

Background: Glutaredoxin-1 (Glrx) inhibits endothelial cell migration by reversing protein-glutathione adducts.
Results: Revascularization after hind limb ischemia was inhibited in Glrx transgenic mice. Glrx overexpression increased soluble VEGF receptor 1 (sFlt) in endothelial cells via NF-κB-dependent Wnt5a production.
Conclusion: Glrx enhances Wnt5a-induced anti-angiogenic sFlt in endothelial cells.
Significance: Up-regulated Glrx inhibits VEGF signaling by increased sFlt causing impaired vascularization.
Keywords: Angiogenesis, Animal Models, Glutathionylation, Ischemia, NF-kappa B (NF-κB), Redox Regulation, Vascular Endothelial Growth Factor (VEGF)
Abstract
Glutaredoxin-1 (Glrx) is a cytosolic enzyme that regulates diverse cellular function by removal of GSH adducts from S-glutathionylated proteins including signaling molecules and transcription factors. Glrx is up-regulated during inflammation and diabetes, and Glrx overexpression inhibits VEGF-induced EC migration. The aim was to investigate the role of up-regulated Glrx in EC angiogenic capacities and in vivo revascularization in the setting of hind limb ischemia. Glrx-overexpressing EC from Glrx transgenic (TG) mice showed impaired migration and network formation and secreted higher levels of soluble VEGF receptor 1 (sFlt), an antagonizing factor to VEGF. After hind limb ischemia surgery Glrx TG mice demonstrated impaired blood flow recovery, associated with lower capillary density and poorer limb motor function compared with wild type littermates. There were also higher levels of anti-angiogenic sFlt expression in the muscle and plasma of Glrx TG mice after surgery. Noncanonical Wnt5a is known to induce sFlt. Wnt5a was highly expressed in ischemic muscles and EC from Glrx TG mice, and exogenous Wnt5a induced sFlt expression and inhibited network formation in human microvascular EC. Adenoviral Glrx-induced sFlt in EC was inhibited by a competitive Wnt5a inhibitor. Furthermore, Glrx overexpression removed GSH adducts on p65 in ischemic muscle and EC and enhanced NF-κB activity, which was responsible for Wnt5a-sFlt induction. Taken together, up-regulated Glrx induces sFlt in EC via NF-κB-dependent Wnt5a, resulting in attenuated revascularization in hind limb ischemia. The Glrx-induced sFlt explains part of the mechanism of redox-regulated VEGF signaling.
Introduction
Excess levels of reactive oxygen and nitrogen species contribute to various pathological conditions. However, physiological levels of oxidants are also essential for cellular signaling in part through modification of redox-sensitive cysteine thiols on proteins. Exposure of reactive cysteine thiols to oxidants in the presence of GSH results in S-glutathionylation, a reversible post-translational modification. GSH adducts may provide signaling mechanism and protect proteins from irreversible oxidation during fluctuations of reactive oxygen and nitrogen species levels (1). This reversible modification can result in functional inhibition (2–4) or activation (5, 6) of a broad range of proteins with redox-sensitive thiols. Thus, regulation of GSH adducts has significance in various clinical conditions (7). Glutaredoxin-1 (Glrx)3 is a cytosolic enzyme that specifically catalyzes the removal of GSH adducts from S-glutathionylated proteins (8). Therefore, Glrx can regulate cellular functions including signal transduction, cytoskeleton dynamics, and gene transcription by reversing S-glutathionylation (9, 10). A well studied example is the NF-κB pathway wherein GSH adducts IKKβ (inactivate inhibitor of κB kinase β) and inhibit DNA binding of p50 (11) and p65 (12) subunits; thus, Glrx promotes NF-κB activation by removing GSH adducts (7, 13, 14). Glrx expression is increased in atherosclerotic human coronary artery (15), allergic mouse airway (16), and diabetic rat retina (13). Plasma Glrx activity is increased in type 2 diabetic patients (17), suggesting an association with oxidative stress. However, because GSH adducts have various effects on protein function, the cellular effects of increased Glrx are more complex than that of an antioxidant enzyme and may differ depending on tissue and pathological conditions.
VEGF A is a major factor in stimulating arteriogenesis and angiogenesis. VEGF binds to VEGF receptor 2 (VEGFR2; Kdr/Flk1), resulting in receptor dimerization and phosphorylation of tyrosine residues. VEGFR2 is the main transducer of VEGF on endothelial proliferation, migration, and network formation (18). However, VEGF binds to VEGFR1 (fms-like tyrosine kinase-1; Flt) with higher affinity than VEGFR2. Flt can function as a negative regulator for VEGFR2 because a full-length membrane-tethered Flt (mFlt) transduces a weaker signal than VEGFR2, and a soluble splice variant (sFlt) can capture the VEGF ligand as a decoy to prevent its binding to VEGFR2 (18, 19). A number of redox-sensitive proteins are involved in VEGF signaling (20). S-Glutathionylation inactivates protein-tyrosine phosphatases (4, 21) and thus promotes tyrosine phosphorylation of VEGF receptor and signaling.
We previously observed that Glrx overexpression inhibited endothelial cells (EC) angiogenic properties in vitro (22). In contrast, a report suggests that Glrx overexpression may be beneficial for recover of cardiac dysfunction after ischemia (23). Therefore, we dissected the mechanism in which Glrx inhibits EC migration and examined whether Glrx up-regulation in vivo may improve or inhibit post-ischemic revascularization using Glrx-overexpressing mice. The overexpression of Glrx was similar to the increase in enzyme activity observed in the onset of diseases such as diabetes.
As a result, we found that Glrx-overexpressing EC produced higher levels of anti-angiogenic sFlt, and inhibiting sFlt expression reversed the anti-angiogenic effects of Glrx in EC. In vivo, the blood flow recovery after femoral artery ligation was significantly impaired in association with poorer motor function in Glrx transgenic (TG) mice compared with WT mice. Interestingly, plasma levels of the inhibitory VEGF receptor sFlt were higher in the Glrx TG mice. Accordingly, we found that Glrx overexpression up-regulated noncanonical Wnt ligand Wnt5a (wingless-type mouse mammary tumor virus (MMTV) integration site family, member 5A) in ischemic muscle and cultured EC. Wnt5a in myeloid cells was shown to inhibit postnatal angiogenesis through Flt induction (24). Wnt5a activates NF-κB via β-catenin-independent signaling in EC (25), whereas inflammatory stimuli induce Wnt5a via NF-κB activation in monocytes (26, 27). We showed that exogenous Wnt5a treatment increased sFlt expression in EC and inhibited endothelial network formation. Our data indicate that Wnt5a regulates sFlt in EC in an autocrine/paracrine fashion, and Glrx up-regulation enhanced the Wnt5a-sFlt pathway, in part through NF-κB activation, and resulted in inhibition of in vivo revascularization in hind limb ischemia.
MATERIALS AND METHODS
Mouse Hind Limb Ischemia (HLI) Model
Animal study protocols were approved by the Institutional Animal Care and Use Committee at Boston University. Glrx TG mice were generated with Human Glrx overexpression driven by the human β-actin promoter at the laboratory of Dr. Y. S. Ho (Wayne State University, Detroit, MI) (23). Heterozygote Glrx TG mice and WT littermates (males 8–9 months old) were subjected to unilateral HLI by surgically excising left femoral artery and vein as previously described (28, 29).
Blood Flow Measurements and the Functional Scores
Blood flow perfusion was measured by LASER Doppler (Moor Instruments UK) on plantar aspects of the feet of anesthetized mice (100 mg/kg ketamine and 10 mg/kg xylazine intraperitoneally) as previously described (28). Severity of ischemia was scored by assessment of limb function and mobility, using the following criteria under blinded conditions: 1) no use of limb, 2) no use of foot, 3) dragging of foot, 4) no toe spreading, and 5) full use of foot. Limb function of all mice was scored at the same time as blood flow measurements prior to sedation. Functional agility was measured on day 10 after surgery by running on a treadmill until exhaustion.
