

Abstract
Significance: Inflammation and immunity can be associated with varying degrees of heme release from hemoproteins, eventually leading to cellular and tissue iron (Fe) overload, oxidative stress, and tissue damage. Presumably, these deleterious effects contribute to the pathogenesis of systemic infections. Recent Advances: Heme release from hemoglobin sensitizes parenchyma cells to undergo programmed cell death in response to proinflammatory cytokines, such as tumor necrosis factor. This cytotoxic effect is driven by a mechanism involving intracellular accumulation of free radicals, which sustain the activation of the c-Jun N-terminal kinase (JNK) signaling transduction pathway. While heme catabolism by heme oxygenase-1 (HO-1) prevents programmed cell death, this cytoprotective effect requires the co-expression of ferritin H (heart/heavy) chain (FTH), which controls the pro-oxidant effect of labile Fe released from the protoporphyrin IX ring of heme. This antioxidant effect of FTH restrains JNK activation, whereas JNK activation inhibits FTH expression, a cross talk that controls metabolic adaptation to cellular Fe overload associated with systemic infections. Critical Issues and Future Directions: Identification and characterization of the mechanisms via which FTH provides metabolic adaptation to tissue Fe overload should provide valuable information to our current understanding of the pathogenesis of systemic infections as well as other immune-mediated inflammatory diseases. Antioxid. Redox Signal. 20, 1754–1769.
Introduction
Iron (Fe) is the metal most commonly used in biologic reduction–oxidation (redox) chemistry (42). Presumably, due to its intrinsic capacity to exchange electrons with a variety of donors/acceptors, Fe became, through evolution, a central component of “core biologic functions,” such as the generation of metabolic energy or sensing and transport of gaseous molecules. However, this same property makes the participation of Fe in the production of free radicals via the Fenton chemistry potentially cytotoxic (50). One of the mechanisms limiting this cytotoxic effect relies on a highly evolutionary conserved multistep enzymatic reaction that incorporates Fe into a protoporphyrin IX carrier ring, giving rise to heme (4, 74). In this manner, the pro-oxidant and cytotoxic effect of Fe can be controlled within the protoporphyrin ring of heme by its incorporation into the “heme pockets” of hemoproteins (4). These play an essential role in a variety of vital cellular functions, based on the capacity of Fe-heme to exchange electrons (4). The heme pockets of hemoproteins are usually composed of aromatic amino acids, including phenylalanine (F), tryptophan (W), or tyrosine (Y) with few or no charged amino acids (108). This confers a relative hydrophobic environment, required for stable heme binding. Five conserved amino acids, that is, histidine (H), methionine (M), cysteine (C), tyrosine (Y), and lysine (K), can act as axial heme ligands (108) with hydrophobic amino acids, such as leucine, (L), isoleucine (I), and valine (V), creating further interactions with the porphyrin structure, whereas positively charged amino acids such as arginine (R) interact with the negatively charged propionate groups (101, 146). The evolutionary success of this strategy is probably best illustrated by the fact that most of the biologic available Fe in mammals exists in the form of heme instead of labile Fe (4). However, this strategy still poses some major biologic challenges in that the eventual release of heme from hemoproteins can be cytotoxic (16, 57, 66, 67, 102, 123, 148). The evolutionary conserved solution that emerged to overcome this problem was to couple heme release to its catabolism, such as afforded by heme oxygenases (HO) (165). These are evolutionary conserved and ubiquitously expressed enzymes that catalyze the degradation of the protoporphyrin ring of heme, producing equimolar amounts of labile Fe, biliverdin, and the gasotransmitter carbon monoxide (CO) (165). However, the salutary effect conferred by this strategy requires yet another regulatory mechanism that controls the redox activity of the labile Fe extracted from the protoporphyrin IX ring of heme. This is accomplished by coupling heme catabolism by HO to Fe extracellular export (17, 54) or to Fe incorporation into ferritin complexes (14, 72). As discussed in the following sections, coupling heme catabolism to Fe storage by ferritin plays an essential role in providing Fe metabolic adaptation in the context of inflammation and immunity, restraining the pathological outcome of systemic infections and possibly other immune-mediated inflammatory diseases.
Fe and Heme Homeostasis
Fe homeostasis is regulated by a series of integrated mechanisms acting at cellular and systemic levels (78). Disruption of Fe homeostasis is often associated with defective erythropoiesis and/or anemia as well as with tissue Fe overload, tissue damage, and disease. As such, mechanisms that control Fe homeostasis are critical to prevent these pathological outcomes (57, 66).
Cellular Fe homeostasis is controlled by several evolutionary conserved genes that act in a concerted manner to regulate intracellular Fe uptake, trafficking, and export (6, 78) (Fig. 1). Mammalian cells acquire Fe bound to transferrin (TF), a plasma homodimeric beta-globulin that binds two ferric Fe molecules with exceedingly high affinity (Kd=10−20 M) (3). Soluble TF-Fe complexes are recognized by transferrin receptor 1 (TFR1) and 2 (TFR2) (81), two transmembrane disulfide-linked glycoproteins encoded by distinct genes sharing 45% homology (90) (Fig. 1). The affinity of TFR2 for diferric TF is 30 times lower to that of TFR1 (90), suggesting that the contribution of TFR2 to intracellular Fe uptake is not as critical compared to TFR1. Cubilin is another TF-Fe receptor expressed in more restricted cellular subsets, including small intestine epithelial cells, renal proximal tubule cells, visceral yolk sac cells, and placenta cytotrophoblasts (Fig. 1) (38).
FIG. 1.

Cellular Fe homeostasis. Fe metabolism is maintained at a cellular level by a series of evolutionary conserved mechanisms regulating intracellular Fe uptake, trafficking, and export. The scheme provides an overlook of these mechanisms without taking into account species and/or cell specificities. Extracellular Fe (yellow circles) exists in plasma mainly bound to transferrin (TF). Fe uptake occurs via a mechanism that involves the recognition of Fe-TF complexes by transferrin receptor 1 (TFR1), transferrin receptor 2 (TFR2), or Cubilin. Upon internalization of Fe-TF complexes by endocytosis, the acidification of the endolysosomal compartment allows for Fe release. Labile Fe is reduced and transported into the cytoplasm by divalent metal transporter 1 (DMT1). In the specific context of Fe absorption by enterocytes, DMT1 is used to absorb Fe from the intestinal lumen. Intracellular labile Fe is directed mainly to the mitochondria, being imported by Fe transporters, which include the mitochondrial Fe importer mitoferrin-2 (Mfrn2). In the mitochondria, Fe is used for heme biosynthesis and Fe-sulfur cluster assembly or stored by mitochondrial ferritin (not depicted in the scheme). When intracellular labile Fe accumulates above a certain threshold level, Fe can be stored and neutralized intracellularly by multimeric ferritin complexes composed of FTH and FTL chains or exported by the Fe transporters ferroportin (FPN). Fe, iron; FTH, ferritin H (heart/heavy) chain; FTL, ferritin liver/light chain. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Fe is extracted from TFR1/TF-Fe or TFR2/TF-Fe complexes through acidification of the endolysosomal compartment, leading to conformational changes in the tertiary structure of TFR1/2. This decreases the affinity for TF-Fe and allows for Fe release from TF (81). Endosomal Fe3+ is reduced into Fe2+ by metalloreductases, including the six-transmembrane epithelial antigen of the prostate family member 3 (STEAP-3) (121) and transported into the cytoplasm by the Fe2+ regulated divalent metal transporter 1 (DMT1) (68). Cytoplasmic Fe2+ can be shuttled to the mitochondria, either bound to chaperone proteins or via interorganelle interactions (139), being internalized by the mitochondria via mitoferrins, of which mitoferrin 2 is ubiquitously expressed (124). In the mitochondria, Fe is used essentially for heme biosynthesis and Fe-sulfur cluster assembly (139) (Fig. 1). Alternatively, Fe can be stored in the mitochondria, by mitochondrial ferritin (105).
When intracellular labile Fe increases above a certain threshold level, its pro-oxidant activity must be controlled to avoid cytotoxicity. This is achieved, in large measure, via a mechanism regulated by iron regulatory proteins (IRPs) 1 and 2 (IRP1/2) (79). Both IRPs act as Fe sensors and bind to short conserved cis-regulatory stem-loop iron responsive elements (IRE) in the untranslated mRNA regions (UTR) of genes, where expression is regulated by IRPs (12, 76, 77). Binding of IRPs to IRE located in the 3′UTR increases mRNA stability and promotes translation (71), whereas translation is repressed upon binding of IRPs to IRE located in the 5′UTR (44, 72, 181).
Ferritin H (heart/heavy) chain (FTH) is a prototypical gene regulated by IRPs, encoding a 21 kDa protein that catalyzes the conversion of Fe2+ into ferric Fe3+ and allowing for intracellular storage of inert Fe3+ (72) (Fig. 1). The FTH gene contains one 5′UTR IRE, recognized under low cellular Fe content by IRP1/2, repressing FTH translation (71). Although TFR1 is also a gene regulated by IRPs, it contains several IRE in its 3′UTR that are recognized under low Fe by IRP1/2, promoting its translation (71). This is not the case for TFR2, where expression is not regulated by Fe (56), consistent with the lack of IRE in TFR2 mRNA (56). Another gene regulated by IRPs is ferroportin (FPN), which encodes a 62 kDa transmembrane Fe transporter that exports Fe2+ from cells (45) (Fig. 1). The FPN gene has a 5′UTR IRE regulated by IRPs that represses its translation under low cellular Fe (79). The regulation of FTH, TFR1, and FPN by Fe, sensed by IRPs, allows for rapid adaptation to changes in intracellular Fe content. Regulated expression of FTH appears to be of particular importance for cellular adaptation to Fe overload in the context of inflammation and immunity, as discussed in the next sections (66, 141, 167, 168, 182).
Ferritins are multimeric complexes (∼450 kDa) made of FTH and FTL (liver/light) chains (72). These form heteropolymeric nanocage-like structures comprising of 24 subunits, which assemble as a hollow shell providing a central 80-Å diameter storage cavity that can incorporate up to 4500 Fe atoms (72), in the form of inorganic ferrihydrite aggregates (94). The proportion of the two FTH and FTL subunits composing ferritin multimeric complexes is regulated in a tissue-specific manner with FTL-rich nanocages being produced mainly by hepatocytes and FTH-rich nanocages by the brain and muscle cells (72). Proportion of FTH and FTL subunits is also regulated in response to inflammation, with proinflammatory cytokines inducing FTH expression and hence enriching its content in ferritin nanocages (141, 168, 182). This creates several isoferritins with different Fe storage capacity as well as other physiological properties (72).
FTH has ferroxidase activity (28, 80), which catalyzes the reaction: 4Fe2++4 H++O2=4Fe3++2H2O, converting reactive Fe2+ into Fe3+ and forming inert ferrihydrite aggregates that do not partake in the production of free radicals via the Fenton chemistry (9, 50). The ferroxidase activity of FTH is provided by a core of evolutionary conserved amino acids, that is, Glu-27, Glu-62, and His-65 (80, 103). These are absent from FTL, which does not have ferroxidase activity (8). FTL, however, is essential to promote Fe nucleation by ferritin nanocages, a process leading to the formation of inorganic ferrihydrite aggregates (107). FTL also confers stability to multimeric ferritin, protecting the nanocage from elevated temperatures and denaturants (145). While FTL can form homopolymers (106), this is not the case for FTH, which promote aggregation and degradation of ferritin nanocage (65).
As mentioned above, FTH and FTL are prototypical Fe-dependent genes, which expression is regulated essentially at post-transcriptional level by the dissociation of IRP1/2 from mRNA 3′UTR IREs (71). In response to high cellular Fe content, IRP1/2 are degraded via the 26S proteasome pathway (71, 86) promoting mRNA stability and hence FTH and FTL translation. Physiological modulators of FTH translation, via the IRP/IRE system, include nitric oxide (NO) as well as reactive oxygen species and hypoxia (77). Expression of FTH is also regulated at the transcriptional level via the activation of the transcription factors nuclear factor kappa B (NF-κB) (100, 127) and NF-E2-related factor 2 (NRF2) (129). More recently, FTH expression was show to be post-transcriptionally regulated by miR-200b, which reduces its expression (151).
