

Abstract
Hepatitis C virus (HCV) is one of the main causes of liver disease worldwide, and alterations of glucose metabolism have reached pandemic proportions in western countries. However, the frequent coexistence between these two conditions is more than simply coincidental, since HCV can induce insulin resistance through several mechanisms. Indeed, the virus interferes with insulin signaling both directly and indirectly, inducing the production of pro-inflammatory cytokines. Furthermore, the entire viral life cycle has strict interconnections with lipid metabolism, and HCV is responsible for a “viral” steatosis which is frequently superimposed to a “metabolic” one. Several evidences suggest that HCV-induced metabolic disorders contribute both to the evolution of liver fibrosis and, likely, to the progression of the other disorders which are typically associated with altered metabolism, in particular atherosclerosis. In the present review, we will examine in depth the links between HCV infection and insulin resistance, liver steatosis and diabetes, and analyze the impact of these interactions on the progression of liver fibrosis and atherosclerosis. Special attention will be focused on the highly debated topic of the relationship between HCV infection and cardiovascular disease. The available clinical literature on this item will be broadly reviewed and all the mechanisms possibly implied will be discussed.
Keywords: Hepatitis C virus, Metabolism, Insulin resistance, Diabetes mellitus, Steatosis, Fibrosis, Atherosclerosis, Cardiovascular risk
Core tip: In this review we will analyze the mechanisms possibly contributing to the relationship between hepatitis C virus (HCV) infection and altered metabolism, as well as the clinical data suggesting that HCV-induced metabolic disorders favour both the progression of liver damage in terms of steatosis/fibrosis and the development of atherosclerosis. Particular attention will be devoted to the highly debated topic concerning the link between HCV infection and cardiovascular disease, a time-related interpretation on the factors impacting cardiovascular risk in the course of HCV infection will be provided, and, finally, the complex virus/host interplay will be graphically synthesized to provide an intuitive picture of the item.
METABOLIC EFFECTS OF HEPATITIS C VIRUS INFECTION
Hepatitis C virus (HCV) is one of the main causes of liver disease worldwide[1], with more than 150 million of persons chronically infected, at risk of developing liver cirrhosis and cancer. Moreover, HCV infection is associated with glucose and lipid metabolism disturbances. Alterations of glucose metabolism, i.e., impaired fasting glucose, impaired glucose tolerance and diabetes mellitus (DM), have reached pandemic proportions in western countries[2]. Their keystone is insulin resistance (IR), they are closely linked to obesity and increase the risk of cardiovascular events. Given the prevalence of HCV infection and of these glucose metabolism disturbances, their frequent relationship is not unexpected: however, physiopathologically, it is not only coincidental. In fact, the virus causes IR and predisposes to DM. Several studies analyzed the frequency of DM in HCV-infected patients and confirmed this association. IR and DM are more prevalent in the course of HCV infection than in other liver diseases, independently from the stage of fibrosis[3,4], and HCV infection increases the incidence of DM after liver transplantation[5]; on the other side, the prevalence of HCV infection among diabetic patients is higher than in the age-matched general population[6].
The coexistence of these metabolic derangements affects the progression and prognosis of liver disease and, at the same time, contributes to the systemic burden of HCV infection. Indeed the virus, through interactions with glucose and lipid homeostasis, via IR and steatosis, adds other mechanisms of liver damage and participates in the pathogenesis of non-liver-related disorders, such as cardiovascular disease[7].
HCV and insulin resistance
Several epidemiological, experimental and clinical studies showed that chronic hepatitis C (CHC), from the early stages of infection, is associated with alterations of glucose metabolism. Indeed, both in retrospective and in longitudinal studies, the risk of developing IR or DM of HCV-infected patients, even when corrected for confounding factors, is approximately two-fold[8]. The most commonly used surrogate measure of insulin-resistance, i.e., the homeostatic model assessment (HOMA) index, is elevated also in early stages of disease[9], and it is higher than in patients with chronic HBV-infection matched for age, body mass index (BMI) and fibrosis[8]. Indeed, although an association between HBV infection and glucose metabolism disorders has been suggested, possibly secondary to HBV-induced pancreatic islet injury, epidemiological data are still highly controversial[10].
In addition, people with HCV infection are predisposed to develop DM approximately one decade earlier that those without the infection[3]. Many studies demonstrated that eradication of HCV infection with antiviral therapy is associated with a decrease of HOMA-index and of the incidence of glucose metabolism alterations[11-13], although these data have not been universally confirmed[14]. Moreover, the relationship between HCV and IR seems to be dependent on viral load[8] and more pronounced in genotypes (G) 1, 2 and 4[15].
The target tissues of HCV-related metabolic disturbances are the liver, the primary site of infection, and the skeletal muscle. It is very interesting to note that the glucidic function of adipose tissue is not affected, unlike what is commonly described in the course of “pure” IR conditions[16]. Indeed, during euglycemic hyperinsulinemic clamp, patients with CHC without fibrosis and metabolic syndrome show an endogenous glucose output more than three times the normal and an abnormal muscle uptake of glucose, with a normal suppression of lipolysis from adipose tissue[17]. The presence of hepatic IR results in increased fasting glucose, while peripheral IR determines a reduced uptake of glucose[17,18], with the impairment of glucose oxidation. In a mouse model transgenic for HCV core protein, Shintani et al[19] showed that during a clamp with tracers infusion, the main site of resistance was the liver, as demonstrated by the capability to stimulate the muscle uptake of glucose but the failure to inhibit the endogenous glucose output. On the contrary, in humans, Milner et al[18] demonstrated that IR is principally peripheral, as evidenced by the decreased glucose disposal in the absence of endogenous glucose production (high dose clamp), without differences compared with healthy patients in glucose output at low dose insulin. In addition, Vanni et al[17] confirmed the predominant role of muscle in the development of IR, with an approximate 80% of peripheral contribution, demonstrating the higher glucose disposal during the clamp in controls compared to HCV patients. Finally, after liver transplantation, HCV-diabetic patients show an improvement in glucose tolerance but a persistent insulin resistance in peripheral tissues, particularly in the skeletal muscle[20,21]. All together, regardless of the prevalent site of IR, whose analysis is likely influenced by the technique used (duration of clamp and dose of insulin) and the population selected, it is evident that during CHC it develops an exclusive insulin resistant state which is different, but often superimposed, to the host metabolic derangements and that the two conditions influence and enhance each other.
Molecular pathways of insulin resistance in HCV infection
Many different mechanisms are associated with the development of IR during chronic liver disease and, in particular, in HCV-infection[10]. The virus directly interacts at different points of the insulin signalling cascade. In liver tissue from HCV-infected patients, Aytug et al[22] firstly reported an inhibition of the ability of insulin receptor substrate (IRS)-1 to associate with insulin receptor, a critical point in the regulation of hepatic gluconeogenesis, mediated by a reduced tyrosine phosphorylation of IRS-1 and a consequent defective downstream phosphorylation of phosphatidylinositol 3-kinase (PI3K) and protein kinase Akt. In addition, the virus may interfere with this pattern also through the up-regulation of the protein phosphatase 2A (PP2A), which dephosphorylates and inhibits Akt[23], although other studies failed to demonstrate a correlation between intrahepatic levels of PP2A and HOMA-IR. It is also interesting to note that, in vitro, HCV leads to over-expression of PP2A by inducing endoplasmic reticulum stress[24]. In experimental models based on the expression of HCV core protein, Kawaguchi et al[25] described the involvement of suppressor of cytokine signalling-3 (SOCS-3), which promotes the ubiquitin-mediated IRS-1 degradation; similarly, HCV may activate the proteasome activator 28 gamma (PA28γ) and, in the transgenic mouse, targeted delection of PA28γ gene restores insulin sensitivity[26]. The HCV core protein inhibits also the peroxisome proliferator activated receptors (PPARs). In particular, an inhibition of PPAR-α expressed in hepatocytes has been shown[27], while the effect of the virus on PPAR-γ has been observed only in G3 infection, inducing an alteration of adiponectin levels[28,29].
On the other side, HCV may also indirectly trigger IR inducing the production of pro-inflammatory cytokines, which contribute to the metabolic derangements not only in infected tissues but also in uninfected ones, such as the striated muscle. IL-18 and tumor necrosis factor (TNF) are some of the main molecules involved[17,18]. The capability of these proinflammatory cytokines to disturb insulin signalling is well recognized in the context of DM and metabolic syndrome but it is also described during chronic viral disease, irrespective of aetiology[30]. CHC is in fact associated with the up-regulation of T helper 1 lymphocyte cytokines[19,31] and, in HCV-infected patients, a relationship between the increased serum levels of soluble TNF receptors and HOMA-IR has been described[32]. In a transgenic mice model expressing the HCV core protein, IR was reverted by anti TNF-α antibodies[19]. In contrast, in a controlled study comparing non diabetic patients with HCV infection to matched uninfected controls, although serum levels of TNF-α were significantly higher in the HCV cohort, correlating with the extent of histological injury, they were not associated with IR in the multivariate model[33]. The association between IL-18 and hepatic IR seems more specific[17]. Indeed, IL-18 suppresses adiponectin expression in adipocytes and stimulates SOCS3 expression in the adipose tissue of obese mice, providing an indirect mechanism of IR.
A key role in the development of liver injury and metabolic disturbances is played by both hepatic and systemic oxidative stress. In addition to chronic inflammation itself, the virus specifically induces reactive oxygen species (ROS) via multiple mechanisms involving the core and other non-structural proteins. The result is the loss of equilibrium between oxidants and antioxidant defenses, which causes oxidative damage to liver cells and interference with the mechanisms of DNA repair, rendering hepatocytes more susceptible to mutagen-induced alterations[34] and favouring fibrogenesis through increased proliferation of hepatic stellate cells[35]. The production of ROS may also be involved in the peroxidation of membrane lipids and structural proteins, such as those involved in lipid trafficking, therefore blocking very low density lipoprotein (VLDL) secretion and leading to steatosis[36].
HCV and steatosis
Steatosis is a typical feature of CHC, with a reported prevalence of 40%-80%[37]. It is so frequent that, in the past, it has been used as a diagnostic tool for the diagnosis of non-A non-B chronic hepatitis[38,39]. Among viral hepatitides, the association between HCV and steatosis seems somehow specific, since, for example, steatosis in HBV infection is as frequent as in the general population and related to metabolic factors[40]. On the contrary, during CHC, steatosis prevalence remains so high also when adjusted for metabolic risk factors (30%-40%)[41]. In fact, although non-alcoholic fatty liver disease (NAFLD) and CHC are both highly prevalent, epidemiological data confirmed that the rate of steatosis in CHC is greater than twice that expected on the basis of a simple random coexistence[42]. A direct viral effect on steatogenesis is relevant, more frequent and severe in G3[43], where a strong association is further supported by two observations: the correlation between steatosis grade and intrahepatic RNA titers and viral core protein expression[44,45]; the reduced or disappeared content of fat in the liver after a successful antiviral treatment[46,47].