Histological Assessment
Capillary density was quantified in non-ischemic and ischemic gastrocnemius muscle by histological assessment, by isolectin B4 (Vector Labs) staining.
ELISA
Murine VEGF-A and soluble VEGFR1 ELISA (R&D Systems) were performed on plasma from WT and Glrx TG mice, and media were collected from EC.
Aortic Sprouting Assay
Aortic rings from WT and Glrx TG mice were placed on growth factor-reduced Matrigel (BD Bioscience) supplemented with DMEM, 2% FBS, and 1% penicillin/streptomycin. Experiments were conducted using four mice from each group and 4–5 rings per mouse, and the area of outgrowth was measured using ImageJ.
Isolation of Mouse Endothelial Cells
Microvascular EC were selectively (CD31; BD Pharmingen) isolated from heart from WT and Glrx TG mice using a MACS automated cell sorter (Miltenyi Biotec) after digestion in gentleMACSTM Dissociator (Miltenyi Biotec).
Endothelial Cell Network Formation
Quiescent EC were seeded (1 × 104 cells/well) in 96-well plates coated with Matrigel (growth factor reduced; BD Biosciences). Network formation was assessed after treatment with or without VEGF (50 ng/ml) in low serum endothelial growth media (Lonza) (22). Network formation was scored or length was measured in a blinded manner as previously described (30).
Endothelial Cell Proliferation
Quiescent EC were seeded (1 × 104 cells/well) in gelatin-coated 96-well plates. After 24 h, EC proliferation was measured in the presence and absence of VEGF (50 ng/ml) in EBM2 media with 0.5% FBS using cell proliferation kit (Biovision).
Endothelial Cell Migration
As previously described (30), EC migration was measured in the presence and absence of VEGF (50 ng/ml) after performing scratch in a confluent monolayer of WT and TG mouse cardiac microvascular endothelial cells (mEC). Migration was quantified at four defined locations by measuring the area of the scratch per well at 0 and 18 h using ImageJ.
Glutaredoxin Activity Assay
Glrx activity in muscle homogenate was determined by the NADPH-dependent reduction of 2-hydroxyethildisulfide (31).
Glrx Adenoviral Overexpression
Adenovirus expressing human Glrx was a generous gift from Dr. Young J. Lee (University of Pittsburgh) (32), and it has been used in previous reports (22, 33). Human cardiac microvascular endothelial cells (hEC; Lonza) were infected in FBS-free EBM2 media with Glrx or β-galactosidase (LacZ) (control) adenoviruses with 10 μg/ml PolybreneTM (Sigma) and subsequently used for 72 h.
siRNA in hECs
After adenoviral overexpression of LacZ or Glrx, p65 knockdown was achieved using on target plus siRNA (Dharmacon). hEC were transfected with siRNA against p65 (200 pmol; Dharmacon) or nonsilencing control using LipofectamineTM 2000 reagent (Invitrogen) following the manufacturer's instructions. The cells were used in experiments 48–72 h after transfection. Specific knockdown of sFLT was achieved by targeting two siRNA sequences to the unique 3′ sequence of sFLT (Dharmacon siGENOME) as previously reported (34).
qRT-PCR
Total RNA was isolated from tissues or cells using TRIzolTM reagent (Invitrogen), and cDNA was generated utilizing a high capacity RNA to cDNA kit (Invitrogen). Quantitative PCR was conducted using inventory mouse (Mm) and human (Hs); gene-specific TaqManTM primers (Invitrogen): Glrx (Mm00728386_m1, Hs00829752_g1), Wnt5a (Mm00437347_m1, Hs00998537_m1), mFLT (Mm00438980_m1), VEGFA (Mm01281449_m1, Hs0090055_m1), Flk/KDR (Mm00440111_m1, Hs00911700_m1), Ror2 (Mm00443470_m1), E-selectin (Hs00950401_m1), IL6 (Hs00985639_m1), β-actin (mouse 4352933, human 4326315). Expression was obtained and analyzed using comparative CT (ΔΔCT) with StepOneTM real-time PCR software (Applied Biosystems), normalized to β-actin. The sequence for the custom designed TaqManTM assay to assess human sFlt transcript was kindly provided by Dr. Stefanie Dimmeler (35). SYBR green primers were used to assess murine mFlt and sFlt according to the transcripts as published (24).
NF-κB Luciferase Activity Assay
NF-κB reporter activity in WT and Glrx TG mEC was assessed by co-transfection with pNF-κB-LUC reporter and Renilla luciferase adenoviruses. The EC were stimulated with TNFα (100 ng/ml; Sigma) for 4 h, and subsequent dual luciferase for Firefly luciferase and Renilla luciferase (Promega) activity was measured using luminometer (TECAN). The NF-κB luciferase activity was normalized to the Renilla luciferase activity (NF-κB/Renilla luciferase activity).
Hypoxia
EC in EBM2 with 0.1% FBS were incubated at 0.5% or 0.1% O2, 70% humidity at 37 °C in a hypoxia chamber (Pathology Devices, Inc.) for 20 h prior to isolation for biotin switch assay.
Biotin Switch Assay
S-Glutathionylation of p65 was assessed as previously described (30, 36), hECs were lysed with maleimide (100 mol/liter) to block free cysteines. Modified thiols were reduced with DTT (20 mm) and subsequently labeled with biotin-HPDP (N-[6-(biotinamido) hexyl]-3′-(2′-pyridyldithio) propionamide) (Pierce). Biotinylated proteins were purified with magnetic streptavidin-agarose beads (Pierce) and biotin-HPDP modified proteins were immunoblotted with anti-p65 antibody (Cell Signaling).
Reagents
Reagents were obtained as follows: recombinant Wnt5a (R&D Systems), Wnt5a antagonist Box5 (Millipore), and rat anti-Wnt5a antibody (R&D Systems). Human and mouse Glrx antibodies were custom ordered by Bethyl Laboratories, Inc.
Statistical Analysis
All group data are reported as means ± S.E. except as otherwise indicated. For the experimental data, statistical analysis comparing two groups was carried out using Student's unpaired t test. Analysis of more than two groups was performed by either one-way ANOVA or two-way ANOVA followed by Tukey post hoc comparison test. Sequential measurements were analyzed by repeated measure one-way ANOVA. Analyses were completed using GraphPad Prism v5.0. p values <0.05 were considered significant.
RESULTS
VEGF-induced Migration and Network Formation Were Diminished in Glrx TG Endothelial Cells
To understand the functional significance of endothelial Glrx on angiogenic function, we explored the function of Glrx-overexpressing EC in vitro and ex vivo. Microvascular endothelial cells (Cd31+) were selectively isolated from WT and Glrx TG hearts. Human Glrx expression was confirmed in Glrx TG mouse EC, and there was no change in endogenous Glrx expression between WT and Glrx TG mEC (Fig. 1A). Furthermore, we assessed mEC network formation and migration. VEGF-induced network formation of mEC seeded on Matrigel was significantly inhibited in Glrx TG compared with WT mEC (Fig. 1, B and C). VEGF-induced migration was also attenuated in Glrx TG mEC (Fig. 1, D and E). In addition, mouse aorta rings were cultured ex vivo on Matrigel to assess EC sprouting. VEGF (50 ng/ml)-induced sprouting was attenuated in Glrx TG aortae compared with WT (Fig. 1, F and G). Together, these data indicate VEGF-induced angiogenic function was impaired in Glrx TG-derived EC.
FIGURE 1.
