

Abstract
Many biochemical traits are recognised as risk factors, which contribute to or predict the development of disease. Only a few are in widespread use, usually to assist with treatment decisions and motivate behavioural change. The greatest effort has gone into evaluation of risk factors for cardiovascular disease and/or diabetes, with substantial overlap as ‘cardiometabolic’ risk. Over the past few years many genome-wide association studies (GWAS) have sought to account for variation in risk factors, with the expectation that identifying relevant polymorphisms would improve our understanding or prediction of disease; others have taken the direct approach of genomic case-control studies for the corresponding diseases. Large GWAS have been published for coronary heart disease and Type 2 diabetes, and also for associated biomarkers or risk factors including body mass index, lipids, C-reactive protein, urate, liver function tests, glucose and insulin. Results are not encouraging for personal risk prediction based on genotyping, mainly because known risk loci only account for a small proportion of risk. Overlap of allelic associations between disease and marker, as found for low density lipoprotein cholesterol and heart disease, supports a causal association, but in other cases genetic studies have cast doubt on accepted risk factors. Some loci show unexpected effects on multiple markers or diseases. An intriguing feature of risk factors is the blurring of categories shown by the correlation between them and the genetic overlap between diseases previously thought of as distinct. GWAS can provide insight into relationships between risk factors, biomarkers and diseases, with potential for new approaches to disease classification.
Introduction
Clinical chemistry has developed from an initial focus on diagnostic tests into a combination of predictive, diagnostic and monitoring roles. Over time, quantitative biochemical tests have played an increasing role in epidemiology and some have been identified as predictors or ‘risk factors’ for disease. Biomarkers or risk factors have also been widely used in genetic research, because the genetics of risk factors should give insight into the genetics of disease. Both for quantitative risk factor studies and for case-control comparisons, identification of genes or loci whose variation is associated with variation in risk should lead to identification of pathways to disease and to opportunities for dietary, lifestyle or pharmacological interventions to reduce the incidence of disease.
This review focuses on polygenic effects on disease risk or quantitative traits related to risk. The term ‘cardiometabolic’ is intended to cover cardiovascular and metabolic disease, including diabetes- and obesity-related traits and biomarkers known to be associated with risk. Genetic variants with large effects, such as those producing familial hypercholesterolaemia, familial combined hyperlipidaemia, or the monogenic forms of diabetes, are not considered in detail because relevant information can be found elsewhere.1–4
A distinction should be made between causative risk factors, which contribute to the disease process and for which interventions which affect the risk factor will change the incidence of disease, and biomarkers which are not necessarily causative but usefully reflect current or future disease. Interventions which change biomarker results may or may not change the incidence of disease. Genetic studies can help to clarify the distinction between causative risk factors and non-causative biomarkers.
One of the earliest and best-known of the studies which have followed cohorts of subjects recruited from the general population over time, and assessed outcomes in relation to initial characteristics, is the Framingham Heart Study. This has been running for over 60 years and is studying grandchildren of the original participants. Their objective has been “to identify the common factors or characteristics that contribute to cardiovascular disease by following its development over a long period of time in a large group of participants who had not yet developed overt symptoms”.5 Success in identifying such ‘common factors’ led to a scoring system and to risk-driven interventions which have made a substantial contribution to decreasing cardiovascular mortality. For example, Australian data show that age-standardised mortality from coronary heart disease has decreased by over 80% in both men and women since about 1970.6 Numerous studies have concluded that around half the decrease in mortality is due to improvement in risk factors (see 7, particularly their Figure 2). Therefore, epidemiological studies can lead not only to understanding or risk prediction, but to successful policies for intervention and disease prevention.
Figure 2.

Type 2 diabetes loci and glycaemic control (from data in 69). Loci are listed in increasing order of p-values for Type 2 diabetes). HOMA-IR measures insulin resistance and HOMA-B measures islet beta-cell function. Filled cells indicate p < 10−3 for the most significant SNP for that locus and phenotype.
Hundreds of characteristics have been implicated as risk factors by prospective epidemiological studies, and the term has entered the language. It is intriguing that quantitative cardiovascular markers have been more successful than biomarkers or risk factors for other conditions such as cancers, susceptibility to infectious diseases, or psychiatric diseases. This may reflect the difference in investment or in the nature of the disease. Biomarkers have also been useful for defining risk of Type 2 diabetes. Here the known risk factors are much more closely linked to the definition of disease because a high glucose (subject to some caveats) defines diabetes. The glucose tolerance test, glycated haemoglobin, and measures of insulin sensitivity are all closer to the core of diabetes than cholesterol is to coronary heart disease.