The exact mechanisms at the base of HCV-induced steatosis are not definitely explained. HCV core protein is able to increase free fatty acids synthesis[48], favours the intracytoplasmic accumulation of lipids and reduces their mechanisms of export and degradation[48,49]. The entire HCV life cycle is in strict contact with lipid metabolism. HCV entry may be mediated by the low density lipoprotein (LDL) receptor[50]; HCV core protein modifies VLDL secretion[51]; the virus diverts the host lipoprotein assembly and secretion pathways for virion export[52]; virions circulate complexed with lipoproteins in low density lipo-viro particles that facilitate reuptake by hepatocytes by fastening to the LDL receptor[53]. In addition, it has been recently demonstrated that HCV-induced overexpression of seipin, a protein implicated in maturation of lipid droplets whose surface is the seat of the virus start of assembly[54], decreases virion export and induces steatosis, possibly representing a defensive mechanism against viral export. If confirmed, this evidence will lead to consider “viral steatosis” a defensive mechanism. The accumulation of fatty acids in the form of triglycerides may in fact represent a mechanism through which render them not available for replication complexes involving HCV. This hypothesis is supported by the evidence that, when the degree of steatosis correlates with virus replication level, viral replication precedes fat accumulation and not viceversa[46,55,56]. On the contrary, in metabolic patients, whose steatosis precedes viral infection, the level of viral replication is not associated with the severity of fatty liver.
Going back to mechanisms specifically involved in triglyceride accumulation, impaired secretion of lipids from the infected hepatocytes has been the first historically considered. In fact, patients with CHC have low levels of total cholesterol and triglycerides[57] and phenotypic similarities with familiar hypobetalipoprotinemia[58]. HCV induced hypobetalipoproteinemia is more commonly seen with G3 infection than with G1[59]. Moreover, in patients with G3 infection, but not in those with G1, sustained virological response (SVR) significantly reduced steatosis[46], and the disappearance of steatosis in patients who responded to therapy was parallel to the normalization of cholesterol and apolipoprotein B levels[59,60]. Experimental models in transgenic mouse showed that HCV core protein interfere with VLDL assembly by reducing the activity of microsomal triglyceride transfer protein (MTTP)[61,62], which is a rate-limiting enzyme in lipoprotein metabolism. These data are confirmed by the reduced intrahepatic levels of MTTP mRNA observed in human liver of patients with CHC, especially in those with G3[61]. Another contribute to the blockage of lipoprotein secretion may be offered by oxidative stress. In fact, the HCV core protein may accumulate in mitochondria and induce liver damage through reactive oxygen species production[63], lipid peroxidation of microsomal membranes and impaired VLDL secretion.
HCV induces steatosis also via ex-novo synthesis of fatty acids. The virus activates the steroid responsive element binding proteins (SREBP 1c e 2) that control expression of enzymes involved in the fatty acid and cholesterol metabolism[64], inducing de novo lipogenesis. The virus can cause steatosis also by impairing metabolism and degradation of fatty acids. Indeed, HCV has been shown to inhibit transcription of the nuclear factor PPAR-α and this inhibition would reduce transcription of enzymes involved in fatty acid oxidation, such as the carnitine palmitoyltransferase-1 (CPT-1), which is the rate-limiting enzyme of mitochondrial beta oxidation[65,66]. Finally, a great attention has been pointed on the cytokines secreted by adipose tissue. For example, serum adiponectin levels are low in patients with CHC, with the lowest value observed in G3 infection[67], and HCV can induce the overexpression of retinol binding protein (RBP)-4 which is a steatogenic adipokine associated with the development of steatosis not related to insulin resistance[68-70].
All these evidences are very important because they highlight different possible meanings of the word “steatosis” in a patient with CHC. The virus can induce two types of steatosis, i.e., metabolic and viral, with different pathogenetic mechanisms, often overlapped. In addition, virus-induced steatosis may exist together with a fatty liver due to other causes. The degree of steatosis does not always have a direct correlation with the degree of IR. It has been shown that patients with G1 and G4 infection have a level of IR, measured by the HOMA-index, greater than that of patients with G3 infection but with a lower degree of steatosis (greater in G3)[8,9,19,57]. In most patients with non-G3 infection the steatosis score is not correlated with HCV-RNA but with BMI[37], and the steatosis is not or it is only partially modified by antiviral treatment. Therefore, in patients with non-G3 infection, steatosis is regarded as more “metabolic” and less “viral”, while in G3 ones, as more “viral” and less “metabolic”. At the same time, it is not possible to assign exclusively a type of steatosis to a specific or to a group of genotypes. Indeed, it is clear that also genotypes non-3 may induce some degree of viral steatosis and, at the same time, also G3 may induce metabolic abnormalities. Many mechanisms, such as oxidative stress induced by core protein, may simultaneously induce steatosis (“viral”) and impair insulin signalling (“metabolic”). In conclusion, the two types of steatosis can be observed in all genotypes but steatosis phenotype, modulated by metabolic abnormalities (primary metabolic dysfunctions and host factors) and by all microheterogeneities in viral genomic regions, will be more “viral” in G3 and more “metabolic” in others.
Insulin resistance and steatosis: synergism with the virus in the progression of liver disease
The clinical relevance of IR and steatosis in CHC resides in the role played by insulin and fat accumulation in the progression of fibrosis, response to antiviral therapy and occurrence of hepatocarcinoma (HCC). While the annual risk of developing HCC in HCV-related cirrhosis has been estimated to be 3% (2%-6%) per year[71], a recent metanalysis calculated that it is increased 17% by overweight and 90%, almost doubled, by obesity[72]. Moreover, this risk is increased 3-fold by the presence of DM, 37-fold by the co-existence of HCV and DM and up to 100-fold by the association between HCV, DM and obesity[42,72]. Despite most studies described an association between steatosis and the degree of fibrosis, at present, data are not univocal. Most of these studies have a low statistical power and often lack multivariate analysis. Moreover, this association may not be causal as both conditions may simply represent the marker and the consequence of the inflammatory activity[73-75]. In this sense, in particular metabolic steatosis would also be a marker of IR, responsible for both steatosis and increasing fibrosis. In fact, by multivariate analysis, it was IR and not steatosis that correlated with fibrosis, also in G3[76,77]. IR represents a link between steatosis and fibrosis through the capability of insulin, glucose, and leptin, whose receptors are expressed on stellate cells, to induce the production of connective tissue growth factor[78]. Although the exact pathogenetic mechanisms are not clearly understood, available data suggest a role also for oxidative stress, lipid peroxidation and the higher levels of proinflammatory cytokines, which are able to activate stellate cells[79,80].
Concerning antiviral treatment, many studies reported that hepatic steatosis is negatively correlated with SVR rates after peg-interferon and ribavirin treatment[69,81]. This association may be explained through mechanisms that involve IR-induced SOCS, which in turn are responsible for a reduced activation of signal transducer and activator of transcription (STAT) proteins involved in interferon signalling[82]. On the other side, hepatic IR increases viral replication[83] and produces lipo-viro particles[84]. Since steatosis observed in G3 patients has not been related to decreased likelihoods of SVR[47], it seems that the central and more specific role is played by metabolic steatosis. At the same time, the rationale of reducing IR to increase response to antiviral treatment is not completely supported. A recent randomized clinical trial (TRIC-1) examined the effect of adding metformin to standard therapy in the treatment of CHC. The study demonstrated that patients infected with G1 and with HOMA index > 2, treated with metformin, showed an early greater drop in viral load and doubled SVR in women[85]. On the other hand, the correction of IR with pioglitazone didn’t improve response to therapy in two different trials[86,87]. The different genotypes and design of the studies could explain, at least in part, the discrepancies between these results. Further evidence is needed in order to define the optimal therapeutic strategy for improving response to therapy in insulin resistant patients undergoing antiviral treatment.
HCV INFECTION AND ATHEROSCLEROSIS
As previously described, HCV is able to directly induce metabolic and inflammatory alterations and is responsible for the occurrence of IR and DM. In view of this complex interplay between HCV infection, metabolic disorders and “classical” cardiovascular risk factors, several studies aimed to evaluate the possible role of HCV in the development and progression of atherosclerosis and in the incidence of vascular events and vascular mortality (Table 1). To note, several retrospective and cross-sectional studies have clearly demonstrated that different infectious agents, such as chlamydia pneumoniae[88,89], cytomegalovirus[90], herpes simplex virus[91], and hepatitis A virus[92], can participate the process of atherosclerosis[93], suggesting that also HCV may play a role through the potentiation of the inflammatory boost, which is a key event in atherosclerosis.
Table 1.
