

Abstract
Mild traumatic brain injury (mTBI) often has long-term effects on cognitive function and social behavior. Altered gene expression may be predictive of long-term psychological effects of mTBI, even when acute clinical effects are minimal or transient. Controlled cortical impact (CCI), which causes concussive, but nonpenetrant, trauma to underlying (non-cortical) brain, resulting in persistent changes in hippocampal synaptic function, was used as a model of mTBI. The hippocampal transcriptomes of sham-operated or injured male rats at 1, 7, and 30 days postinjury were examined using microarrays comprising a comprehensive set of expressed genes, subsequently confirmed by quantitative reverse-transcriptase polymerase chain reaction. Transcripts encoding the chemokines, chemokine (C-C motif) ligand (Ccl)2 and Ccl7, inflammatory mediators lipocalin-2 (Lcn2) and tissue inhibitor of metalloproteinase 1 (Timp1), immunocyte activators C-C chemokine receptor type 5 (Ccr5) and Fc fragment of IgG, low affinity IIb, receptor (CD32) (Fcgr2b), the major histocompatibility complex II immune response-related genes, Cd74 and RT1 class II, locus Da (RT1-Da), the complement component, C3, and the transcription factor, Kruppel-like factor 4 (Klf4), were identified as early (Ccl2, Ccl7, Lcn2, and Timp1), intermediate (Ccr5, Fcgr2b, Cd74, RT1-Da, and C3), and late (Klf4) markers for bilateral hippocampal response to CCI. Ccl2 and Ccl7 transcripts were up-regulated within 24 h after CCI, and their elevation subsided within 1 week of injury. Other transcriptional changes occurred later and were more stable, some persisting for at least 1 month, suggesting that short-term inflammatory responses trigger longer-term alteration in the expression of genes previously associated with injury, aging, and neuronal function in the brain. These transcriptional responses to mTBI may underlie long-term changes in excitatory and inhibitory neuronal imbalance in hippocampus, leading to long-term behavioral consequences of mTBI.
Key words: : Ccl2, Ccl7, controlled cortical injury (CCI), Lcn2, mild traumatic brain injury (mTBI)
Introduction
Traumatic brain injury (TBI) has a high incidence in both the civilian and military populations, albeit distributed somewhat differently between age and gender. The CDC (Centers for Disease Control and Prevention) estimates that, each year, 1.7 million people in the United States sustain a TBI. Most TBI occurs as a result of falls (35.2% of total), followed by vehicular accidents (17.3%), in the civilian population1 and as a result of injuries from both penetrating and nonpenetrating ordnance among soldiers. A significant fraction of TBI (up to 80%) is considered mild (mTBI) and is the result of concussive nonpenetrating insult, such as that caused by blast overpressure subsequent to exposure to improvised explosive devices, or by a high-impact blow to the head. mTBI is characterized by a brief period of unconsciousness, with no signs of brain injury upon neuroimaging, but is associated with subsequent cognitive, emotional, or memory dysfunction. mTBI is a hypothesized cause of post-traumatic stress disorder (PTSD) in both civilian2 and military3 populations.
Models for mTBI are not easily established and include focused exposure to pressurized air (blast overpressure; BoP),4 actual blast exposure,5 closed-head injury (CHI),6 fluid percussion in which injury is administered by a column of liquid,7 and controlled cortical injury (CCI). The CCI model aims to overcome the limitations of CHI, including lack of reproducibility of actual impact effects resulting from irregularities in rodent cranial surface features, by the administration of a partially penetrating injury to the surface of the brain through an aperture in the skull. In CCI, injury is administered by a metal piston8 to the cortical surface. Methodological considerations may favor one model or the other to address a specific research question. The major concern about the applicability of each type of CCI is that it represents a continuum of penetrating (to the region of the brain closest to the source of impact) and nonpenetrating (regions of the brain distant from the site of impact) insult to the brain. It is therefore critical to specify whether the sequelae of injury examined are direct (i.e., modeling TBI) or indirect (i.e., modeling mTBI).
In the study described in this article, we have employed metal piston CCI (which we will henceforth refer to simply as CCI) as a model for mTBI as transmitted to the hippocampus. We have first established that the degree of insult is indeed nonpenetrating to the hippocampus and then examined altered expression of all known transcripts encoded within the hippocampus at 1, 7, and 30 days after injury. The patterns of altered gene expression, and their bilaterality after unilateral CCI, suggest that CCI is indeed an appropriate model for mTBI. They further indicate that neuroinflammatory mechanisms after concussive injury might contribute to long-term changes in the brain leading to impairment in the hippocampus, a major brain center for memory and cognitive processing.
Methods
Animals
Experiments were performed using male Sprague-Dawley rats (Taconic Farms, Rockville, MD), 5–6 weeks old, weighing 150–200 g at the start of the experiments. Animals were pair-housed until the day of the surgery and then housed individually in an animal facility approved by the Association for Assessment and Accreditation of Laboratory Animal Care, in an environmentally controlled room (20–23°C, 12-h light/dark cycle, lights on 7:00 AM), with food and water available ad libitum. All animal use procedures were in accord with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the animal care and use committee of the Uniformed Services University of the Health Sciences (Bethesda, MD). All efforts were made to minimize the number of animals used and their suffering.
