml glucose oxidase, 13 μg/ml catalase, 60 units/ml SOD, and 2 mm glucose). Spectra were recorded every 10 min (arrow depicts the direction of absorption changes). Inset, reaction time course (dotted line) and its best fit (full line) to a single exponential (k′ = 0.32 h−1).
NO• Binding and Dissociation from Ferrous CBS
The kinetics of NO• binding to ferrous CBS was investigated at 25 °C by stopped-flow absorption spectroscopy, again using authentic NO• gas. In these assays, CBS (1.6 μm in heme before mixing) was reduced by addition of 90 μm sodium dithionite and anaerobically mixed in the stopped-flow apparatus against solutions of NO• at different concentrations (50 μm to 1.5 mm, before mixing). As a representative dataset, we report in Fig. 3A the time-resolved absorption spectra acquired after mixing ferrous CBS with 100 μm NO• (before mixing). The data show that conversion of ferrous CBS into the Fe(II)-NO• adduct is complete within 10 s. Global fit analysis of this kinetic dataset shows that the reaction is associated to a single optical transition (from Fe(II) to Fe(II)-NO• CBS, inset to Fig. 3B), with no evidence for optically detectable intermediates. The reaction follows a slightly biphasic time course with a major kinetic phase proceeding at ∼ 0.6 s−1 (Fig. 3B). Identical results were obtained by monitoring the reaction at single wavelength, i.e. 449 nm, where NO• binding to ferrous CBS yields maximal absorption changes (Fig. 4A). Repeating these measurements at various NO• concentrations and with different protein preparations, we observed a linear dependence of the observed rate constant on NO• concentration, yielding a second-order rate constant k ∼ 8 × 103 m−1 s−1 (inset to Fig. 4A and Table 1). By mixing CBS at higher concentration (8 μm before mixing) with substoichiometric, equimolar or excess NO•, again we observed direct conversion of ferrous CBS to the final penta-coordinate NO•-bound form of the enzyme, with no detectable intermediates (data not shown).
FIGURE 3.
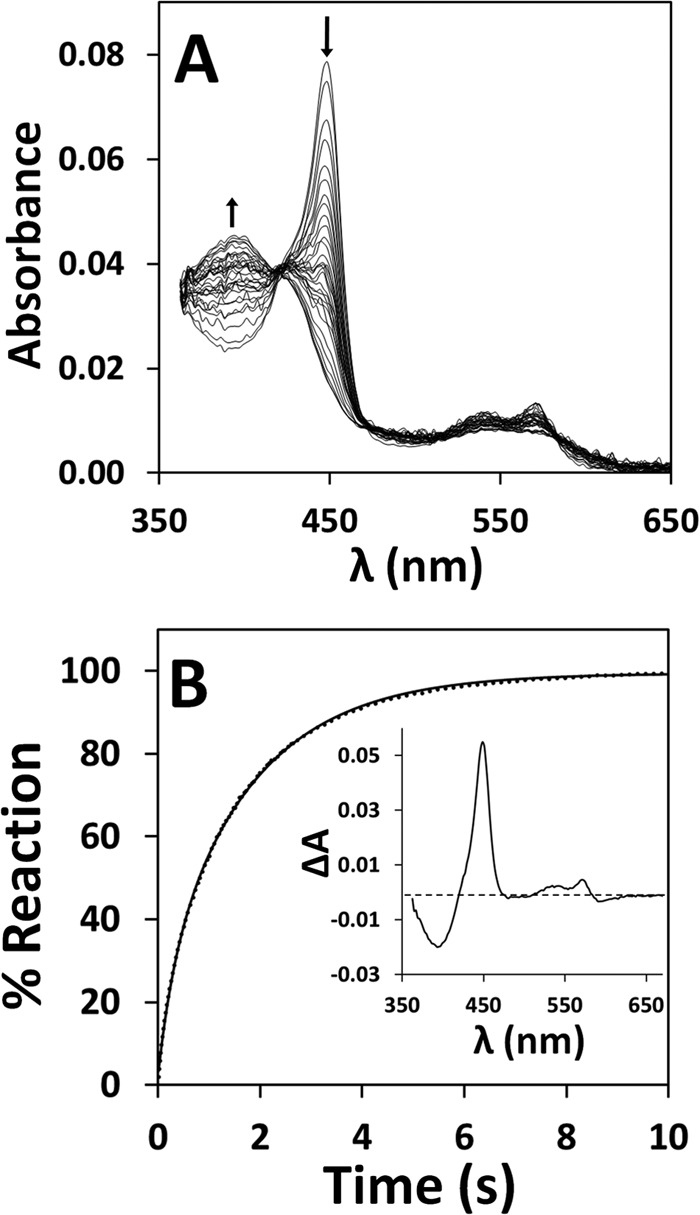
Kinetics of NO• binding to ferrous CBS. A, spectra collected over 10 s after mixing 1.6 μm ferrous CBS (in heme content) with 100 μm NO• at 25 °C, in 50 mm potassium phosphate, 300 mm KCl, 10% glycerol, 100 μm EDTA, pH 7.0, containing 4 units/ml glucose oxidase, 13 μg/ml catalase, 60 units/ml SOD, and 2 mm glucose. Arrows depict the direction of absorption changes. B, reaction time course (dotted line) and its best fit (full line) to the sum of two exponentials (k1 = 3.2 s−1 and k2 = 0.56 s−1, accounting for 20 and 80% of the overall amplitude, respectively). Inset, optical transition as obtained by global fit analysis.