Overview of the main studies assessing the association between hepatitis C virus infection and the prevalence or incidence of cardio-cerebrovascular disease
| Ref. | Study design | Country-setting | Total patients (%HCV+) | Main results |
| Prevalence of cardio- or cerebrovascular disease | ||||
| Ishizaka et al[94], 2002 | Cross-sectional | Japan-general health screening | 4784 (2.1) | HCV+ independently associated with increased IMT [OR = 2.9 (2.3-3.6)] and CP [OR = 1.9 (1.6-2.4)] |
| Bilora et al[121], 2002 | Case-control | Italy-hepatitis outpatient clinic | 98 (49) | HCV+ have lower prevalence of CP, no significant difference of FP |
| Ishizaka et al[95], 2003 | Cross-sectional | Japan-general health screening | 1992 (1.3) | HCV+ associated with CP [OR = 5.5 (2.4-12.8)] and IMT [OR = NA] |
| HCV+ independently associated with CP [OR = 5.6 (2.1-15.3)], but not with IMT | ||||
| Fukui et al[96], 2003 | Cross-sectional | Japan-ultrasound carotid screening | 210 (14.8) | HCV+ have higher prevalence of increased IMT and CP. HCV+ is independently associated with CP [OR = NA] |
| Volzke et al[107], 2004 | Cross-sectional | Germany-population registry data | 4266 (5.5) | HCV+ or HBV+ not associated with IMT, CP, MI or S |
| Vassalle et al[102], 2004 | Case-control | Not specified | 686 (5.1) | HCV+ independently associated with CAD [OR = 4.2 (1.4-13)] |
| Arcari et al[108], 2006 | Case-control | United States-United States army | 582 (8.9) | HCV+ not associated with MI |
| Targher et al[97], 2007 | Cross-sectional | United Kingdom-outpatient clinic | 120 (50) | HCV+ independently associated with IMT [OR = 1.6 (1.1-2.5)] |
| Boddi et al[98], 2007 | Cross-sectional | Italy-cardiovascular risk factor centre | 151 (20.5) | HCV+ independently associated with IMT [OR = 4.4 (1.4-13.9)] , but not with CP |
| Alyan et al[103], 2008 | Case-control | Turkey-cardiology unit | 364 (38.2) | HCV+ independently associated with CAD [OR = 2.0 (1.6-2.6)] |
| Tien et al[119], 20095 | Cross-sectional | United States-women’s interagency HIV study | 503 (10.5) | HCV+ not associated with IMT or CP |
| Mostafa et al[120], 2010 | Cross-sectional | Egypt-village metabolic study | 494 (37.9) | IMT and CP not different in HCV+; HCV+ independently associated with IMT and CP [OR = 3.5 (1.2-9.9)] |
| Adinolfi et al[100], 2012 | Case-control | Italy-liver outpatient clinic and general population screening | 803 (40.6) | Increased IMT and CP more prevalent in HCV+; HCV-RNA independently associated with CP [OR = 5.2 (2.6-10.5)] |
| Petta et al[99], 2012 | Case-control | Italy-liver and cardiology outpatient unit | 348 (50) | Increased IMT and CP more prevalent in HCV+; HCV+ independently associated with IMT [OR = NA]. In HCV patient, older age [OR = 1.04 (1.01-1.08)] and severe fibrosis [OR = 2.18 (1.04-4.54)] are independently associated with CP |
| Younossi et al[125], 2013 | Cross-sectional | United States-NHANES database | 19741 (0.9) | HCV+ independently associated with CHF [OR = 2.5 (1.1-6)], but not with CHD |
| Miyajima et al[123], 2013 | Cross-sectional | Japan-seven country study | 1908 (2.1) | IMT significantly reduced in HCV+ |
| Incidence of cardio- or cerebrovascular disease | ||||
| Younossi et al[116], 19996 | Retrospective 24.6 yr FU | United States-transplant centre | 54 (22.2) | HCV+ associated with CHD mortality [HR NA], but not with CHD |
| Haji et al[104], 20047 | Retrospective 4.2 yr FU | United States-transplant centre | 417 (8.2) | HCV+ independently associated with increased mortality [HR 2.8 (1.3-5.7)] and CAD [HR 3.1 (1.5-6.2)] |
| Amin et al[111], 20061 | Retrospective | Australia-Australian national death index | 117547 (66.7) | HCV+ independently associated with cardiovascular mortality [HR 1.3 (1.2-1.5)] |
| Neal et al[117], 2007 | Prospective 6.7 yr FU | United Kingdom-trent hepatitis C cohort | 22858 | HCV+ not associated with cardiovascular mortality |
| Bilora et al[122], 2008 | Case-control prospective 5 yr FU | Italy-not specified | 67 (50.7) | HCV+ have lower prevalence of CP, no difference in FP |
| Caliskan et al[133], 20092 | Prospective 59 mo FU | Turkey-hemodialysis unit | 72 (50) | HCV+ have lower increase of IMT, not significant difference in increase of CP and FP No differences in IMT, FMD and CP in HCV+ |
| Butt et al[109], 2009 | Prospective 5 yr FU | United States-ERCHIVES database | 171665 (47.8) | HCV+ not associated with CHD in univariate analysis, but independently associated with CHD [HR 1.25 (1.2-1.3)], in adjusted models |
| Lee et al[113], 2010 | Prospective 16.9 yr FU | Taiwan-general population | 23665 (5.5) | HCV+ independently associated with CVD mortality [HR 2.2 (1.5-3.2)]. CVD risk increases with HCV-RNA |
| Bedimo et al[105], 20103 | Retrospective 3.9 yr FU | United States-HIV infected United States veterans | 19424 (31.6) | HCV+ independently associated with CVD [HR 1.2 (1.1-1.4)], but not MI |
| Ohsawa et al[114], 20112 | Prospective 5 yr FU | Japan-KAREN Study | 1077 (10.1) | HCV+ independently associated with cardiovascular mortality [HR 1.8 (1.1-3)] |
| Freiberg et al[106], 20114 | Retrospective 7.5 yr FU | United States-veterans aging cohort study | 8579 (16.8) | HCV+/HIV+ independently associated with incident CHD as compared with HCV-/HIV+ or controls. HCV+ not associated with incident CHD |
| Kristiansen et al[118], 2011 | Prospective 7 yr FU | Norway-population registry data | 10108 | HCV+ not associated with increased risk of cardiovascular mortality |
| Forde et al[110], 2012 | Retrospective 3.9 yr FU | United Kingdom-health improvement network | 4809 (6.3) | HCV+ not associated with incident MI |
| Lee et al[115], 2012 | Prospective 16.9 yr FU | Taiwan-general population | 1095 (5.6) | HCV+ associated with vascular mortality [HR 1.5 (1.1-2)] |
| Hsu et al[124], 2013 | Retrospective case-control 5 yr FU | Taiwan-Taiwan national health insurance program | 3113 (20) | HCV+ independently associated with S [HR 1.2 (1.1-1.4)] |
Carried out in hepatitis C virus (HCV) and/or hepatitis B virus patient;
Carried out in patients in hemodialysis;
Carried out in human immunodeficiency virus (HIV) mono-infected and HIV/HCV coinfected veterans;
Carried out in HIV and/or HCV United States veterans (only males);
Carried out in women HIV and/or HCV positive: 220 HCV+/HIV+; 53 HCV+; 950 HIV+; 452 controls;
Carried out in kidney transplant recipients with allografts functioning over 20 years;
Carried out in cardiac transplant recipients who received hearths from HCV+ or - donors;
Carried out on HCV patients compared to their reference general population. IMT: Intima-media thickness; CP: Carotid plaques; FP: Femoral plaques; FMD: Flow-mediated-dilation; FU: Follow-up; CAD: Coronary artery disease, angiographically documented; MI: Myocardial infarction; S: Stroke; CHD: Coronary hearth disease (myocardial infarction, unstable angina, need for revascularization procedures); CHF: Congestive hearth failure; CVD: Cerebrovascular disease (transient ischemic attack or stroke); NA: Not available.
Clinical evidences of the association between HCV and atherosclerosis
In 2002 and 2003, Ishizaka et al[94,95] firstly described the association between the presence of anti-HCV antibodies and/or serum HCV core protein and an increased risk of carotid artery plaques. These findings were corroborated by other studies, which found intima-media thickness (IMT) and the prevalence of carotid plaques to be increased in HCV patients[96-100], and in which HCV genomic and antigenomic RNA strands were identified within carotid plaques tissue of HCV-positive patients (even in three patients positive for anti-HCV antibodies but with undetectable HCV-RNA in serum)[98,101], suggesting a possible direct local pathogenetic role of HCV in atherosclerotic plaque formation. More recently, HCV seropositivity was identified as an independent predictor of increased coronary atherosclerosis[102-104], even though an increased incidence of acute myocardial infarction (AMI) was demonstrated only in HIV/HCV coinfected patients[105,106], but not in HCV mono infected ones[107-110]. Furthermore, the incidences of vascular events and of cardiovascular mortality of HCV-positive patients were reported to be either higher[104,105,111-116] or, at least, comparable to those observed in the general population[117,118].
In contrast with these data, other studies failed to demonstrate any significant difference in IMT and in the prevalence of carotid plaques between HCV-positive and HCV-negative patients[107,119,120], and some others reported an even lower risk of atherosclerosis in HCV patients with respects to controls[121-123]. Three large population studies gave conflicting results concerning the association between HCV-infection and the incidence of stroke[107,113,124] and, recently, Younossi et al[125] found HCV infection to be independently associated with IR, hypertension and congestive heart failure, but not with ischemic heart disease and stroke.
Many possible confounding elements should be considered while comparing these different studies and trying to interpret their sometimes divergent results. First of all, the study populations were recruited from different contexts, namely hepatitis or cardiology outpatient clinics, population registries or general health screening programs. Some studies included and some others excluded HIV and HBV coinfected patients. Moreover, not in all of these studies multivariate models were created in order to analyze if the association between HCV-infection and markers of subclinical atherosclerosis or incidences of vascular events/cardiovascular mortality was independent from the other metabolic risk factors. In this regard, it should also be stressed that data on the duration of HCV infection and of DM are not available in any of these works, and that liver histology of HCV patients, which gives the opportunity to correlate vascular outcomes with the histological grading and staging of the hepatic disease, was available from only one study[99]. Finally, another important point to be considered is that cirrhotic patients were frequently excluded or poorly represented in the study populations. Notwithstanding epidemiological data are very limited, cirrhosis is currently considered a condition associated with a decreased risk of cardiovascular events[126]. Indeed, although clearly predisposing to DM, cirrhosis is characterized by an overall favourable risk profile (low blood pressure, low cholesterol, impaired procoagulative cascade and low platelet count).
In conclusion, even if literature on this topic is scant and sometimes ambiguous, current evidence seems to support an association between HCV and atherosclerosis, which can account for the increased prevalence and incidence of vascular disease in patients with HCV infection. As supported by some studies, it seems reasonable to speculate that the contribution of HCV to the atherogenic process, either direct or indirect, or both, could increase with the duration of the infection, the development of IR and eventually DM, and the increase of circulating products of oxidative stress and inflammation. On the contrary, once cirrhosis has developed, several mechanisms determining a reduction of the cardiovascular risk progressively come into play (Figure 1).
Figure 1.
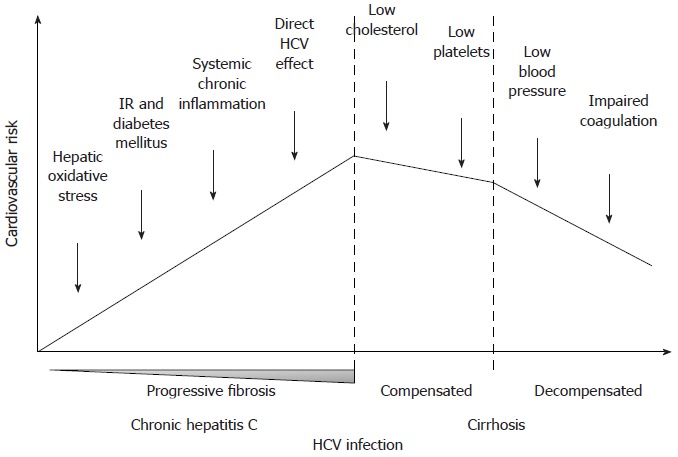
Hypothetical trend of cardiovascular risk during the natural history of hepatitis C virus infection, from chronic hepatitis to decompensated cirrhosis. IR: Insulin resistance; HCV: Hepatitis C virus.