Initial risk factors recognised for development of cardiovascular disease were lipids and blood pressure, and for diabetes fasting or post-challenge glucose results. Obesity is associated with increased risk of both. Many of the known quantitative risk factors for ‘cardiometabolic’ disease are not obviously associated with atherosclerosis or glucose homeostasis but they nevertheless predict mortality, cardiovascular disease or Type 2 diabetes. In particular, common liver function tests (gamma-glutamyl transferase (GGT),8,9 alanine and aspartate aminotransferases (ALT, AST),10–12 butyrylcholinesterase,13–15 bilirubin16) predict mortality or onset of disease although they do not directly cause it. Similarly, triglycerides,17–19 urate20,21 and homocysteine22 are associated with cardiometabolic risk, although their associations may become non-significant when other risk factors are included as covariates in data analysis.
The search for novel biomarkers which might add value to the Framingham score, or increase the number of epidemiologically proven risk factors, continues,23 but attempts to improve prediction with quantitative tests on serum have not been effective.24 If common diseases are heritable, then identification of genetic markers would add to the range of potential biomarkers and might improve our ability to assess risk.
Evidence for Genetic Effects on Risk Factors and on Disease
Most common diseases and their risk factors have been the subject of twin or family studies that have demonstrated significant heritability. Exact estimates vary between studies, with typical findings including 0.61–0.83 (or 61% to 83% of variance) for low-density lipoprotein cholesterol (LDL-C), 0.62–0.75 for high-density lipoprotein cholesterol (HDL-C), 0.48–0.71 for triglycerides,25 0.52 and 0.49 for GGT,26,27 0.30–0.61 for C-reactive protein (CRP),28–30 0.73 for urate31 and 0.44–0.63 for homocysteine.32–34 The existence of significant genetic influences on risk factor and biomarker values implies either that the risk of disease is heritable, or that there is heritable variation in the risk factor which is not related to risk of disease.
The former is certainly true; most common diseases have a genetic component, usually demonstrated by the risk for siblings of patients being greater than for the general population. Studies on coronary heart disease have included analysis of data on first-degree relatives,35 twin pairs,36,37 parents and offspring,38 and similarity of offspring to their natural versus adoptive parents.39 Each of these has shown a genetic component to coronary heart disease risk, with heritability estimates around 0.4 to 0.6 and greater for disease occurring at younger ages. For Type 2 diabetes, concordance rates in monozygotic twin pairs reached 75% after 15 years observation,40 and heritability estimates of around 0.7 have been reported.41,42
The genetic effects on risk factors can of course differ from those for actual disease, so conclusions based on risk factors alone must be approached with caution. The possibility that there is genetic variation affecting only the risk factor (and not the disease) is also relevant from a laboratory perspective. Such variation can, if it is substantial enough, be taken into account by genotype-specific reference ranges.
The heritability estimates vary between studies and have a degree of uncertainty, both because of limited sample size and because of variation with age or due to interactions with unmeasured demographic factors. However, demonstration of significant heritability in both biomarkers and disease risk has justified the search for genes or loci where variation contributes to the overall genetic effect.
Genome-Wide Association Studies (GWAS)
The principles of genetic association studies are well-known and many reviews or commentaries on this approach are available.43 Very briefly, a sub-set of the known polymorphisms, in practice of single-nucleotide polymorphisms (SNPs), across the entire genome is selected for their ability to ‘tag’ regions of each chromosome which constitute linkage disequilibrium blocks. These SNPs (initially about 300,000, now up to 5 million) are genotyped for each study participant using allele-specific probes immobilised on a genotyping chip. As a rough guide, the cost of genotyping chips was initially around $1000 per sample and is now around $100, depending on the number of SNPs included. With increasing knowledge of the patterns of linkage disequilibrium across the genome, and of common haplotypes, the genotypes of many untyped SNPs can be imputed and sometimes this results in discovery of loci which did not show significant results for the set of genotyped SNPs. Associations between the genotype (or more commonly the allele count) at each SNP and the phenotype (a quantitative characteristic of each subject, or their case/control status, adjusted where necessary for covariates) are computed. Because a very large number of possibilities for association are tested, a stringent p-value for significance (usually 5 × 10–8, the usual p <0.05 divided by a million for the estimated number of independent loci) is applied. This means that substantial numbers of subjects (several thousand) are needed to give adequate power to detect small effects (such as 1% of variance for a quantitative phenotype and a relative risk around 1.2 for a disease).