Presumably, when the levels of cellular Fe are reduced, redistribution of the Fe sequestered inside the ferritin nanocages occurs through a mechanism regulated at least partially by FTH and FTL proteolytic degradation (10). The mechanisms involved in Fe release from ferritins are not well defined, but are thought to involve cysteine and/or serine proteases acting in acidic or autophagic lysosomes (11, 99, 166). Additional mechanisms involved in ferritin disassembly and Fe release from ferritin include FTH ubiquitin-dependent 26S proteasomal degradation (117, 143), a notion in keeping with our own observation that pharmacological inhibition of the 26s proteasome increases FTH expression in cells exposed in vitro to heme plus tumor necrosis factor (TNF) (Raffaella Gozzelino, unpublished).
More than 80% of bioavailable Fe in mammals is contained within the hydrophobic methane-bridged tetrapyrrole ring of heme, where it is used within the prosthetic groups of hemoproteins (Fig. 2). The most abundant hemoproteins in mammals are hemoglobin (Hb) and myoglobin, expressed in red blood cells (RBCs) and muscle cells, respectively (67) (Fig. 2). Another significant pool of hemoproteins is formed by ubiquitously expressed cytochromes (67) (Fig. 2).
FIG. 2.

Bioavailable Fe. Only an estimated 20%–25% of the bioavailable Fe in mammals exists in the form of nonheme (labile) Fe, either bound to TF, stored by ferritin, as part of a Fe-Sulfur cluster or transiently bound to Fe chaperones and transporters. The remaining 75%–80% of the bioavailable Fe is contained inside the protoporphyn IX ring of heme as prosthetic groups of hemoproteins. The major hemoproteins compartments in mammals are hemoglobin (Hb) in red blood cells (RBCs), myoglobin in muscle cells, and cytochromes in virtual all cell types. Hb contains an estimated 70% of the total pool of heme, myoglobin 5%–10%, and cytochromes 25%–20%, respectively. Fe can transit from the “nonheme” to the “heme” compartment through de novo heme synthesis, a process driven by a sequence of eight enzymatic steps, the last being catalyzed by ferrochelatase, which inserts Fe inside the proptoporphyrin IX ring giving rise to heme. Fe can also transit from the heme to the nonheme pool via the catabolism of heme by HO enzymes or via heme oxidation, which releases Fe from the proptoporphyrin IX ring of heme. HO, heme oxygenases. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Under conditions of oxidative stress, noncovalently bound heme can be released from hemoproteins, as demonstrated for Hb (32, 123). The redox activity of nonhemoprotein-bound heme, referred hereby as “free heme,” is no longer controlled by the heme pockets of hemoproteins, and thus, free heme becomes pro-oxidant and cytotoxic (66, 67, 102, 148). This deleterious effect is restrained by two proteins expressed at high levels in human plasma, namely haptoglobin (36–195 mg/dL) and hemopexin (40–150 mg/dL). Binding of haptoglobin to cell-free Hb (Kd≈10−15) prevents heme release from Hb, with the resulting heterodimeric complex being recognized by the macrophage (Mø) transmembrane receptor CD163 (98). Hemopexin scavenges free heme (Kd<10−12 M in humans), forming a heterodimeric complex recognized by the macrophage transmembrane low-density lipoprotein (LDL) receptor-related protein 1 (LRP1/CD91) (82) (Fig. 3). Cellular Hb uptake can also occur irrespectively of haptoglobin or hemopexin via engulfment of senescent RBC by hemophagocytic Mø in the spleen (88).
FIG. 3.

Cellular heme homeostasis. When released from Hb, nonprotein bound heme, that is, free heme, is captured in plasma by hemopexin (HPX) or by albumin (Alb), forming heme-hemopexin and heme-Alb complexes, respectively. Cell-free Hb can also be captured in plasma by haptoglobin (HP), forming Hb-haptoglobin (HP) complexes, from which heme release is inhibited. Plasma Hb-haptoglobin and heme-hemopexin complexes are recognized by the macrophage transmembrane CD163 and the low-density lipoprotein receptor-related protein 1 (CD91) receptors, respectively, and internalized into endolysosomes. Possibly, extracellular heme-Alb or eventually “free heme” can also be internalized via the heme transporters feline leukemia virus C receptor 2 (FLVCR2), the heme responsive gene-1 (HRG-1), and the heme carrier protein 1 (HCP1), although this remains to be formally established. Intracellular heme transits from endolysosomes to the cytoplasm via a mechanism involving HRG-1, giving rise to intracytoplasmic heme, which can be transported into the mitochondria via a heme transporter that remains to be identified (?). A possible involvement of ATP-binding cassette subfamily B member 6 (ABCB6) in heme transfer into mitochondria has been proposed. Mitochondrial heme can be exported to the cytoplasm via the FLVCR1β isoform. Intracytoplasmic heme can be catabolized by HO, a process assisted by FTH, which neutralizes the Fe extracted from the protoporphyrin ring of heme, storing it into multimeric ferritin complexes. Alternatively, intracytoplasmic heme can be exported from cells by the ATP-binding cassette subfamily G member 2 (ABCG2) and the FLVCR1α. Whether heme is transported to the nucleus is likely to be the case, although this has not been established. For a recent and comprehensive review on heme transport, see reference (189). To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
In a similar manner to TF-Fe, cellular uptake of haptoglobin/Hb and hemopexin/heme complexes converges at the endolysosomal compartment, where heme is extracted (82, 98) (Fig. 3). Subsequent transport of heme into the cytoplasm is mediated by the heme transporter heme responsive gene-1 (HRG-1) (137), as demonstrated in hemophagocytic Mø (184) (Fig. 3). Hrg-1 gene deletion impairs erythroid development, as demonstrated in zebrafish (137) and attenuates heme transport from the phagolysosomal compartment of mouse erythrophagocytic Mø (184), illustrating the nonredundant role of these heme transporter in heme/Fe homeostasis (184).
Presumably, most of the cytoplasmic heme transits to the mitochondria via a mechanism involving specific heme chaperones and mitochondrial transporters (189). Several other genes are known to control the fate of cytoplasmic heme, including HO, which catabolize heme degradation (165). There are two HO isoforms, namely HO-1 (≈32 kDa) and HO-2 (≈36 kDa), encoded by the HMOX1 and HMOX2 genes respectively (25, 113, 171). HO-2 is constitutively expressed in most cells (171), whereas excess intracellular heme induces the expression of the stress-responsive HO-1 isoform, which is constitutively expressed by hemophagocytic Mø as well as by natural regulatory T cells (29, 190).
Intracellular heme is exported from cells via the feline leukemia virus C receptor 1 (FLVCR1) (91) (Fig. 3). There are two FLVCR1 isoforms, namely FLCVR1α (≈60 kDa) and FLVCR1β (≈28 kDa), generated via alternative splicing of the same FLCVR gene (35, 91). FLCVR1α drives heme extracellular transport, whereas FLVCR1β mediates mitochondria heme export (35). At least two other ABC transporters can regulate intracellular heme trafficking, namely ABCG2 (≈72 kDa), which promotes heme extracellular transport (96) and ABCB6 (≈94 kDa), which is involved in intracellular heme import (92, 97) (Fig. 3). The heme carrier protein 1 (HCP1) is another putative heme transporter, probably involved in intracellular heme import (149).
A functional system ensuring heme transport and catabolism is essential to sustain systemic Fe homeostasis, as demonstrated unequivocally for HO-1 deficiency in mice (131, 132) and humans (188). In both cases, the lack of HO-1 expression is associated with impaired Fe recycling, anemia, vascular damage, and depletion of hemophagocytic Mø (95), presumably driven by heme cytotoxicity (59). Moreover, HO-1 deficiency is also associated with increased expression of FLVCR in the kidneys (160), suggesting that mechanisms regulating heme catabolism and transport cross-regulate each other to maintain Fe homeostasis. Flvcr gene deficiency is lethal in mice, due to defective erythropoiesis and severe macrocytic anemia (91). This is probably owed to the lack of mitochondrial FLVCR1 expression, given that specific deletion of the Flvcr1α gene variant is dispensable for erythropoiesis, although necessary to prevent edema and hemorrhage (35). FLVCR1 mutations in humans are associated with the development of posterior column ataxia and retinitis pigmentosa (136), whereas FLVCR2 mutations have been associated to proliferative brain vasculopathy, such as observed in the Fowler syndrome (47). Conversely, mutations in ABCG2 and ABCB6 do not impair erythroid differentiation, suggesting a redundant role for these transporters in erythropoiesis (75, 144, 189).
Systemic Fe homeostasis relies on continuous Fe intake from diet (Fig. 4), that is, average of 1–2 mg of Fe per day in humans (70). Fe is absorbed in the form of Fe3+, which is then reduced by cytochrome B reductase (Dcytb) expressed in gut epithelial cells, that is, enterocytes (114). This produces Fe2+, which is transported into the cytoplasm by DMT1 (68) and exported from enterocytes by FPN (45) (Fig. 1). Expression of FTH in enterocytes is required to sustain this process (176) (Fig. 1). Extracellular Fe2+ is delivered to TF and consequently to TFR1-expressing erythroblasts (81), where it is used in heme biosynthesis and loaded into nascent Hb in the process of erythropoiesis (Fig. 4). Dietary Fe can also be extracted from heme, which involves most probably the catabolism of heme by HO, allowing for Fe extraction and transport.
FIG. 4.

Systemic Fe homeostasis in mammals. Systemic Fe homeostasis is maintained in mammals through a series of mechanisms regulating Fe absorption, its mobilization, and storage into different compartments [adapted from Hentze et al. (78)]. Considering that daily Fe excretion is minimal and absorption is residual compared to the amount required to sustain erythropoiesis and other vital functions, almost all the Fe used in mammals must be recycled within different compartments of the organism. The main pathways of Fe uptake and trafficking involved in the maintenance of Fe homeostasis at a systemic level are illustrated. Normal values for Fe content in different human tissues are also shown [adapted from Hentze et al. (78)]. Notice that 75%–80% of the bioavailable Fe exists in the form of heme as a prosthetic group of Hb. Fe recycling via phagocytosis of senescent RBCs by hemophagocytic macrophages (Mø) in the red pulp of the spleen insures that Fe is extracted from heme and recycled back via TF to provide a steady-state Fe supply for erythropoiesis in the bone marrow. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Recycling of the Fe extracted from the prosthetic heme groups of Hb in senescent RBCs is essential to maintain Fe homeostasis (Fig. 4). This is achieved by hemophagocytic Mø in the red pulp of the spleen, via a mechanism involving the heme transporters HRG-1 (137) and presumably FLVCR2 (189). These deliver heme from the phagocytic endosomes or plasma membranes, respectively, into the cytoplasm allowing its catabolism by HO-1, which extracts the Fe that is then exported by FPN (45). The Fe captured by TF in plasma is incorporated into TFR1-expressing erythroblasts, sustaining erythropoiesis (81) (Fig. 4). Expression of FTH by hemophagocytic Mø is most probably not required to this process, as suggested by the deletion of the mouse Fth allele specifically Mø (data not shown) (66).
Fe concentration in plasma is regulated by hepcidin (62), an acute-phase protein produced mainly by hepatocytes (46, 64). Hepcidin is also produced by innate immune cells, for example, neutrophils and Mø (126). This occurs in response to pathogen recognition by pattern recognition receptors (PRR), for example, toll-like receptor (TLR) 2 or 4, or in response to cytokines, for example interleukin-6 (IL-6), IL-22, and type I interferon or IL-1β (46, 64, 126). Hepcidin expression occurs via a mechanism regulated essentially at transcriptional level by the transcription factor signal transducer and activator of transcription 3 (STAT3) (46) as well as by SMAD proteins (64, 138), adjusting the requirement of Fe for erythropoiesis (122).
Hepcidin binds to and triggers FPN degradation (119), thus suppressing cellular Fe export and dietary Fe uptake, while retaining intracellular labile Fe systemically (119). When the levels of hepcidin in plasma increase above a certain threshold level, such as observed during inflammatory and immune reactions, cellular Fe export by FPN is no longer an available option to reduce cellular Fe overload (46, 61, 183). Presumably, this explains the central role played by FTH in preventing the deleterious effects of cellular Fe overload in the context of inflammation and immunity (66). The role of hepcidin in the control of Fe homeostasis will not be discussed in further detail hereby, and the reader is kindly directed to recent reviews on the subject (46, 55, 63, 64, 128).