Hypothetical pathogenic processes “directly” or “indirectly” linking HCV to atherosclerosis
Nowadays, it is widely accepted that infective agents contribute to the progression of chronic immuno-mediated cell inflammation underlying atherosclerosis through the inflammatory response elicited in the host[127]. They can accelerate the occurrence of several key steps in the plaque formation since they can promote endothelial dysfunction, potentiate the recruitment and activation of T-lympho-monocytes and enhance the proliferation and migration of smooth-muscle cells. However, the detection of HCV-RNA in carotid atherosclerotic plaques, predominantly in patients with G2 HCV-infection, strongly suggested also a direct local role of HCV in atherogenesis[98,101]. Consistent with this finding, viral load has been recently associated with carotid atherosclerosis[100], and with the risk of cerebrovascular mortality[113]. This hypothesis is also supported by several experimental studies. For instance, some HCV proteins can enhance local oxidative stress[128], and increase the concentration of soluble intracellular adhesion molecules[129]. HCV particles have affinity with circulating lipoproteins in the blood and HCV G2 seems to be the most closely associated with these lipoproteins[130]. In addition to hepatocytes, HCV can also infect lymphocytes and through these can induce vasculitis and the production of anti-endothelial antibodies[131].
HCV may also “indirectly” favour atherosclerosis, via liver damage or virus-induced, metabolic disorders. Accordingly, in biopsy-proven chronic hepatitis C patients, IMT and prevalence of carotid plaques were recently found to be associated with the severity of fibrosis[99]. It can be speculated that oxidative stress and inflammation, which are associated with the evolution of liver fibrosis, can be associated or directly contribute also to the atherogenic process. After all, the well-known relationship between NAFLD and cardiovascular disease has already demonstrated how liver damage could be directly involved in the pathogenesis of cardiovascular disease, through the systemic release of proatherogenic mediators from the steatotic and inflamed liver or through the worsening of IR and of atherogenic dyslipidemia[132]. However, in contrast with NAFLD, HCV is associated with a favourable lipoprotein profile, namely hypobetalipoproteinemia. The net effect of increased IR and favourable lipoprotein profile on the cardiovascular risk was recently investigated by Mostafa et al[120], who found IMT and carotid plaques to be significantly associated with HCV-infection only after adjustment for “classical” cardiovascular risk factors, particularly LDL cholesterol and systolic blood pressure. Accordingly, in a larger prospective study, including HCV infected patient owning better cardiovascular risk profile (lower prevalence of DM and lower cholesterol), HCV-infection was found to be associated with coronary heart disease only after correction for potential metabolic confounders[109]. These results may suggest that HCV affects the cardiovascular risk mainly via non-conventional pathways, and not by virus-induced metabolic modifications, i.e., IR and good lipoprotein profile, which possibly balance each other. In agreement with this hypothesis, in studies where HCV was found to be independently associated with vascular disease, the relationship between HCV infection and vascular outcomes was generally adjusted for metabolic risk factors, in contrast to what has been done in the majority of studies failing to demonstrate this association[108,117,118,123,133]. One exception is a big retrospective population study in which HCV was not associated with AMI even after adjusting for all the metabolic confounders[110].
HCV INFECTION AND METABOLIC DISORDERS: SUMMARY OF A COMPLEX INTERPLAY
All the data provided above suggest a strong interrelationship between HCV infection and metabolic disorders, which is likely implicated in the progression both of liver damage and of the atherogenic process (Figure 2). HCV can directly interact with intracellular mediators of insulin activity, such as PP2A, SOCS3, IRS and PPARs[22-29], or indirectly hamper the insulin message by inducing hepatic and low-grade systemic inflammation[17,18]. Moreover, the virus increases the synthesis of free fatty acids and reduces their mechanisms of export and degradation[48,49], therefore inducing a “viral” steatosis which is often superimposed to a “metabolic” one. Progression of liver damage is favoured by the steatosis-induced hepatic reduction of antioxidant defenses and by a direct stimulatory effect of hyperinsulinemia, oxidative stress and lipid peroxidation on hepatic fibrogenic cells. At the same time, HCV-induced alterations of glucose metabolism and the systemic release of inflammatory and procoagulative mediators by the diseased liver may well contribute to the atherogenic process. Moreover, experimental evidences support a direct role of HCV proteins, which, for example, can enhance oxidative stress and increase the concentration of soluble intracellular adhesion molecules at the atherosclerotic plaque level[128,129].
Figure 2.
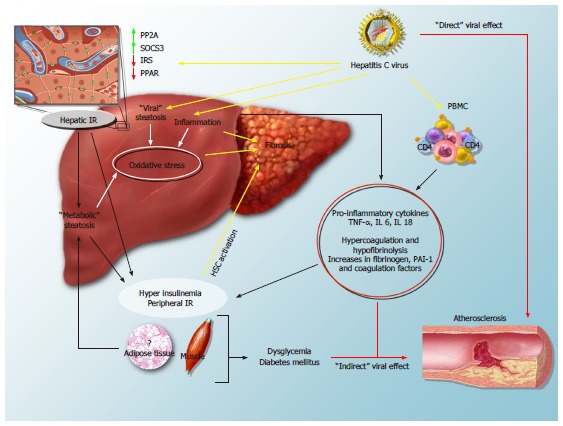
Mechanisms of hepatitis C virus-induced insulin resistance and steatosis and their impact on the progression of fibrosis and cardiovascular disease. In the hepatocyte, the virus interferes with insulin signalling, leads to overexpression of protein phosphatase 2A (PP2A) and suppressor of cytokine signalling-3 (SOCS-3), and down-regulates the expression of peroxisome proliferator activated receptors (PPAR) and of insulin receptor substrate (IRS): all these mechanisms lead to hepatic insulin resistance (IR). By inducing hepatic injury and activating peripheral blood mononuclear cells (PBMC), HCV increases circulating levels of proinflammatory cytokines, such as tumor necrosis factor-α (TNF-α), interleukin-6 and -18 (IL-6 and IL-18), and leads to peripheral IR and hyperinsulinemia. “Viral” and “metabolic” steatosis, together with the direct stimulus of increased insulin levels on hepatic stellate cells (HSCs), likely stimulate the progression of fibrosis. Furthermore, systemic inflammation, the procoagulative state and direct viral effects may contribute to the atherogenic process.
CONCLUSION
In the present manuscript, an overview of the mechanisms which link HCV infection with insulin resistance and metabolic disorders has been provided, as well as the clinical data confirming that this association may contribute both to the progression of liver damage and to atherosclerosis. All together, a complex scenario emerges where the multiple interactions between the host and the virus determine much more complications than those possibly induced only by the virus itself. Together with cryoglobulinemia, HCV-related arthritis and keratoconjunctivitis sicca, these evidences prompt to consider CHC a systemic disease rather than a simple infection of the liver.
ACKNOWLEDGMENTS
We thank Roberto Piccinocchi for his kind technical assistance in the production of Figure 2.
Footnotes
P- Reviewers: Cucchetti A, Wang CC S- Editor: Cui XM L- Editor: A E- Editor: Zhang DN
References
- 1.Lavanchy D. The global burden of hepatitis C. Liver Int. 2009;29 Suppl 1:74–81. doi: 10.1111/j.1478-3231.2008.01934.x. [DOI] [PubMed] [Google Scholar]
- 2.Zimmet P. The burden of type 2 diabetes: are we doing enough? Diabetes Metab. 2003;29:6S9–618. doi: 10.1016/S1262-3636(03)72783-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mehta SH, Brancati FL, Sulkowski MS, Strathdee SA, Szklo M, Thomas DL. Prevalence of type 2 diabetes mellitus among persons with hepatitis C virus infection in the United States. Ann Intern Med. 2000;133:592–599. doi: 10.7326/0003-4819-133-8-200010170-00009. [DOI] [PubMed] [Google Scholar]
- 4.Mehta SH, Brancati FL, Strathdee SA, Pankow JS, Netski D, Coresh J, Szklo M, Thomas DL. Hepatitis C virus infection and incident type 2 diabetes. Hepatology. 2003;38:50–56. doi: 10.1053/jhep.2003.50291. [DOI] [PubMed] [Google Scholar]
- 5.Chen T, Jia H, Li J, Chen X, Zhou H, Tian H. New onset diabetes mellitus after liver transplantation and hepatitis C virus infection: meta-analysis of clinical studies. Transpl Int. 2009;22:408–415. doi: 10.1111/j.1432-2277.2008.00804.x. [DOI] [PubMed] [Google Scholar]
- 6.Simó R, Lecube A, Genescà J, Esteban JI, Hernández C. Sustained virological response correlates with reduction in the incidence of glucose abnormalities in patients with chronic hepatitis C virus infection. Diabetes Care. 2006;29:2462–2466. doi: 10.2337/dc06-0456. [DOI] [PubMed] [Google Scholar]
- 7.Deltenre P, Louvet A, Lemoine M, Mourad A, Fartoux L, Moreno C, Henrion J, Mathurin P, Serfaty L. Impact of insulin resistance on sustained response in HCV patients treated with pegylated interferon and ribavirin: a meta-analysis. J Hepatol. 2011;55:1187–1194. doi: 10.1016/j.jhep.2011.03.010. [DOI] [PubMed] [Google Scholar]