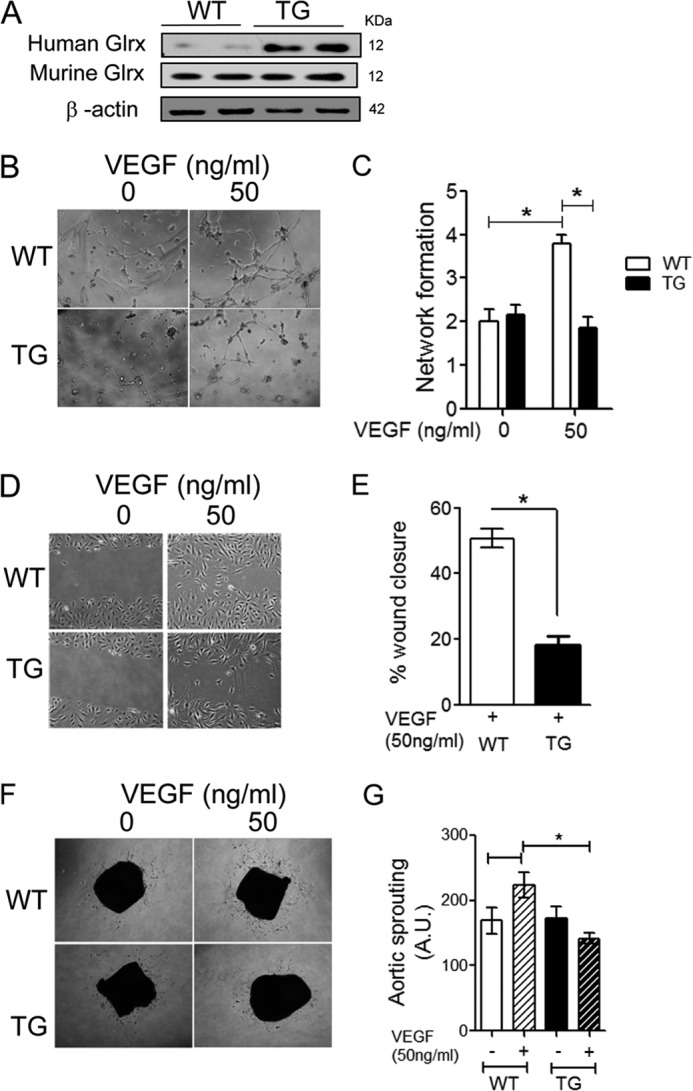
Glrx overexpression attenuates EC migration and network formation, as well as aortic sprouting. A, Western blot analysis of human Glrx transgene or endogenous Glrx in WT and TG mouse microvascular EC (mEC). B and C, representative (B) and quantitative (C) blinded scoring of EC network formation in WT and TG mEC seeded onto Matrigel in the presence and absence of VEGF (50 ng/ml) (n = 6; *, p < 0.05). D and E, representative (D) and percentage (E) wound closure of WT and TG mEC after 18h VEGF (50 ng/ml) treatment, measured at four predetermined locations (n = 3; *, p < 0.05). F and G, representative (F) and quantitative (G) assessment of sprouting of EC from isolated WT and TG aortae after 6 days cultured on Matrigel in DMEM 2% FBS with and without VEGF (50 ng/ml) (n = 4; *, p < 0.05).
Soluble Flt Isoform Was Induced by Glrx Overexpression
Alternative splicing of the anti-angiogenic Flt gene results in the membrane anchored isoform (mFlt) and a soluble isoform that is secreted extracellularly (sFlt). In seeking a mechanism for decreased angiogenic behavior of Glrx TG EC, we used primers specific for different Flt isoforms and found both sFlt and mFlt mRNA expression were increased in mEC isolated from Glrx TG mice compared with cells from WT control (Fig. 2, A and B). Hypoxia induces sFLT expression in EC. After 24 h of hypoxic treatment, sFlt and mFlt expression remained significantly higher in TG EC. To determine whether EC could secrete sFlt, we collected media (after 24 h of quiescence) from mEC isolated from Glrx TG and WT mice under normoxic and hypoxic conditions. sFlt levels were markedly greater in media from Glrx TG mEC compared with WT mEC. Interestingly, a marked increase was observed with hypoxic treatment in TG mEC (Fig. 2C).
FIGURE 2.

Glrx attenuates EC function via the anti-angiogenic receptor, soluble FLT. A and B, mRNA levels of sFlt (A) and mFlt (B) isoforms in WT and TG mEC under normoxia and hypoxia (0.5% oxygen, 20 h, n = 5; *, p < 0.05). C, ELISA; sFLT level in the media from WT and TG mEC in normoxic and hypoxic (0.5% oxygen, 20 h) conditions (n = 5; *, p < 0.05; #, p < 0.01). D, sFlt mRNA expression by qPCR in hEC treated with siRNA to sFlt (si-sFlt) or off target scramble RNA (scr) after AdGlrx or AdLacZ infection (n = 3; *, p < 0.05). E, proliferation of hEC in the presence and absence of VEGF (50 ng/ml, 24 h) after adenoviral Glrx or LacZ overexpression and subsequent sFlt knockdown by siRNA compared with scr control (n = 4; *, p < 0.05). F and G, quantitative data (F) and representative images (G) of AdGlrx and AdLacZ treated hEC after 18 h VEGF (50 ng/ml) treatment, measured at four predetermined locations. The effects of si-sFlt are shown compared with scr (n = 5; *, p < 0.05). H and I, quantitative measurement (H) and representative images (I) of EC network formation in AdGlrx and AdLacZ treated hEC with siRNA sFLT or scr control in the presence of VEGF (50 ng/ml) (n = 6; *, p < 0.05; ***, p < 0.001).
Inhibiting sFlt Reverses the Anti-angiogenic Effect of Glrx
To elucidate the contribution of sFlt in the anti-angiogenic role of Glrx, we specifically knocked down sFlt in human EC using siRNA designed to target the unique C-terminal region of the sFlt isoform as previously reported for human EC (34).
To be able to utilize the reported sFLT siRNA, we used hEC and induced Glrx overexpression by adenoviral infection. Glrx adenoviral overexpression in human EC (22) caused the same phenotype we observed in mEC isolated from Glrx mice, i.e., inhibition of migration and network formation. Quantitative PCR showed significant suppression of sFlt mRNA with siRNA for sFlt (Fig. 2D). Proliferation was decreased in hEC overexpressing Glrx; however, knockdown of sFlt reinstated the level of proliferation observed in LacZ-treated EC (Fig. 2E). Likewise, sFlt knockdown rescued Glrx-induced attenuation of hEC migration (Fig. 2, F and G). Furthermore, hEC network formation that was inhibited by Glrx overexpression was reversed with sFlt knockdown (Fig. 2, H and I). Together these data confirm that Glrx inhibits endothelial function through sFlt.
Wnt5a Is Increased by Glrx Overexpression and Regulates sFlt in EC
It has recently been reported that Wnt5a inhibits developmental angiogenesis through Flt regulation in myeloid cells (24). Therefore, we investigated whether Glrx increased sFlt via Wnt5a induction. Wnt5a expression was increased in mEC isolated from Glrx TG mice (Fig. 3A). In addition, receptor tyrosine kinase-like orphan receptor 2 (Ror2), a receptor for the Wnt5a ligand (37) was also up-regulated in Glrx TG mEC (Fig. 3B). To explore the control of sFlt expression further, hEC were treated with recombinant Wnt5a. Wnt5a (100 ng/ml) induced a marked but transient increase in mRNA expression of sFlt and mFlt within 3 h (Fig. 3, C and D). Next, hEC network formation and migration were evaluated in response to Wnt5a treatment. After 24 h of incubation with Wnt5a, hEC network formation was inhibited (Fig. 3, E and F) similar to the observations in Glrx TG mEC. Interestingly, Glrx overexpression in hEC resulted in increased Wnt5a mRNA expression, which was associated with increased sFlt expression (Fig. 3, G and H). To determine the role of Wnt5a on the expression of sFlt in these cells, the Wnt5a competitive antagonist Box5 was utilized to block noncanonical Wnt5a signaling (38). hEC infected with Glrx or LacZ were treated with or without Box5 (100 μm, 18 h). Box5 prevented Glrx-induced sFlt expression in hEC (Fig. 3I). Together, our data indicate that Wnt5a increases sFlt in human EC as shown previously in myeloid cells (24) and confirm that Glrx induces sFlt via Wnt5a in EC.