Controlled cortical impact surgery
After 5 days of acclimatization, rats were submitted to unilateral cortical contusion using the CCI model of TBI. Animals were anesthetized with isoflurane (2.5%) delivered by nose cone, with the skull shaved and the head placed in a stereotaxic frame. The skull was exposed using a small surgical incision over the scalp. A 4.0-mm hole was drilled in the skull at 3.0 mm lateral from the mid-line and 4.0 mm posterior to the bregma over the left tempoparietal cortex. The overlying portion of bone was carefully removed with no disruption of the dura. Each animal received a single impact of 2.0 mm depth of deformation using an impact tip of 3 mm in diameter with an impact velocity of 3.5 m/sec and a dwell time of 200 ms. After impact, the bone was replaced and fixed using bone wax, and the incision was closed with absorbable sutures. After surgery, animals received Ringer's solution (5 mL) subcutaneously for rehydration. A group of sham-injured rats underwent identical craniotomy procedures without CCI injury. Body temperature was maintained at 37±0.5°C using a heating pad coupled to a rectal probe.
Fixation and tissue processing
Rats subjected to CCI or sham surgery were used for histological studies as follows. One, 7, or 30 days after surgery, rats were deeply anesthetized using Nembutal (75–100 mg/kg, intraperitoneally) and transcardially perfused with phosphate-buffered saline (PBS; 100 mL), followed by 4% paraformaldehyde (PFA; 250 mL). Brains were removed and postfixed in 4% PFA overnight at 4°C, transferred to a solution of 30% sucrose in PBS for 72 h, and frozen in Dry Ice before storage at −80°C until sectioning. A series of sections containing the rostrocaudal extent of the amygdala was cut at 40-μm intervals on a sliding microtome (SM2000R; Leica Microsystems, Wetzlar, Germany), as previously described.9,10 Every fifth section was placed in a cryoprotectant solution (30% ethylene glycol and 30% glycerol in 0.05 M of sodium phosphate buffer) and stored at −20°C until processing. Free-floating sections were collected from the cryoprotectant solution and washed three times for 5 min each. Slices were mounted on a slide, air-dried overnight, and processed for Nissl staining with cresyl violet. Sections shown in Figure 2 are representative of sections examined from 2 CCI animals at 1, 7, and 30 days and 1 sham animal at 1, 7, and 30 days. Eight CCI and 8 sham animals euthanized at 7 days postsurgery were used to collect the data for quantitative assessment of cell number in the CA1 region of the hippocampus (see Stereological Quantification, below).
FIG. 2.

Regulatory networks for CCI-dependent transcripts in rat hippocampal tissue. Transcripts with an abundance of 1.5 or greater in cortically injured (CCI) male rats at 1, 7 and 30 days post-injury compared to sham-operated by microarray analysis (see Supplementary Table 2) (see online supplementary material at http://www.liebertpub.com) were used as the input for analysis of potential networks using the signal transduction knowledge environment of Ingenuity (http://www.ingenuity.com). A, B, and C are the highest score networks involving the CCI-up-regulated transcripts at day one, seven and 30 respectively. CCI-regulated transcripts are depicted in gray and linked components in white. Circular lines above symbols indicate auto-regulation; connecting lines without arrows indicate direct protein interaction; dashed and solid arrows indicate indirect (e.g. regulation of messenger RNA levels) and direct (e.g. enzymatic) activation. CCI, controlled cortical impact.
Stereological quantification
CA1 is the part of the hippocampus in closest proximity to the area of cortical impact in our CCI model and was therefore used as a representative subdomain of the hippocampus in which a valid assessment of cell number within the entire region, as a function of injury, could be obtained using design-based stereology in Nissl-stained sections.10 Sections were viewed with an Axioplan 2ie fluorescent microscope (Zeiss, Oberkochen, Germany) with a motorized stage, interfaced with a computer running StereoInvestigator software (9.0; MicroBrightField, Williston, VT). The CA1 region was identified on slide-mounted sections and delineated for each slide of each animal, under a 2.5×objective, based on the atlas of Paxinos and Watson.11 All sampling was done under a 63×oil immersion objective. Nissl-stained neurons were distinguished from glial cells by their larger size and pale nuclei surrounded by darkly stained cytoplasm containing Nissl bodies. The total number of Nissl-stained neurons was estimated by using the optional fractionator probe and, along with the coefficient of error (CE), was calculated by using StereoInvestigator software (9.0; MicroBrightField). The CE was calculated using MicroBrightField software according to the equations of Gundersen and colleagues (m=1)12 and Schmitz and Hof (second estimation).13 For Nissl-stained neurons in the CA1, a 1-in-10 series of sections was analyzed (seven sections, on average). The counting frame was 20×20 μm, the counting grid was 250×250 μm, and the dissector height was 10 μm. Nuclei were counted when the cell body came into focus within the dissector, which was placed 2 μm below the section surface. Section thickness was measured at every counting site (average mounted section thickness, 25.7 μm). An average of 234 neurons per hemisphere per rat were counted. Eight rats per group were analyzed, and the average CE was 0.07 for both Gunderson and colleagues and Schmitz and Hof equations.
RNA extraction, microarray procedure and analysis
Left and right hemispheric dorsal hippocampus was removed from the brains of 200-g male rats subjected to CCI or sham surgery (24 experimental animals comprising six groups—CCI day 1, CCI day 7, CCI day 30, sham day 1, sham day 7, and sham day 30—with n=3 per group for Affymetrix array and n=4 per group for Illumina array). Tissues were snap-frozen on Dry Ice and stored until RNA isolation. Total RNA was extracted using an ultrasonic processor (GE 130PB; Hielscher Systems, Ringwood, NJ) and a commercial kit (RNAqueous; Ambion, Inc., Austin, TX), according to the manufacturer's instructions, and quantified by spectrophotometry. RNA was processed for Illumina and Affymetrix array after Bioanalyzer (Agilent, Inc., Santa Clara, CA) determination of quantity and quality. All samples used had RNA integrity number greater than 7.3; Illumina RatRef-12 Expression BeadChip (catalog no.: BD-27-303) and Affymetrix GeneChip Rat Gene 1.0 ST (catalog no.: 901173) arrays were used for hybridization.