FIGURE 4.
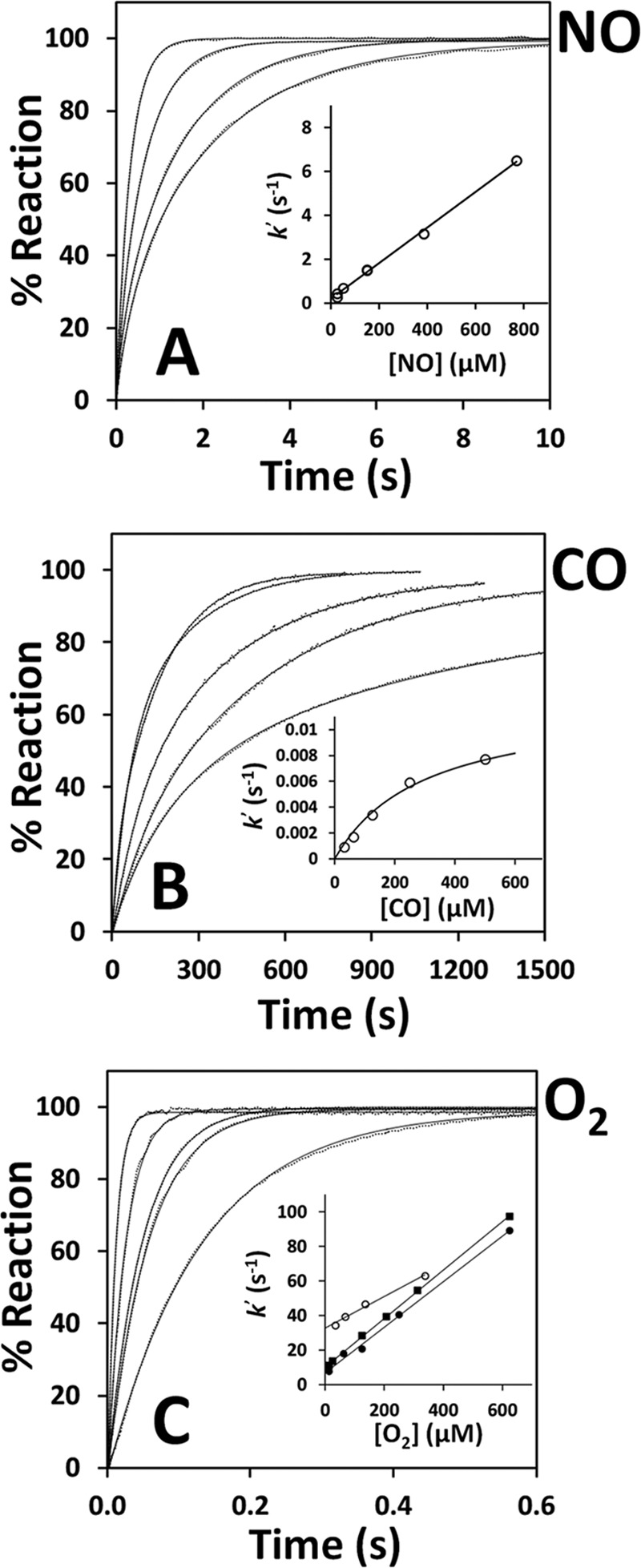
Reaction of ferrous CBS with NO•, CO, and O2. Reactions were investigated at 25 °C in 50 mm potassium phosphate, 300 mm KCl, 10% glycerol, 100 μm EDTA, pH 7.0, containing 2 mm glucose, 4 units/ml glucose oxidase, 13 μg/ml catalase, and SOD (6 units/ml for NO• and CO assays and 6–90 units/ml for O2 assays). A, normalized absorption changes measured at 449 nm after mixing ferrous CBS (1.6 μm in heme, before mixing) with NO• (770, 300, 100, and 50 μm before mixing, from left to right). Traces (dotted lines) are shown with their best fit (full lines) to the sum of two exponentials. Inset, [NO•] dependence of the rate constant relative to the major kinetic phase, yielding k ∼ 8 × 103 m−1 s−1. B, reaction of ferrous CBS (2.6 μm in heme, before mixing) with CO (1000, 500, 250, 125 and 62.5 μm before mixing, from left to right). Traces (dotted lines) are shown with their best fit (full lines) to the sum of two exponentials. Inset, [CO] dependence of the rate constant relative to the major kinetic phase, yielding k′lim = 0.012 s−1. C, normalized absorption changes measured at 449 nm after mixing ferrous CBS (1.4 μm in heme, before mixing) with O2 (1250, 500, 250, 125, and 25 μm before mixing, from left to right). Traces (dotted lines) are shown with their best fit (full lines) to a single exponential. Inset, [O2] dependence of the rate constant measured at [SOD] = 6 units/ml (open circles, k ∼ 9.2 × 104 m−1 s−1, k′0 ∼ 32.6 s−1), 60 units/ml (filled squares, k ∼ 1.4 × 105 m−1 s−1, k′0 ∼ 10.1 s−1), and 90 units/ml (filled circles, k ∼ 1.3 × 105 m−1 s−1, k′0 ∼ 6.8 s−1).