Heme Cytotoxicity
Inflammation and immunity are associated with the production of free radicals, aimed at destroying evading pathogens and providing host resistance to infections (116). The evolutionary trade-off of this defense strategy is that free radicals can lead to oxidative stress in host tissues and eventually cause tissue damage and disease (116). Under oxidative stress, noncovalently bound heme can be released from hemoproteins, producing free heme (32, 66, 123) (Fig. 5). This phenomenon is well illustrated for Hb, which can donate its prosthetic heme groups to albumin or hemopexin (32) as well as to LDL (15, 87). Many factors associated with inflammation and immunity can act directly or indirectly to promote varying degrees of RBC lyses and as such the generation of cell-free Hb and the subsequent release of its prosthetic heme groups (Fig. 5). Moreover, other hemoproteins containing noncovalently bound prosthetic heme groups, such as myoglobin, can probably act in a similar manner, although this remains to be established.
FIG. 5.

Heme release from Hb. Pathogens, products associated with the activation of innate and adaptive immunity, changes in plasma pH and osmolarity, microvascular clotting, vasoconstriction, and molecules released in the context of tissue damage can act directly or indirectly to trigger varying levels of RBC lyses and concomitant Hb leakage into plasma. Upon release from RBC, Hb tetramers are dissociated into dimers favoring oxidation of their prosthetic heme groups and promoting heme release. The shear number of RBC (2–3×1013 in humans), their high Hb (3×106 molecules/RBC), and heme (1.2×107 molecules/RBC) content make that lysis of a small fraction of RBC not detectable by standard hematological analyzes can lead to the release of significant amount of heme into plasma. Presumably for this reason, Hb is the main source of free heme involved in the pathogenesis of immune-mediated inflammatory diseases, such as severe sepsis and malaria. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
While not cytotoxic per se, free heme can sensitize nonhematopoietic cells to undergo programmed cell death in response to proinflammatory agonists, such as demonstrated for TNF (148), among others (67, 102) (Fig. 6). This deleterious effect is driven by Fe (66), although it is not clear if Fe must be released from heme to become cytotoxic or whether cytotoxicity is exerted by Fe within the context of the protoporphyrin ring of heme or both (67).
FIG. 6.

Protective effect of FTH against systemic infection. Disruption of RBCs produces cell-free Hb, that upon oxidation releases heme. Non-Hb (free) heme sensitizes hepatocytes to undergo programmed cell death in response to TNF. This cytotoxic effect is mediated via a mechanism involving the production of free radicals (reactive oxygen species; ROS) that sustain JNK activation, leading to caspase activation and ultimately to programmed cell death by apoptosis. Labile Fe released from the protoporphyrin ring of heme catalyzes the production of free radicals via the Fenton chemistry, sustaining JNK activation and leading to programmed cell death. Labile Fe, however, also induces FTH expression, which neutralizes its pro-oxidant effects, suppressing JNK activation and programmed cell death. Expression of FTH can also be induced via NF-κB activation in response to TNF. Expression of FTH is inhibited by JNK activation, promoting the accumulation of labile Fe and the production of ROS, leading to programmed cell death. This pathological process can compromise host survival when confronted with systemic infections, as demonstrated for severe forms of malaria. JNK, c-Jun N-terminal kinase; NF-κB, nuclear factor kappa B; TNF, tumor necrosis factor. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
The mechanism underlying Fe-heme-mediated cytotoxicity involves the sustained activation of the c-Jun N-terminal kinase (JNK) signaling transduction pathway (66). This is in keeping with the notion that JNK activation plays a central role in the mechanism via which cytotoxic agonists associated with the generation of free radicals induce programmed cell death, as illustrated for TNF (111) and UV radiation (170), among others (110). However, under pathophysiological conditions, TNF cytotoxicity is prevented via a mechanism involving the activation of NF-κB, a transcription factor that uncouples inflammation from programmed cell death (20). The cytoprotective action of NF-κB is exerted via the expression of the so-called protective genes (13), which include manganese superoxide dismutase (186), A20 (40, 51, 104), GADD45 (43, 125), and FTH (127), all of which control cellular accumulation of free radicals in response to TNF (89). The antioxidant effect of these NF-κB-dependent genes restrains JNK activation and as such the induction of programmed cell death in response to TNF (30, 164). The mechanism underlying this cytoprotective effect involves the activation of redox-sensitive phosphatases inhibiting JNK activation, via the conversion of their catalytic cysteine residues to sulfenic acid upon exposure to free radicals (89, 127). This explains why restraining the participation of Fe in the Fenton chemistry prevents sustained activation of JNK and programmed cell death in response to TNF (21, 66, 127) (Fig. 6).
The cytoprotective effect exerted by NF-κB-dependent genes and in particular by FTH was so far only observed under nonphysiological conditions, such as when specific components of the NF-κB family of transcription factors and/or when NF-κB-responsive genes are deleted in vitro (127). Our recent findings that the pro-oxidant effect of intracellular heme can bypass this protective pathway, sustaining JNK activation and triggering programmed cell death (66), provides evidence for a pathophysiological relevance for this phenomenon. This cytotoxic effect of free heme is strictly dependent on the accumulation of free radicals (66), most likely produced by the mitochondria. These observations provide a physiological context in support of the notion that tissue Fe overload can drive the oxidative activation of JNK and promote programmed cell death (66), a deleterious effect that plays a central role in the pathogenesis of inflammatory diseases as illustrated for severe malaria (52, 102, 148) as well as for severe sepsis (102) (Fig. 6).
Heme sensitization to TNF-mediated programmed cell death is partially suppressed by pharmacological inhibition of caspase-3 (66, 101, 148), indicating that heme sensitizes cells to undergo a caspase-dependent form of programmed cell death, that is, apoptosis (49). However, heme sensitization to TNF-mediated programmed cell death is also suppressed by pharmacological inhibition of the receptor interacting serine/threonine kinases 1 (RIPK1) as well as by deletion of the Rip3 gene (Raffaella Gozzelino and Ana Ribeiro unpublished), suggesting that heme sensitizes cells to undergo programmed cell death by necrosis, that is, necroptosis (175). This is in keeping with the recent finding that heme can signal via TLR-4 to induce TNF-mediated necroptosis in Mø (59) as well as with the notion that sustained JNK activation shifts TNF-mediated programmed cell death from apoptosis to necrosis (175, 177). The cytotoxic effect of free heme and in particular its ability to trigger necroptosis may be particularly relevant in the context of intracellular pathogens, as illustrated for Mycobacterium tuberculosis (140, 153). We suggest that heme sensitization to TNF-mediated programmed cell death acts in a rather peculiar way, in that it can probably drive, simultaneously, the activation of two antagonistic cytotoxic signal transduction pathways, that is, apoptosis and necroptosis. The mechanisms via which this occurs remain to be elucidated and are currently subjects of our studies.
Cytoprotective Effect of Heme Catabolism by HO-1
Although heme oxidation can release Fe from its protoporphyrin ring (18), this process is strongly catalyzed by HO (165). Of particular relevance to this process is the stress-responsive HO-1 isoform whose expression is regulated essentially at the transcriptional level via several stress-responsive transcription factors (36), including NRF2 (5). Under steady-state conditions, NRF2 is constitutively targeted for proteolytic degradation by the 26s proteasomal pathway, driven by its binding to the Kelch-like ECH-associated protein (Keap1) (84). Keap1 has several redox-sensitive cysteine residues that form disulfide bounds when exposed to free radicals, altering its tertiary structure and disrupting its interaction with NRF2 (85, 93). While this allows for NRF2 nuclear translocation, it is not sufficient per se to promote the transcription of the HMOX1 gene encoding HO-1 (84). The reason for this is that HMOX1 transcription is constitutively repressed by the binding of BRCA-1 associated carboxy C-terminal helicase (BACH-1) to DNA Maf responsive elements (MARE) present in its promoter (163). BACH-1, however, is a heme sensor targeted for 26s proteosomal degradation upon cognate heme binding (5, 83). This releases the MARE in the HMOX1 promoter and allows nuclear NRF2 to drive HMOX1 transcription (5, 83). Although NRF2 appears to play a central role in the transcriptional regulation of the HMOX1 gene, several other stress-responsive transcription factors also contribute to the regulation of HO-1 expression, as reviewed elsewhere (36, 67). More recently, HO-1 expression was also shown to be under the control of several micro-RNAs, including miR-155 and miR122, which promote HO-1 expression by inhibiting BACH-1 (133, 134), whereas miR-217, miR-377, and miR-378 inhibit HMOX1 translation via direct interaction with the 3′UTR in HMOX1 mRNA (19, 154).
HO-1 confers cytoprotection to different forms of programmed cell death (157, 179), including apoptosis and/or necroptosis driven by heme and TNF (67, 102, 148). This cytoprotective effect is driven by heme degradation per se as well as by its end products, including the gasotransmitter CO (27) and the antioxidants biliverdin/bilirubin (147). CO can exert cytoprotective effects via the modulation of cellular signal transduction pathways, including the p38 mitogen activating protein kinase (MAPK) (26, 27, 152, 159). In addition, CO can bind Fe in the heme pockets of hemoproteins, inhibiting heme release and preventing the its cytotoxic effects, as illustrated for Hb, to which CO binds avidly to suppress its oxidation as well as heme release (73, 123). While the conversion of biliverdin into bilirubin, catalyzed by biliverdin reductase, may contribute to the antioxidant and cytoprotective effect of heme catabolism by HO-1 (147), this is probably not sufficient to per se to override the pro-oxidant effect of the labile Fe produced via heme catabolism. It is also possible that the cytoprotective effect of HO-1 is mediated to some extent, in the absence of its catalytic activity, via regulation of gene transcription consequent to C-terminal cleavage and nuclear translocation (39, 109).
We have argued that cytoprotective effects of HO-1 contribute critically to its salutary effects exerted in a variety of immune-mediated inflammatory diseases (156, 158), including the rejection of transplanted organs (155), autoimmune diseases (37), subclinical recurrent abortions (191), or infectious diseases, such as severe malaria (52, 123, 148) and sepsis (102). In the context of systemic infections, induction of HO-1 expression by the infected host provides protection against the cytotoxic effects of free heme released from Hb (52, 67, 123, 148). This cytoprotective effect is essential to support host survival and yet it does not appear to exert a negative impact on the pathogen, a phenomenon referred to as disease tolerance (116).
The relative importance of this interplay, between heme and HO-1, to the pathogenesis of severe forms of malaria is strongly supported by the finding that sickle Hb, a mutation in the beta chain Hb that confers protection against Plasmodium infection in human populations (31), does so via a mechanism that relies on the induction of HO-1 expression, through the activation of the transcription factor NRF2 (Fig. 7) (53, 142). Moreover, the production of the gasotransmitter CO, such as induced by sickle Hb, prevents heme release from Hb, conferring protection against severe forms of malaria (53, 123, 142). This protective effect acts irrespectively of pathogen load, and as such is said to confer disease tolerance to malaria (53, 116) (Fig. 7).
FIG. 7.

Induction of heme catabolism by sickle cell trait confers disease tolerance to malaria. Sickle cell disease is a molecular disease caused by a single-point mutation in the β chain of Hb (β6Glu>Val). When present in the heterozygous form, the Hb β6Glu>Val sickle mutation is not pathogenic, conferring a survival advantage against malaria (sickle cell trait). This protective effect acts via the accumulation of low (noncytotoxic) levels of free heme in plasma that induce the expression of HO-1 via a mechanism involving the activation of the transcription factor NF-E2-related factor 2 (NRF2) (not illustrated). The CO produced via heme catabolism by HO-1 binds to cell-free Hb and prevents the accumulation of free heme following Plasmodium infection, thus suppressing the pathogenesis of severe forms of malaria. This protective effect does not interfere with parasite load revealing that sickle Hb confers disease tolerance to malaria. CO, carbon monoxide. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Fe, Heme, and Infection
Microorganisms, including invading pathogens, are sensed by host PRR, which trigger inflammatory responses aimed at restricting microbial growth and when necessary achieve their clearance, while limiting tissue damage (115). Through the perspective of a microbial organism, a successful infection depends strictly on its capacity to divert components of host metabolism into its own metabolic pathways (88). One of the strategies via which inflammatory responses limit the growth of invading pathogens is through the deployment of mechanisms limiting microbial access to key metabolic components such as Fe, which are essential for microbial growth (33, 183). These host defense mechanisms act in a concerted manner to provide systemic (i) neutralization of circulating Fe, (ii) inhibition of cellular Fe delivery, and (iii) intracellular Fe retention (7).