FIGURE 3.

Glrx causes a Wnt5a-dependent increase in sFLT expression in human ECs. A and B, qRT-PCR analysis for Wnt5a (A) and Ror2 (B) mRNA in mEC isolated from WT and TG mice (n = 5; *, p < 0.05). C and D, qRT-PCR analysis of sFlt (C) and mFlt (D) in hEC treated with rhWnt5a (100 ng/ml) over time indicated (n = 5; *, p < 0.05). E and F, representative images (E) and quantitative assessment (F) of hEC network formation in the presence and absence of Wnt5a (100 ng/ml, 24 h), with and without VEGF treatment (50 ng/ml, 18 h, n = 4; *, p < 0.05). G and H, qRT-PCR for Wnt5a (G) and sFlt (H) in hEC overexpressing Glrx or LacZ by adenovirus treatment. (n = 6; *, p < 0.05). I, sFlt mRNA expression in AdLacZ and AdGlrx treated hEC in the presence and absence of Box5 (Wnt5a antagonist, 100 μm, 24 h; n = 6; *, p < 0.05).
Glrx Overexpression Impairs in Vivo Blood Recovery after Femoral Artery Ligation
To investigate the in vivo role of Glrx up-regulation on post-ischemia revascularization, Glrx TG mice were utilized in which overexpression of the human Glrx transgene was achieved by the β-actin promoter (Fig. 4A) (27). Expression of the human Glrx observed in TG gastrocnemius muscle had no effect on endogenous Glrx expression because murine Glrx protein was similar between Glrx TG and WT mice (Fig. 4B). The additional Glrx expression from transgene resulted in a significant 3-fold increase in Glrx activity in the TG muscle and heart homogenate compared with WT homogenate (Fig. 4C), which was a similar level of increase in Glrx activity to that observed in diabetic animals (13). Hind limb ischemic blood flow recovery was compared in Glrx TG mice to WT littermate controls. Blood flow was assessed serially after HLI surgery (days 0, 3, 7, and 14) by LASER Doppler speckle tracking in the plantar aspect of the paws (Fig. 4E). In WT mice blood flow recovery was evident on day 3, whereas Glrx TG blood flow was consistently lower over the 2-week measurement period (Fig. 4, D and E; p < 0.05, repeated measured ANOVA). Glrx TG mice often had necrotic toes and feet (Fig. 4F). The functional impact of impaired revascularization was assessed by examining ambulatory foot movement. Scoring of motor function showed that Glrx TG mice were less able to use the ischemic limb, whereas WT mice generally had full use of the limb, and their toes had grip reflex 7 days after the surgery (Fig. 4G). To further examine the consequence of ischemia on limb motor function, mice were placed on a treadmill, and exercise function was assessed by calculating the time required to run until exhaustion (akin to the clinical walk test conducted on peripheral artery disease patients). Ten days after surgery Glrx TG mice showed impairment of limb function as they ran for a shorter time period compared with WT mice (Fig. 4H). Because blood flow recovery in part corresponds to increased tissue capillary density, we examined the capillary density in gastrocnemius muscle. As expected, in WT mice, capillary density increased in the ischemic gastrocnemius muscle; however in the ischemic Glrx TG muscle, there was a marked attenuation of the capillary increase (Fig. 4, I and J). There was no significant difference in capillary density in non-ischemic limbs.
FIGURE 4.

Glrx overexpression attenuates revascularization, limb motor function, and capillary density after HLI surgery. A, the structure of the human Glrx transgene driven by the β-actin promoter. CAP, transcriptional initiation site of the human β-actin gene. B, Western blot analysis of human Glrx transgene (hGlrx) and endogenous Glrx (mGlrx) in WT and TG gastrocnemius muscle. C, Glrx activity in heart and skeletal muscle homogenate from WT and Glrx TG mice. Activity was normalized to protein weight (n = 5; *, p < 0.05). D, quantitative serial assessment of LASER Doppler measuring blood flow recovery to the plantar aspect of WT and TG paws after HLI surgery (n = 15; *, p < 0.05). E, representative LASER Doppler images obtained after surgery and on day 14. F, representative images of ischemic paws from WT and TG mice 14 days after surgery. G, scoring of motor function of ischemic limb from WT and TG mice on day 14 (n = 12; *, p < 0.05). H, duration of time on treadmill 10 days after HLI surgery running at 5 m/min with 5 m/min increments every 5 min (n = 6; *, p < 0.05). I and J, representative images (I, scale, 100 μm) and quantitative analysis (J) of isolectin B4 staining for capillaries in non-ischemic (NI) and ischemic (ISC) gastrocnemius muscle in WT and TG mice 14 days after HLI surgery (n = 5; *, p < 0.05).
Soluble Flt Expression Is Increased in Glrx TG Mouse Muscle and Plasma
In accordance with the increase in sFlt in Glrx-overexpressing EC, we found both sFlt and mFlt mRNA expression were higher in Glrx TG ischemic muscle compared with WT control 14 days after HLI surgery (Fig. 5, A and B), whereas no difference was observed in VEGF and VEGFR2 expression (Fig. 5, C and D). Furthermore, the level of sFlt, assessed by ELISA was significantly higher in plasma from Glrx TG mice on both 4 and 14 days after HLI (Fig. 5E). We analyzed VEGF levels in mouse plasma 4 and 14 days after the HLI surgery by ELISA and found no difference in circulating VEGF levels (Fig. 5F). Interestingly, Wnt5a expression was markedly increased in Glrx TG ischemic muscle compared with WT. In contrast, Wnt5a levels were low in non-ischemic limb from both WT and Glrx TG mice (Fig. 5G). These data suggest that Wnt5a and sFlt induction in Glrx-overexpressing EC might occur in Glrx TG mice.
FIGURE 5.

Up-regulation of the anti-angiogenic receptor, soluble FLT and Wnt5a in Glrx-overexpressing mice post HLI. A–D, mRNA expression of soluble (A) and membrane-bound (B) Flt isoforms, VEGF (C), and VEGFR2 (D) in non-ischemic (NI) and ischemic (ISC) gastrocnemius muscles from WT and Glrx TG mice 14 days after HLI (n = 6, p < 0.05). E and F, ELISA for sFLT1 (E) and VEGF (F) in plasma from WT and Glrx TG mice at 4 days (n = 6) and 14 days (n = 12) after HLI (*, p < 0.05). G, representative immunoblot of Wnt5a in non-ischemic and ischemic gastrocnemius muscle normalized to tubulin.
Glrx Removes p65-NF-κB GSH Adducts in Ischemic Muscle and EC
We used a biotin switch assay to quantify reversible cysteine modifications. This assay first irreversibly blocks free cysteines, and after reduction with DTT selectively labels reversible cysteine modifications with biotin-HPDP. The HPDP-modified proteins are then precipitated with streptavidin and blotted for the protein of interest to indicate the level of thiol modification of that protein. Interestingly, we found that thiol adducts on p65 were increased in the ischemic muscle of WT mice (Fig. 6, A and B) but were significantly attenuated in that of Glrx TG mice, consistent with a decrease in GSH adducts. This also indicates significantly higher Glrx activity (removing GSH adducts) in vivo in Glrx TG mice, thus confirming that the modest increase observed in Glrx activity (Fig. 4C) has an in vivo biological consequence. We also found that adenoviral overexpression of Glrx lowered GSH adducts of p65 compared with LacZ-infected EC (Fig. 6C). Furthermore, Glrx overexpression prevented the increase in GSH adducts on p65 under hypoxia (0.1%, 20 h) (Fig. 6, C and D).
FIGURE 6.