TABLE 1.
Rates and dissociation constants for the reactions of ferrous CBS with NO, CO, and O2
Data from this work are in bold. * indicates that, at variance from NO and O2, CO binding to ferrous CBS is reportedly limited by the low off-rate (k′lim) of Cys-52.
Measurements of the off-rate of NO• from ferrous CBS were attempted in ligand displacement experiments, according to the method established by Moore and Gibson (29), i.e. by anaerobically mixing Fe(II)-NO• CBS with a CO-equilibrated solution containing 45 mm dithionite (giving 500 μm CO and 22.5 mm dithionite after mixing). Monitoring the reaction at single wavelength (422 nm) to minimize possible ligand photodissociation, we observed a slightly biphasic reaction, with a major component proceeding at k′ = 0.003 ± 0.001 s−1 (line a in Fig. 5A). As a control, an identical rate was measured at 431 nm, the isosbestic wavelength for CO binding to CBS (data not shown).
FIGURE 5.
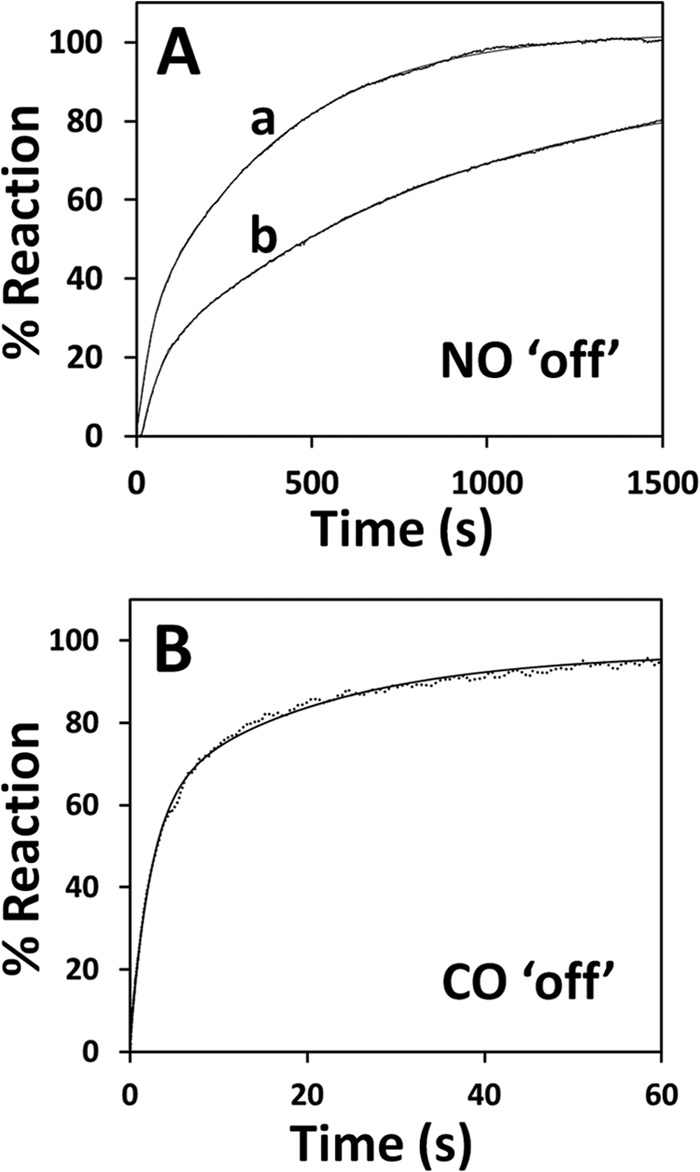
Dissociation of NO• and CO from ferrous CBS. Reactions were investigated at 25 °C in 50 mm potassium phosphate, 300 mm KCl, 10% glycerol, 100 μm EDTA, pH 7.0, containing 2 mm glucose, 4 units/ml glucose oxidase, 13 μg/ml catalase, and SOD (60 units/ml for NO• dissociation and 6 units/ml for CO dissociation). A, NO• dissociation measured after mixing NO•-bound ferrous CBS (2 μm in heme, before mixing) with CO-equilibrated buffer containing 45 mm dithionite (line a, 422 nm) or O2-equilibrated buffer (line b, 426 nm). Fitted rate constant (percentage of the overall amplitude): line a, k1 = 0.05 s−1 (20%) and k2 = 0.002 s−1 (80%); line b, k1 = 0.016 s−1 (25%) and k2 = 0.001 s−1 (75%). B, CO dissociation measured after mixing CO-bound ferrous CBS (1.8 μm in heme, before mixing) with 1 mm NO• (before mixing; λ = 415 nm). Fitted rate constant (percentage of the overall amplitude): k1 = 0.44 s−1 (60%) and k2 = 0.05 s−1 (40%).