Systemic neutralization of circulating Fe in response to infection is mediated via a mechanism involving lactoferrin, a potent soluble Fe chelator secreted by activated neutrophils, which limits Fe availability to pathogens while exerting antimicrobial activity (24, 173, 174, 180). Pathogens evolved strategies to counter this host defense mechanism, via the expression of siderophores, that is, low-molecular-weight Fe-binding complexes that can extract Fe from lactoferrin as well as from TF (120). These virulence factors are themselves countered by siderocalin/lipocalin-2, a host acute-phase Fe-binding protein that sequesters siderophores and decreases susceptibility to infection (58). Pathogens also evolved mechanisms to circumvent this host defense strategy, including the production of stealth siderophores, precluding siderocalin binding (1, 135).
Expression of natural resistance-associated Mø protein 1 (Nramp-1) in late phagolysosomes is another host defense strategy that reduces Fe availability to intracellular bacteria (34, 88, 172, 178), conferring resistance to infection (60). Moreover, Nramp-1 modulates immune responses to microbial organisms, regulating cytokine production and recruitment of phagocytic cells to the site of the infection (60).
Pathogens can also acquire Fe from host heme, using HO-1 homologues, including ChuS (161), HemO (192), HugZ (69), and HmuO (185), for Fe extraction. While essential to the establishment of host–microbe interaction, the strategies employed by microbes to acquire Fe from the host and those used by the host to starve microbes from Fe will not be reviewed in further detail hereby as these have been recently reviewed elsewhere (33).
Systemic intracellular Fe retention by the infected host is achieved through the action of hepcidin, via a mechanism involving the phosphorylation, internalization, and subsequent lysosomal degradation of the Fe cellular exporter FPN (46, 63). The trade-off to this host defense strategy is deregulation of host Fe homeostasis with cellular Fe retention interrupting Fe recycling and reducing the levels of circulating Fe available not only to microbes but also to host erythropoiesis (88). In keeping with this notion, high levels of hepcidin in plasma and low levels of FPN are associated with impaired erythropoiesis, anemia, and hypoferrimia (46), as well as with cellular and tissue Fe overload (88). Moreover, systemic inhibition of FPN expression also causes intracellular Fe accumulation leading to oxidative stress and tissue damage (7). It should be noted that deregulated Fe metabolism, such as driven by the sustained production of hepcidin is not specific to infection, but rather to inflammation. As such it can be associated to a variety of immune-mediated inflammatory diseases as well as with cancer progression (162, 169), where it leads to poor prognosis, unfavorable outcome, and metastasis (130).
The deleterious effects of sustained hepcidin expression are exacerbated under inflammatory conditions associated with hemolysis and the generation of cell-free Hb. While cell-free Hb can provide some level of resistance against bacterial pathogens (23), the evolutionary trade-off of this defense mechanism is the release of its prosthetic heme groups (32). This leads to systemic heme loading into host cells, eventually causing oxidative tissue damage and disease (52, 67).
Heme induces the expression of HO-1, which inhibits hepcidin expression via a mechanism mediated by CO (150), presumably restoring FPN expression and hence Fe cellular export. NO can also induce FPN expression (118), contributing to the antimicrobial activity of this gasotransmitter (22, 118), while allowing Fe to recycle as to be used in erythropoiesis. Despite these salvage pathways, high levels of hepcidin still promote the systemic accumulation of intracellular Fe. Moreover, while cytoprotective per se, heme catabolism by HO-1 produces intracellular labile Fe (14, 16, 50, 67, 102, 123, 148), which cannot be readily exported due to inhibition of FPN expression enforced by hepcidin (46). Therefore, under inflammatory conditions, the intracellular pool of labile Fe must be neutralized by the induction of FtH expression (14, 21, 48, 167).
FTH and Infection
Ferritin is an acute-phase protein induced during systemic infections, via a mechanism regulated at both transcriptional and post-transcriptional levels (167) (see Fe and Heme Homeostasis section). FTH expression is regulated at a transcriptional level by NF-κB (100, 127), a transcription factor that plays a central role in the regulation of inflammation and immunity. This argues strongly for the integration of Fe metabolic adaptation as an intrinsic component of inflammation and immunity, presumably contributing to host protection against infection (46, 116). FTH transcription is also regulated by the transcription factor NRF2 (112, 129), suggesting yet another level of integration between cellular adaptation to oxidative stress and Fe metabolic adaptation during infection. The transcription factor hypoxia-inducible factor alpha and heat shock factor 1 also regulate FTH expression in Caenorhabditis elegans (2), arguing for yet a broader level of integration between Fe metabolic adaptation and cellular responses to different forms of stress associated with inflammation and immunity. Whether this level of integration is evolutionary conserved, as to be extrapolated from C. elegans to mammals, remains to be established.
A functional effect of ferritin in the outcome of systemic infections has only recently started to be elucidated. FTH acts via a mechanism involving its ferroxidase activity to provide metabolic adaptation to tissue Fe overload during systemic infections, as demonstrated for malaria (66) and severe sepsis (Sebastian Weiss and Rasmus Larsen, unpublished) in mice. Expression of FTH prevents intracellular Fe accumulation from sustaining JNK activation and hence from sensitizing nonhematopoietic cells to undergo programmed cell death (66). This finding is in line with previous demonstration of a cytoprotective effect of FTH in vitro (14, 41, 127, 187) and in vivo (21). The cytoprotective effect of FTH against heme is apparently at odds with the notion that FTH cannot target Fe inside heme (66). An alternative interpretation, however, is that the cytotoxic effect of heme acts via a mechanism driven by labile Fe released from, which can be targeted by FTH, and not by the Fe contained within its protoporphyrin ring, which cannot be targeted by FTH (67). This is in keeping with the notion that labile Fe fuels the activation of JNK in response to TNF (127), a cytotoxic effect repressed by the ferroxidase activity of FTH (66, 127).
Deletion of the Fth allele in mice promotes tissue Fe overload, oxidative stress and damage in response to systemic infections, despite normal levels of HO-1 expression (67). This suggests that FTH is required to support the salutary effects of HO-1 in the context of systemic infection (148), uncoupling heme catabolism from Fe-driven cytotoxicity (14, 66). Moreover, when HO-1 expression is deleted, induction of FTH expression can be impaired (66), which may explain the high lethality of these mice in response to systemic infections (66, 148). To what extent the protective effect of FTH is required to sustain the salutary effects of HO-1 against other immune-mediated inflammatory diseases remains to be established.
The combination of heme and TNF produced during systemic infections inhibits the expression of FTH via a mechanism that involves JNK activation, as demonstrated in vitro (66, 127) as well as in vivo (66) (Fig. 8). This reduction in FTH expression leads to cellular Fe overload and oxidative stress, ultimately involved in the mechanism via which JNK triggers programmed cell death during systemic infections (66, 127). This observation reveals the existence of a functional link between JNK activation and metabolic adaptation to cellular Fe overload during infection, in which TNF acts via JNK activation to promote tissue Fe overload (66) (Fig. 8). Moreover, inhibition of FTH expression also contributes to explain how JNK activation in response to TNF reinforces the accumulation of free radicals, an effect that can shift programmed cell death from apoptosis to necrosis (177). The molecular mechanism via which JNK activation inhibits FTH expression and deregulates Fe homeostasis remains, however, to be elucidated (Fig. 8).
FIG. 8.
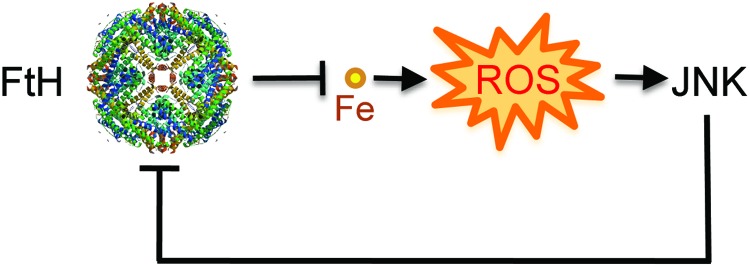
FTH prevents pro-oxidant labile Fe from sustaining JNK activation. FTH controls JNK activation indirectly via a mechanism that prevents labile iron from partaking in the production of free radicals via the Fenton chemistry. This is consistent with previous studies showing that reducing free radical production is sufficient per se to control JNK activity, via inhibition of redox-sensitive phosphatases regulating JNK activity (not illustrated). Presumably, the mechanism underlying the cytotoxic effect of JNK activation involves the inhibition of FTH expression, which promotes cellular Fe overload, accumulation of free radicals, and programmed cell death. This functional cross talk between FTH and JNK controls the host metabolic adaptation to tissue Fe overload during systemic infections. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Conclusion
Host defense strategies limiting Fe availability to pathogens should be considered as an integral component of inflammation and immunity providing host resistance to infection. Moreover, metabolic adaptation to tissue Fe overload, as conferred by the expression of FTH should be taken into account as an integral component of host protection against infection, promoting disease tolerance to infection (66, 116).
Abbreviations Used
- BACH-1
BRCA-1 associated carboxy C-terminal helicase
- CO
carbon monoxide
- Dcytb
cytochrome B reductase
- DMT1
divalent metal transporter 1
- Fe
iron
- FLVCR
feline leukemia virus C receptor
- FPN
ferroportin
- FTH
ferritin H (heart/heavy) chain
- FTL
ferritin liver/light chain
- Hb
hemoglobin
- HO
heme oxygenases
- HRG-1
heme responsive gene-1
- IL
interleukin
- IRE
iron responsive elements
- IRP
iron regulatory protein
- JNK
c-Jun N-terminal kinase
- Keap1
Kelch-like ECH-associated protein
- LRP-1
low-density lipoprotein receptor-related protein 1
- MAPK
mitogen activating protein kinase
- MARE
Maf responsive elements
- Mø
macrophage
- NF-κB
nuclear factor kappa B
- Nramp-1
natural resistance-associated macrophage protein 1
- NRF2
NF-E2-related factor 2
- RBC
red blood cell
- RIPK
receptor interacting serine/threonine kinase
- STEAP
six-transmembrane epithelial antigen of the prostate
- TF
transferrin
- TFR
transferrin receptor
- TLR
toll-like receptor
- TNF
tumor necrosis factor
- UTR
untranslated mRNA regions
Acknowledgments
Authors are indebted to Birte Blankenhaus and Sebastian Weis for critical review of the article as well as all other members of the inflammation laboratory for intellectual input over the years. Authors work is supported by Fundação para a Ciência e Tecnologia (Portugal), SFRH/BPD/44256/2008 to R.G. and PTDC/BIA-BCM/101311/2008, PTDC/SAU-TOX/116627/2010, HMSP-ICT/0022/2010, RECI/IMI-IMU/0038/2012, European Research Council ERC-2011-Advanced Grant no. 294709 DAMAGECONTROL to M.P.S.