Glrx decreases p65-glutathione adducts in ischemic muscle and endothelial cells. A–D, biotin switch assay showing the biotin labeled S-glutathionylated cysteines within p65 (pull down) in WT (A) and Glrx TG non-ischemic (NI) and ischemic (ISC) muscle 4 days after HLI, and in hEC overexpressing Glrx or LacZ (C) under normoxic or hypoxic (0.1% 24 h) conditions with respective quantification of GSH adducts (B and D) normalized to p65 input, samples prior to incubation with streptavidin beads (n = 3–4; *, p < 0.05; **, p < 0.01). IB, immunoblot.
Glrx Enhances Wnt5a and sFlt in a NF-κB-dependent Manner
To confirm that removal of glutathione adducts enhanced NF-κB activity in Glrx TG mEC, an NF-κB reporter assay was used in isolated mEC treated with TNFα, a well described NF-κB activator. TNFα stimulated greater NF-κB activity in Glrx TG mEC compared with WT mEC (Fig. 7A). Next, we investigated whether the inhibition of NF-κB activity could prevent Glrx-induced Wnt5a expression. We used hEC overexpressing Glrx by adenovirus infection. First, hEC were treated with MG132, a proteasome inhibitor, which stabilizes IκBα and inhibits NF-κB. MG132 prevented the transcriptional up-regulation of Wnt5a by Glrx overexpression (Fig. 7B). Likewise, we assessed whether siRNA knockdown of p65 could prevent Wnt5a expression. siRNA treatment was effective after 72 h, inhibiting p65 protein expression (Fig. 7C) and preventing TNFα-induced E-selectin (Fig. 7D) and AdGlrx induced IL-6 expression (Fig. 7E), which validated p65 knockdown inactivating NF-κB. Furthermore, siRNA knockdown of p65 attenuated the AdGlrx-induced increase in Wnt5a and sFlt observed in hEC (Fig. 7, F and G). AdGlrx expression had no effect on VEGFR2 expression, whereas p65 knockdown increased VEGFR2 expression (Fig. 7H). Together these data indicate that induction of Wnt5a expression by Glrx is mediated via NF-κB activation and promotes sFlt production in EC, summarized in Fig. 8.
FIGURE 7.

Glrx enhances Wnt5a and sFLT in a NF-κB-dependent manner. A, NF-κB activity measured by luciferase reporter assay in the presence and absence of TNFα (50 ng/ml, 4 h). NF-κB reporter activity normalized to the co-transfected Renilla luciferase reporter in WT and Glrx TG mEC (*, p < 0.05; ***, p < 0.001; n = 3). B, Wnt5a mRNA expression in AdGlrx infected hEC incubated with and without MG132 (1 μm, 24 h; p < 0.05, n = 4). C–H, siRNA knockdown of p65 NFκB subunit on Glrx-overexpressing hEC. C, immunoblot for p65 and β-actin of hEC treated with a concentration range (10, 25, 50, and 100 nm for 72 h) of siRNA to p65 or equivalent off target control (scr). D, hEC mRNA for E-selectin after p65 knockdown (25 nm) in the presence and absence of TNFα (40 ng/ml) stimulation. E–H, IL-6 (E), Wnt5a (F), sFlt (G), and VEGFR2 (H) mRNA expression assessed by qRT-PCR after p65-siRNA or scrambled control treatment in AdGlrx- or AdLacZ-infected hEC (p < 0.05, n = 6).
FIGURE 8.
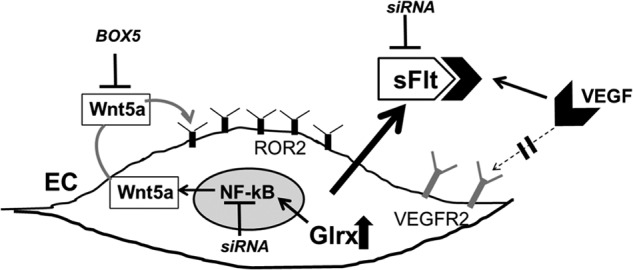
Up-regulated EC Glrx increases NF-κB-dependent Wnt5a, which promotes sFlt production. Summary of this study is shown. Glrx overexpression increased Wnt5a and sFlt mRNA and secretion of sFlt from EC; thus VEGF-induced vascularization is impaired. NF-κB inactivation (siRNA) suppressed Glrx-induced sFlt and Wnt5a. Wnt5a antagonist (BOX5) inhibited Glrx-induced sFlt. Exogenous Wnt5a increased sFlt in EC. Inhibiting sFlt (siRNA) improved angiogenic phenotype of EC overexpressing Glrx.
DISCUSSION
Glutathione adducts on redox-sensitive cysteine thiols are becoming increasingly recognized as an important mechanism for cellular signaling (39). Glrx specifically reverses protein-bound glutathione adducts, thus acting as a signaling switch on a range of important proteins (7, 40). Up-regulated Glrx under oxidative stress (e.g., ischemia) might accelerate turnover of GSH adducts, thus promoting pathological signaling. Our data report for the first time that Glrx regulates anti-angiogenic sFlt production through the noncanonical Wnt5a pathway that may have important implications in ischemic vascular disease, because up-regulation of Glrx impaired ischemic limb revascularization.
Up-regulated Glrx resulted in impaired ischemic limb revascularization and exercise function. In addition, capillary density in the ischemic muscle from Glrx mice was lower compared with WT mice. Interestingly, the alternative splice variant of VEGFR1 (Flt), sFlt, was increased in plasma after HLI surgery from Glrx TG mice. In addition, we detected higher sFlt in the media of EC isolated from Glrx TG mice. VEGFR2 mainly transduces VEGF-induced angiogenic responses, whereas Flt negatively regulates angiogenic signals by sequestering VEGF with high affinity to prevent the ligand binding to VEGFR2, and the membrane-tethered Flt (mFlt) appears to have weaker tyrosine kinase activity than VEGFR2 in angiogenic signaling (18). Adenoviral sFlt gene transfer inhibited revascularization in murine HLI (41). Elevated sFlt during pregnancy has been implicated in pre-eclampsia (42) and peripartum cardiomyopathy (43) by anti-angiogenic mechanisms. Soluble Flt expression was elevated in the serum of diabetic rats (44) and the ischemic muscle of diet-induced diabetic mice in association with impaired blood flow after femoral artery ligation (45). Glrx expression is associated with various disease processes. Glrx levels were shown to be higher in diabetic rat (13) and in plasma from diabetic patients (17). In this study, we used mice that overexpressed Glrx to a similar degree to the pathological level (13). Our findings may help to explain why diabetic patients have poor revascularization capability and a higher incidence of vascular ischemia, intermittent claudication, and lower limb amputations (45).
Soluble Flt production is regulated by alternative splicing (35), and ectodomain shedding of Flt by proteolytic cleavage has also been suggested as a mechanism of sFlt production in cancer cells (46). Using primers that distinguish the splice variants, we found that Glrx overexpression increased mFlt as well as sFlt in human and mouse EC. At this point, it is not clear whether splicing factors and/or ectodomain shedding are regulated by Glrx. VEGF gene expression and VEGF levels in plasma were not different by Glrx TG, and VEGFR2 expression was not altered by Glrx overexpression in EC.
Previously, Stefater et al. (24) reported that the noncanonical Wnt ligand, Wnt5a, in myeloid cells inhibited postnatal angiogenesis by inducing sFlt. The importance of Wnt ligands as developmental regulators of cell proliferation, migration, differentiation, and polarity are well known. In addition, recent reports suggest inhibitory roles of myeloid derived Wnt5a in developmental angiogenesis and wound repair (24, 47). Surprisingly, Wnt5a was highly expressed in ischemic hind limb muscle of Glrx TG compared with WT control. Also, Wnt5a expression was significantly higher in EC from Glrx TG mice, and Glrx overexpression increased Wnt5a as well as sFlt mRNA in hEC, suggesting that dysregulation of Wnt5a in the adult may have pathological significance. Recombinant Wnt5a induced sFlt in EC and inhibited EC network formation extending the previous finding in myeloid cells (24) to a different cell type. Ror2 is involved in noncanonical Wnt5a signaling and is a negative regulator of canonical Wnt/β-catenin pathway (37, 48). Elevation of Wnt5a levels causes increases in Ror2 expression (49). We found that Ror2 expression was significantly higher in Glrx TG-derived EC, consistent with Wnt5a responsiveness being increased in Glrx TG mice. Furthermore, the competitive Wnt5a inhibitor Box5 blocked Glrx-induced sFlt mRNA in EC, suggesting Glrx-induced sFlt in EC is Wnt5a-dependent.