We also mixed Fe(II)-NO• CBS with O2-equilibrated buffer (625 μm O2 after mixing) in the stopped-flow apparatus and monitored the enzyme oxidation at 426 nm. In this case, a strongly biphasic time course was observed (line b in Fig. 5A), with a faster phase proceeding at ∼0.02 s−1 and a major slower phase proceeding at ∼0.001 s−1 (accounting for 20 and 80% of the overall absorption changes, respectively). This kinetic behavior is more complex than observed under anaerobic conditions, which may not be surprising given that several reactions, including the formation of superoxide anion and peroxynitrite, may take place upon reaction of Fe(II)-NO• CBS with O2.
Reactivity of Ferrous CBS with CO and O2
As internal control, the same CBS samples used in the experiments described above were probed for their reactivity with CO and O2 in the same experimental conditions of the NO• assays. Using static titrations followed by visible spectroscopy, we obtained dissociation constants for CO binding Kd1 = 0.8 ± 0.4 μm and Kd2 = 50 ± 10 μm, fully compatible with the previously reported values for full-length CBS (Ref. 16 and Table 1). The kinetic behavior observed after anaerobically mixing ferrous CBS with 1 mm CO in the stopped-flow apparatus was identical to what has been reported in the literature (15, 19). In agreement with previous reports (19), CO binding was very slow, causing the expected spectral changes (blueshift of the Soret band from 449 to 422 nm and flattening of the visible bands). As inferred by global fit analysis, the reaction was associated with a single optical transition (from Fe(II) to Fe(II)-CO CBS) and followed a biphasic time course with a major kinetic phase proceeding at ∼0.007 s−1 at 500 μm CO (Fig. 4B). The observed rate constant showed a hyperbolic dependence on [CO] with a plateau value (k′lim) of 0.012 s−1 (inset to Fig. 4B and Table 1).
The off-rate of CO was measured in ligand displacement experiments in which Fe(II)-CO CBS was anaerobically mixed with NO• (varied between 100 μm and 1.9 mm before mixing). By monitoring the conversion of CO-bound CBS to the NO•-bound species at 415 nm (the isosbestic for NO• binding to ferrous CBS), a major kinetic phase was detected proceeding at 0.5 ± 0.1 s−1, independent of NO• concentration within the tested concentration range (Fig. 5B). The reaction followed a biphasic time course, in agreement with the proposal (19) that the two hemes within a CBS dimer bind CO anticooperatively and release the ligand at different rates.
As reported previously (22), a fast reaction was observed after mixing unliganded ferrous CBS with air-equilibrated buffer, with the spectrum of ferric CBS being restored within ∼100 ms (data not shown). By varying the O2 concentration (from 25 μm to 1.25 mm before mixing) and monitoring CBS oxidation at 449 nm, we observed a linear dependence of the observed rate constant on [O2] (Fig. 4C). This reaction was studied in the presence of varying SOD concentrations (6, 60, and 90 units/ml) and 13 μg/ml catalase (concentrations of both scavengers after mixing). In full agreement with Ref. 22, regression analysis of the data collected in the presence of 90 units/ml SOD yielded a second-order rate constant k ∼ 1.3 × 105 m−1 s−1 and an extrapolated observed rate constant at zero oxygen k′0 ∼ 6.8 s−1. At lower [SOD], there was an increase in k′0 (∼32.6 and ∼10.1 s−1 for 6 units/ml and 60 units/ml SOD, respectively), in line with the information that heme oxidation by O2 in CBS leads to the generation of superoxide anion (22).
DISCUSSION
In this study we aimed to analyze the reactivity of the heme moiety in human CBS toward NO•, with the purpose of shedding light on CBS regulation by NO•, given its relevance in human (patho)physiology (23, 30, 31). We report that NO• binds to ferrous CBS tightly with Kd ≤0.23 μm (Fig. 1), in contrast to previous studies where a much lower affinity (Kd of 281 μm (20) and 30 μm (21)) was reported (Table 1). In the present study we employed authentic NO• gas and a relatively low dithionite concentration (11.3 μm) to reduce CBS. In contrast, in previous studies Kd determination was attempted by using the NO• releaser DEA-NONOate and either much higher dithionite concentrations (in the low millimolar range) (20) or NADPH and methionine synthase reductase (21) to reduce CBS. It should be noted that the Kd of 0.23 μm measured here may be an overestimate because we observed that increasing dithionite proportionally affects the Kd for NO•, likely because of the direct reaction of the reductant with NO• in solution over the time of the titrations. Altogether, these observations show that NO• binds to ferrous CBS more tightly than CO, which binds to the protein with Kd1= 0.8–3.9 μm and Kd2 = 50–68 μm (this study and Refs. 16, 19); the two Kd values measured for CO binding have been previously suggested to originate from differences in the heme microenvironment (16) or from heme anticooperativity within a CBS dimer (19).