References
- 1.Abergel RJ, Wilson MK, Arceneaux JE, Hoette TM, Strong RK, Byers BR, and Raymond KN. Anthrax pathogen evades the mammalian immune system through stealth siderophore production. Proc Natl Acad Sci U S A 103: 18499–18503, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ackerman D. and Gems D. Insulin/IGF-1 and hypoxia signaling act in concert to regulate iron homeostasis in Caenorhabditis elegans. PLoS Genet 8: e1002498, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aisen P, Leibman A, and Zweier J. Stoichiometric and site characteristics of the binding of iron to human transferrin. J Biol Chem 253: 1930–1937, 1978 [PubMed] [Google Scholar]
- 4.Ajioka RS, Phillips JD, and Kushner JP. Biosynthesis of heme in mammals. Biochim Biophys Acta 1763: 723–736, 2006 [DOI] [PubMed] [Google Scholar]
- 5.Alam J, Stewart D, Touchard C, Boinapally S, Choi AM, and Cook JL. NRF2, a Cap'n'Collar transcription factor, regulates induction of the heme oxygenase-1 gene. J Biol Chem 274: 26071–26078, 1999 [DOI] [PubMed] [Google Scholar]
- 6.Andrews NC. Forging a field: the golden age of iron biology. Blood 112: 219–230, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Andrews NC. and Schmidt PJ. Iron homeostasis. Annu Rev Physiol 69: 69–85, 2007 [DOI] [PubMed] [Google Scholar]
- 8.Andrews SC, Arosio P, Bottke W, Briat JF, von Darl M, Harrison PM, Laulhere JP, Levi S, Lobreaux S, and Yewdall SJ. Structure, function, and evolution of ferritins. J Inorg Biochem 47: 161–174, 1992 [DOI] [PubMed] [Google Scholar]
- 9.Arosio P. and Levi S. Ferritin, iron homeostasis, and oxidative damage. Free Radic Biol Med 33: 457–463, 2002 [DOI] [PubMed] [Google Scholar]
- 10.Arosio P. and Levi S. Cytosolic and mitochondrial ferritins in the regulation of cellular iron homeostasis and oxidative damage. Biochim Biophys Acta 1800: 783–792, 2010 [DOI] [PubMed] [Google Scholar]
- 11.Asano T, Komatsu M, Yamaguchi-Iwai Y, Ishikawa F, Mizushima N, and Iwai K. Distinct mechanisms of ferritin delivery to lysosomes in iron-depleted and iron-replete cells. Mol Cell Biol 31: 2040–2052, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aziz N. and Munro HN. Iron regulates ferritin mRNA translation through a segment of its 5′ untranslated region. Proc Natl Acad Sci U S A 84: 8478–8482, 1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bach FH, Hancock WW, and Ferran C. Protective genes expressed in endothelial cells: a regulatory response to injury. Immunol Today 18: 483–486, 1997 [DOI] [PubMed] [Google Scholar]
- 14.Balla G, Jacob HS, Balla J, Rosenberg M, Nath K, Apple F, Eaton JW, and Vercellotti GM. Ferritin: a cytoprotective antioxidant strategem of endothelium. J Biol Chem 267: 18148–18153, 1992 [PubMed] [Google Scholar]
- 15.Balla G, Jacob HS, Eaton JW, Belcher JD, and Vercellotti GM. Hemin: a possible physiological mediator of low density lipoprotein oxidation and endothelial injury. Arterioscler Thromb 11: 1700–1711, 1991 [DOI] [PubMed] [Google Scholar]
- 16.Balla G, Vercellotti GM, Eaton JW, and Jacob HS. Iron loading of endothelial cells augments oxidant damage. J Lab Clin Med 116: 546–554, 1990 [PubMed] [Google Scholar]
- 17.Baranano DE, Wolosker H, Bae BI, Barrow RK, Snyder SH, and Ferris CD. A mammalian iron ATPase induced by iron. J Biol Chem 275: 15166–15173, 2000 [DOI] [PubMed] [Google Scholar]
- 18.Battistuzzi G, Bellei M, Bortolotti CA, and Sola M. Redox properties of heme peroxidases. Arch Biochem Biophys 500: 21–36, 2010 [DOI] [PubMed] [Google Scholar]
- 19.Beckman JD, Chen C, Nguyen J, Thayanithy V, Subramanian S, Steer CJ, and Vercellotti GM. Regulation of heme oxygenase-1 protein expression by miR-377 in combination with miR-217. J Biol Chem 286: 3194–3202, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beg AA. and Baltimore D. An essential role for NF-kappaB in preventing TNF-alpha-induced cell death. Science 274: 782–784, 1996 [DOI] [PubMed] [Google Scholar]
- 21.Berberat PO, Katori M, Kaczmarek E, Anselmo D, Lassman C, Ke B, Shen X, Busuttil RW, Yamashita K, Csizmadia E, Tyagi S, Otterbein LE, Brouard S, Tobiasch E, Bach FH, Kupiec-Weglinski JW, and Soares MP. Heavy chain ferritin acts as an antiapoptotic gene that protects livers from ischemia reperfusion injury. FASEB J 17: 1724–1726, 2003 [DOI] [PubMed] [Google Scholar]
- 22.Bogdan C. Nitric oxide and the immune response. Nat Immunol 2: 907–916, 2001 [DOI] [PubMed] [Google Scholar]
- 23.Bogdan C. Oxidative burst without phagocytes: the role of respiratory proteins. Nat Immunol 8: 1029–1031, 2007 [DOI] [PubMed] [Google Scholar]
- 24.Borregaard N, Sorensen OE, and Theilgaard-Monch K. Neutrophil granules: a library of innate immunity proteins. Trends Immunol 28: 340–345, 2007 [DOI] [PubMed] [Google Scholar]
- 25.Braggins PE, Trakshel GM, Kutty RK, and Maines MD. Characterization of two heme oxygenase isoforms in rat spleen: comparison with the hematin-induced and constitutive isoforms of the liver. Biochem Biophys Res Commun 141: 528–533, 1986 [DOI] [PubMed] [Google Scholar]
- 26.Brouard S, Berberat PO, Tobiasch E, Seldon MP, Bach FH, and Soares MP. Heme oxygenase-1-derived carbon monoxide requires the activation of transcription factor NF-kappa B to protect endothelial cells from tumor necrosis factor-alpha-mediated apoptosis. J Biol Chem 277: 17950–17961, 2002 [DOI] [PubMed] [Google Scholar]
- 27.Brouard S, Otterbein LE, Anrather J, Tobiasch E, Bach FH, Choi AM, and Soares MP. Carbon monoxide generated by heme oxygenase 1 suppresses endothelial cell apoptosis. J Exp Med 192: 1015–1026, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Broxmeyer HE, Cooper S, Levi S, and Arosio P. Mutated recombinant human heavy-chain ferritins and myelosuppression in vitro and in vivo: a link between ferritin ferroxidase activity and biological function. Proc Natl Acad Sci U S A 88: 770–774, 1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brusko TM, Wasserfall CH, Agarwal A, Kapturczak MH, and Atkinson MA. An integral role for heme oxygenase-1 and carbon monoxide in maintaining peripheral tolerance by CD4+CD25+ regulatory T cells. J Immunol 174: 5181–5186, 2005 [DOI] [PubMed] [Google Scholar]
- 30.Bubici C, Papa S, Pham CG, Zazzeroni F, and Franzoso G. The NF-kappaB-mediated control of ROS and JNK signaling. Histol Histopathol 21: 69–80, 2006 [DOI] [PubMed] [Google Scholar]
- 31.Bunn HF. The triumph of good over evil: protection by the sickle gene against malaria. Blood 121: 20–25, 2013 [DOI] [PubMed] [Google Scholar]
- 32.Bunn HF. and Jandl JH. Exchange of heme among hemoglobins and between hemoglobin and albumin. J Biol Chem 243: 465–475, 1968 [PubMed] [Google Scholar]
- 33.Cassat JE. and Skaar EP. Iron in infection and immunity. Cell Host Microbe 13: 509–519, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cellier MF, Courville P, and Campion C. Nramp1 phagocyte intracellular metal withdrawal defense. Microbes Infect 9: 1662–1670, 2007 [DOI] [PubMed] [Google Scholar]
- 35.Chiabrando D, Marro S, Mercurio S, Giorgi C, Petrillo S, Vinchi F, Fiorito V, Fagoonee S, Camporeale A, Turco E, Merlo GR, Silengo L, Altruda F, Pinton P, and Tolosano E. The mitochondrial heme exporter FLVCR1b mediates erythroid differentiation. J Clin Invest 122: 4569–4579, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Choi AM. and Alam J. Heme oxygenase-1: function, regulation, and implication of a novel stress-inducible protein in oxidant-induced lung injury. Am J Respir Cell Mol Biol 15: 9–19, 1996 [DOI] [PubMed] [Google Scholar]
- 37.Chora AA, Fontoura P, Cunha A, Pais TF, Cardoso S, Ho PP, Lee LY, Sobel RA, Steinman L, and Soares MP. Heme oxygenase-1 and carbon monoxide suppress autoimmune neuroinflammation. J Clin Invest 117: 438–447, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Christensen EI. and Birn H. Megalin and cubilin: multifunctional endocytic receptors. Nat Rev Mol Cell Biol 3: 256–266, 2002 [DOI] [PubMed] [Google Scholar]
- 39.Collinson EJ, Wimmer-Kleikamp S, Gerega SK, Yang YH, Parish CR, Dawes IW, and Stocker R. The yeast homolog of heme oxygenase-1 affords cellular antioxidant protection via the transcriptional regulation of known antioxidant genes. J Biol Chem 286: 2205–2214, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cooper JT, Stroka DM, Brostjan C, Palmetshofer A, Bach FH, and Ferran C. A20 blocks endothelial cell activation through a NF-kappaB-dependent mechanism. J Biol Chem 271: 18068–18073, 1996 [DOI] [PubMed] [Google Scholar]
- 41.Cozzi A, Levi S, Corsi B, Santambrogio P, Campanella A, Gerardi G, and Arosio P. Role of iron and ferritin in TNFalpha-induced apoptosis in HeLa cells. FEBS Lett 537: 187–192, 2003 [DOI] [PubMed] [Google Scholar]
- 42.Crichton RR. and Charloteaux-Wauters M. Iron transport and storage. Eur J Biochem 164: 485–506, 1987 [DOI] [PubMed] [Google Scholar]
- 43.De Smaele E, Zazzeroni F, Papa S, Nguyen DU, Jin R, Jones J, Cong R, and Franzoso G. Induction of gadd45beta by NF-kappaB downregulates pro-apoptotic JNK signalling. Nature 414: 308–313, 2001 [DOI] [PubMed] [Google Scholar]
- 44.DeRusso PA, Philpott CC, Iwai K, Mostowski HS, Klausner RD, and Rouault TA. Expression of a constitutive mutant of iron regulatory protein 1 abolishes iron homeostasis in mammalian cells. J Biol Chem 270: 15451–15454, 1995 [DOI] [PubMed] [Google Scholar]
- 45.Donovan A, Brownlie A, Zhou Y, Shepard J, Pratt SJ, Moynihan J, Paw BH, Drejer A, Barut B, Zapata A, Law TC, Brugnara C, Lux SE, Pinkus GS, Pinkus JL, Kingsley PD, Palis J, Fleming MD, Andrews NC, and Zon LI. Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature 403: 776–781, 2000 [DOI] [PubMed] [Google Scholar]
- 46.Drakesmith H. and Prentice AM. Hepcidin and the iron-infection axis. Science 338: 768–772, 2012 [DOI] [PubMed] [Google Scholar]
- 47.Duffy SP, Shing J, Saraon P, Berger LC, Eiden MV, Wilde A, and Tailor CS. The Fowler syndrome-associated protein FLVCR2 is an importer of heme. Mol Cell Biol 30: 5318–5324, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eisenstein RS, Garcia-Mayol D, Pettingell W, and Munro HN. Regulation of ferritin and heme oxygenase synthesis in rat fibroblasts by different forms of iron. Proc Natl Acad Sci U S A 88: 688–692, 1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ellis HM. and Horvitz HR. Genetic control of programmed cell death in the nematode C. elegans. Cell 44: 817–829, 1986 [DOI] [PubMed] [Google Scholar]
- 50.Fenton HJH. Oxidation of tartaric acid in presence of iron. J Chem Soc 65: 899–910, 1894 [Google Scholar]
- 51.Ferran C, Stroka DM, Badrichani AZ, Cooper JT, Wrighton CJ, Soares M, Grey ST, and Bach FH. A20 inhibits NF-kappaB activation in endothelial cells without sensitizing to tumor necrosis factor-mediated apoptosis. Blood 91: 2249–2258, 1998 [PubMed] [Google Scholar]