Because a locus may contain more than one independent effect, conditional analysis (repeating the association analysis but including one or many SNPs already known to be significant as covariates) may reveal more variants. For many of the conditions or phenotypes discussed, rare gene variants or mutations with large effects were known before the GWAS era. GWAS has often detected smaller effects associated with common variants in the same genes.
GWAS can identify a chromosomal location or a linkage disequilibrium block, but the block will often cover multiple genes and it can be difficult to decide which is the relevant one. If the association is found near an obvious gene, such as variation at CRP affecting serum C-reactive protein or variation near TF affecting serum transferrin, there is little problem. Otherwise, it may be necessary to type more SNPs across the region to see whether more significant and possibly more biologically relevant results are achieved, or to test whether variants affect gene expression by direct experiment or by searching published data.
Combination of data from multiple studies through meta-analysis, sometimes including over 100,000 subjects, allows detection of small effects which would not be found by any single study. This is illustrated by Figure 1. Because of the small contributions of individual loci to heritability, meta-analysis has become an indispensable tool in genetic association studies. The realisation that individual studies would have no hope of discovering the range of loci accessible through combining data has led to a cultural shift towards collaboration and towards deposition of data for other researchers to use.
Figure 1.
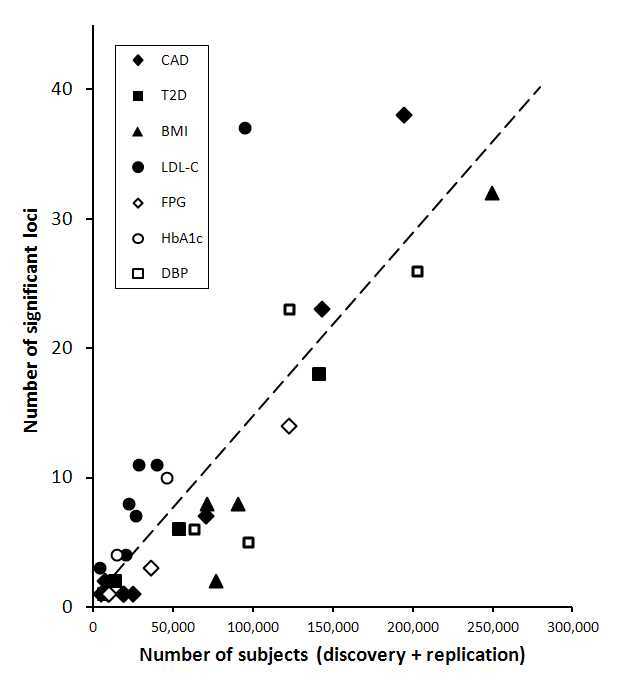
Relationship between study size and number of loci shown to be genome-wide significant, for coronary artery disease (CAD), type 2 diabetes (T2D), and their risk factors body mass index (BMI), LDL cholesterol (LDL-C), fasting plasma glucose (FPG), glycated haemoglobin (HbA1c) and diastolic blood pressure (DBP).
Some technical issues are relevant to an understanding of GWAS results. Low-frequency SNPs (with minor allele frequency below about 5%) were not selected for inclusion in the first generation of GWAS chips, but this is changing. However the effects associated with low-frequency SNPs will not be detectable unless either their effect sizes or the number of subjects are large. Genome-wide-significant SNPs discovered so far only account for a few percent of variation, giving rise to a ‘missing heritability’ problem, but there are strong indications that most uncharacterised genetic variation is due to multiple SNPs of individually small effect which studies are under-powered to detect.
Another consideration, particularly relevant for a review, is that later studies tend to include all data from earlier studies and it is therefore most relevant to cite and discuss recent ones. Because of the widespread use of stringent p-values, and the requirement for replication of novel results in independent cohorts, later studies nearly always confirm results from earlier ones and therefore displace them.