Samples for Illumina microarray were processed by preparing biotinylated complementary RNA (cRNA) using an Illumina Total Prep RNA Amplification Kit (Ambion). Briefly, 5 μg of total RNA from the ipsilateral dorsal hippocampus was converted to double-stranded cDNA using T7-oligo (dT) primers, followed by an in vitro transcription reaction to amplify biotinylated cRNA, as described in the manufacturer's instructions (Illumina Inc., San Diego, CA). The biotinylated cRNA was hybridized to a RatRef-12 Expression BeadChip platform that contains 22,523 probes (Illumina Inc.). Hybridization, washing, and scanning were performed according to the manufacturer's instructions. Chips were scanned using an Illumina Bead Chip Scanner.
For Affymetrix microarray chip processing, samples were prepared according to Affymetrix protocols (Affymetrix, Inc., Santa Clara, CA). Total RNA (300 ng) from the ipsilateral dorsal hippocampus was used per labeling reaction, in conjunction with the Affymetrix-recommended protocol, “One-Cycle Target Labeling and Control Reagents.” Hybridizations were performed with three sets of arrays with labeled RNA from either untreated samples (sham) or from treated samples (CCI). The hybridization cocktail containing the fragmented and labeled complementary DNAs (cDNAs) was added to the Affymetrix GeneChip, and chips were washed and stained by the Affymetrix Fluidics Station using the standard format and protocols as described by Affymetrix. Probe arrays were stained with streptavidin phycoerythrin solution (Molecular Probes, Carlsbad, CA) and enhanced by using an antibody solution containing 0.5 mg/mL of biotinylated anti-streptavidin (Vector Laboratories, Burlingame, CA). An Affymetrix Gene Chip Scanner 3000 was used to scan the probe arrays.
Affymetrix and Illumina scanned arrays were analyzed using the Partek Genomic suite following the vendors' recommendations. For Affymetrix arrays, robust multi-chip analysis normalization was used, followed by Student's t-test to determine significantly up- or down-regulated genes. A subset of genes was selected whose expression was altered after CCI at least or greater than 1.5-fold (p≤0.05, Student's t-test; n=4 for each group for Illumina array; n=3 for each group for Affymetrix array) at 1, 7, and 30 days postinjury and annotated using the latest annotation files (from http://www.affymetrix.com).
Bioinformatic analysis
Using the Ingenuity Pathway Analysis (IPA) tool (http://www.ingenuity.com), the transcripts up-regulated 1.5-fold or more after CCI in rat hippocampal tissue were clustered based on their functions. Genes were also clustered based on their distribution in significant canonical pathways. Both indirect and direct networks were built using the default settings to identify interacting gene products.
Quantitative reverse-transcription polymerase chain reaction
Microarray results were validated by quantitative reverse-transcription polymerase chain reaction (qRT-PCR) using the same RNA samples as those used for the microarray. Approximately 2 μg of total RNA from both ipsi- and contralateral dorsal hippocampus was submitted to DNase I (RNase-free; Invitrogen, Carlsbad, CA) digestion and reverse transcribed using random hexamers pdN6 (Invitrogen) and SuperScript II RNase H¯ reverse transcriptase (Invitrogen). Splign (http://www.ncbi.nlm.nih.gov/sutils/splign/splign.cgi?textpage=online@level=form) was used to identify the exon and intron boundaries. Then, gene-specific forward and reverse primers (listed in Supplementary Table 1) (see online supplementary material at http://www.liebertpub.com) were designed using Primer 3 input (http:///frodo.wi.mit.edu) across exon-exon junctions. Amplicon specificity was confirmed by single, sharp melting curves, and predicted amplicon size by gel electrophoresis, at the end of qRT-PCR. qRT-PCR was performed in a premade reaction mix in the presence of the transcribed cDNA and 10 nM of each of the specific primers, using the SYBR green chemistry and an iCycler real-time detection system (Bio-Rad, Hercules, CA). Fold changes in messenger RNA (mRNA) levels were determined by normalizing against a nonvariable control transcript (glyceraldehyde 3-phosphate dehydrogenase (Gapdh)), using the ΔΔCt method.14
Statistical analysis
For comparisons between two groups, a t-test was performed. A p value of<0.05 was taken as statistically significant.
Results
Analysis of traumatic brain injury severity
To characterize the injury severity caused by our CCI conditions (velocity, 3.5 m/sec; deformation level, 2.0 mm), Nissl staining was performed on coronal brain slices and design-based stereology was used to count neurons in ipsi- and contralateral CA1, the region of the hippocampus in closest proximity to the area of cortical impact in our CCI model. Our results reveal that the hippocampus did not show signs of injury or deformation, compared to the sham, either ipsi- or contralateral to the site of injury induced in cortex by CCI in either gross aspect (Fig. 1A), at high power (Fig. 1B) or upon neuronal quantification using blinded, computer-assisted stereology (Fig. 1C). These data indicate that the CCI conditions used here result in a model for mTBI, a head trauma that usually does not present with discernible cell death in the brain.15
FIG. 1.