- 52.Ferreira A, Balla J, Jeney V, Balla G, and Soares MP. A central role for free heme in the pathogenesis of severe malaria: the missing link? J Mol Med 86: 1097–1111, 2008 [DOI] [PubMed] [Google Scholar]
- 53.Ferreira A, Marguti I, Bechmann I, Jeney V, Chora A, Palha NR, Rebelo S, Henri A, Beuzard Y, and Soares MP. Sickle hemoglobin confers tolerance to Plasmodium infection. Cell 145: 398–409, 2011 [DOI] [PubMed] [Google Scholar]
- 54.Ferris CD, Jaffrey SR, Sawa A, Takahashi M, Brady SD, Barrow RK, Tysoe SA, Wolosker H, Baranano DE, Dore S, Poss KD, and Snyder SH. Haem oxygenase-1 prevents cell death by regulating cellular iron. Nat Cell Biol 1: 152–157, 1999 [DOI] [PubMed] [Google Scholar]
- 55.Fleming MD. The regulation of hepcidin and its effects on systemic and cellular iron metabolism. Hematology Am Soc Hematol Educ Program 151–158, 2008 [DOI] [PubMed] [Google Scholar]
- 56.Fleming RE, Migas MC, Holden CC, Waheed A, Britton RS, Tomatsu S, Bacon BR, and Sly WS. Transferrin receptor 2: continued expression in mouse liver in the face of iron overload and in hereditary hemochromatosis. Proc Natl Acad Sci U S A 97: 2214–2219, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fleming RE. and Ponka P. Iron overload in human disease. N Engl J Med 366: 348–359, 2012 [DOI] [PubMed] [Google Scholar]
- 58.Flo TH, Smith KD, Sato S, Rodriguez DJ, Holmes MA, Strong RK, Akira S, and Aderem A. Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature 432: 917–921, 2004 [DOI] [PubMed] [Google Scholar]
- 59.Fortes GB, Alves LS, de Oliveira R, Dutra FF, Rodrigues D, Fernandez PL, Souto-Padron T, De Rosa MJ, Kelliher M, Golenbock D, Chan FK, and Bozza MT. Heme induces programmed necrosis on macrophages through autocrine TNF and ROS production. Blood 119: 2368–2375, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fritsche G, Nairz M, Libby SJ, Fang FC, and Weiss G. Slc11a1 (Nramp1) impairs growth of Salmonella enterica serovar typhimurium in macrophages via stimulation of lipocalin-2 expression. J Leukoc Biol 92: 353–359, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ganz T. Iron in innate immunity: starve the invaders. Curr Opin Immunol 21: 63–67, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ganz T. Hepcidin and iron regulation, 10 years later. Blood 117: 4425–4433, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ganz T. and Nemeth E. Hepcidin and disorders of iron metabolism. Annu Rev Med 62: 347–360, 2011 [DOI] [PubMed] [Google Scholar]
- 64.Ganz T. and Nemeth E. Hepcidin and iron homeostasis. Biochim Biophys Acta 1823: 1434–1443, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Goralska M, Holley BL, and McGahan MC. Identification of a mechanism by which lens epithelial cells limit accumulation of overexpressed ferritin H-chain. J Biol Chem 278: 42920–42926, 2003 [DOI] [PubMed] [Google Scholar]
- 66.Gozzelino R, Andrade BB, Larsen R, Luz NF, Vanoaica L, Seixas E, Coutinho A, Cardoso S, Rebelo S, Poli M, Barral-Netto M, Darshan D, Kuhn LC, and Soares MP. Metabolic adaptation to tissue iron overload confers tolerance to malaria. Cell Host Microbe 12: 693–704, 2012 [DOI] [PubMed] [Google Scholar]
- 67.Gozzelino R, Jeney V, and Soares MP. Mechanisms of cell protection by heme oxygenase-1. Annu Rev Pharmacol Toxicol 50: 323–354, 2010 [DOI] [PubMed] [Google Scholar]
- 68.Gunshin H, Mackenzie B, Berger UV, Gunshin Y, Romero MF, Boron WF, Nussberger S, Gollan JL, and Hediger MA. Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature 388: 482–488, 1997 [DOI] [PubMed] [Google Scholar]
- 69.Guo Y, Guo G, Mao X, Zhang W, Xiao J, Tong W, Liu T, Xiao B, Liu X, Feng Y, and Zou Q. Functional identification of HugZ, a heme oxygenase from Helicobacter pylori. BMC Microbiol 8: 226, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hallberg L. Bioavailable nutrient density: a new concept applied in the interpretation of food iron absorption data. Am J Clin Nutr 34: 2242–2247, 1981 [DOI] [PubMed] [Google Scholar]
- 71.Harford JB. and Klausner RD. Coordinate post-transcriptional regulation of ferritin and transferrin receptor expression: the role of regulated RNA-protein interaction. Enzyme 44: 28–41, 1990 [DOI] [PubMed] [Google Scholar]
- 72.Harrison PM. and Arosio P. The ferritins: molecular properties, iron storage function and cellular regulation. Biochim Biophys Acta 1275: 161–203, 1996 [DOI] [PubMed] [Google Scholar]
- 73.Hebbel RP, Morgan WT, Eaton JW, and Hedlund BE. Accelerated autoxidation and heme loss due to instability of sickle hemoglobin. Proc Natl Acad Sci U S A 85: 237–241, 1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Heinemann IU, Jahn M, and Jahn D. The biochemistry of heme biosynthesis. Arch Biochem Biophys 474: 238–251, 2008 [DOI] [PubMed] [Google Scholar]
- 75.Helias V, Saison C, Ballif BA, Peyrard T, Takahashi J, Takahashi H, Tanaka M, Deybach JC, Puy H, Le Gall M, Sureau C, Pham BN, Le Pennec PY, Tani Y, Cartron JP, and Arnaud L. ABCB6 is dispensable for erythropoiesis and specifies the new blood group system Langereis. Nat Genet 44: 170–173, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hentze MW, Caughman SW, Rouault TA, Barriocanal JG, Dancis A, Harford JB, and Klausner RD. Identification of the iron-responsive element for the translational regulation of human ferritin mRNA. Science 238: 1570–1573, 1987 [DOI] [PubMed] [Google Scholar]
- 77.Hentze MW. and Kuhn LC. Molecular control of vertebrate iron metabolism: mRNA-based regulatory circuits operated by iron, nitric oxide, and oxidative stress. Proc Natl Acad Sci U S A 93: 8175–8182, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hentze MW, Muckenthaler MU, and Andrews NC. Balancing acts: molecular control of mammalian iron metabolism. Cell 117: 285–297, 2004 [DOI] [PubMed] [Google Scholar]
- 79.Hentze MW, Muckenthaler MU, Galy B, and Camaschella C. Two to tango: regulation of Mammalian iron metabolism. Cell 142: 24–38, 2010 [DOI] [PubMed] [Google Scholar]
- 80.Honarmand Ebrahimi K, Bill E, Hagedoorn PL, and Hagen WR. The catalytic center of ferritin regulates iron storage via Fe(II)-Fe(III) displacement. Nat Chem Biol 8: 941–948, 2012 [DOI] [PubMed] [Google Scholar]
- 81.Huebers HA. and Finch CA. The physiology of transferrin and transferrin receptors. Physiol Rev 67: 520–582, 1987 [DOI] [PubMed] [Google Scholar]
- 82.Hvidberg V, Maniecki MB, Jacobsen C, Hojrup P, Moller HJ, and Moestrup SK. Identification of the receptor scavenging hemopexin-heme complexes. Blood 106: 2572–2579, 2005 [DOI] [PubMed] [Google Scholar]
- 83.Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, Hatayama I, Yamamoto M, and Nabeshima Y. An NRF2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun 236: 313–322, 1997 [DOI] [PubMed] [Google Scholar]
- 84.Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, and Yamamoto M. Keap1 represses nuclear activation of antioxidant responsive elements by NRF2 through binding to the amino-terminal Neh2 domain. Genes Dev 13: 76–86, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Itoh K, Wakabayashi N, Katoh Y, Ishii T, O'Connor T, and Yamamoto M. Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of NRF2 in response to electrophiles. Genes Cells 8: 379–391, 2003 [DOI] [PubMed] [Google Scholar]
- 86.Iwai K, Drake SK, Wehr NB, Weissman AM, LaVaute T, Minato N, Klausner RD, Levine RL, and Rouault TA. Iron-dependent oxidation, ubiquitination, and degradation of iron regulatory protein 2: implications for degradation of oxidized proteins. Proc Natl Acad Sci U S A 95: 4924–4928, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jeney V, Balla J, Yachie A, Varga Z, Vercellotti GM, Eaton JW, and Balla G. Pro-oxidant and cytotoxic effects of circulating heme. Blood 100: 879–887, 2002 [DOI] [PubMed] [Google Scholar]
- 88.Johnson EE. and Wessling-Resnick M. Iron metabolism and the innate immune response to infection. Microbes Infect 14: 207–216, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kamata H, Honda S, Maeda S, Chang L, Hirata H, and Karin M. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell 120: 649–661, 2005 [DOI] [PubMed] [Google Scholar]
- 90.Kawabata H, Yang R, Hirama T, Vuong PT, Kawano S, Gombart AF, and Koeffler HP. Molecular cloning of transferrin receptor 2. A new member of the transferrin receptor-like family. J Biol Chem 274: 20826–20832, 1999 [DOI] [PubMed] [Google Scholar]
- 91.Keel SB, Doty RT, Yang Z, Quigley JG, Chen J, Knoblaugh S, Kingsley PD, De Domenico I, Vaughn MB, Kaplan J, Palis J, and Abkowitz JL. A heme export protein is required for red blood cell differentiation and iron homeostasis. Science 319: 825–828, 2008 [DOI] [PubMed] [Google Scholar]
- 92.Kiss K, Brozik A, Kucsma N, Toth A, Gera M, Berry L, Vallentin A, Vial H, Vidal M, and Szakacs G. Shifting the paradigm: the putative mitochondrial protein ABCB6 resides in the lysosomes of cells and in the plasma membrane of erythrocytes. PLoS One 7: e37378, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kobayashi A, Kang MI, Okawa H, Ohtsuji M, Zenke Y, Chiba T, Igarashi K, and Yamamoto M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of NRF2. Mol Cell Biol 24: 7130–7139, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Koorts AM. and Viljoen M. Ferritin and ferritin isoforms I: structure-function relationships, synthesis, degradation and secretion. Arch Physiol Biochem 113: 30–54, 2007 [DOI] [PubMed] [Google Scholar]
- 95.Kovtunovych G, Eckhaus MA, Ghosh MC, Ollivierre-Wilson H, and Rouault TA. Dysfunction of the heme recycling system in heme oxygenase 1-deficient mice: effects on macrophage viability and tissue iron distribution. Blood 116: 6054–6062, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Krishnamurthy P. and Schuetz JD. The ABC transporter Abcg2/Bcrp: role in hypoxia mediated survival. Biometals 18: 349–358, 2005 [DOI] [PubMed] [Google Scholar]
- 97.Krishnamurthy PC, Du G, Fukuda Y, Sun D, Sampath J, Mercer KE, Wang J, Sosa-Pineda B, Murti KG, and Schuetz JD. Identification of a mammalian mitochondrial porphyrin transporter. Nature 443: 586–589, 2006 [DOI] [PubMed] [Google Scholar]
- 98.Kristiansen M, Graversen JH, Jacobsen C, Sonne O, Hoffman HJ, Law SK, and Moestrup SK. Identification of the haemoglobin scavenger receptor. Nature 409: 198–201, 2001 [DOI] [PubMed] [Google Scholar]
- 99.Kurz T, Gustafsson B, and Brunk UT. Cell sensitivity to oxidative stress is influenced by ferritin autophagy. Free Radic Biol Med 50: 1647–1658, 2011 [DOI] [PubMed] [Google Scholar]
- 100.Kwak EL, Larochelle DA, Beaumont C, Torti SV, and Torti FM. Role for NF-kappa B in the regulation of ferritin H by tumor necrosis factor-alpha. J Biol Chem 270: 15285–15293, 1995 [DOI] [PubMed] [Google Scholar]