The location of GWAS findings, relative to genes, has attracted some attention. Genome-wide significance is often found, because of linkage disequilibrium, across a considerable region but it is the location (and possible functional significance) of the most significant SNP which is of interest. Lead SNPs might be concentrated in gene exons and introns, or in 5′ and 3′ regions close to genes, or away from any gene. Examples of all these are found, but there is an enrichment of significant SNP associations in or near known genes, particularly in the 5′ untranslated region, and a below-average occurrence in inter-genic regions.44 Usually, each of the lead SNPs only contributes 1 or 2% of the overall variance but there are several examples of what might be called ‘oligogenic’ effects. These often occur at a locus coding for a protein whose plasma concentration is the phenotype analysed, such as butyrylcholinesterase45 and transferrin,46 but it may also occur at a rate-limiting step in the metabolism of the phenotype molecule, such as UGT1A1 variation affecting bilirubin concentration.47
The first generation of GWAS for most common diseases or their risk factors and biomarkers is drawing to a close. Genotyping of selected SNPs may perhaps be replaced by whole-genome sequencing, but chip-based SNP genotyping is robust and cheap and imputation of uncommon variants continues to improve. Some chips have been designed to emphasise dense genotyping near genes identified in early GWAS or candidate genes for groups of related conditions, while others concentrate on the whole exome. The initial focus on variants found in people of European descent has decreased, and many studies on people of African or Asian descent are being published. Apart from the need to extend any benefits from GWAS to people of all ancestries, comparison of results across populations with differing polymorphisms or differing patterns of linkage disequilibrium can help to identify relevant genes within a locus and has identified additional loci as relevant for disease.
A useful compilation of GWAS results can be found at the website of the National Human Genome Research Institute.48 This can be searched by SNP, gene or chromosomal location, or by the disease or trait of interest. A karyogram summarising all GWAS hits, and hits for selected conditions, is available and periodically updated.49
Very large amounts of money and time have been invested in GWAS for many diseases. The expectations were that this would lead to discovery of novel loci, genes and pathways which contribute to disease and that prediction of disease risk could be improved by adding genetic data to existing risk assessment algorithms. On the whole, the discovery expectations are being met but risk predictions for common polygenic disease are not usefully improved by adding genetic information. One outcome which was not appreciated is the value of genetic information for addressing the traditional epidemiological question of distinguishing between correlation and causation.
Gene-Disease Associations
Coronary Artery Disease
Coronary artery disease was one of seven conditions included in an early case-control GWAS by the Wellcome Trust Case-Control Consortium (WTCCC).50 With approximately 2000 cases for each disease and 3000 controls free from any of the diseases (or 15,000 controls if a disease-specific perspective is taken), it was powered to have an 80% chance of detecting loci conferring a relative risk of 1.5 or more. The results for coronary heart disease showed only one significant locus, near CDKN2A and CDKN2B on chromosome 9. The most significant SNP, identified as rs1333049, showed p = 1.8×10−14, with odds ratios (relative to homozygotes for the non-risk allele) of 1.47 for heterozygotes and 1.9 for homozygotes for the risk-increasing allele. This locus was also associated with risk of Type 2 diabetes; subsequent reports soon replicated the coronary heart disease association and showed significant associations in the same region (but not always for the same SNPs) for a wide range of diseases including aneurysm, heart failure, stroke, Type 2 diabetes, melanoma and glioma.
Subsequent findings about this 9p21 region are instructive, and as the authors of one of the papers addressing its functional significance say, they “demonstrate the utility of genome-wide association study findings in directing studies to novel genomic loci and biological processes important for disease aetiology”.51 It was not obvious how CDKN2A or CDKN2B variation could affect coronary heart disease or the other diseases for which associations were found in this region. There is no association between this locus and known risk factors, and the most significant SNP is about 100 kilobases from CDKN2B, the closer of the two genes. It was subsequently found that the CDKN2A/CDKN2B region containing the significant SNPs for coronary heart disease affects expression of both these genes, and also of ANRIL or CDKN2B-AS1 (which overlaps with CDKN2B and with the coronary heart disease locus, and codes for a long non-coding RNA).51–53 The proposal is that variation in the coronary heart disease SNPs affects the response of CDKN2B to interferon signalling and therefore changes the response of endothelial cells to inflammation,51 though this is still open to question.54
Returning to GWAS for coronary heart disease, combination of data for large meta-analyses has now identified many more loci. Analysis of data from around 22,000 cases and 65,000 controls, followed by genotyping of another 56,000 people, confirmed 10 reported loci and identified 13 new ones.55 A further increase in meta-analysis size to include 64,000 cases and 130,000 controls found 15 novel loci,56 for a total of 46. Many of these loci contained independent effects from SNPs which were not strongly associated with each other (low linkage disequilibrium between them). Despite the substantial number of significant loci, they only account for a small proportion of the genetic variation in risk; about 6–10% depending on criteria used.