The relatively high affinity for NO• measured here prompted us to investigate by stopped-flow absorption spectroscopy the kinetics of NO• binding and dissociation from ferrous CBS. All reactions were investigated under anaerobic conditions. Maintenance of these conditions was of paramount importance to avoid heme damaging by reactive oxygen species, particularly superoxide anion and hydrogen peroxide, possibly generated by the reaction of dithionite with residual O2 (22). To this end, the reaction buffer contained the O2-scavenging system glucose/glucose oxidase in the presence of catalase and SOD. Prior to analyzing the reactivity of ferrous CBS with gaseous ligands (NO•, CO, and O2), we tested the possibility of light-induced artifacts because of the high intensity light employed in the stopped-flow apparatus, when used in the multiwavelength mode. Indeed, with full white light intensity, we observed photo-oxidation of the CBS heme (data not shown), which could optically and chemically interfere with the binding reactions. This prompted us to reduce the intensity of the incident light beam and use a UV filter cutting light at λ <360 nm that eliminated the observed artifacts. Moreover, our adventitious observation that sulfite, a product of dithionite decomposition, slowly binds to and oxidizes ferrous CBS (k ∼ 1.7 × 101 m−1 s−1, Fig. 1A), led us to avoid prolonged incubation of the protein with excess dithionite prior to testing CBS reactivity with gaseous ligands.
In line with the tight binding of NO• to CBS, we found that NO• binds to ferrous CBS quickly (seconds, Figs. 3 and 4A), yielding the previously characterized Fe(II)-NO• adduct (20), and it dissociates from the protein very slowly (several minutes, Fig. 5A). At variance from CO, NO• was interestingly found to react with ferrous CBS with observed rate constants linearly dependent on NO• concentration, according to a second-order rate constant k ∼ 8 × 103 m−1 s−1 (Fig. 4A). NO• binding is therefore >100-fold faster than CO binding and apparently not limited by the slow dissociation of Cys-52 from the heme iron, as occurs for CO (15, 19) (compare Fig. 4, A and B). In line with the much higher affinity of NO• (compared with CO), we found that NO• dissociation from ferrous CBS is much slower than CO dissociation (∼0.003 s−1 versus ∼0.5 s−1, Fig. 5 and Table 1).
NO•-bound CBS is optically similar to the penta-coordinate nitrosyl heme adduct of other proteins, such as the eukaryotic initiation factor 2α kinase, the carbon monoxide oxidation activator CooA and cytochrome c′ from denitrifying bacteria (32). Interestingly, it also resembles the penta-coordinate heme species obtained by displacement of the Cys-52 thiolate ligand upon incubation of ferric CBS with mercurium chloride (33). In analogy with other hexa-coordinate heme proteins, such as soluble guanylate cyclase, bacterial cytochrome c′, or heme nitric oxide/oxygen sensor (34–36), conversion of ferrous CBS into the penta-coordinate Fe(II)-NO• adduct is therefore expected to take place via the transient formation of a hexa-coordinate NO complex intermediate with characteristic optical features (Fig. 6). In the case of CBS, however, no evidence for such intermediate was obtained upon mixing the ferrous protein with substoichiometric, equimolar or excess NO•.
FIGURE 6.
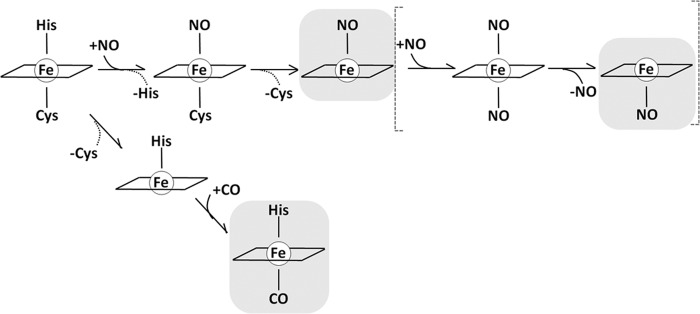
Mechanisms for NO• and CO binding to CBS.
As a control, we checked that the protein samples employed in the NO• binding experiments displayed canonical reactivity toward CO and O2 (Fig. 4, B and C). Indeed, in full agreement with the literature (15, 19, 22), CO was found to bind to ferrous CBS very slowly (k′lim ∼ 0.012 s−1, Fig. 4B), and O2 proved to oxidize the CBS heme much faster, with k ∼ 1.3 × 105 m−1 s−1 (Fig. 4C). Oxygen was previously proposed to react with ferrous CBS via an outer sphere mechanism, not involving direct binding to the metal center (22).
From a mechanistic viewpoint, the fact that NO• binds to CBS much faster than CO, and thus much faster than Cys-52 dissociation from the heme iron, is somewhat unexpected. As outlined in Ref. 37, in hexa-coordinate heme proteins, dissociation of the endogenous ligand at the sixth iron coordination position is expected to equally affect (and possibly rate-limit) the binding of exogenous ligands to the heme iron. This has been documented for hexa-coordinate globins, and particularly for neuroglobin, where O2, CO, and NO have all been shown to bind to the ferrous protein at the same low dissociation rate of distal histidine from heme iron (38, 39). The finding that, upon binding to CBS, NO• displaces both axial endogenous ligands (see the Introduction), without being limited by the slow dissociation of Cys-52, favors the possibility that NO• binds initially on the His-65 side of the heme, causing fast dissociation of Cys-52, and thus formation of a penta-coordinate Fe(II)-NO• species (Fig. 6). It remains to be determined whether such adduct (with NO• bound on the histidine side and Cys-52 dissociated) represents the end point species in the reaction or whether the binding of a second NO• molecule occurs, leading to formation of a transient bis-NO• intermediate, eventually resulting into a penta-coordinate species with NO• bound on the cysteine side (Fig. 6). In this regard, it is worth mentioning that in some heme proteins (soluble guanylate cyclase, bacterial cytochrome c′ and heme nitric oxide/oxygen sensor), high affinity NO• binding is achieved by multiple NO• binding steps leading to formation of a highly stable penta-coordinate NO• complex (34–36). This may be the case also for human CBS, although direct spectroscopic evidence supporting a multistep NO• binding is yet missing.