- 101.Larsen R, Gouveia Z, Soares MP, and Gozzelino R. Heme cytotoxicity and the pathogenesis of immune-mediated inflammatory diseases. Front Pharmacol 3: 77, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Larsen R, Gozzelino R, Jeney V, Tokaji L, Bozza FA, Japiassu AM, Bonaparte D, Cavalcante MM, Chora A, Ferreira A, Marguti I, Cardoso S, Sepulveda N, Smith A, and Soares MP. A central role for free heme in the pathogenesis of severe sepsis. Sci Transl Med 2: 51ra71, 2010 [DOI] [PubMed] [Google Scholar]
- 103.Lawson DM, Treffry A, Artymiuk PJ, Harrison PM, Yewdall SJ, Luzzago A, Cesareni G, Levi S, and Arosio P. Identification of the ferroxidase centre in ferritin. FEBS Lett 254: 207–210, 1989 [DOI] [PubMed] [Google Scholar]
- 104.Lee EG, Boone DL, Chai S, Libby SL, Chien M, Lodolce JP, and Ma A. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science 289: 2350–2354, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Levi S, Corsi B, Bosisio M, Invernizzi R, Volz A, Sanford D, Arosio P, and Drysdale J. A human mitochondrial ferritin encoded by an intronless gene. J Biol Chem 276: 24437–24440, 2001 [DOI] [PubMed] [Google Scholar]
- 106.Levi S, Santambrogio P, Cozzi A, Rovida E, Corsi B, Tamborini E, Spada S, Albertini A, and Arosio P. The role of the L-chain in ferritin iron incorporation. Studies of homo and heteropolymers. J Mol Biol 238: 649–654, 1994 [DOI] [PubMed] [Google Scholar]
- 107.Levi S, Yewdall SJ, Harrison PM, Santambrogio P, Cozzi A, Rovida E, Albertini A, and Arosio P. Evidence of H- and L-chains have co-operative roles in the iron-uptake mechanism of human ferritin. Biochem J 288 (Pt 2): 591–596, 1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Li T, Bonkovsky HL, and Guo JT. Structural analysis of heme proteins: implications for design and prediction. BMC Struct Biol 11: 13, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lin Q, Weis S, Yang G, Weng YH, Helston R, Rish K, Smith A, Bordner J, Polte T, Gaunitz F, and Dennery PA. Heme oxygenase-1 protein localizes to the nucleus and activates transcription factors important in oxidative stress. J Biol Chem 282: 20621–20633, 2007 [DOI] [PubMed] [Google Scholar]
- 110.Liu J. and Lin A. Role of JNK activation in apoptosis: a double-edged sword. Cell Res 15: 36–42, 2005 [DOI] [PubMed] [Google Scholar]
- 111.Liu ZG, Hsu H, Goeddel DV, and Karin M. Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis while NF-kappaB activation prevents cell death. Cell 87: 565–576, 1996 [DOI] [PubMed] [Google Scholar]
- 112.MacKenzie EL, Ray PD, and Tsuji Y. Role and regulation of ferritin H in rotenone-mediated mitochondrial oxidative stress. Free Radic Biol Med 44: 1762–1771, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Maines MD, Trakshel GM, and Kutty RK. Characterization of two constitutive forms of rat liver microsomal heme oxygenase. Only one molecular species of the enzyme is inducible. J Biol Chem 261: 411–419, 1986 [PubMed] [Google Scholar]
- 114.McKie AT, Barrow D, Latunde-Dada GO, Rolfs A, Sager G, Mudaly E, Mudaly M, Richardson C, Barlow D, Bomford A, Peters TJ, Raja KB, Shirali S, Hediger MA, Farzaneh F, and Simpson RJ. An iron-regulated ferric reductase associated with the absorption of dietary iron. Science 291: 1755–1759, 2001 [DOI] [PubMed] [Google Scholar]
- 115.Medzhitov R. Origin and physiological roles of inflammation. Nature 454: 428–435, 2008 [DOI] [PubMed] [Google Scholar]
- 116.Medzhitov R, Schneider DS, and Soares MP. Disease tolerance as a defense strategy. Science 335: 936–941, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mehlhase J, Sandig G, Pantopoulos K, and Grune T. Oxidation-induced ferritin turnover in microglial cells: role of proteasome. Free Radic Biol Med 38: 276–285, 2005 [DOI] [PubMed] [Google Scholar]
- 118.Nairz M, Schleicher U, Schroll A, Sonnweber T, Theurl I, Ludwiczek S, Talasz H, Brandacher G, Moser PL, Muckenthaler MU, Fang FC, Bogdan C, and Weiss G. Nitric oxide-mediated regulation of ferroportin-1 controls macrophage iron homeostasis and immune function in Salmonella infection. J Exp Med 210: 855–873, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, Ganz T, and Kaplan J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 306: 2090–2093, 2004 [DOI] [PubMed] [Google Scholar]
- 120.Noinaj N, Cornelissen CN, and Buchanan SK. Structural insight into the lactoferrin receptors from pathogenic Neisseria. J Struct Biol 184: 83–92, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ohgami RS, Campagna DR, Greer EL, Antiochos B, McDonald A, Chen J, Sharp JJ, Fujiwara Y, Barker JE, and Fleming MD. Identification of a ferrireductase required for efficient transferrin-dependent iron uptake in erythroid cells. Nat Genet 37: 1264–1269, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Pak M, Lopez MA, Gabayan V, Ganz T, and Rivera S. Suppression of hepcidin during anemia requires erythropoietic activity. Blood 108: 3730–3735, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Pamplona A, Ferreira A, Balla J, Jeney V, Balla G, Epiphanio S, Chora A, Rodrigues CD, Gregoire IP, Cunha-Rodrigues M, Portugal S, Soares MP, and Mota MM. Heme oxygenase-1 and carbon monoxide suppress the pathogenesis of experimental cerebral malaria. Nat Med 13: 703–710, 2007 [DOI] [PubMed] [Google Scholar]
- 124.Paradkar PN, Zumbrennen KB, Paw BH, Ward DM, and Kaplan J. Regulation of mitochondrial iron import through differential turnover of mitoferrin 1 and mitoferrin 2. Mol Cell Biol 29: 1007–1016, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Patton GW, Paciga JE, and Shelley SA. NR8383 alveolar macrophage toxic growth arrest by hydrogen peroxide is associated with induction of growth-arrest and DNA damage-inducible genes GADD45 and GADD153. Toxicol Appl Pharmacol 147: 126–134, 1997 [DOI] [PubMed] [Google Scholar]
- 126.Peyssonnaux C, Zinkernagel AS, Datta V, Lauth X, Johnson RS, and Nizet V. TLR4-dependent hepcidin expression by myeloid cells in response to bacterial pathogens. Blood 107: 3727–3732, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Pham CG, Bubici C, Zazzeroni F, Papa S, Jones J, Alvarez K, Jayawardena S, De Smaele E, Cong R, Beaumont C, Torti FM, Torti SV, and Franzoso G. Ferritin heavy chain upregulation by NF-kappaB inhibits TNFalpha-induced apoptosis by suppressing reactive oxygen species. Cell 119: 529–542, 2004 [DOI] [PubMed] [Google Scholar]
- 128.Pietrangelo A. Hepcidin in human iron disorders: therapeutic implications. J Hepatol 54: 173–181, 2011 [DOI] [PubMed] [Google Scholar]
- 129.Pietsch EC, Chan JY, Torti FM, and Torti SV. NRF2 mediates the induction of ferritin H in response to xenobiotics and cancer chemopreventive dithiolethiones. J Biol Chem 278: 2361–2369, 2003 [DOI] [PubMed] [Google Scholar]
- 130.Pinnix ZK, Miller LD, Wang W, D'Agostino R, Jr., Kute T, Willingham MC, Hatcher H, Tesfay L, Sui G, Di X, Torti SV, and Torti FM. Ferroportin and iron regulation in breast cancer progression and prognosis. Sci Transl Med 2: 43ra56, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Poss KD. and Tonegawa S. Heme oxygenase 1 is required for mammalian iron reutilization. Proc Natl Acad Sci U S A 94: 10919–10924, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Poss KD. and Tonegawa S. Reduced stress defense in heme oxygenase 1-deficient cells. Proc Natl Acad Sci U S A 94: 10925–10930, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Pulkkinen KH, Yla-Herttuala S, and Levonen AL. Heme oxygenase 1 is induced by miR-155 via reduced BACH1 translation in endothelial cells. Free Radic Biol Med 51: 2124–2131, 2011 [DOI] [PubMed] [Google Scholar]
- 134.Qiu L, Fan H, Jin W, Zhao B, Wang Y, Ju Y, Chen L, Chen Y, Duan Z, and Meng S. miR-122-induced down-regulation of HO-1 negatively affects miR-122-mediated suppression of HBV. Biochem Biophys Res Commun 398: 771–777, 2010 [DOI] [PubMed] [Google Scholar]
- 135.Raffatellu M, George MD, Akiyama Y, Hornsby MJ, Nuccio SP, Paixao TA, Butler BP, Chu H, Santos RL, Berger T, Mak TW, Tsolis RM, Bevins CL, Solnick JV, Dandekar S, and Baumler AJ. Lipocalin-2 resistance confers an advantage to Salmonella enterica serotype Typhimurium for growth and survival in the inflamed intestine. Cell Host Microbe 5: 476–486, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Rajadhyaksha AM, Elemento O, Puffenberger EG, Schierberl KC, Xiang JZ, Putorti ML, Berciano J, Poulin C, Brais B, Michaelides M, Weleber RG, and Higgins JJ. Mutations in FLVCR1 cause posterior column ataxia and retinitis pigmentosa. Am J Hum Genet 87: 643–654, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Rajagopal A, Rao AU, Amigo J, Tian M, Upadhyay SK, Hall C, Uhm S, Mathew MK, Fleming MD, Paw BH, Krause M, and Hamza I. Haem homeostasis is regulated by the conserved and concerted functions of HRG-1 proteins. Nature 453: 1127–1131, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ramos E, Kautz L, Rodriguez R, Hansen M, Gabayan V, Ginzburg Y, Roth MP, Nemeth E, and Ganz T. Evidence for distinct pathways of hepcidin regulation by acute and chronic iron loading in mice. Hepatology 53: 1333–1341, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Richardson DR, Lane DJ, Becker EM, Huang ML, Whitnall M, Suryo Rahmanto Y, Sheftel AD, and Ponka P. Mitochondrial iron trafficking and the integration of iron metabolism between the mitochondrion and cytosol. Proc Natl Acad Sci U S A 107: 10775–10782, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Roca FJ. and Ramakrishnan L. TNF dually mediates resistance and susceptibility to mycobacteria via mitochondrial reactive oxygen species. Cell 153: 521–534, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Rogers JT, Andriotakis JL, Lacroix L, Durmowicz GP, Kasschau KD, and Bridges KR. Translational enhancement of H-ferritin mRNA by interleukin-1 beta acts through 5′ leader sequences distinct from the iron responsive element. Nucleic Acids Res 22: 2678–2686, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Rosenthal PJ. Lessons from sickle cell disease in the treatment and control of malaria. N Engl J Med 364: 2549–2551, 2011 [DOI] [PubMed] [Google Scholar]
- 143.Rudeck M, Volk T, Sitte N, and Grune T. Ferritin oxidation in vitro: implication of iron release and degradation by the 20S proteasome. IUBMB Life 49: 451–456, 2000 [DOI] [PubMed] [Google Scholar]
- 144.Saison C, Helias V, Ballif BA, Peyrard T, Puy H, Miyazaki T, Perrot S, Vayssier-Taussat M, Waldner M, Le Pennec PY, Cartron JP, and Arnaud L. Null alleles of ABCG2 encoding the breast cancer resistance protein define the new blood group system Junior. Nat Genet 44: 174–177, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Santambrogio P, Levi S, Arosio P, Palagi L, Vecchio G, Lawson DM, Yewdall SJ, Artymiuk PJ, Harrison PM, Jappelli R, et al. Evidence that a salt bridge in the light chain contributes to the physical stability difference between heavy and light human ferritins. J Biol Chem 267: 14077–14083, 1992 [PubMed] [Google Scholar]