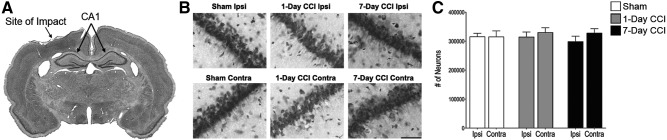
Characterization of injury severity caused by CCI. Mild CCI does not cause a significant loss of neurons in the CA1 region 24 hours or 7 days after injury. (A) Photomicrograph of Nissl-stained brain coronal slice. Indicated are the sites of impact and the ipsilateral (Ipsi) and contralateral (Contra) CA1. (B) Representative photomicrographs from Nissl-stained sections showing BLA cells from the ipsilateral (top) and contralateral (bottom) sides of sham, 1-day CCI, and 7-day CCI animals, respectively. Total magnification is 630×; scale bar, 50 μm. (C) Group data (mean±standard error; n=8 for each group) of stereological estimation of the total number of Nissl-stained neurons in the CA1. There were no significant differences in neuronal number between sham and 1-day or 7-day CCI, on either ipsilateral or contralateral sides, using Student's t-test. CCI, controlled cortical impact.
Transcriptome response to controlled cortical impact
To assess the effect of CCI injury on hippocampal neuron gene transcription, we analyzed gene expression changes using two different platforms: a rat microbead Illumina array targeting the 3′ ends of approximately 22,000 genes and a rat gene chip Affymetrix array containing approximately 27,000 genes represented by about 20 probes spread across the full length of the gene. The use of two separate microarray platforms, with differential probe and detection technologies, allowed us to identify, with high confidence, those transcripts that were unchanged by CCI (i.e., those transcripts whose abundance was unchanged in either microarray platform). Transcripts whose abundance was significantly changed by CCI, but less than 1.5-fold, were discarded (not evaluated) because this represents the group of genes with the highest likelihood of both false-positive and false-negative results. Transcripts whose abundance was significantly changed by CCI at 1, 7, and 30 days postinjury with change magnitude 1.5-fold or more, on either array, were selected for further analysis (see Supplementary Table 2 greatest fold-change in either array is listed) (see online supplementary material at http://www.liebertpub.com).
Using IPA analysis, the genes changed in each condition (listed in Supplementary Table 2) (see online supplementary material at http://www.liebertpub.com) were tiled according to their function. Our results show that, by day 1, approximately one third of the up-regulated genes fell into a cluster related to inflammatory response. Among these, two chemokines, Ccl2 and Ccl7, known to be increased in the central nervous system (CNS) in several neurodegenerative diseases16,17 were highly up-regulated (more than 5-fold) at 1 day post-CCI. Chemokines are a family of structurally related cytokines that activate and recruit leukocytes into areas of inflammation.18–20 Both microarrays also show that lipocalin 2 (Lcn2) and tissue inhibitor of metalloproteinase-1 (Timp1) mRNA were significantly up-regulated 24 h after CCI. Both transcripts encode acute-phase proteins up-regulated by various proinflammatory stimuli, including tumor necrosis factor alpha (TNF-α).19,20 Lcn2 is induced by TNF-α in cortical neurons solely by its type 1 receptor and is significantly decreased in cerebrospinal fluid (CSF) of patients with mild cognitive impairment (MCI) and Alzheimer's disease (AD) and increased in regions associated with AD pathology in the human brain.21
By days 7 and 30, the set of transcripts characteristic of acute inflammation response was no longer up-regulated and instead a new set of genes associated with complement system, dendritic cell maturation, T-cell differentiation, and with the major histocompatibilty complex (MHC) II pathway, such as complement components (C3 and Serping1), immune cell-activating factors (C-C chemokine receptor type 5 [Ccr5] and Fc fragment of IgG, low affinity IIb, receptor (CD32) [Fcgr2b]), and MHC II components (RT1 class II, locus Da [Rt1-Da] and Cd74) were up-regulated in CCI animals, compared to sham. Along with prolonged elevation of MHC II–related transcripts, there occurs a stably altered expression of mRNAs encoding several neuron-specific transcripts, such as regulator of axon guidance (Nav3), neuropeptide CART (Cartpt), monoamine transporter VMAT2 (Slc18a2), and GABAergic receptors (Gabrg1 and Gabrb2; see Supplementary Table 2) (see online supplementary material at http://www.liebertpub.com), with implications for the balance of excitatory and inhibitory transmission in hippocampus.
Clustering of the down-regulated genes, on the other hand, seems to be more disparate. It is noteworthy that several transcripts that are down-regulated after 1 day of injury, such as those encoding laminin alpha 2, thrombospondin 2, nescient helix loop helix 1, growth differentiation factor 15, neurotrophin 3 (Ntf3), and synuclein alpha interacting protein, are involved in development and function of the nervous system. At days 7 and 30, transcripts encoding transcription factors, such as POU domain, class 3, transcription factor 1 (Pou3f1) and POU domain, class 4, transcription factor 1 (Pou4f1; day 7) or Nupr1, Kruppel-like factor (Klf)4, Klf2, and Gabpa (day 30), are mainly tiled in a cluster related to cell survival or death.
IPA was used to determine whether the sets of transcripts that were increased at days 1, 7, and 30 after CCI were related within any known signaling networks. At day 1, a group of nuclear factor kappa B (NF-κB)-responsive genes made up the most prominent cohort of regulated transcripts, with several being known targets of signaling by TNF-α acting through the TNFR1 receptor (Fig. 2A; TNF-α-responsive genes indicated with bold arrows). At day 7, the highest-scoring network was also associated with inflammation and revealed prominent contributions to gene regulation by signaling through signal transducer and activator of transcription 1 (Stat1), extracellular signal-regulated kinase ERK, and interferon regulatory factor 8 (Irf8; Fig. 2B). By day 30, no inflammation-associated networks were significantly represented, and the network scoring highest was one for cell-cell interaction (Fig. 2C).