Regardless of the detailed NO• binding mechanism, it remains to be explained why in CBS CO preferentially binds to the cysteine side, whereas NO• seems to bind (at least initially) to the histidine side. Despite the lack of an unequivocal explanation for this, we point out that, at equilibrium, binding of NO• and CO on opposite sides of the heme is not unprecedented, having been reported for bacterial cytochrome c′ (40–42). According to current views, affinity for NO•, CO, and O2 in heme proteins is largely affected by the chemical nature and geometry of the endogenous heme axial ligand(s), as well as by a number of features of the heme pocket, including electrostatic interactions, H-bonding and steric hindrance (34–36). Whereas steric constraints affect the affinity of the three ligands uniformly, changes in the electrostatic field have been reported to affect O2 affinity preferentially (43).
As a general observation, heme proteins with histidine as the proximal ligand and an apolar distal site follow the so-called “sliding scale rule,” such that the affinity for NO• is ∼103-fold greater than for CO, which in turn binds ∼103-fold more tightly than O2 (36). In other words, in heme proteins following this rule the ratios for NO•:CO:O2 dissociation constants are invariantly 1:∼103:∼106 (36). However, a leveling effect on the ratio between NO• and CO affinities has been observed for heme proteins with strong field heme ligands, such as cysteine thiolates (36). It is therefore not surprising that human CBS apparently does not follow the sliding scale rule, based on the Kd values determined for NO• and CO (this work and Refs. 16 and 19).
In conclusion, our results show that NO• binds to CBS at a much higher rate than CO and with much higher affinity than reported in the literature. These observations shed new light on CBS regulation by NO• and its (patho)physiological relevance, enforcing the growing evidence for an interplay among the gasotransmitters NO•, CO, and H2S.
This work was supported in part by PEst-OE/SAU/UI4013/2011, SFRH/BD/43934/2008 (to M. I. S. M.), by Ministero dell'Istruzione, dell'Università e della Ricerca of Italy (PNR-CNR Aging Program 2012–2014 and FIRB RBFR08F41U_001 (to A. G.), FIRB RBIN06E9Z8 and PRIN 20107Z8XBW_005 (to P. S.)), and by a bilateral grant award by Consiglio Nazionale delle Ricerche of Italy and Fundação para a Ciência e Tecnologia of Portugal (to A. G. and J. B. V.).
- CBS
- cystathionine β-synthase
- DEA-NONOate
- diethylammonium (Z)-1-(N,N-diethylamino)diazen-1-ium-1,2-diolate
- SOD
- superoxide dismutase.
REFERENCES
- 1. Kabil O., Banerjee R. (2014) Enzymology of H2S biogenesis, decay and signaling. Antioxid. Redox Signal. 20, 770–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kajimura M., Fukuda R., Bateman R. M., Yamamoto T., Suematsu M. (2010) Interactions of multiple gas-transducing systems: hallmarks and uncertainties of CO, NO, and H2S gas biology. Antioxid. Redox Signal. 13, 157–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mustafa A. K., Gadalla M. M., Snyder S. H. (2009) Signaling by gasotransmitters. Sci. Signal. 2, re2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Li L., Hsu A., Moore P. K. (2009) Actions and interactions of nitric oxide, carbon monoxide and hydrogen sulphide in the cardiovascular system and in inflammation: a tale of three gases. Pharmacol. Ther. 123, 386–400 [DOI] [PubMed] [Google Scholar]
- 5. Kabil O., Banerjee R. (2010) Redox biochemistry of hydrogen sulfide. J. Biol. Chem. 285, 21903–21907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Singh S., Padovani D., Leslie R. A., Chiku T., Banerjee R. (2009) Relative contributions of cystathionine β-synthase and γ-cystathionase to H2S biogenesis via alternative trans-sulfuration reactions. J. Biol. Chem. 284, 22457–22466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yadav P. K., Yamada K., Chiku T., Koutmos M., Banerjee R. (2013) Structure and kinetic analysis of H2S production by human mercaptopyruvate sulfurtransferase. J. Biol. Chem. 288, 20002–20013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pey A. L., Majtan T., Sanchez-Ruiz J. M., Kraus J. P. (2013) Human cystathionine β-synthase (CBS) contains two classes of binding sites for S-adenosylmethionine (SAM): complex regulation of CBS activity and stability by SAM. Biochem. J. 449, 109–121 [DOI] [PubMed] [Google Scholar]