Wnt5a causes p65 nuclear translocation and inflammation in EC (25), but also Wnt5a can be induced via NF-κB activation in monocytes (26, 27). As previously mentioned, Glrx promotes NF-κB activation through reversing S-glutathionylation of NF-κB components. We demonstrated higher TNFα-induced NF-κB activity in Glrx-overexpressing EC. Moreover, using a biotin switch assay we showed Glrx decreased reversible thiol modifications on p65 in ischemic muscle and EC. Specific DNA binding of NF-κB (p50, p65) is inhibited by GSH adducts (11, 12, 23). We speculate IKKβ and p50 might be S-glutathionylated as well and contribute to NF-κB activation by Glrx, although we were unable to show that by the biotin switch method. Inhibition of NF-κB by MG132 or siRNA to p65 blocked Wnt5a induction by Glrx overexpression. In addition, Glrx-induced sFlt was attenuated in EC through NF-κB inhibition. These data indicate that Glrx-induced Wnt5a and sFlt is related to activation of NF-κB. Glrx expression itself is induced by NF-κB activity (50) and up-regulated by inflammation and diabetes (13), suggesting deleterious positive feedback of the Glrx-NF-κB-Wnt5a pathway. In accordance with our data, inhibition of NF-κB with mutant IkBα increased disorganized vasculature in ischemic mouse hind limb (51) and enhanced tumor growth (52), suggesting that tight regulation of NF-κB is required to maintain normal vascularization (53).
A recent report suggests that after coronary artery ligation, Glrx TG mice may have improved neovascularization and cardiac remodeling because of less apoptosis (23). This report showed that α-actin staining was increased in the heart and VEGF expression was elevated, which may be insufficient to conclude that Glrx TG promotes neovascularization in heart. Higher VEGF expression does not necessarily correlate with angiogenesis (45). They also showed hyperactivation of NF-κB that may contribute to the protection of myocytes from apoptosis. Glrx may be protective in ischemic heart because of inhibition of apoptosis. We conducted a more extensive study of angiogenic properties in Glrx-overexpressing EC and mice. Our results clearly demonstrated that up-regulated Glrx inhibits EC migration and network formation in vitro and impairs hind limb revascularization in vivo.
In myeloid cells, Wnt5a induction of sFlt is likely through nuclear factor of activated T cells (NFAT) activation (47). Interestingly, the interaction between NFAT and NF-κB p65 promotes synergistic activation of NFAT (54). Also, many transcription factors are known to be redox-sensitive including AP-1/c-Jun (55), p53 (56), Nrf-2, and its regulatory protein, Keap-1 (57). Therefore, other transcriptional regulation might be involved in Glrx-induced Wnt5a and sFlt. Furthermore, we assume there may be multiple Glrx targets involved in VEGF and angiogenesis signaling because many related proteins are known to be S-glutathionylated, such as sarco/endoplasmic reticulum Ca2+-ATPase (5, 22), H-Ras (33, 36), PTP1B (4), β-actin (10, 58), eNOS (59), and sirtuin-1 (2, 60), all of which could be involved. Further studies will elucidate redox-sensitive regulation of sFlt induction.
In summary, we report a novel pathway by which Glrx overexpression induces sFlt through increasing Wnt5a in EC, resulting in impaired in vivo revascularization after limb ischemia. In addition, we extended the importance of the Wnt5a-sFlt pathway to endothelial cells. These results are of significant importance because Glrx is up-regulated in pathological conditions including diabetes in which ischemic conditions are worsened.
Acknowledgments
We thank Leah Mycoff and Pratibha Chauhan for technical assistance.
This work was supported, in whole or in part, by National Institutes of Health Grants PO1 HL 068758, R37 HL104017 (to R. A. C.), and HL081587 (to K. W.) and NHLBI Contract HHSN268201000031C (to R. A. C.). This work was also supported by Robert Dawson Evans Scholar Award from the Department of Medicine, Boston University School of Medicine (to R. A. C.).
- Glrx
- glutaredoxin-1
- EC
- endothelial cell(s)
- mEC
- mouse cardiac microvascular endothelial cell(s)
- hEC
- human cardiac microvascular endothelial cell(s)
- HLI
- hind limb ischemia
- sFlt
- soluble fms-like tyrosine kinase-1, VEGF receptor-1
- mFlt
- membrane-bound VEGF receptor-1
- Ror2
- receptor tyrosine kinase-like orphan receptor 2
- Wnt5a
- wingless-type MMTV integration site family, member 5A
- MMTV
- mouse mammary tumor virus
- TG
- transgenic
- VEGFR
- VEGF receptor
- qRT-PCR
- quantitative real-time polymerase chain reaction
- HPDP
- N-[6-(biotinamido) hexyl]-3′-(2′-pyridyldithio) propionamide
- ANOVA
- analysis of variance
- NF-κB
- nuclear factor of κB
- NFAT
- nuclear factor of activated T cells.