Analysis of messenger RNA levels in hippocampus after CCI
We then confirmed the validity of positive microarray results by measuring independently, with qRT-PCR, the effect of CCI on Ccl2 and Ccl7 mRNA expression. As observed with microarray, our results show that transcriptome changes after CCI were multi-phasic. Within 24 h after CCI, levels of transcripts Ccl2 and Ccl7 were elevated and were no longer up-regulated by days 7 and 30. Significantly, up-regulation of both Ccl2 and Ccl7 occurred on both sides of the injured brain, suggesting a response to global concussive injury, rather than local unilateral physical impact at the site of injury (Fig. 3A–D). To assess the intermediate and later effect of CCI, we expanded quantification to genes that significantly changed at 7 (Cd74, C3, and Rt1-Da) and 30 days (Cd74, C3, and Klf4) and measured the mRNA changes of these transcripts by qRT-PCR (Fig. 4). Our data show that Cd74, C3, and Rt1-Da were significantly stimulated, in a range from 9- to 20-fold, 7 days postinjury (Fig. 4A, B, and C) and remained persistently up-regulated at 30 days post-CCI (Fig. 4A and B), whereas Klf4 mRNA levels were significantly down-regulated (Fig. 4D). Transcripts for Ccr5 and Fcgrb2 were also significantly up-regulated at day 7 after CCI (data not shown).
FIG. 3.

Chemokine transcript up-regulation in hippocampal tissue from the ipsilateral (i.d.) dorsal or contralateral (c.d.) dorsal side of sham-operated (Sham) or cortically injured (CCI) male rats at 1, 7 and 30 days post-injury. Ccl2 (A, i.d. and C, c.d.) and Ccl7 (B, i.d. and D, c.d.) mRNA levels, determined by qRT-PCR, are expressed as fold increase over corresponding control values and represent means±standard error of the mean of 3 samples (animals) for each condition. *p<0.05; **p<0.01; versus the corresponding control (Student's t-test). CCI, controlled cortical impact; mRNA, messenger RNA; Ccl2, chemokine (C-C motif) ligand.
FIG. 4.

Intermediary and longer-term transcriptome changes after CCI. Cd74 (A), C3 (B), RT1-Da (C), and Klf4 (D) mRNA levels, determined by quantitative reverse-transcription polymerase chain reaction, are expressed as fold increase over corresponding control values and represent means±standard error of the mean of 3 samples (animals) for each condition. *p<0.05; **p<0.01; versus the corresponding control (Student's t-test). CCI, controlled cortical impact; mRNA, messenger RNA; RT1-Da, RT1 class II, locus Da; Klf4, Kruppel-like factor 4.
We examined, in particular, the expression of transcipts from the brain-derived neurotrophic factor (BNDF) gene, because BNDF expression has been implicated in recovery from various insults to the hippocampus.22 The BDNF transcript is one that is expressed by multiple promoters in the CNS, and thus it is possible that a specific BDNF transcript from one of these, in a particular cell type in the hippocampus, would be elevated, but that this increase would be lost in the measurement of total BDNF mRNA changes after CCI. Because gene expression can be examined in single-exon detail using the data obtained from the Affymetrix array platform, this analysis was also performed. No single 5′ or 3′ untranslated exons of the BDNF gene were elevated in the hippocampus after CCI.
Discussion
The present study highlights the hippocampal transcriptome changes that can occur after an acute exposure to mTBI. mTBI is the most common form of TBI worldwide and is defined as a biomechanically induced brain injury characterized by the absence of gross anatomic lesions and without causing any neuronal loss.23–25 Our data show that the energy of impact applied in our model of CCI provokes a mild deformation of the rat cortex with the impact lesion fairly localized to the immediate cortical area and without structurally affecting other areas of the brain, such as the hippocampus. Thus, CCI, under these conditions, appears to be an appropriate model for mTBI, as manifested clinically. The results from our study show that mTBI pathophysiology involves alterations in the expression levels of transcripts coding for molecules involved in immune response, intracellular signaling, and cell death and survival. This is consistent with the likely complex signaling events that occur after mTBI, that are likely to involve, at least initially, inflammatory events.
TBI has been reported to cause elevation in mRNA encoding the neurotrophin, BDNF, in the hippocampus,26,27 consistent with a contribution to survival and morphological plasticity of hippocampal neurons by this neurotrophic molecule.28 In our study, no changes in abundance of BDNF mRNA occurred after mTBI, although we cannot rule out very transient alterations immediately after TBI. These results highlight a qualitative difference between the expression profile after mild, compared to moderate or severe,29 TBI. On the other hand, microarray analysis indicated that transcripts for neurotrophin-3 (Ntf3) are decreased within 1 day of CCI, suggesting the possibility that decreased expression of this neurotrophin may be a part of the deleterious sequelae of mTBI.
Neuroinflammation is implicated in the cognitive deficits that accompany neurodegenerative conditions and brain insults (e.g., TBI and sepsis).30–33 This damage can be progressive, evolving from hours to days after the initial trauma. It may be associated with compromise of the blood–brain barrier (BBB), permitting extravasation of circulating neutrophils, monocytes, and lymphocytes into the brain parenchyma. Inflammatory factors released by these infiltrating immune cells as well as resident microglia can increase cell death and secondary tissue damage, but also lead to less-obvious changes in brain function.34,35 The proinflammatory cytokines, interleukin (IL)-1, TNF-α, and IL-6, have been reported to be involved in TBI and are released within minutes after challenge,34–37 triggering inflammatory reactions by up-regulation of adhesion molecules, release of chemotactic chemokines, and activation of inflammatory cells.38 Our microarray analysis did not show any increase in proinflammatory cytokine mRNAs in the hippocampus, perhaps because these genes are very transiently expressed in mTBI. However, transcripts encoding mediators, such as Ccl2, Ccl7, Lcn2, and Timp1, known to be involved in acute inflammation under the control of TNF-α, are substantially increased after CCI. Importantly, increases in the mRNAs encoding these inflammatory factors were elevated bilaterally after CCI. This further confirms that unilateral cortical impact is likely transmitted by a concussive energy transfer, as occurs typically in mTBI, rather than by spreading of inflammatory signaling directly from the site of the impact injury as a result of penetration of the BBB, glial activation, or macrophage infiltration specifically at that location.