The potential for false negative results from GWAS can be appreciated in the association between variation at the LPA locus, (coding for lipoprotein (a)) and coronary heart disease. This locus has long been known to affect the concentration of Lp(a) in plasma, and several reports of association with cardiovascular risk have shown that SNPs affecting Lp(a) concentration are associated with substantial variation in coronary heart disease risk.57–59 A combination of low minor allele frequency and poor tagging of the relevant variants by SNPs included on GWAS chips led to failure to identify this locus in early genome-wide studies.
Several approaches have been used to extract information from the accumulated body of information on allelic associations with coronary heart disease risk (as opposed to examination of individual loci). These have included comparisons between the loci for coronary heart disease and those for diabetes or for known risk factors for coronary heart disease. Another approach is to examine the list of significant, suggestive, or possibly true associations (selected using varying thresholds of statistical significance) for common features related to gene functions, or association with known pathways or processes, in the hope of confirming or discovering precursors of disease. For coronary heart disease, the most recent GWAS publication56 took both these routes. Genes whose variation affects coronary heart disease also tend to have reported associations with lipids and blood pressure, but not with diabetes or glucose homeostasis. Associating coronary heart disease-related genes to cellular or biochemical pathways, using a more relaxed p-value to include more of the potentially relevant genes, showed positive and biologically plausible results for lipid metabolism, morphology of atherosclerotic lesions, immune cell migration or adhesion, and inflammation.
Other Cardiovascular Conditions
Other cardiovascular diseases, which overlap with coronary heart disease in their conventional risk factor profile, show only limited genomic overlap. GWAS for ischaemic stroke60,61 have shown a distinction between sub-types, with different genes being implicated in large-vessel disease (HDAC9, an intergenic region at chromosome 6p21.1, and the chromosome 9p21 CDKN2B-AS1 locus discussed above) and cardioembolic stroke (PITX2 and ZFHX3, also associated with atrial fibrillation). Other loci have been reported as significant but not replicated. Ischaemic stroke provides an interesting example of sub-classification improving the outcome of genetic association studies, and conversely of GWAS reinforcing the existence of subtypes of a disease.
Large studies on hypertension, or on continuous variation in blood pressure, have now identified 29 independent effects at 28 loci on either systolic or diastolic blood pressure.62 Compared to other GWAS results, the 29 effects accounted for a rather small proportion of variation (<1%) in either diastolic or systolic blood pressure. Most loci (22 out of 28) were not near genes which might have been expected on the basis of previous knowledge about their biology. Nearly all loci affect both systolic and diastolic pressures, although three have been shown to affect them in opposite directions.63 Overlap between loci affecting blood pressure and other conditions was assessed by computing a genomic risk score from genotypes at the significant loci for blood pressure, and this score was significantly associated not only with hypertension but with left ventricular wall thickness, stroke and coronary heart disease, though not with kidney disease.62
One study has identified loci affecting incident heart failure using combined data from four prospective studies,64 with different associations in European-ancestry and African-ancestry groups. Only the association with USP3 in the European group reached the standard threshold of p < 5 × 10−8.
Several reports have appeared on abdominal aortic aneurysm, with four significant loci identified. The 9p21 CDKN2BAS1 locus showed significant results for abdominal aortic aneurysm and (unlike the other loci) suggestive association with intracranial aneurysm.65 Other loci include an LDL-receptor-associated protein, LRP1; this locus did not show associations with coronary heart disease or lipids but there was evidence for a functional role in aortic tissue.66 Another was in the region of FBN1, which is associated with Marfan’s syndrome,67 while the fourth within DAB2IP was associated with coronary heart disease and peripheral arterial disease but not with conventional coronary heart disease risk factors.68
A number of genes known for effects on other diseases or biochemical characteristics have been found among those significant for cardiovascular conditions. For coronary heart disease, the lipid-related loci are assumed to act through effects on the classical risk factor LDL, but the presence of the ABO blood group locus (which has been shown to affect a surprisingly wide range of characteristics) is unexplained. For blood pressure, MTHFR and HFE are well-known for affecting homocysteine- and iron-related phenotypes. However the MTHFR effect may well be due to variation in the nearby gene NPPB, which codes for natriuretic peptide precursor. The reported SNP for the HFE effect on blood pressure62 was rs1799945 (H63D), rather than rs1800562 (C282Y) which has larger effects on iron, lipids and coronary heart disease.