- 8.Moucari R, Asselah T, Cazals-Hatem D, Voitot H, Boyer N, Ripault MP, Sobesky R, Martinot-Peignoux M, Maylin S, Nicolas-Chanoine MH, et al. Insulin resistance in chronic hepatitis C: association with genotypes 1 and 4, serum HCV RNA level, and liver fibrosis. Gastroenterology. 2008;134:416–423. doi: 10.1053/j.gastro.2007.11.010. [DOI] [PubMed] [Google Scholar]
- 9.Hui JM, Sud A, Farrell GC, Bandara P, Byth K, Kench JG, McCaughan GW, George J. Insulin resistance is associated with chronic hepatitis C virus infection and fibrosis progression [corrected] Gastroenterology. 2003;125:1695–1704. doi: 10.1053/j.gastro.2003.08.032. [DOI] [PubMed] [Google Scholar]
- 10.Picardi A, D’Avola D, Gentilucci UV, Galati G, Fiori E, Spataro S, Afeltra A. Diabetes in chronic liver disease: from old concepts to new evidence. Diabetes Metab Res Rev. 2006;22:274–283. doi: 10.1002/dmrr.636. [DOI] [PubMed] [Google Scholar]
- 11.Romero-Gómez M, Fernández-Rodríguez CM, Andrade RJ, Diago M, Alonso S, Planas R, Solá R, Pons JA, Salmerón J, Barcena R, et al. Effect of sustained virological response to treatment on the incidence of abnormal glucose values in chronic hepatitis C. J Hepatol. 2008;48:721–727. doi: 10.1016/j.jhep.2007.11.022. [DOI] [PubMed] [Google Scholar]
- 12.Kawaguchi T, Ide T, Taniguchi E, Hirano E, Itou M, Sumie S, Nagao Y, Yanagimoto C, Hanada S, Koga H, et al. Clearance of HCV improves insulin resistance, beta-cell function, and hepatic expression of insulin receptor substrate 1 and 2. Am J Gastroenterol. 2007;102:570–576. doi: 10.1111/j.1572-0241.2006.01038.x. [DOI] [PubMed] [Google Scholar]
- 13.Arase Y, Suzuki F, Suzuki Y, Akuta N, Kobayashi M, Kawamura Y, Yatsuji H, Sezaki H, Hosaka T, Hirakawa M, et al. Sustained virological response reduces incidence of onset of type 2 diabetes in chronic hepatitis C. Hepatology. 2009;49:739–744. doi: 10.1002/hep.22703. [DOI] [PubMed] [Google Scholar]
- 14.Giordanino C, Bugianesi E, Smedile A, Ciancio A, Abate ML, Olivero A, Pellicano R, Cassader M, Gambino R, Bo S, et al. Incidence of type 2 diabetes mellitus and glucose abnormalities in patients with chronic hepatitis C infection by response to treatment: results of a cohort study. Am J Gastroenterol. 2008;103:2481–2487. doi: 10.1111/j.1572-0241.2008.02002.x. [DOI] [PubMed] [Google Scholar]
- 15.Mihm S. Hepatitis C virus, diabetes and steatosis: clinical evidence in favor of a linkage and role of genotypes. Dig Dis. 2010;28:280–284. doi: 10.1159/000282103. [DOI] [PubMed] [Google Scholar]
- 16.Sheikh MY, Choi J, Qadri I, Friedman JE, Sanyal AJ. Hepatitis C virus infection: molecular pathways to metabolic syndrome. Hepatology. 2008;47:2127–2133. doi: 10.1002/hep.22269. [DOI] [PubMed] [Google Scholar]
- 17.Vanni E, Abate ML, Gentilcore E, Hickman I, Gambino R, Cassader M, Smedile A, Ferrannini E, Rizzetto M, Marchesini G, et al. Sites and mechanisms of insulin resistance in nonobese, nondiabetic patients with chronic hepatitis C. Hepatology. 2009;50:697–706. doi: 10.1002/hep.23031. [DOI] [PubMed] [Google Scholar]
- 18.Milner KL, van der Poorten D, Trenell M, Jenkins AB, Xu A, Smythe G, Dore GJ, Zekry A, Weltman M, Fragomeli V, et al. Chronic hepatitis C is associated with peripheral rather than hepatic insulin resistance. Gastroenterology. 2010;138:932–941.e1-3. doi: 10.1053/j.gastro.2009.11.050. [DOI] [PubMed] [Google Scholar]
- 19.Shintani Y, Fujie H, Miyoshi H, Tsutsumi T, Tsukamoto K, Kimura S, Moriya K, Koike K. Hepatitis C virus infection and diabetes: direct involvement of the virus in the development of insulin resistance. Gastroenterology. 2004;126:840–848. doi: 10.1053/j.gastro.2003.11.056. [DOI] [PubMed] [Google Scholar]
- 20.Perseghin G, Mazzaferro V, Sereni LP, Regalia E, Benedini S, Bazzigaluppi E, Pulvirenti A, Leão AA, Calori G, Romito R, et al. Contribution of reduced insulin sensitivity and secretion to the pathogenesis of hepatogenous diabetes: effect of liver transplantation. Hepatology. 2000;31:694–703. doi: 10.1002/hep.510310320. [DOI] [PubMed] [Google Scholar]
- 21.Tietge UJ, Selberg O, Kreter A, Bahr MJ, Pirlich M, Burchert W, Müller MJ, Manns MP, Böker KH. Alterations in glucose metabolism associated with liver cirrhosis persist in the clinically stable long-term course after liver transplantation. Liver Transpl. 2004;10:1030–1040. doi: 10.1002/lt.20147. [DOI] [PubMed] [Google Scholar]
- 22.Aytug S, Reich D, Sapiro LE, Bernstein D, Begum N. Impaired IRS-1/PI3-kinase signaling in patients with HCV: a mechanism for increased prevalence of type 2 diabetes. Hepatology. 2003;38:1384–1392. doi: 10.1016/j.hep.2003.09.012. [DOI] [PubMed] [Google Scholar]
- 23.Bernsmeier C, Duong FH, Christen V, Pugnale P, Negro F, Terracciano L, Heim MH. Virus-induced over-expression of protein phosphatase 2A inhibits insulin signalling in chronic hepatitis C. J Hepatol. 2008;49:429–440. doi: 10.1016/j.jhep.2008.04.007. [DOI] [PubMed] [Google Scholar]
- 24.Christen V, Treves S, Duong FH, Heim MH. Activation of endoplasmic reticulum stress response by hepatitis viruses up-regulates protein phosphatase 2A. Hepatology. 2007;46:558–565. doi: 10.1002/hep.21611. [DOI] [PubMed] [Google Scholar]
- 25.Kawaguchi T, Yoshida T, Harada M, Hisamoto T, Nagao Y, Ide T, Taniguchi E, Kumemura H, Hanada S, Maeyama M, et al. Hepatitis C virus down-regulates insulin receptor substrates 1 and 2 through up-regulation of suppressor of cytokine signaling 3. Am J Pathol. 2004;165:1499–1508. doi: 10.1016/S0002-9440(10)63408-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miyamoto H, Moriishi K, Moriya K, Murata S, Tanaka K, Suzuki T, Miyamura T, Koike K, Matsuura Y. Involvement of the PA28gamma-dependent pathway in insulin resistance induced by hepatitis C virus core protein. J Virol. 2007;81:1727–1735. doi: 10.1128/JVI.01683-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dharancy S, Malapel M, Perlemuter G, Roskams T, Cheng Y, Dubuquoy L, Podevin P, Conti F, Canva V, Philippe D, et al. Impaired expression of the peroxisome proliferator-activated receptor alpha during hepatitis C virus infection. Gastroenterology. 2005;128:334–342. doi: 10.1053/j.gastro.2004.11.016. [DOI] [PubMed] [Google Scholar]
- 28.de Gottardi A, Pazienza V, Pugnale P, Bruttin F, Rubbia-Brandt L, Juge-Aubry CE, Meier CA, Hadengue A, Negro F. Peroxisome proliferator-activated receptor-alpha and -gamma mRNA levels are reduced in chronic hepatitis C with steatosis and genotype 3 infection. Aliment Pharmacol Ther. 2006;23:107–114. doi: 10.1111/j.1365-2036.2006.02729.x. [DOI] [PubMed] [Google Scholar]
- 29.Pazienza V, Vinciguerra M, Andriulli A, Mangia A. Hepatitis C virus core protein genotype 3a increases SOCS-7 expression through PPAR-{gamma} in Huh-7 cells. J Gen Virol. 2010;91:1678–1686. doi: 10.1099/vir.0.020644-0. [DOI] [PubMed] [Google Scholar]
- 30.Picardi A, Gentilucci UV, Zardi EM, Caccavo D, Petitti T, Manfrini S, Pozzilli P, Afeltra A. TNF-alpha and growth hormone resistance in patients with chronic liver disease. J Interferon Cytokine Res. 2003;23:229–235. doi: 10.1089/107999003321829944. [DOI] [PubMed] [Google Scholar]
- 31.Nelson DR, Lim HL, Marousis CG, Fang JW, Davis GL, Shen L, Urdea MS, Kolberg JA, Lau JY. Activation of tumor necrosis factor-alpha system in chronic hepatitis C virus infection. Dig Dis Sci. 1997;42:2487–2494. doi: 10.1023/a:1018804426724. [DOI] [PubMed] [Google Scholar]
- 32.Lecube A, Hernández C, Genescà J, Simó R. Proinflammatory cytokines, insulin resistance, and insulin secretion in chronic hepatitis C patients: A case-control study. Diabetes Care. 2006;29:1096–1101. doi: 10.2337/diacare.2951096. [DOI] [PubMed] [Google Scholar]
- 33.Cua IH, Hui JM, Bandara P, Kench JG, Farrell GC, McCaughan GW, George J. Insulin resistance and liver injury in hepatitis C is not associated with virus-specific changes in adipocytokines. Hepatology. 2007;46:66–73. doi: 10.1002/hep.21703. [DOI] [PubMed] [Google Scholar]
- 34.Farinati F, Cardin R, Bortolami M, Burra P, Russo FP, Rugge M, Guido M, Sergio A, Naccarato R. Hepatitis C virus: from oxygen free radicals to hepatocellular carcinoma. J Viral Hepat. 2007;14:821–829. doi: 10.1111/j.1365-2893.2007.00878.x. [DOI] [PubMed] [Google Scholar]
- 35.Poli G. Pathogenesis of liver fibrosis: role of oxidative stress. Mol Aspects Med. 2000;21:49–98. doi: 10.1016/s0098-2997(00)00004-2. [DOI] [PubMed] [Google Scholar]
- 36.Clément S, Pascarella S, Negro F. Hepatitis C virus infection: molecular pathways to steatosis, insulin resistance and oxidative stress. Viruses. 2009;1:126–143. doi: 10.3390/v1020126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Adinolfi LE, Gambardella M, Andreana A, Tripodi MF, Utili R, Ruggiero G. Steatosis accelerates the progression of liver damage of chronic hepatitis C patients and correlates with specific HCV genotype and visceral obesity. Hepatology. 2001;33:1358–1364. doi: 10.1053/jhep.2001.24432. [DOI] [PubMed] [Google Scholar]
- 38.Dienes HP, Popper H, Arnold W, Lobeck H. Histologic observations in human hepatitis non-A, non-B. Hepatology. 1982;2:562–571. doi: 10.1002/hep.1840020509. [DOI] [PubMed] [Google Scholar]