Ccl2 and Ccl7 (also called monocyte chemottractant protein (MCP)-1 and MCP-3, for monocyte/macrophage chemoattractant proteins) are reported to be produced by astrocytes, and, in some cases, neurons, within the injured brain.39–41 In chronic degenerative diseases, they may be associated with recruitment of mononuclear cell infiltrates into the brain.17,42,43 However, here, the return to normal levels of the bilateral increase of Ccl2 and Ccl7 mRNA levels within 1 week after injury suggests a short-term inflammatory response with subsequent triggering of longer-term alteration of brain function.
The expression of a discrete set of inflammatory mediators is increased, but only after several days, in CCI/mTBI. These include the neuroinflammatory markers, Cd74, and the MHC II–associated genes, C3, Ccr5, Fcgr2b, and Rt1-Da. This pattern of expression suggests that up-regulation of these genes by earlier chemokine expression may represent a mechanism whereby a transient insult is registered as a permanent, or at least long-term, effect on the hippocampus. It is noteworthy that each of these transcripts is reported to be increased in the aging hippocampus.44 A linkage between the expression of Cd74 and MHC II–associated transcripts, and regulation of transcription factors, such as Klf4, and other neuron-specific transcripts in the hippocampus (this study) on the one hand, and functional alterations in inhibitory neurotransmission in the hippocampus and amygdala, on the other,45 has yet to be established.
This study suggests an underlying mechanism for long-term effects on hippocampal function mediated by endogenous chemokine expression in the hippocampus. Although the initial mediator of this effect has not been identified, the strong up-regulation of the chemokines, Ccl2 and Ccl7, in addition to Lcn2 and Timp1, suggests regulation by NF-κB likely in response to TNF-α in neurons and/or astrocytes, as noted in other studies.21,41,46 This is consistent with the report that injury associated with TBI is ameliorated by administration of an inhibitor of TNF-α biosynthesis, 3,6-dithiothalidomide.47 Further investigation of this potential cytokine/chemokine cascade in propagation of damage caused by concussive injury to the brain is a promising avenue for understanding and treatment of the long-term sequelae of mTBI.
Supplementary Material
Acknowledgments
The authors thank Charisse Winston (NIMH-IRP Technical IRTA) and Dr. Babru Samal (NIMH Bioinformatics Core Specialist), for contributions to early phases of this work, and Dr. Abdel Eklahloun (Core Manager, NIHGRI-NIMH-NINCDS Microarray Core Facility) for invaluable assistance and consultation throughout the duration of these experiments. Work reported here was funded, in part, by the NIMH Intramural Research Program and by a grant from the Department of Defense administered through the Center for Neuroscience and Regenerative Medicine. The contributions of Djida Ait-Ali, who participated in the project as a Henry M. Jackson Foundation Research Fellow, are acknowledged here in lieu of co-authorship for which her written permission could not be obtained.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Coronado V.G., Xu L., Basavaraju S.V., McGuire L.C., Wald M.M., Faul M.D., Guzman B.R., and Hemphill J.D. (2011). Surveillance for traumatic brain injury-related deaths—United States, 1997–2007. MMWR Surveill. Summ. 60, 1–32 [PubMed] [Google Scholar]
- 2.Harvey A.G., and Bryant R.A. (2000). Two-year prospective evaluation of the relationship between acute stress disorder and posttraumatic stress disorder following mild traumatic brain injury. Am. J. Psychiatry 157, 626–628 [DOI] [PubMed] [Google Scholar]
- 3.Hoge C.W., McGurk D., Thomas J.L., Cox A.L., Engel C.C., and Castro C.A. (2008). Mild traumatic brain injury in U.S. Soldiers returning from Iraq. N. Engl. J. Med. 358, 453–463 [DOI] [PubMed] [Google Scholar]
- 4.Chavko M., Watanabe T., Adeeb S., Lankasky J., Ahlers S.T., and McCarron R.M. (2011). Relationship between orientation to a blast and pressure wave propagation inside the rat brain. J. Neurosci. Methods 195, 61–66 [DOI] [PubMed] [Google Scholar]
- 5.Rubovitch V., Ten-Bosch M., Zohar O., Harrison C.R., Tempel-Brami C., Stein E., Hoffer B.J., Balaban C.D., Schreiber S., Chiu W.T., and Pick C.G. (2011). A mouse model of blast-induced mild traumatic brain injury. Exp. Neurol. 232, 280–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Flierl M.A., Stahel P.F., Beauchamp K.M., Morgan S.J., Smith W.R., and Shohami E. (2009). Mouse closed head injury model induced by a weight-drop device. Nat. Protoc. 4, 1328–1337 [DOI] [PubMed] [Google Scholar]
- 7.Dixon C.E., Lyeth B.G., Povlishock J.T., Findling R.L., Hamm R.J., Marmarou A., Young H.F., and Hayes R.L. (1987). A fluid percussion model of experimental brain injury in the rat. J. Neurosurg. 67, 110–119 [DOI] [PubMed] [Google Scholar]