- 9. Koutmos M., Kabil O., Smith J. L., Banerjee R. (2010) Structural basis for substrate activation and regulation by cystathionine β-synthase (CBS) domains in cystathionine β-synthase. Proc. Natl. Acad. Sci. U.S.A. 107, 20958–20963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Meier M., Janosik M., Kery V., Kraus J. P., Burkhard P. (2001) Structure of human cystathionine β-synthase: a unique pyridoxal 5′-phosphate-dependent heme protein. EMBO J. 20, 3910–3916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Taoka S., Lepore B. W., Kabil O., Ojha S., Ringe D., Banerjee R. (2002) Human cystathionine β-synthase is a heme sensor protein: evidence that the redox sensor is heme and not the vicinal cysteines in the CXXC motif seen in the crystal structure of the truncated enzyme. Biochemistry 41, 10454–10461 [DOI] [PubMed] [Google Scholar]
- 12. Su Y., Majtan T., Freeman K. M., Linck R., Ponter S., Kraus J. P., Burstyn J. N. (2013) Comparative study of enzyme activity and heme reactivity in Drosophila melanogaster and Homo sapiens cystathionine β-synthases. Biochemistry 52, 741–751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Singh S., Madzelan P., Stasser J., Weeks C. L., Becker D., Spiro T. G., Penner-Hahn J., Banerjee R. (2009) Modulation of the heme electronic structure and cystathionine β-synthase activity by second coordination sphere ligands: the role of heme ligand switching in redox regulation. J. Inorg. Biochem. 103, 689–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kabil O., Weeks C. L., Carballal S., Gherasim C., Alvarez B., Spiro T. G., Banerjee R. (2011) Reversible heme-dependent regulation of human cystathionine β-synthase by a flavoprotein oxidoreductase. Biochemistry 50, 8261–8263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Carballal S., Cuevasanta E., Marmisolle I., Kabil O., Gherasim C., Ballou D. P., Banerjee R., Alvarez B. (2013) Kinetics of reversible reductive carbonylation of heme in human cystathionine β-synthase. Biochemistry 52, 4553–4562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Taoka S., West M., Banerjee R. (1999) Characterization of the heme and pyridoxal phosphate cofactors of human cystathionine β-synthase reveals nonequivalent active sites. Biochemistry 38, 2738–2744 [DOI] [PubMed] [Google Scholar]
- 17. Weeks C. L., Singh S., Madzelan P., Banerjee R., Spiro T. G. (2009) Heme regulation of human cystathionine β-synthase activity: insights from fluorescence and Raman spectroscopy. J. Am. Chem. Soc. 131, 12809–12816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Smith A. T., Su Y., Stevens D. J., Majtan T., Kraus J. P., Burstyn J. N. (2012) Effect of the disease-causing R266K mutation on the heme and PLP environments of human cystathionine β-synthase. Biochemistry 51, 6360–6370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Puranik M., Weeks C. L., Lahaye D., Kabil O., Taoka S., Nielsen S. B., Groves J. T., Banerjee R., Spiro T. G. (2006) Dynamics of carbon monoxide binding to cystathionine β-synthase. J. Biol. Chem. 281, 13433–13438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Taoka S., Banerjee R. (2001) Characterization of NO binding to human cystathionine β-synthase: possible implications of the effects of CO and NO binding to the human enzyme. J. Inorg. Biochem. 87, 245–251 [DOI] [PubMed] [Google Scholar]
- 21. Gherasim C., Yadav P. K., Kabil O., Niu W. N., Banerjee R. (2014) Nitrite reductase activity and inhibition of H2S biogenesis by human cystathionine β-synthase. PLoS One 9, e85544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Carballal S., Madzelan P., Zinola C. F., Graña M., Radi R., Banerjee R., Alvarez B. (2008) Dioxygen reactivity and heme redox potential of truncated human cystathionine β-synthase. Biochemistry 47, 3194–3201 [DOI] [PubMed] [Google Scholar]
- 23. Prathapasinghe G. A., Siow Y. L., Xu Z., O., K. (2008) Inhibition of cystathionine-β-synthase activity during renal ischemia-reperfusion: role of pH and nitric oxide. Am. J. Physiol. Renal Physiol. 295, F912–F922 [DOI] [PubMed] [Google Scholar]
- 24. Morikawa T., Kajimura M., Nakamura T., Hishiki T., Nakanishi T., Yukutake Y., Nagahata Y., Ishikawa M., Hattori K., Takenouchi T., Takahashi T., Ishii I., Matsubara K., Kabe Y., Uchiyama S., Nagata E., Gadalla M. M., Snyder S. H., Suematsu M. (2012) Hypoxic regulation of the cerebral microcirculation is mediated by a carbon monoxide-sensitive hydrogen sulfide pathway. Proc. Natl. Acad. Sci. U.S.A. 109, 1293–1298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mendes M. I., Colaço H. G., Smith D. E., Ramos R. J., Pop A., van Dooren S. J., de Almeida I. T., Kluijtmans L. A., Janssen M. C., Rivera I., Salomons G. S., Leandro P., Blom H. J. (2014) Reduced response of cystathionine β-synthase (CBS) to S-adenosylmethionine (SAM): identification and functional analysis of CBS gene mutations in homocystinuria patients. J. Inherit. Metab. Dis. 10.1007/s10545-013-9647-6 [DOI] [PubMed] [Google Scholar]