Type 2 Diabetes and Metabolic Syndrome
Type 2 diabetes was one of the conditions covered in the early (and in retrospect under-powered) WTCCC study.50 It found significant associations for Type 2 diabetes at only one location, TCF7L2, although two previously recognised loci showed supportive results. Since then several rounds of meta-analyses have expanded the number of Type 2 diabetes loci to 63, estimated to account for about 6% of variance in disease risk.69 Many of these loci (though not necessarily the same SNPs70) are associated with other metabolic traits, particularly glucose and insulin but also with adiposity, lipids and CRP. A meta-analysis focusing on glucose and insulin found or confirmed 53 loci of which 33 also showed evidence (at a false discovery rate of 0.05) for affecting Type 2 diabetes.71 The diabetes loci showed a mix of effects on beta-cell function and insulin resistance, with more of the former.70,71 Figure 2 summarises the overlap of loci for Type 2 diabetes, glucose, beta-cell function (HOMA-B) and insulin resistance (HOMA-IR).
Overlap of genetic loci for Type 2 diabetes with those for Type 1 has been sought by a number of studies. On the whole, there is little overlap of susceptibility loci and SNPs associated with Type 2 diabetes do not predict development of Type 1.72,73 Variants near GLIS3 are associated with both, probably through increased beta-cell apoptosis.74 On the other hand, the genes whose variation can cause maturity-onset diabetes of the young (MODY) are well-represented among the Type 2 diabetes GWAS findings; hepatic nuclear factors (HNF1A, HNF1B, HNF4A) and PAX4 contribute to Type 2 diabetes risk while GCK and PDX1 are associated with fasting plasma glucose and also (for GCK) with glycated haemoglobin and metabolic syndrome.
The related condition of metabolic syndrome has been subject to fewer studies. One difficulty is knowing whether it is best to define the condition as present or absent according to the IDF75 or earlier criteria, and perform a case-control study, or to attempt a multivariate assessment based on the underlying quantitative measures. A moderately large study combining these approaches76 found no genome-wide significant results for the syndrome but many loci were significant for pairs of the underlying traits. Interpretation of such associations when the pairings are already known from conventional epidemiology, for example HDL-C and triglycerides, is difficult. A subsequent study comparing metabolic syndrome (but non-diabetic) cases with controls found one locus, APOA1/C3/A4/A5, to be significantly associated with the syndrome itself and with multiple lipid phenotypes.77 Many loci affected one or two of the metabolic syndrome phenotypes (adiposity, dyslipidaemia, impaired glycaemic control, blood pressure) but there was a lack of loci crossing all these domains. The issues of how far metabolic syndrome overlaps genetically with Type 2 diabetes, and whether it is a single genetic entity, remain open.
The issue of genetic factors affecting risk of complications of diabetes is potentially important but significant results have only been reported for Type 1 diabetes, perhaps because such patients are typically at risk for a longer time than those with Type 2. One case-control study, involving some 11,000 patients with or without end-stage renal disease, found two significant loci and a number of others whose effects did not reach significance with the numbers available.78 Such studies on specific complications of common disease, or on penetrance of disease in conditions where a monogenic precondition for disease is known, are likely to increase. Very large numbers of people are monitored for chronic conditions so the phenotype data are potentially available, and if DNA can be collected systematically then the cost of genotyping is small in relation to the costs of diabetes complications and other chronic conditions.
Biomarker Associations
Results for GWAS or GWAS meta-analyses of the most relevant risk factors or biomarkers for cardiometabolic conditions are summarised in Table 1. To some extent, variation in the number of known significant SNPs and the proportion of variation explained is due to variation in the number of people included, which in turn reflects the cost and perceived importance of assessing the phenotype.
Table 1.