- 8.Dixon C.E., Clifton G.L., Lighthall J.W., Yaghmai A.A., and Hayes R.L. (1991). A controlled cortical impact model of traumatic brain injury in the rat. J. Neurosci. Methods 39, 253–262 [DOI] [PubMed] [Google Scholar]
- 9.Figueiredo T.H., Aroniadou-Anderjaska V., Qashu F., Apland J.P., Pidoplichko V., Stevens D., Ferrara T.M., and Braga M.F. (2011). Neuroprotective efficacy of caramiphen against soman and mechanisms of its action. Br. J. Pharmacol. 164, 1495–1505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Figueiredo T.H., Qashu F., Apland J.P., Aroniadou-Anderjaska V., Souza A.P., and Braga M.F. (2011). The GluK1 (GluR5) Kainate/{alpha}-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor antagonist LY293558 reduces soman-induced seizures and neuropathology. J. Pharmacol. Exp. Ther 336, 303–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Paxinos G., and Watson C. (1986). The Rat Brain in Stereotaxic Coordinates. Vol 2nd ed.. Academic Press: Sydney [Google Scholar]
- 12.Gundersen H.J., Jensen E.B., Kieu K., and Nielsen J. (1999). The efficiency of systematic sampling in stereology—reconsidered. J. Microsc. 193, 199–211 [DOI] [PubMed] [Google Scholar]
- 13.Schmitz C., and Hof P.R. (2000). Recommendations for straightforward and rigorous methods of counting neurons based on a computer simulation approach. J. Chem. Neuroanat. 20, 93–114 [DOI] [PubMed] [Google Scholar]
- 14.Livak K.J., and Schmittgen T.D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- 15.Signoretti S., Vagnozzi R., Tavazzi B., and Lazzarino G. (2010). Biochemical and neurochemical sequelae following mild traumatic brain injury: summary of experimental data and clinical implications. Neurosurg. Focus 29, E1. [DOI] [PubMed] [Google Scholar]
- 16.Conductier G., Blondeau N., Guyon A., Nahon J.L., and Rovere C. (2010). The role of monocyte chemoattractant protein MCP1/CCL2 in neuroinflammatory diseases. J. Neuroimmunol. 224, 93–100 [DOI] [PubMed] [Google Scholar]
- 17.Renner N.A., Ivey N.S., Redmann R.K., Lackner A.A., and MacLean A.G. (2011). MCP-3/CCL7 production by astrocytes: implications for SIV neuroinvasion and AIDS encephalitis. J. Neurovirol. 17, 146–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang N., and Oppenheim J.J. (2005). Crosstalk between chemokines and neuronal receptors bridges immune and nervous systems. J. Leukoc. Biol. 78, 1210–1214 [DOI] [PubMed] [Google Scholar]
- 19.Gardner J., and Ghorpade A. (2003). Tissue inhibitor of metalloproteinase (TIMP)-1: the TIMPed balance of matrix metalloproteinases in the central nervous system. J. Neurosci. Res. 74, 801–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu Q., and Nilsen-Hamilton M. (1995). Identification of a new acute phase protein. J. Biol. Chem. 270, 22565–22570 [DOI] [PubMed] [Google Scholar]
- 21.Naude P.J., Nyakas C., Eiden L.E., Ait-Ali D., van der Heide R., Engelborghs S., Luiten P.G., De Deyn P.P., den Boer J.A., and Eisel U.L. (2012). Lipocalin 2: Novel component of proinflammatory signaling in Alzheimer's disease. FASEB J. 26, 2811–2823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Skoglosa Y., Lewen A., Takei N., Hillered L., and Lindholm D. (1999). Regulation of pituitary adenylate cyclase activating polypeptide and its receptor type 1 after traumatic brain injury: comparison with brain-derived neurotrophic factor and the induction of neuronal cell death. Neuroscience 90, 235–247 [DOI] [PubMed] [Google Scholar]
- 23.Bruns J., Jr., and Hauser W.A. (2003). The epidemiology of traumatic brain injury: a review. Epilepsia 44 Suppl 10, 2–10 [DOI] [PubMed] [Google Scholar]
- 24.van der Naalt J. (2001). Prediction of outcome in mild to moderate head injury: a review. J. Clin. Exp. Neuropsychol. 23, 837–851 [DOI] [PubMed] [Google Scholar]
- 25.Vos P.E., Battistin L., Birbamer G., Gerstenbrand F., Potapov A., Prevec T., Stepan Ch A., Traubner P., Twijnstra A., Vecsei L., and von Wild K. (2002). EFNS guideline on mild traumatic brain injury: report of an EFNS task force. Eur. J. Neurol. 9, 207–219 [DOI] [PubMed] [Google Scholar]
- 26.Hicks R.R., Numan S., Dhillon H.S., Prasad M.R., and Seroogy K.B. (1997). Alterations in BDNF and NT-3 mRNAs in rat hippocampus after experimental brain trauma. Brain Res. Mol. Brain Res. 48, 401–406 [DOI] [PubMed] [Google Scholar]
- 27.Matzilevich D.A., Rall J.M., Moore A.N., Grill R.J., and Dash P.K. (2002). High-density microarray analysis of hippocampal gene expression following experimental brain injury. J. Neurosci. Res. 67, 646–663 [DOI] [PubMed] [Google Scholar]