REFERENCES
- 1. Klatt P., Lamas S. (2000) Regulation of protein function by S-glutathiolation in response to oxidative and nitrosative stress. Eur. J. Biochem. 267, 4928–4944 [DOI] [PubMed] [Google Scholar]
- 2. Zee R. S., Yoo C. B., Pimentel D. R., Perlman D. H., Burgoyne J. R., Hou X., McComb M. E., Costello C. E., Cohen R. A., Bachschmid M. M. (2010) Redox regulation of sirtuin-1 by S-glutathiolation. Antioxid. Redox Signal. 13, 1023–1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Reynaert N. L., van der Vliet A., Guala A. S., McGovern T., Hristova M., Pantano C., Heintz N. H., Heim J., Ho Y.-S., Matthews D. E., Wouters E. F., Janssen-Heininger Y. M. (2006) Dynamic redox control of NF-κB through glutaredoxin-regulated S-glutathionylation of inhibitory κB kinase β. Proc. Natl. Acad. Sci. U.S.A. 103, 13086–13091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Barrett W. C., DeGnore J. P., Keng Y. F., Zhang Z. Y., Yim M. B., Chock P. B. (1999) Roles of superoxide radical anion in signal transduction mediated by reversible regulation of protein-tyrosine phosphatase 1B. J. Biol. Chem. 274, 34543–34546 [DOI] [PubMed] [Google Scholar]
- 5. Adachi T., Weisbrod R. M., Pimentel D. R., Ying J., Sharov V. S., Schöneich C., Cohen R. A. (2004) S-Glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat. Med. 10, 1200–1207 [DOI] [PubMed] [Google Scholar]
- 6. Clavreul N., Bachschmid M. M., Hou X., Shi C., Idrizovic A., Ido Y., Pimentel D., Cohen R. A. (2006) S-Glutathiolation of p21ras by peroxynitrite mediates endothelial insulin resistance caused by oxidized low-density lipoprotein. Arterioscler. Thromb. Vasc. Biol. 26, 2454–2461 [DOI] [PubMed] [Google Scholar]
- 7. Mieyal J. J., Gallogly M. M., Qanungo S., Sabens E. A., Shelton M. D. (2008) Molecular mechanisms and clinical implications of reversible protein S-glutathionylation. Antioxid. Redox Signal. 10, 1941–1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chrestensen C. A., Starke D. W., Mieyal J. J. (2000) Acute cadmium exposure inactivates thioltransferase (glutaredoxin), inhibits intracellular reduction of protein-glutathionyl-mixed disulfides, and initiates apoptosis. J. Biol. Chem. 275, 26556–26565 [DOI] [PubMed] [Google Scholar]
- 9. Shelton M. D., Mieyal J. J. (2008) Minireview molecules and regulation by reversible S-glutathionylation. Molecular targets implicated in inflammatory diseases. Mol. Cells 25, 332–346 [PMC free article] [PubMed] [Google Scholar]
- 10. Bachschmid M. M., Xu S., Maitland-Toolan K. A., Ho Y.-S., Cohen R. A., Matsui R. (2010) Attenuated cardiovascular hypertrophy and oxidant generation in response to angiotensin II infusion in glutaredoxin-1 knockout mice. Free Radic. Biol. Med. 49, 1221–1229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pineda-Molina E., Klatt P., Vázquez J., Marina A., García de Lacoba M., Pérez-Sala D., Lamas S. (2001) Glutathionylation of the p50 subunit of NF-κB. A mechanism for redox-induced inhibition of DNA binding. Biochemistry 40, 14134–14142 [DOI] [PubMed] [Google Scholar]
- 12. Qanungo S., Starke D. W., Pai H. V., Mieyal J. J., Nieminen A.-L. (2007) Glutathione supplementation potentiates hypoxic apoptosis by S-glutathionylation of p65-NFκB. J. Biol. Chem. 282, 18427–18436 [DOI] [PubMed] [Google Scholar]
- 13. Shelton M. D., Kern T. S., Mieyal J. J. (2007) Glutaredoxin regulates nuclear factor κ-B and intercellular adhesion molecule in Müller cells. Model of diabetic retinopathy. J. Biol. Chem. 282, 12467–12474 [DOI] [PubMed] [Google Scholar]
- 14. Shelton M. D., Distler A. M., Kern T. S., Mieyal J. J. (2009) Glutaredoxin regulates autocrine and paracrine proinflammatory responses in retinal glial (Muller) cells. J. Biol. Chem. 284, 4760–4766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Okuda M., Inoue N., Azumi H., Seno T., Sumi Y., Hirata Ki, Kawashima S., Hayashi Y., Itoh H., Yodoi J., Yokoyama M. (2001) Expression of glutaredoxin in human coronary arteries. Its potential role in antioxidant protection against atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 21, 1483–1487 [DOI] [PubMed] [Google Scholar]
- 16. Reynaert N. L., Wouters E. F., Janssen-Heininger Y. M. (2007) Modulation of glutaredoxin-1 expression in a mouse model of allergic airway disease. Am. J. Respir. Cell Mol. Biol. 36, 147–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Du Y., Zhang H., Montano S., Hegestam J., Ekberg N. R., Holmgren A., Brismar K., Ungerstedt J. S. (2013) Plasma glutaredoxin activity in healthy subjects and patients with abnormal glucose levels or overt type 2 diabetes. Acta Diabetol. 10.1007/s00592-013-0498-2 [DOI] [PubMed] [Google Scholar]
- 18. Koch S., Claesson-Welsh L. (2012) Signal transduction by vascular endothelial growth factor receptors. Cold Spring Harbor Perspect. Med. 2, a006502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kendall R. L., Thomas K. A. (1993) Inhibition of vascular endothelial cell growth factor activity by an endogenously encoded soluble receptor. Proc. Natl. Acad. Sci. U.S.A. 90, 10705–10709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ushio-Fukai M. (2006) Redox signaling in angiogenesis. Role of NADPH oxidase. Cardiovasc. Res. 71, 226–235 [DOI] [PubMed] [Google Scholar]
- 21. Abdelsaid M. A., El-Remessy A. B. (2012) S-Glutathionylation of LMW-PTP regulates VEGF-mediated FAK activation and endothelial cell migration. J. Cell Sci. 125, 4751–4760 [DOI] [PubMed] [Google Scholar]
- 22. Evangelista A. M., Thompson M. D., Weisbrod R. M., Pimental D. R., Tong X., Bolotina V. M., Cohen R. A. (2012) Redox regulation of SERCA2 is required for vascular endothelial growth factor-induced signaling and endothelial cell migration. Antioxid. Redox Signal. 17, 1099–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Adluri R. S., Thirunavukkarasu M., Zhan L., Dunna N. R., Akita Y., Selvaraju V., Otani H., Sanchez J. A., Ho Y.-S., Maulik N. (2012) Glutaredoxin-1 overexpression enhances neovascularization and diminishes ventricular remodeling in chronic myocardial infarction. PLoS One 7, e34790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Stefater J. A., 3rd, Lewkowich I., Rao S., Mariggi G., Carpenter A. C., Burr A. R., Fan J., Ajima R., Molkentin J. D., Williams B. O., Wills-Karp M., Pollard J. W., Yamaguchi T., Ferrara N., Gerhardt H., Lang R. A. (2011) Regulation of angiogenesis by a non-canonical Wnt-Flt1 pathway in myeloid cells. Nature 474, 511–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kim J., Kim J., Kim D. W., Ha Y., Ihm M. H., Kim H., Song K., Lee I. (2010) Wnt5a induces endothelial inflammation via β-catenin-independent signaling. J. Immunol. 185, 1274–1282 [DOI] [PubMed] [Google Scholar]
- 26. Blumenthal A., Ehlers S., Lauber J., Buer J., Lange C., Goldmann T., Heine H., Brandt E., Reiling N. (2006) The Wingless homolog WNT5A and its receptor Frizzled-5 regulate inflammatory responses of human mononuclear cells induced by microbial stimulation. Blood 108, 965–973 [DOI] [PubMed] [Google Scholar]
- 27. Nanbara H., Wara-aswapati N., Nagasawa T., Yoshida Y., Yashiro R., Bando Y., Kobayashi H., Khongcharoensuk J., Hormdee D., Pitiphat W., Boch J. A., Izumi Y. (2012) Modulation of Wnt5a expression by periodontopathic bacteria. PLoS One 7, e34434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Limbourg A., Korff T., Napp L. C., Schaper W., Drexler H., Limbourg F. P. (2009) Evaluation of postnatal arteriogenesis and angiogenesis in a mouse model of hind-limb ischemia. Nat. Protoc. 4, 1737–1746 [DOI] [PubMed] [Google Scholar]
- 29. Ohashi K., Ouchi N., Higuchi A., Shaw R. J., Walsh K. (2010) LKB1 deficiency in Tie2-Cre-expressing cells impairs ischemia-induced angiogenesis. J. Biol. Chem. 285, 22291–22298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Haeussler D. J., Pimentel D. R., Hou X., Burgoyne J. R., Cohen R. A., Bachschmid M. M. (2013) Endomembrane H-Ras controls vascular endothelial growth factor-induced nitric-oxide synthase-mediated endothelial cell migration. J. Biol. Chem. 288, 15380–15389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Luthman M., Holmgren A. (1982) Glutaredoxin from calf thymus. J. Biol. Chem. 257, 6686–6690 [PubMed] [Google Scholar]