Allelic associations at p < 5 × 10−8, from the largest dataset for each phenotype, sorted alphabetically by gene name.
| Phenotype | Source | Genes or locations | Percent of variance |
|---|---|---|---|
| Lipids | 80 | LDL-C: ABCG5-ABCG8, ABO, ANGPTL3-DOCK7, APOA1-APOC3-APOA4-APOA5, APOB, APOE-APOC1-APOC2, BRAP, CBLN3-KIAA0323, CELSR2-PSRC1-SORT1, CETP, CSPG3-CILP2-PBX4, CYP7A1, DNAH11, FADS1-FADS2-FADS3, FRK, GPAM, HFE-HIST1H4C, HLA, HMGCR, HNF1A, HP-HPR-DHX38, IDOL, IRF2BP2-TOMM20, LDLR, LPA, MAFB, MOSC1, NPC1L1, OSBPL7, PCSK9, PLEC1, PPP1R3B, ST3GAL4, TIMD4-HAVCR1, TMEM57-LDLRAP1, TOP1, TRIB1 |
12% |
| HDL-C: ABCA1, ABCA8, ADM-AMPD3, ANGPTL4, APOA1-APOC3-APOA4-APOA5, APOB, APOE-APOC1-APOC2, ARL15, C6orf106, CCDC92-ZNF664, CETP, CITED2, CMIP, COBLL1, DOCK6-LOC55908, FADS1-FADS2-FADS3, GALNT2, HNF4A, IRS1, KLF14, LACTB, LCAT, LILRA3-LILRB2, LIPC, LIPG, LPA, LPL, LRP1, LRP4-NR1H3, MACF1-PABPC4, MLXIPL, MMAB-MVK, PDE3A, PGS1, PLTP, PPP1R3B, RPS3A-MC4R, SBNO1, SCARB1, SLC39A8, STARD3, TRIB1, TRPS1, TTC39B, UBASH3B, UBE2L3, ZNF648 |
12% | ||
| Triglycerides: AFF1-KLHL8, ANGPTL3-DOCK7, ANKRD55-MAP3K1, APOA1-APOC3-APOA4-APOA5, APOB, APOE-APOC1-APOC2, CAPN3, CCDC92-ZNF664, CETP, COBLL1, CSPG3-CILP2-PBX4, CTF1, CYP26A1, FADS1-FADS2-FADS3, FRMD5, GALNT2, GCKR, HLA, IRS1, JMJD1C, LIPC, LPL, LRP1, MLXIPL, MSL2L1, NAT2, PINX1-XKR6, PLA2G6, PLTP, TIMD4-HAVCR1, TRIB1, TYW1B |
10% | ||
| CRP | 117 | APOC1, ASCL1, BCL7B, CRP, GCKR, GPRC6A, HNF1A, HNF4A, IL1F10, IL6R, IRF1, LEPR, NLRP3, PABPC4, PPP1R3B, PTPN2, RORA, SALL1 | 5% |
| Liver enzymes: GGT | 118 | GGT: ATP8B1, C14orf73, C2orf16-GCKR, CCBL2-PKN2, CD276, CDH6, CEPT1-DENND2D, DDT-DDTL-GSTT1-GSTT2B-MIF, DLG5, DPM3-EFNA1-PKLR, DYNLRB2, EFHD1-LOC100129166, FLJ37644-SOX9, FUT2, GGT1-GGTLC2, HNF1A-C12orf27, ITGA1, MICAL3, MLIP, MLXIPL, MYO1B-STAT4, NEDD4L, RORA, RSG1-EPHA2, SLC2A2, ZNF827 |
2% |
| Liver enzymes: ALT | 118 | ALT: CPN1, HSD17B13-MAPK10, PNPLA3-SAMM50, TRIB1 |
0.1% |
| Liver enzymes: BCHE | 45 | BCHE: BCHE, PPP1R3B-TNKS, RAPH1-ABI2, RNPEP, UGT1A1 |
22% |
| Bilirubin | 47 | SLCO1B3,UGT1A1 | 19% |
| Urate | 119 | A1CF-ASAH2, ABCG2, ACVR1B-ACVRL1, ATXN2-PTPN11, B3GNT4, BAZ1B-MLXIPL, BCAS3-C17orf82, GCKR, HLF, HNF4G, IGF1R, INHBB, INHBC-INHBE, MAF, NFAT5, NRXN2-SLC22A12, ORC4L-ACVR2A, OVOL1-LTBP3, PDZK1, PRKAG2, RREB1, SFMBT1-MUSTN1, SLC16A9, SLC17A1-SLC17A3, SLC22A11, SLC2A9, STC1, TMEM171, TRIM46-PKLR, UBE2Q2-NRG4, VEGFA | 7% |
| Homocysteine | 120 | CBS, CPS1, CUBN, DPEP1, FUT2, GTPB10, HNF1A, MMACHC, MTHFR, MTR, MUT, NOX4, SLC17A3 | 3% |
| BMI | 121 | BDNF, CADM2, ETV5, FAIM2, FANCL, FLJ35779-HMGCR, FTO, GNPDA2, GPRC5B-IQCK, KCTD15, LRP1B, LRRN6C, MAP2K5-LBXCOR1, MC4R, MTCH2-NDUFS3-CUGBP1, MTIF3-GTF3A, NEGR1, NRXN3, NUDT3-HMGA1, PRKD1, PTBP2, QPCTL-GIPR, RBJ-ADCY3-POMC, RPL27A-TUB, SEC16B, SH2B1-APOB48R-SULT1A2-AC138894.2-ATXN2L-TUFM, SLC39A8, TFAP2B, TMEM160-ZC3H4, TMEM18, TNNI3K, ZNF608 | 1.5% |
| Glucose | 71 | AMT, ARAP1, CDKAL1, CDKN2B, DNLZ, FOXA2, GIPR, GLS2, GRB10, IGF2BP2, IKBKAP, KL, P2RX2, PCSK1*, PDX1, PPP1R3B*, RREB1, TOP1, WARS, ZBED3 | 5% |
| Glycated haemoglobin | 122 | ANK1, ANK1, ATP11A-TUBGCP3, FN3K, G6PC2-ABCB11, GCK, HFE, HK1, MTNR1B, SPTA1, TMPRSS6 | 2% |