- 28.Xu B., Gottschalk W., Chow A., Wilson R.I., Schnell E., Zang K., Wang D., Nicoll R.A., Lu B., and Reichardt L.F. (2000). The role of brain-derived neurotrophic factor receptors in the mature hippocampus: modulation of long-term potentiation through a presynaptic mechanism involving TrkB. J. Neurosci. 20, 6888–6897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yu F., Wang Z., Tchantchou F., Chiu C.T., Zhang Y., and Chuang D.M. (2012). Lithium ameliorates neurodegeneration, suppresses neuroinflammation, and improves behavioral performance in a mouse model of traumatic brain injury. J. Neurotrauma 29, 362–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brooks W.M., Stidley C.A., Petropoulos H., Jung R.E., Weers D.C., Friedman S.D., Barlow M.A., Sibbitt W.L., Jr., and Yeo R.A. (2000). Metabolic and cognitive response to human traumatic brain injury: a quantitative proton magnetic resonance study. J. Neurotrauma 17, 629–640 [DOI] [PubMed] [Google Scholar]
- 31.Dash P.K., Orsi S.A., Zhang M., Grill R.J., Pati S., Zhao J., and Moore A.N. (2010). Valproate administered after traumatic brain injury provides neuroprotection and improves cognitive function in rats. PLoS One 5, e11383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Griffin W.S. (2013). Neuroinflammatory cytokine signaling and Alzheimer's disease. N. Engl. J. Med. 368, 770–771 [DOI] [PubMed] [Google Scholar]
- 33.Khandaker G.M., and Jones P.B. (2011). Cognitive and functional impairment after severe sepsis. JAMA 305, 673–674; author reply, 674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lenzlinger P.M., Morganti-Kossmann M.C., Laurer H.L., and McIntosh T.K. (2001). The duality of the inflammatory response to traumatic brain injury. Mol. Neurobiol. 24, 169–181 [DOI] [PubMed] [Google Scholar]
- 35.Ziebell J.M., and Morganti-Kossmann M.C. (2010). Involvement of pro- and anti-inflammatory cytokines and chemokines in the pathophysiology of traumatic brain injury. Neurotherapeutics 7, 22–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morganti-Kossmann M.C., Rancan M., Stahel P.F., and Kossmann T. (2002). Inflammatory response in acute traumatic brain injury: a double-edged sword. Curr. Opin. Crit. Care 8, 101–105 [DOI] [PubMed] [Google Scholar]
- 37.Morganti-Kossmann M.C., Satgunaseelan L., Bye N., and Kossmann T. (2007). Modulation of immune response by head injury. Injury 38, 1392–1400 [DOI] [PubMed] [Google Scholar]
- 38.Cederberg D., and Siesjo P. (2010). What has inflammation to do with traumatic brain injury? Childs Nerv. Syst. 26, 221–226 [DOI] [PubMed] [Google Scholar]
- 39.Hayashi M., Luo Y., Laning J., Strieter R.M., and Dorf M.E. (1995). Production and function of monocyte chemoattractant protein-1 and other beta-chemokines in murine glial cells. J. Neuroimmunol. 60, 143–150 [DOI] [PubMed] [Google Scholar]
- 40.Rhodes J.K., Sharkey J., and Andrews P.J. (2009). The temporal expression, cellular localization, and inhibition of the chemokines MIP-2 and MCP-1 after traumatic brain injury in the rat. J. Neurotrauma 26, 507–525 [DOI] [PubMed] [Google Scholar]
- 41.Thompson W.L., and Van Eldik L.J. (2009). Inflammatory cytokines stimulate the chemokines CCL2/MCP-1 and CCL7/MCP-3 through NFkB and MAPK dependent pathways in rat astrocytes. Brain Res. 1287, 47–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McManus C., Berman J.W., Brett F.M., Staunton H., Farrell M., and Brosnan C.F. (1998). MCP-1, MCP-2 and MCP-3 expression in multiple sclerosis lesions: an immunohistochemical and in situ hybridization study. J. Neuroimmunol 86, 20–29 [DOI] [PubMed] [Google Scholar]
- 43.Wang X., Li X., Yaish-Ohad S., Sarau H.M., Barone F.C., and Feuerstein G.Z. (1999). Molecular cloning and expression of the rat monocyte chemotactic protein-3 gene: a possible role in stroke. Brain Res. Mol. Brain Res. 71, 304–312 [DOI] [PubMed] [Google Scholar]
- 44.VanGuilder H.D., Bixler G.V., Brucklacher R.M., Farley J.A., Yan H., Warrington J.P., Sonntag W.E., and Freeman W.M. (2011). Concurrent hippocampal induction of MHC II pathway components and glial activation with advanced aging is not correlated with cognitive impairment. J. Neuroinflammation 8, 138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Almeida C.P., Pidoplichko V., Marini A.M., Li Z., Eiden L.E., and Braga M.F.M. (2011). Pathophysiological alterations in temporal lobe structures after TBI and their role in the development of epilepsy. Society for Neuroscience, 2011 online. 2011 Neuroscience Meeting Planner, Washington, DC, 6011./EE6017 [Google Scholar]
- 46.Bugno M., Witek B., Bereta J., Bereta M., Edwards D.R., and Kordula T. (1999). Reprogramming of TIMP-1 and TIMP-3 expression profiles in brain microvascular endothelial cells and astrocytes in response to proinflammatory cytokines. FEBS Lett. 448, 9–14 [DOI] [PubMed] [Google Scholar]
- 47.Baratz R., Tweedie D., Rubovitch V., Luo W., Yoon J.S., Hoffer B.J., Greig N.H., and Pick C.G. (2011). Tumor necrosis factor-alpha synthesis inhibitor, 3,6'-dithiothalidomide, reverses behavioral impairments induced by minimal traumatic brain injury in mice. J. Neurochem. 118, 1032–1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.