- 39.Wiese M, Haupt R. [Histomorphologic picture of chronic non-A, non-B hepatitis] Dtsch Z Verdau Stoffwechselkr. 1985;45:101–110. [PubMed] [Google Scholar]
- 40.Machado MV, Oliveira AG, Cortez-Pinto H. Hepatic steatosis in hepatitis B virus infected patients: meta-analysis of risk factors and comparison with hepatitis C infected patients. J Gastroenterol Hepatol. 2011;26:1361–1367. doi: 10.1111/j.1440-1746.2011.06801.x. [DOI] [PubMed] [Google Scholar]
- 41.Lonardo A, Adinolfi LE, Loria P, Carulli N, Ruggiero G, Day CP. Steatosis and hepatitis C virus: mechanisms and significance for hepatic and extrahepatic disease. Gastroenterology. 2004;126:586–597. doi: 10.1053/j.gastro.2003.11.020. [DOI] [PubMed] [Google Scholar]
- 42.Chen CL, Yang HI, Yang WS, Liu CJ, Chen PJ, You SL, Wang LY, Sun CA, Lu SN, Chen DS, et al. Metabolic factors and risk of hepatocellular carcinoma by chronic hepatitis B/C infection: a follow-up study in Taiwan. Gastroenterology. 2008;135:111–121. doi: 10.1053/j.gastro.2008.03.073. [DOI] [PubMed] [Google Scholar]
- 43.Mihm S, Fayyazi A, Hartmann H, Ramadori G. Analysis of histopathological manifestations of chronic hepatitis C virus infection with respect to virus genotype. Hepatology. 1997;25:735–739. doi: 10.1002/hep.510250340. [DOI] [PubMed] [Google Scholar]
- 44.Fujie H, Yotsuyanagi H, Moriya K, Shintani Y, Tsutsumi T, Takayama T, Makuuchi M, Matsuura Y, Miyamura T, Kimura S, et al. Steatosis and intrahepatic hepatitis C virus in chronic hepatitis. J Med Virol. 1999;59:141–145. doi: 10.1002/(sici)1096-9071(199910)59:2<141::aid-jmv3>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 45.Rubbia-Brandt L, Quadri R, Abid K, Giostra E, Malé PJ, Mentha G, Spahr L, Zarski JP, Borisch B, Hadengue A, et al. Hepatocyte steatosis is a cytopathic effect of hepatitis C virus genotype 3. J Hepatol. 2000;33:106–115. doi: 10.1016/s0168-8278(00)80166-x. [DOI] [PubMed] [Google Scholar]
- 46.Kumar D, Farrell GC, Fung C, George J. Hepatitis C virus genotype 3 is cytopathic to hepatocytes: Reversal of hepatic steatosis after sustained therapeutic response. Hepatology. 2002;36:1266–1272. doi: 10.1053/jhep.2002.36370. [DOI] [PubMed] [Google Scholar]
- 47.Poynard T, Ratziu V, McHutchison J, Manns M, Goodman Z, Zeuzem S, Younossi Z, Albrecht J. Effect of treatment with peginterferon or interferon alfa-2b and ribavirin on steatosis in patients infected with hepatitis C. Hepatology. 2003;38:75–85. doi: 10.1053/jhep.2003.50267. [DOI] [PubMed] [Google Scholar]
- 48.Abid K, Pazienza V, de Gottardi A, Rubbia-Brandt L, Conne B, Pugnale P, Rossi C, Mangia A, Negro F. An in vitro model of hepatitis C virus genotype 3a-associated triglycerides accumulation. J Hepatol. 2005;42:744–751. doi: 10.1016/j.jhep.2004.12.034. [DOI] [PubMed] [Google Scholar]
- 49.Negro F. Mechanisms and significance of liver steatosis in hepatitis C virus infection. World J Gastroenterol. 2006;12:6756–6765. doi: 10.3748/wjg.v12.i42.6756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Agnello V, Abel G, Elfahal M, Knight GB, Zhang QX. Hepatitis C virus and other flaviviridae viruses enter cells via low density lipoprotein receptor. Proc Natl Acad Sci USA. 1999;96:12766–12771. doi: 10.1073/pnas.96.22.12766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Perlemuter G, Sabile A, Letteron P, Vona G, Topilco A, Chrétien Y, Koike K, Pessayre D, Chapman J, Barba G, et al. Hepatitis C virus core protein inhibits microsomal triglyceride transfer protein activity and very low density lipoprotein secretion: a model of viral-related steatosis. FASEB J. 2002;16:185–194. doi: 10.1096/fj.01-0396com. [DOI] [PubMed] [Google Scholar]
- 52.Syed GH, Amako Y, Siddiqui A. Hepatitis C virus hijacks host lipid metabolism. Trends Endocrinol Metab. 2010;21:33–40. doi: 10.1016/j.tem.2009.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Negro F. HCV infection and metabolic syndrome: which is the chicken and which is the egg? Gastroenterology. 2012;142:1288–1292. doi: 10.1053/j.gastro.2011.12.063. [DOI] [PubMed] [Google Scholar]
- 54.Clément S, Fauvelle C, Branche E, Kaddai V, Conzelmann S, Boldanova T, Bartosch B, Minehira K, Negro F. Role of seipin in lipid droplet morphology and hepatitis C virus life cycle. J Gen Virol. 2013;94:2208–2214. doi: 10.1099/vir.0.054593-0. [DOI] [PubMed] [Google Scholar]
- 55.Rubbia-Brandt L, Leandro G, Spahr L, Giostra E, Quadri R, Malé PJ, Negro F. Liver steatosis in chronic hepatitis C: a morphological sign suggesting infection with HCV genotype 3. Histopathology. 2001;39:119–124. doi: 10.1046/j.1365-2559.2001.01208.x. [DOI] [PubMed] [Google Scholar]
- 56.Rubbia-Brandt L, Giostra E, Mentha G, Quadri R, Negro F. Expression of liver steatosis in hepatitis C virus infection and pattern of response to alpha-interferon. J Hepatol. 2001;35:307. doi: 10.1016/s0168-8278(01)00087-3. [DOI] [PubMed] [Google Scholar]
- 57.Negro F. Abnormalities of lipid metabolism in hepatitis C virus infection. Gut. 2010;59:1279–1287. doi: 10.1136/gut.2009.192732. [DOI] [PubMed] [Google Scholar]
- 58.Tarugi P, Averna M, Di Leo E, Cefalù AB, Noto D, Magnolo L, Cattin L, Bertolini S, Calandra S. Molecular diagnosis of hypobetalipoproteinemia: an ENID review. Atherosclerosis. 2007;195:e19–e27. doi: 10.1016/j.atherosclerosis.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 59.Serfaty L, Andreani T, Giral P, Carbonell N, Chazouillères O, Poupon R. Hepatitis C virus induced hypobetalipoproteinemia: a possible mechanism for steatosis in chronic hepatitis C. J Hepatol. 2001;34:428–434. doi: 10.1016/s0168-8278(00)00036-2. [DOI] [PubMed] [Google Scholar]
- 60.Hofer H, Bankl HC, Wrba F, Steindl-Munda P, Peck-Radosavljevic M, Osterreicher C, Mueller C, Gangl A, Ferenci P. Hepatocellular fat accumulation and low serum cholesterol in patients infected with HCV-3a. Am J Gastroenterol. 2002;97:2880–2885. doi: 10.1111/j.1572-0241.2002.07056.x. [DOI] [PubMed] [Google Scholar]
- 61.Mirandola S, Realdon S, Iqbal J, Gerotto M, Dal Pero F, Bortoletto G, Marcolongo M, Vario A, Datz C, Hussain MM, et al. Liver microsomal triglyceride transfer protein is involved in hepatitis C liver steatosis. Gastroenterology. 2006;130:1661–1669. doi: 10.1053/j.gastro.2006.02.035. [DOI] [PubMed] [Google Scholar]
- 62.Bugianesi E, Salamone F, Negro F. The interaction of metabolic factors with HCV infection: does it matter? J Hepatol. 2012;56 Suppl 1:S56–S65. doi: 10.1016/S0168-8278(12)60007-5. [DOI] [PubMed] [Google Scholar]
- 63.Okuda M, Li K, Beard MR, Showalter LA, Scholle F, Lemon SM, Weinman SA. Mitochondrial injury, oxidative stress, and antioxidant gene expression are induced by hepatitis C virus core protein. Gastroenterology. 2002;122:366–375. doi: 10.1053/gast.2002.30983. [DOI] [PubMed] [Google Scholar]
- 64.Su AI, Pezacki JP, Wodicka L, Brideau AD, Supekova L, Thimme R, Wieland S, Bukh J, Purcell RH, Schultz PG, et al. Genomic analysis of the host response to hepatitis C virus infection. Proc Natl Acad Sci USA. 2002;99:15669–15674. doi: 10.1073/pnas.202608199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yamaguchi A, Tazuma S, Nishioka T, Ohishi W, Hyogo H, Nomura S, Chayama K. Hepatitis C virus core protein modulates fatty acid metabolism and thereby causes lipid accumulation in the liver. Dig Dis Sci. 2005;50:1361–1371. doi: 10.1007/s10620-005-2788-1. [DOI] [PubMed] [Google Scholar]
- 66.Cheng Y, Dharancy S, Malapel M, Desreumaux P. Hepatitis C virus infection down-regulates the expression of peroxisome proliferator-activated receptor alpha and carnitine palmitoyl acyl-CoA transferase 1A. World J Gastroenterol. 2005;11:7591–7596. doi: 10.3748/wjg.v11.i48.7591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Petit JM, Minello A, Jooste V, Bour JB, Galland F, Duvillard L, Verges B, Olsson NO, Gambert P, Hillon P. Decreased plasma adiponectin concentrations are closely related to steatosis in hepatitis C virus-infected patients. J Clin Endocrinol Metab. 2005;90:2240–2243. doi: 10.1210/jc.2004-1266. [DOI] [PubMed] [Google Scholar]
- 68.Larter CZ, Farrell GC. Insulin resistance, adiponectin, cytokines in NASH: Which is the best target to treat? J Hepatol. 2006;44:253–261. doi: 10.1016/j.jhep.2005.11.030. [DOI] [PubMed] [Google Scholar]
- 69.Cammà C, Bruno S, Di Marco V, Di Bona D, Rumi M, Vinci M, Rebucci C, Cividini A, Pizzolanti G, Minola E, et al. Insulin resistance is associated with steatosis in nondiabetic patients with genotype 1 chronic hepatitis C. Hepatology. 2006;43:64–71. doi: 10.1002/hep.20983. [DOI] [PubMed] [Google Scholar]
- 70.Petta S, Tripodo C, Grimaudo S, Cabibi D, Cammà C, Di Cristina A, Di Marco V, Di Vita G, Ingrao S, Mazzola A, et al. High liver RBP4 protein content is associated with histological features in patients with genotype 1 chronic hepatitis C and with nonalcoholic steatohepatitis. Dig Liver Dis. 2011;43:404–410. doi: 10.1016/j.dld.2010.12.013. [DOI] [PubMed] [Google Scholar]