- 146.Schneider S, Marles-Wright J, Sharp KH, and Paoli M. Diversity and conservation of interactions for binding heme in b-type heme proteins. Nat Prod Rep 24: 621–630, 2007 [DOI] [PubMed] [Google Scholar]
- 147.Sedlak TW, Saleh M, Higginson DS, Paul BD, Juluri KR, and Snyder SH. Bilirubin and glutathione have complementary antioxidant and cytoprotective roles. Proc Natl Acad Sci U S A 106: 5171–5176, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Seixas E, Gozzelino R, Chora A, Ferreira A, Silva G, Larsen R, Rebelo S, Penido C, Smith NR, Coutinho A, and Soares MP. Heme oxygenase-1 affords protection against noncerebral forms of severe malaria. Proc Natl Acad Sci U S A 106: 15837–15842, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Shayeghi M, Latunde-Dada GO, Oakhill JS, Laftah AH, Takeuchi K, Halliday N, Khan Y, Warley A, McCann FE, Hider RC, Frazer DM, Anderson GJ, Vulpe CD, Simpson RJ, and McKie AT. Identification of an intestinal heme transporter. Cell 122: 789–801, 2005 [DOI] [PubMed] [Google Scholar]
- 150.Shin DY, Chung J, Joe Y, Pae HO, Chang KC, Cho GJ, Ryter SW, and Chung HT. Pretreatment with CO-releasing molecules suppresses hepcidin expression during inflammation and endoplasmic reticulum stress through inhibition of the STAT3 and CREBH pathways. Blood 119: 2523–2532, 2012 [DOI] [PubMed] [Google Scholar]
- 151.Shpyleva SI, Tryndyak VP, Kovalchuk O, Starlard-Davenport A, Chekhun VF, Beland FA, and Pogribny IP. Role of ferritin alterations in human breast cancer cells. Breast Cancer Res Treat 126: 63–71, 2011 [DOI] [PubMed] [Google Scholar]
- 152.Silva G, Cunha A, Gregoire IP, Seldon MP, and Soares MP. The antiapoptotic effect of heme oxygenase-1 in endothelial cells involves the degradation of p38 alpha MAPK isoform. J Immunol 177: 1894–1903, 2006 [DOI] [PubMed] [Google Scholar]
- 153.Silva-Gomes S, Appelberg R, Larsen R, Soares MP, and Gomes MS. Heme catabolism by heme oxygenase-1 confers host resistance to mycobacterium infection. Infect Immun 2013. [Epub ahead of print]; DOI: 10.1128/IAI.00251-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Skrzypek K, Tertil M, Golda S, Ciesla M, Weglarczyk K, Collet G, Guichard A, Kozakowska M, Boczkowski J, Was H, Gil T, Kuzdzal J, Muchova L, Vitek L, Loboda A, Jozkowicz A, Kieda C, and Dulak J. Interplay between heme oxygenase-1 and miR-378 affects non-small cell lung carcinoma growth, vascularization, and metastasis. Antioxid Redox Signal 19: 644–660, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Soares MP. and Bach FH. Heme oxygenase-1 in organ transplantation. Front Biosci 12: 4932–4945, 2007 [DOI] [PubMed] [Google Scholar]
- 156.Soares MP. and Bach FH. Heme oxygenase-1: from biology to therapeutic potential. Trends Mol Med 15: 50–58, 2009 [DOI] [PubMed] [Google Scholar]
- 157.Soares MP, Lin Y, Anrather J, Csizmadia E, Takigami K, Sato K, Grey ST, Colvin RB, Choi AM, Poss KD, and Bach FH. Expression of heme oxygenase-1 can determine cardiac xenograft survival. Nat Med 4: 1073–1077, 1998 [DOI] [PubMed] [Google Scholar]
- 158.Soares MP, Marguti I, Cunha A, and Larsen R. Immunoregulatory effects of HO-1: how does it work? Curr Opin Pharmacol 9: 482–489, 2009 [DOI] [PubMed] [Google Scholar]
- 159.Soares MP, Usheva A, Brouard S, Berberat PO, Gunther L, Tobiasch E, and Bach FH. Modulation of endothelial cell apoptosis by heme oxygenase-1-derived carbon monoxide. Antioxid Redox Signal 4: 321–329, 2002 [DOI] [PubMed] [Google Scholar]
- 160.Starzynski RR, Canonne-Hergaux F, Lenartowicz M, Krzeptowski W, Willemetz A, Stys A, Bierla J, Pietrzak P, Dziaman T, and Lipinski P. Ferroportin expression in haem oxygenase 1-deficient mice. Biochem J 449: 69–78, 2013 [DOI] [PubMed] [Google Scholar]
- 161.Suits MD, Jaffer N, and Jia Z. Structure of the Escherichia coli O157:H7 heme oxygenase ChuS in complex with heme and enzymatic inactivation by mutation of the heme coordinating residue His-193. J Biol Chem 281: 36776–36782, 2006 [DOI] [PubMed] [Google Scholar]
- 162.Sun CC, Vaja V, Babitt JL, and Lin HY. Targeting the hepcidin-ferroportin axis to develop new treatment strategies for anemia of chronic disease and anemia of inflammation. Am J Hematol 87: 392–400, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Sun J, Hoshino H, Takaku K, Nakajima O, Muto A, Suzuki H, Tashiro S, Takahashi S, Shibahara S, Alam J, Taketo MM, Yamamoto M, and Igarashi K. Hemoprotein Bach1 regulates enhancer availability of heme oxygenase-1 gene. EMBO J 21: 5216–5224, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Tang G, Minemoto Y, Dibling B, Purcell NH, Li Z, Karin M, and Lin A. Inhibition of JNK activation through NF-kappaB target genes. Nature 414: 313–317, 2001 [DOI] [PubMed] [Google Scholar]
- 165.Tenhunen R, Marver HS, and Schmid R. The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc Natl Acad Sci U S A 61: 748–755, 1968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Terman A. and Kurz T. Lysosomal iron, iron chelation, and cell death. Antioxid Redox Signal 18: 888–898, 2013 [DOI] [PubMed] [Google Scholar]
- 167.Torti FM. and Torti SV. Regulation of ferritin genes and protein. Blood 99: 3505–3516, 2002 [DOI] [PubMed] [Google Scholar]
- 168.Torti SV, Kwak EL, Miller SC, Miller LL, Ringold GM, Myambo KB, Young AP, and Torti FM. The molecular cloning and characterization of murine ferritin heavy chain, a tumor necrosis factor-inducible gene. J Biol Chem 263: 12638–12644, 1988 [PubMed] [Google Scholar]
- 169.Torti SV. and Torti FM. Ironing out cancer. Cancer Res 71: 1511–1514, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Tournier C, Hess P, Yang DD, Xu J, Turner TK, Nimnual A, Bar-Sagi D, Jones SN, Flavell RA, and Davis RJ. Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science 288: 870–874, 2000 [DOI] [PubMed] [Google Scholar]
- 171.Trakshel GM, Kutty RK, and Maines MD. Purification and characterization of the major constitutive form of testicular heme oxygenase. The noninducible isoform. J Biol Chem 261: 11131–11137, 1986 [PubMed] [Google Scholar]
- 172.Valdez Y, Grassl GA, Guttman JA, Coburn B, Gros P, Vallance BA, and Finlay BB. Nramp1 drives an accelerated inflammatory response during Salmonella-induced colitis in mice. Cell Microbiol 11: 351–362, 2009 [DOI] [PubMed] [Google Scholar]
- 173.Valenti P. and Antonini G. Lactoferrin: an important host defence against microbial and viral attack. Cell Mol Life Sci 62: 2576–2587, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Valenti P, Catizone A, Pantanella F, Frioni A, Natalizi T, Tendini M, and Berlutti F. Lactoferrin decreases inflammatory response by cystic fibrosis bronchial cells invaded with Burkholderia cenocepacia iron-modulated biofilm. Int J Immunopathol Pharmacol 24: 1057–1068, 2011 [DOI] [PubMed] [Google Scholar]
- 175.Vandenabeele P, Galluzzi L, Vanden Berghe T, and Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol 11: 700–714, 2010 [DOI] [PubMed] [Google Scholar]
- 176.Vanoaica L, Darshan D, Richman L, Schumann K, and Kuhn LC. Intestinal ferritin H is required for an accurate control of iron absorption. Cell Metab 12: 273–282, 2010 [DOI] [PubMed] [Google Scholar]
- 177.Ventura JJ, Cogswell P, Flavell RA, Baldwin AS, Jr., and Davis RJ. JNK potentiates TNF-stimulated necrosis by increasing the production of cytotoxic reactive oxygen species. Genes Dev 18: 2905–2915, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Vidal SM, Malo D, Vogan K, Skamene E, and Gros P. Natural resistance to infection with intracellular parasites: isolation of a candidate for Bcg. Cell 73: 469–485, 1993 [DOI] [PubMed] [Google Scholar]
- 179.Vile GF, Basu-Modak S, Waltner C, and Tyrrell RM. Heme oxygenase 1 mediates an adaptive response to oxidative stress in human skin fibroblasts. Proc Natl Acad Sci U S A 91: 2607–2610, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Vogel HJ. Lactoferrin, a bird's eye view. Biochem Cell Biol 90: 233–244, 2012 [DOI] [PubMed] [Google Scholar]
- 181.Walden WE, Patino MM, and Gaffield L. Purification of a specific repressor of ferritin mRNA translation from rabbit liver. J Biol Chem 264: 13765–13769, 1989 [PubMed] [Google Scholar]
- 182.Wei Y, Miller SC, Tsuji Y, Torti SV, and Torti FM. Interleukin 1 induces ferritin heavy chain in human muscle cells. Biochem Biophys Res Commun 169: 289–296, 1990 [DOI] [PubMed] [Google Scholar]
- 183.Weiss G. Iron and immunity: a double-edged sword. Eur J Clin Invest 32Suppl 1: 70–78, 2002 [DOI] [PubMed] [Google Scholar]
- 184.White C, Yuan X, Schmidt PJ, Bresciani E, Samuel TK, Campagna D, Hall C, Bishop K, Calicchio ML, Lapierre A, Ward DM, Liu P, Fleming MD, and Hamza I. HRG1 is essential for heme transport from the phagolysosome of macrophages during erythrophagocytosis. Cell Metab 17: 261–270, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Wilks A. and Schmitt MP. Expression and characterization of a heme oxygenase (Hmu O) from Corynebacterium diphtheriae. Iron acquisition requires oxidative cleavage of the heme macrocycle. J Biol Chem 273: 837–841, 1998 [DOI] [PubMed] [Google Scholar]
- 186.Wong GH. and Goeddel DV. Induction of manganous superoxide dismutase by tumor necrosis factor: possible protective mechanism. Science 242: 941–944, 1988 [DOI] [PubMed] [Google Scholar]
- 187.Xie C, Zhang N, Zhou H, Li J, Li Q, Zarubin T, Lin SC, and Han J. Distinct roles of basal steady-state and induced H-ferritin in tumor necrosis factor-induced death in L929 cells. Mol Cell Biol 25: 6673–6681, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Yachie A, Niida Y, Wada T, Igarashi N, Kaneda H, Toma T, Ohta K, Kasahara Y, and Koizumi S. Oxidative stress causes enhanced endothelial cell injury in human heme oxygenase-1 deficiency. J Clin Invest 103: 129–135, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Yuan X, Fleming MD, and Hamza I. Heme transport and erythropoiesis. Curr Opin Chem Biol 17: 204–211, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Zelenay S, Chora A, Soares MP, and Demengeot J. Heme oxygenase-1 is not required for mouse regulatory T cell development and function. Int Immunol 19: 11–18, 2007 [DOI] [PubMed] [Google Scholar]
- 191.Zenclussen ML, Casalis PA, El-Mousleh T, Rebelo S, Langwisch S, Linzke N, Volk HD, Fest S, Soares MP, and Zenclussen AC. Haem oxygenase-1 dictates intrauterine fetal survival in mice via carbon monoxide. J Pathol 225: 293–304, 2011 [DOI] [PubMed] [Google Scholar]
- 192.Zhu W, Hunt DJ, Richardson AR, and Stojiljkovic I. Use of heme compounds as iron sources by pathogenic neisseriae requires the product of the hemO gene. J Bacteriol 182: 439–447, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]