- 26. Stubauer G., Giuffrè A., Brunori M., Sarti P. (1998) Cytochrome c oxidase does not catalyze the anaerobic reduction of NO. Biochem. Biophys. Res. Commun. 245, 459–465 [DOI] [PubMed] [Google Scholar]
- 27. Dixon M. (1971) The acceptor specificity of flavins and flavoproteins. I. Techniques for anaerobic spectrophotometry. Biochim. Biophys. Acta 226, 241–258 [DOI] [PubMed] [Google Scholar]
- 28. Henry E. R., Hofrichter J. (1992) Singular value decomposition: application to analysis of experimental data. Methods Enzymol. 210, 129–192 [Google Scholar]
- 29. Moore E. G., Gibson Q. H. (1976) Cooperativity in the dissociation of nitric oxide from hemoglobin. J. Biol. Chem. 251, 2788–2794 [PubMed] [Google Scholar]
- 30. Kwiatkoski M., Soriano R. N., Francescato H. D., Batalhao M. E., Coimbra T. M., Carnio E. C., Branco L. G. (2012) Hydrogen sulfide as a cryogenic mediator of hypoxia-induced anapyrexia. Neuroscience 201, 146–156 [DOI] [PubMed] [Google Scholar]
- 31. Tang X. Q., Fang H. R., Zhou C. F., Zhuang Y. Y., Zhang P., Gu H. F., Hu B. (2013) A novel mechanism of formaldehyde neurotoxicity: inhibition of hydrogen sulfide generation by promoting overproduction of nitric oxide. PLoS One 8, e54829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Igarashi J., Sato A., Kitagawa T., Yoshimura T., Yamauchi S., Sagami I., Shimizu T. (2004) Activation of heme-regulated eukaryotic initiation factor 2α kinase by nitric oxide is induced by the formation of a five-coordinate NO-heme complex: optical absorption, electron spin resonance, and resonance Raman spectral studies. J. Biol. Chem. 279, 15752–15762 [DOI] [PubMed] [Google Scholar]
- 33. Ojha S., Hwang J., Kabil O., Penner-Hahn J. E., Banerjee R. (2000) Characterization of the heme in human cystathionine β-synthase by x-ray absorption and electron paramagnetic resonance spectroscopies. Biochemistry 39, 10542–10547 [DOI] [PubMed] [Google Scholar]
- 34. Martin E., Berka V., Sharina I., Tsai A. L. (2012) Mechanism of binding of NO to soluble guanylyl cyclase: implication for the second NO binding to the heme proximal site. Biochemistry 51, 2737–2746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tsai A. L., Martin E., Berka V., Olson J. S. (2012) How do heme-protein sensors exclude oxygen? Lessons learned from cytochrome c′, Nostoc puntiforme heme nitric oxide/oxygen-binding domain, and soluble guanylyl cyclase. Antioxid. Redox Signal. 17, 1246–1263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tsai A. L., Berka V., Martin E., Olson J. S. (2012) A “sliding scale rule” for selectivity among NO, CO, and O2 by heme protein sensors. Biochemistry 51, 172–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kakar S., Hoffman F. G., Storz J. F., Fabian M., Hargrove M. S. (2010) Structure and reactivity of hexa-coordinate hemoglobins. Biophys. Chem. 152, 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Brunori M., Giuffrè A., Nienhaus K., Nienhaus G. U., Scandurra F. M., Vallone B. (2005) Neuroglobin, nitric oxide, and oxygen: functional pathways and conformational changes. Proc. Natl. Acad. Sci. U.S.A. 102, 8483–8488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dewilde S., Kiger L., Burmester T., Hankeln T., Baudin-Creuza V., Aerts T., Marden M. C., Caubergs R., Moens L. (2001) Biochemical characterization and ligand binding properties of neuroglobin, a novel member of the globin family. J. Biol. Chem. 276, 38949–38955 [DOI] [PubMed] [Google Scholar]
- 40. Lawson D. M., Stevenson C. E., Andrew C. R., Eady R. R. (2000) Unprecedented proximal binding of nitric oxide to heme: implications for guanylate cyclase. EMBO J. 19, 5661–5671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Antonyuk S. V., Rustage N., Petersen C. A., Arnst J. L., Heyes D. J., Sharma R., Berry N. G., Scrutton N. S., Eady R. R., Andrew C. R., Hasnain S. S. (2011) Carbon monoxide poisoning is prevented by the energy costs of conformational changes in gas-binding haemproteins. Proc. Natl. Acad. Sci. U.S.A. 108, 15780–15785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Andrew C. R., Green E. L., Lawson D. M., Eady R. R. (2001) Resonance Raman studies of cytochrome c′ support the binding of NO and CO to opposite sides of the heme: implications for ligand discrimination in heme-based sensors. Biochemistry 40, 4115–4122 [DOI] [PubMed] [Google Scholar]
- 43. Garton E. M., Pixton D. A., Petersen C. A., Eady R. R., Hasnain S. S., Andrew C. R. (2012) A distal pocket Leu residue inhibits the binding of O2 and NO at the distal heme site of cytochrome c′. J Am Chem. Soc. 134, 1461–1463 [DOI] [PubMed] [Google Scholar]