- 71.Gomez EV, Rodriguez YS, Bertot LC, Gonzalez AT, Perez YM, Soler EA, Garcia AY, Blanco LP. The natural history of compensated HCV-related cirrhosis: a prospective long-term study. J Hepatol. 2013;58:434–444. doi: 10.1016/j.jhep.2012.10.023. [DOI] [PubMed] [Google Scholar]
- 72.Larsson SC, Wolk A. Overweight, obesity and risk of liver cancer: a meta-analysis of cohort studies. Br J Cancer. 2007;97:1005–1008. doi: 10.1038/sj.bjc.6603932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Asselah T, Boyer N, Guimont MC, Cazals-Hatem D, Tubach F, Nahon K, Daïkha H, Vidaud D, Martinot M, Vidaud M, et al. Liver fibrosis is not associated with steatosis but with necroinflammation in French patients with chronic hepatitis C. Gut. 2003;52:1638–1643. doi: 10.1136/gut.52.11.1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ghany MG, Kleiner DE, Alter H, Doo E, Khokar F, Promrat K, Herion D, Park Y, Liang TJ, Hoofnagle JH. Progression of fibrosis in chronic hepatitis C. Gastroenterology. 2003;124:97–104. doi: 10.1053/gast.2003.50018. [DOI] [PubMed] [Google Scholar]
- 75.Ryder SD, Irving WL, Jones DA, Neal KR, Underwood JC. Progression of hepatic fibrosis in patients with hepatitis C: a prospective repeat liver biopsy study. Gut. 2004;53:451–455. doi: 10.1136/gut.2003.021691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bugianesi E, Marchesini G, Gentilcore E, Cua IH, Vanni E, Rizzetto M, George J. Fibrosis in genotype 3 chronic hepatitis C and nonalcoholic fatty liver disease: Role of insulin resistance and hepatic steatosis. Hepatology. 2006;44:1648–1655. doi: 10.1002/hep.21429. [DOI] [PubMed] [Google Scholar]
- 77.Svegliati-Baroni G, Bugianesi E, Bouserhal T, Marini F, Ridolfi F, Tarsetti F, Ancarani F, Petrelli E, Peruzzi E, Lo Cascio M, et al. Post-load insulin resistance is an independent predictor of hepatic fibrosis in virus C chronic hepatitis and in non-alcoholic fatty liver disease. Gut. 2007;56:1296–1301. doi: 10.1136/gut.2006.107946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Aleffi S, Petrai I, Bertolani C, Parola M, Colombatto S, Novo E, Vizzutti F, Anania FA, Milani S, Rombouts K, et al. Upregulation of proinflammatory and proangiogenic cytokines by leptin in human hepatic stellate cells. Hepatology. 2005;42:1339–1348. doi: 10.1002/hep.20965. [DOI] [PubMed] [Google Scholar]
- 79.Kitase A, Hino K, Furutani T, Okuda M, Gondo T, Hidaka I, Hara Y, Yamaguchi Y, Okita K. In situ detection of oxidized n-3 polyunsaturated fatty acids in chronic hepatitis C: correlation with hepatic steatosis. J Gastroenterol. 2005;40:617–624. doi: 10.1007/s00535-005-1596-x. [DOI] [PubMed] [Google Scholar]
- 80.Gochee PA, Jonsson JR, Clouston AD, Pandeya N, Purdie DM, Powell EE. Steatosis in chronic hepatitis C: association with increased messenger RNA expression of collagen I, tumor necrosis factor-alpha and cytochrome P450 2E1. J Gastroenterol Hepatol. 2003;18:386–392. doi: 10.1046/j.1440-1746.2003.02984.x. [DOI] [PubMed] [Google Scholar]
- 81.Petta S, Amato M, Cabibi D, Cammà C, Di Marco V, Giordano C, Galluzzo A, Craxì A. Visceral adiposity index is associated with histological findings and high viral load in patients with chronic hepatitis C due to genotype 1. Hepatology. 2010;52:1543–1552. doi: 10.1002/hep.23859. [DOI] [PubMed] [Google Scholar]
- 82.Walsh MJ, Jonsson JR, Richardson MM, Lipka GM, Purdie DM, Clouston AD, Powell EE. Non-response to antiviral therapy is associated with obesity and increased hepatic expression of suppressor of cytokine signalling 3 (SOCS-3) in patients with chronic hepatitis C, viral genotype 1. Gut. 2006;55:529–535. doi: 10.1136/gut.2005.069674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ishida H, Li K, Yi M, Lemon SM. p21-activated kinase 1 is activated through the mammalian target of rapamycin/p70 S6 kinase pathway and regulates the replication of hepatitis C virus in human hepatoma cells. J Biol Chem. 2007;282:11836–11848. doi: 10.1074/jbc.M610106200. [DOI] [PubMed] [Google Scholar]
- 84.Nielsen SU, Bassendine MF, Burt AD, Martin C, Pumeechockchai W, Toms GL. Association between hepatitis C virus and very-low-density lipoprotein (VLDL)/LDL analyzed in iodixanol density gradients. J Virol. 2006;80:2418–2428. doi: 10.1128/JVI.80.5.2418-2428.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Romero-Gómez M, Diago M, Andrade RJ, Calleja JL, Salmerón J, Fernández-Rodríguez CM, Solà R, García-Samaniego J, Herrerías JM, De la Mata M, et al. Treatment of insulin resistance with metformin in naïve genotype 1 chronic hepatitis C patients receiving peginterferon alfa-2a plus ribavirin. Hepatology. 2009;50:1702–1708. doi: 10.1002/hep.23206. [DOI] [PubMed] [Google Scholar]
- 86.Overbeck K, Genné D, Golay A, Negro F. Pioglitazone in chronic hepatitis C not responding to pegylated interferon-alpha and ribavirin. J Hepatol. 2008;49:295–298. doi: 10.1016/j.jhep.2008.03.033. [DOI] [PubMed] [Google Scholar]
- 87.Conjeevaram H, Burant CF, McKenna Harsh D, Kang H, Das AK, Everett L, White D, Lok ASF. A randomized, double-blind, placebo-controlled study of PPAR-gamma agonist pioglitazone given in combination with peginterferon and ribavirin in patients with genotype-1 chronic hepatitis C. Hepatology. 2008:384A. [Google Scholar]
- 88.Muhlestein JB, Anderson JL, Hammond EH, Zhao L, Trehan S, Schwobe EP, Carlquist JF. Infection with Chlamydia pneumoniae accelerates the development of atherosclerosis and treatment with azithromycin prevents it in a rabbit model. Circulation. 1998;97:633–636. doi: 10.1161/01.cir.97.7.633. [DOI] [PubMed] [Google Scholar]
- 89.Shimada K, Daida H, Mokuno H, Watanabe Y, Sawano M, Iwama Y, Seki E, Kurata T, Sato H, Ohashi S, et al. Association of seropositivity for antibody to Chlamydia-specific lipopolysaccharide and coronary artery disease in Japanese men. Jpn Circ J. 2001;65:182–187. doi: 10.1253/jcj.65.182. [DOI] [PubMed] [Google Scholar]
- 90.Adam E, Melnick JL, Probtsfield JL, Petrie BL, Burek J, Bailey KR, McCollum CH, DeBakey ME. High levels of cytomegalovirus antibody in patients requiring vascular surgery for atherosclerosis. Lancet. 1987;2:291–293. doi: 10.1016/s0140-6736(87)90888-9. [DOI] [PubMed] [Google Scholar]
- 91.Yamashiroya HM, Ghosh L, Yang R, Robertson AL. Herpesviridae in the coronary arteries and aorta of young trauma victims. Am J Pathol. 1988;130:71–79. [PMC free article] [PubMed] [Google Scholar]
- 92.Zhu J, Quyyumi AA, Norman JE, Costello R, Csako G, Epstein SE. The possible role of hepatitis A virus in the pathogenesis of atherosclerosis. J Infect Dis. 2000;182:1583–1587. doi: 10.1086/317613. [DOI] [PubMed] [Google Scholar]
- 93.Espinola-Klein C, Rupprecht HJ, Blankenberg S, Bickel C, Kopp H, Victor A, Hafner G, Prellwitz W, Schlumberger W, Meyer J. Impact of infectious burden on progression of carotid atherosclerosis. Stroke. 2002;33:2581–2586. doi: 10.1161/01.str.0000034789.82859.a4. [DOI] [PubMed] [Google Scholar]
- 94.Ishizaka N, Ishizaka Y, Takahashi E, Tooda Ei, Hashimoto H, Nagai R, Yamakado M. Association between hepatitis C virus seropositivity, carotid-artery plaque, and intima-media thickening. Lancet. 2002;359:133–135. doi: 10.1016/s0140-6736(02)07339-7. [DOI] [PubMed] [Google Scholar]
- 95.Ishizaka Y, Ishizaka N, Takahashi E, Unuma T, Tooda E, Hashimoto H, Nagai R, Yamakado M. Association between hepatitis C virus core protein and carotid atherosclerosis. Circ J. 2003;67:26–30. doi: 10.1253/circj.67.26. [DOI] [PubMed] [Google Scholar]
- 96.Fukui M, Kitagawa Y, Nakamura N, Yoshikawa T. Hepatitis C virus and atherosclerosis in patients with type 2 diabetes. JAMA. 2003;289:1245–1246. doi: 10.1001/jama.289.10.1245-b. [DOI] [PubMed] [Google Scholar]
- 97.Targher G, Bertolini L, Padovani R, Rodella S, Arcaro G, Day C. Differences and similarities in early atherosclerosis between patients with non-alcoholic steatohepatitis and chronic hepatitis B and C. J Hepatol. 2007;46:1126–1132. doi: 10.1016/j.jhep.2007.01.021. [DOI] [PubMed] [Google Scholar]
- 98.Boddi M, Abbate R, Chellini B, Giusti B, Solazzo V, Soft F, Pratesi G, Pratesi C, Gensini G, Zignego AL. HCV infection facilitates asymptomatic carotid atherosclerosis: preliminary report of HCV RNA localization in human carotid plaques. Dig Liver Dis. 2007;39 Suppl 1:S55–S60. doi: 10.1016/s1590-8658(07)80012-0. [DOI] [PubMed] [Google Scholar]
- 99.Petta S, Torres D, Fazio G, Cammà C, Cabibi D, Di Marco V, Licata A, Marchesini G, Mazzola A, Parrinello G, et al. Carotid atherosclerosis and chronic hepatitis C: a prospective study of risk associations. Hepatology. 2012;55:1317–1323. doi: 10.1002/hep.25508. [DOI] [PubMed] [Google Scholar]
- 100.Adinolfi LE, Restivo L, Zampino R, Guerrera B, Lonardo A, Ruggiero L, Riello F, Loria P, Florio A. Chronic HCV infection is a risk of atherosclerosis. Role of HCV and HCV-related steatosis. Atherosclerosis. 2012;221:496–502. doi: 10.1016/j.atherosclerosis.2012.01.051. [DOI] [PubMed] [Google Scholar]
- 101.Boddi M, Abbate R, Chellini B, Giusti B, Giannini C, Pratesi G, Rossi L, Pratesi C, Gensini GF, Paperetti L, et al. Hepatitis C virus RNA localization in human carotid plaques. J Clin Virol. 2010;47:72–75. doi: 10.1016/j.jcv.2009.10.005. [DOI] [PubMed] [Google Scholar]