- 32. Song J. J., Lee Y. J. (2003) Differential role of glutaredoxin and thioredoxin in metabolic oxidative stress-induced activation of apoptosis signal-regulating kinase 1. Biochem. J. 373, 845–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Adachi T., Pimentel D. R., Heibeck T., Hou X., Lee Y. J., Jiang B., Ido Y., Cohen R. A. (2004) S-Glutathiolation of Ras mediates redox-sensitive signaling by angiotensin II in vascular smooth muscle cells. J. Biol. Chem. 279, 29857–29862 [DOI] [PubMed] [Google Scholar]
- 34. Ahmad S., Hewett P. W., Al-Ani B., Sissaoui S., Fujisawa T., Cudmore M. J., Ahmed A. (2011) Autocrine activity of soluble Flt-1 controls endothelial cell function and angiogenesis. Vasc. Cell 3, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Boeckel J.-N., Guarani V., Koyanagi M., Roexe T., Lengeling A., Schermuly R. T., Gellert P., Braun T., Zeiher A., Dimmeler S. (2011) Jumonji domain-containing protein 6 (Jmjd6) is required for angiogenic sprouting and regulates splicing of VEGF-receptor 1. Proc. Natl. Acad. Sci. U.S.A. 108, 3276–3281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Clavreul N., Adachi T., Pimental D. R., Ido Y., Schöneich C., Cohen R. A. (2006) S-glutathiolation by peroxynitrite of p21ras at cysteine-118 mediates its direct activation and downstream signaling in endothelial cells. FASEB J. 20, 518–520 [DOI] [PubMed] [Google Scholar]
- 37. Mikels A., Minami Y., Nusse R. (2009) Ror2 receptor requires tyrosine kinase activity to mediate Wnt5A signaling. J. Biol. Chem. 284, 30167–30176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jenei V., Sherwood V., Howlin J., Linnskog R., Säfholm A., Axelsson L., Andersson T. (2009) A t-butyloxycarbonyl-modified Wnt5a-derived hexapeptide functions as a potent antagonist of Wnt5a-dependent melanoma cell invasion. Proc. Natl. Acad. Sci. U.S.A. 106, 19473–19478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dulce R. A., Schulman I. H., Hare J. M. (2011) S-Glutathionylation. A redox-sensitive switch participating in nitroso-redox balance. Circ. Res. 108, 531–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Fernandes A. P., Holmgren A. (2004) Glutaredoxins. Glutathione-dependent redox enzymes with functions far beyond a simple thioredoxin backup system. Antioxid. Redox Signal. 6, 63–74 [DOI] [PubMed] [Google Scholar]
- 41. Jacobi J., Tam B. Y., Wu G., Hoffman J., Cooke J. P., Kuo C. J. (2004) Adenoviral gene transfer with soluble vascular endothelial growth factor receptors impairs angiogenesis and perfusion in a murine model of hindlimb ischemia. Circulation 110, 2424–2429 [DOI] [PubMed] [Google Scholar]
- 42. Ahmad S., Ahmed A. (2004) Elevated placental soluble vascular endothelial growth factor receptor-1 inhibits angiogenesis in preeclampsia. Circ. Res. 95, 884–891 [DOI] [PubMed] [Google Scholar]
- 43. Patten I. S., Rana S., Shahul S., Rowe G. C., Jang C., Liu L., Hacker M. R., Rhee J. S., Mitchell J., Mahmood F., Hess P., Farrell C., Koulisis N., Khankin E. V., Burke S. D., Tudorache I., Bauersachs J., del Monte F., Hilfiker-Kleiner D., Karumanchi S. A., Arany Z. (2012) Cardiac angiogenic imbalance leads to peripartum cardiomyopathy. Nature 485, 333–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Khazaei M., Fallahzadeh A. R., Sharifi M. R., Afsharmoghaddam N., Javanmard S. H., Salehi E. (2011) Effects of diabetes on myocardial capillary density and serum angiogenesis biomarkers in male rats. Clinics 66, 1419–1424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hazarika S., Dokun A. O., Li Y., Popel A. S., Kontos C. D., Annex B. H. (2007) Impaired angiogenesis after hindlimb ischemia in type 2 diabetes mellitus. Differential regulation of vascular endothelial growth factor receptor 1 and soluble vascular endothelial growth factor receptor 1. Circ. Res. 101, 948–956 [DOI] [PubMed] [Google Scholar]
- 46. Rahimi N., Golde T. E., Meyer R. D. (2009) Identification of ligand-induced proteolytic cleavage and ectodomain shedding of VEGFR-1/FLT1 in leukemic cancer cells. Cancer Res. 69, 2607–2614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Stefater J. A., 3rd, Rao S., Bezold K., Aplin A. C., Nicosia R. F., Pollard J. W., Ferrara N., Lang R. A. (2013) Macrophage Wnt-Calcineurin-Flt1 signaling regulates mouse wound angiogenesis and repair. Blood 121, 2574–2578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kikuchi A., Yamamoto H., Sato A., Matsumoto S. (2012) Wnt5a. Its signalling, functions and implication in diseases. Acta Physiol. (Oxf.) 204, 17–33 [DOI] [PubMed] [Google Scholar]
- 49. O'Connell M. P., Fiori J. L., Xu M., Carter A. D., Frank B. P., Camilli T. C., French A. D., Dissanayake S. K., Indig F. E., Bernier M., Taub D. D., Hewitt S. M., Weeraratna A. T. (2010) The orphan tyrosine kinase receptor, ROR2, mediates Wnt5A signaling in metastatic melanoma. Oncogene 29, 34–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Aesif S. W., Kuipers I., van der Velden J., Tully J. E., Guala A. S., Anathy V., Sheely J. I., Reynaert N. L., Wouters E. F., van der Vliet A., Janssen-Heininger Y. M. (2011) Activation of the glutaredoxin-1 gene by nuclear factor κB enhances signaling. Free Radic. Biol. Med. 51, 1249–1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tirziu D., Jaba I. M., Yu P., Larrivée B., Coon B. G., Cristofaro B., Zhuang Z. W., Lanahan A. A., Schwartz M. A., Eichmann A., Simons M. (2012) Endothelial nuclear factor-κB-dependent regulation of arteriogenesis and branching. Circulation 126, 2589–2600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kisseleva T., Song L., Vorontchikhina M., Feirt N., Kitajewski J., Schindler C. (2006) NF-κB regulation of endothelial cell function during LPS-induced toxemia and cancer. J. Clin. Invest. 116, 2955–2963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tabruyn S. P., Griffioen A. W. (2007) A new role for NF-κB in angiogenesis inhibition. Cell Death Differ. 14, 1393–1397 [DOI] [PubMed] [Google Scholar]
- 54. Liu Q., Chen Y., Auger-Messier M., Molkentin J. D. (2012) Interaction between NFκB and NFAT coordinates cardiac hypertrophy and pathological remodeling. Circ. Res. 110, 1077–1086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Klatt P., Molina E. P., Lamas S. (1999) Nitric oxide inhibits c-Jun DNA binding by specifically targeted S-glutathionylation. J. Biol. Chem. 274, 15857–15864 [DOI] [PubMed] [Google Scholar]
- 56. Velu C. S., Niture S. K., Doneanu C. E., Pattabiraman N., Srivenugopal K. S. (2007) Human p53 is inhibited by glutathionylation of cysteines present in the proximal DNA-binding domain during oxidative stress. Biochemistry 46, 7765–7780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lukosz M., Jakob S., Büchner N., Zschauer T.-C., Altschmied J., Haendeler J. (2010) Nuclear redox signaling. Antioxid. Redox Signal. 12, 713–742 [DOI] [PubMed] [Google Scholar]
- 58. Wang J., Boja E. S., Tan W., Tekle E., Fales H. M., English S., Mieyal J. J., Chock P. B. (2001) Reversible glutathionylation regulates actin polymerization in A431 cells. J. Biol. Chem. 276, 47763–47766 [DOI] [PubMed] [Google Scholar]
- 59. Chen C.-A., Wang T.-Y., Varadharaj S., Reyes L. A., Hemann C., Talukder M. A., Chen Y.-R., Druhan L. J., Zweier J. L. (2010) S-Glutathionylation uncouples eNOS and regulates its cellular and vascular function. Nature 468, 1115–1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Shao D., Fry J. L., Han J., Hou X., Pimentel D. R., Matsui R., Cohen R. A., Bachschmid M. M. (January 22, 2014) A redox-resistant sirtuin-1 protects against hepatic metabolic and oxidant stress. J. Biol. Chem. 10.1074/jbc.M113.520403 [DOI] [PMC free article] [PubMed] [Google Scholar]