- 102.Vassalle C, Masini S, Bianchi F, Zucchelli GC. Evidence for association between hepatitis C virus seropositivity and coronary artery disease. Heart. 2004;90:565–566. doi: 10.1136/hrt.2003.018937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Alyan O, Kacmaz F, Ozdemir O, Deveci B, Astan R, Celebi AS, Ilkay E. Hepatitis C infection is associated with increased coronary artery atherosclerosis defined by modified Reardon severity score system. Circ J. 2008;72:1960–1965. doi: 10.1253/circj.cj-08-0459. [DOI] [PubMed] [Google Scholar]
- 104.Haji SA, Starling RC, Avery RK, Mawhorter S, Tuzcu EM, Schoenhagen P, Cook DJ, Ratliff NB, McCarthy PM, Young JB, et al. Donor hepatitis-C seropositivity is an independent risk factor for the development of accelerated coronary vasculopathy and predicts outcome after cardiac transplantation. J Heart Lung Transplant. 2004;23:277–283. doi: 10.1016/S1053-2498(03)00148-7. [DOI] [PubMed] [Google Scholar]
- 105.Bedimo R, Westfall AO, Mugavero M, Drechsler H, Khanna N, Saag M. Hepatitis C virus coinfection and the risk of cardiovascular disease among HIV-infected patients. HIV Med. 2010;11:462–468. doi: 10.1111/j.1468-1293.2009.00815.x. [DOI] [PubMed] [Google Scholar]
- 106.Freiberg MS, Chang CC, Skanderson M, McGinnis K, Kuller LH, Kraemer KL, Rimland D, Goetz MB, Butt AA, Rodriguez Barradas MC, et al. The risk of incident coronary heart disease among veterans with and without HIV and hepatitis C. Circ Cardiovasc Qual Outcomes. 2011;4:425–432. doi: 10.1161/CIRCOUTCOMES.110.957415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Völzke H, Schwahn C, Wolff B, Mentel R, Robinson DM, Kleine V, Felix SB, John U. Hepatitis B and C virus infection and the risk of atherosclerosis in a general population. Atherosclerosis. 2004;174:99–103. doi: 10.1016/j.atherosclerosis.2004.01.010. [DOI] [PubMed] [Google Scholar]
- 108.Arcari CM, Nelson KE, Netski DM, Nieto FJ, Gaydos CA. No association between hepatitis C virus seropositivity and acute myocardial infarction. Clin Infect Dis. 2006;43:e53–e56. doi: 10.1086/507031. [DOI] [PubMed] [Google Scholar]
- 109.Butt AA, Xiaoqiang W, Budoff M, Leaf D, Kuller LH, Justice AC. Hepatitis C virus infection and the risk of coronary disease. Clin Infect Dis. 2009;49:225–232. doi: 10.1086/599371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Forde KA, Haynes K, Troxel AB, Trooskin S, Osterman MT, Kimmel SE, Lewis JD, Lo Re V. Risk of myocardial infarction associated with chronic hepatitis C virus infection: a population-based cohort study. J Viral Hepat. 2012;19:271–277. doi: 10.1111/j.1365-2893.2011.01545.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Amin J, Law MG, Bartlett M, Kaldor JM, Dore GJ. Causes of death after diagnosis of hepatitis B or hepatitis C infection: a large community-based linkage study. Lancet. 2006;368:938–945. doi: 10.1016/S0140-6736(06)69374-4. [DOI] [PubMed] [Google Scholar]
- 112.Guiltinan AM, Kaidarova Z, Custer B, Orland J, Strollo A, Cyrus S, Busch MP, Murphy EL. Increased all-cause, liver, and cardiac mortality among hepatitis C virus-seropositive blood donors. Am J Epidemiol. 2008;167:743–750. doi: 10.1093/aje/kwm370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lee MH, Yang HI, Wang CH, Jen CL, Yeh SH, Liu CJ, You SL, Chen WJ, Chen CJ. Hepatitis C virus infection and increased risk of cerebrovascular disease. Stroke. 2010;41:2894–2900. doi: 10.1161/STROKEAHA.110.598136. [DOI] [PubMed] [Google Scholar]
- 114.Ohsawa M, Kato K, Tanno K, Itai K, Fujishima Y, Okayama A, Turin TC, Onoda T, Suzuki K, Nakamura M, et al. Seropositivity for anti-HCV core antigen is independently associated with increased all-cause, cardiovascular, and liver disease-related mortality in hemodialysis patients. J Epidemiol. 2011;21:491–499. doi: 10.2188/jea.JE20100187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lee MH, Yang HI, Lu SN, Jen CL, You SL, Wang LY, Wang CH, Chen WJ, Chen CJ. Chronic hepatitis C virus infection increases mortality from hepatic and extrahepatic diseases: a community-based long-term prospective study. J Infect Dis. 2012;206:469–477. doi: 10.1093/infdis/jis385. [DOI] [PubMed] [Google Scholar]
- 116.Younossi ZM, Braun WE, Protiva DA, Gifford RW, Straffon RA. Chronic viral hepatitis in renal transplant recipients with allografts functioning for more than 20 years. Transplantation. 1999;67:272–275. doi: 10.1097/00007890-199901270-00015. [DOI] [PubMed] [Google Scholar]
- 117.Neal KR, Ramsay S, Thomson BJ, Irving WL. Excess mortality rates in a cohort of patients infected with the hepatitis C virus: a prospective study. Gut. 2007;56:1098–1104. doi: 10.1136/gut.2006.113217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kristiansen MG, Løchen ML, Gutteberg TJ, Mortensen L, Eriksen BO, Florholmen J. Total and cause-specific mortality rates in a prospective study of community-acquired hepatitis C virus infection in northern Norway. J Viral Hepat. 2011;18:237–244. doi: 10.1111/j.1365-2893.2010.01290.x. [DOI] [PubMed] [Google Scholar]
- 119.Tien PC, Schneider MF, Cole SR, Cohen MH, Glesby MJ, Lazar J, Young M, Mack W, Hodis HN, Kaplan RC. Association of hepatitis C virus and HIV infection with subclinical atherosclerosis in the women’s interagency HIV study. AIDS. 2009;23:1781–1784. doi: 10.1097/QAD.0b013e32832d7aa8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Mostafa A, Mohamed MK, Saeed M, Hasan A, Fontanet A, Godsland I, Coady E, Esmat G, El-Hoseiny M, Abdul-Hamid M, et al. Hepatitis C infection and clearance: impact on atherosclerosis and cardiometabolic risk factors. Gut. 2010;59:1135–1140. doi: 10.1136/gut.2009.202317. [DOI] [PubMed] [Google Scholar]
- 121.Bilora F, Rinaldi R, Boccioletti V, Petrobelli F, Girolami A. Chronic viral hepatitis: a prospective factor against atherosclerosis. A study with echo-color Doppler of the carotid and femoral arteries and the abdominal aorta. Gastroenterol Clin Biol. 2002;26:1001–1004. [PubMed] [Google Scholar]
- 122.Bilora F, Campagnolo E, Rinaldi R, Rossato A, Arzenton M, Petrobelli F. Carotid and femoral atherosclerosis in chronic hepatitis C: a 5-year follow-up. Angiology. 2008;59:717–720. doi: 10.1177/0003319707311536. [DOI] [PubMed] [Google Scholar]
- 123.Miyajima I, Kawaguchi T, Fukami A, Nagao Y, Adachi H, Sasaki S, Imaizumi T, Sata M. Chronic HCV infection was associated with severe insulin resistance and mild atherosclerosis: a population-based study in an HCV hyperendemic area. J Gastroenterol. 2013;48:93–100. doi: 10.1007/s00535-012-0610-3. [DOI] [PubMed] [Google Scholar]
- 124.Hsu CS, Kao JH, Chao YC, Lin HH, Fan YC, Huang CJ, Tsai PS. Interferon-based therapy reduces risk of stroke in chronic hepatitis C patients: a population-based cohort study in Taiwan. Aliment Pharmacol Ther. 2013;38:415–423. doi: 10.1111/apt.12391. [DOI] [PubMed] [Google Scholar]
- 125.Younossi ZM, Stepanova M, Nader F, Younossi Z, Elsheikh E. Associations of chronic hepatitis C with metabolic and cardiac outcomes. Aliment Pharmacol Ther. 2013;37:647–652. doi: 10.1111/apt.12234. [DOI] [PubMed] [Google Scholar]
- 126.Marchesini G, Ronchi M, Forlani G, Bugianesi E, Bianchi G, Fabbri A, Zoli M, Melchionda N. Cardiovascular disease in cirrhosis--a point-prevalence study in relation to glucose tolerance. Am J Gastroenterol. 1999;94:655–662. doi: 10.1111/j.1572-0241.1999.00931.x. [DOI] [PubMed] [Google Scholar]
- 127.Stoll G, Bendszus M. Inflammation and atherosclerosis: novel insights into plaque formation and destabilization. Stroke. 2006;37:1923–1932. doi: 10.1161/01.STR.0000226901.34927.10. [DOI] [PubMed] [Google Scholar]
- 128.Tardif KD, Waris G, Siddiqui A. Hepatitis C virus, ER stress, and oxidative stress. Trends Microbiol. 2005;13:159–163. doi: 10.1016/j.tim.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 129.Peng YS, Chiang CK, Hsu SP, Pai MF, Hung KY, Kao JH. Influence of hepatitis C virus infection on soluble cellular adhesion molecules in hemodialysis patients. Blood Purif. 2005;23:106–112. doi: 10.1159/000083204. [DOI] [PubMed] [Google Scholar]
- 130.Kono Y, Hayashida K, Tanaka H, Ishibashi H, Harada M. High-density lipoprotein binding rate differs greatly between genotypes 1b and 2a/2b of hepatitis C virus. J Med Virol. 2003;70:42–48. doi: 10.1002/jmv.10372. [DOI] [PubMed] [Google Scholar]
- 131.Zignego AL, Craxì A. Extrahepatic manifestations of hepatitis C virus infection. Clin Liver Dis. 2008;12:611–636, ix. doi: 10.1016/j.cld.2008.03.012. [DOI] [PubMed] [Google Scholar]
- 132.Targher G, Day CP, Bonora E. Risk of cardiovascular disease in patients with nonalcoholic fatty liver disease. N Engl J Med. 2010;363:1341–1350. doi: 10.1056/NEJMra0912063. [DOI] [PubMed] [Google Scholar]
- 133.Caliskan Y, Oflaz H, Pusuroglu H, Boz H, Yazici H, Tamer S, Karsidag K, Yildiz A. Hepatitis C virus infection in hemodialysis patients is not associated with insulin resistance, inflammation and atherosclerosis. Clin Nephrol. 2009;71:147–157. doi: 10.5414/cnp71147. [DOI] [PubMed] [Google Scholar]