Note that some estimates of the percentage of variance accounted for are based on the discovery cohorts and may be over-estimates. Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase; BCHE, butyrylcholinesterase; BMI, body mass index; CRP, C-reactive protein; GGT, gamma-glutamyl transferase; HDL-C, high-density lipoprotein cholesterol; LDL-C, low-density lipoprotein cholesterol.
Some patterns can be seen in the lists of loci. The genes coding for the protein feature for butyrylcholinesterase, CRP and GGT; many genes for apolipoproteins, their receptors or enzymes of lipid metabolism are seen for the lipids; and some genes show significance for unexpected phenotypes. For lipids in particular, many of the genes known from studies on monogenic disease occur (for common SNPs) among the GWAS findings, and it is notable that two genes which have already been exploited as drug targets for treatment of high LDL-C (HMGCR and PCSK9) would have been revealed as important to cholesterol and lipoprotein metabolism by GWAS.
The loci identified for lipids account for about 10–12% of the phenotypic variance, based on data from about 100,000 people. The data for body mass index (BMI) are based on about 250,000 people but the proportion of variation explained is low, possibly because variants of large effect have been selected against. The phenotypes with substantial proportions of variance explained, bilirubin and butyrylcholinesterase, each have one variant with a substantial effect (>15%) and others with much smaller effects.
There are several intriguing aspects to these results. LDL-C is unexpectedly associated with SNPs at the ABO blood group locus and at BRCA2, HFE and UGT1A1 (more readily associated with breast cancer, haemochromatosis and bilirubin, respectively). The ABO locus has been associated with a wide range of biomarker and disease phenotypes, including myocardial infarction and coronary heart disease,55,79 which tends to support the LDL-C association. The most significant HFE SNP is rs1800562, which codes for the C282Y variant associated with haemochromatosis and with variation in iron status in the general population, and the iron-increasing A allele is associated with decreased LDL-C.80 Other nearby variants within the HLA region are significant for both LDL-C and triglycerides. The UGT1A1 locus, which controls conjugation of bilirubin, has a significant effect on butyrylcholinesterase activity45 as well as LDL-C. The BRCA2 locus recently reported to be associated with total and LDL cholesterol81 is known for its association with breast and ovarian cancer but the variant affecting total and LDL cholesterol, rs9534275, is intronic, extremely common, and not likely to affect cancer risk. The way in which it affects cholesterol is not known. Two of the loci affecting glycated haemoglobin, HFE and TMPRSS6, are known to affect iron and erythrocyte measures so the association may be with erythrocyte characteristics rather than glycaemia.
Genetic Loci Affecting Multiple Risk or Disease Phenotypes
Phenotypic Correlation Between Biomarkers
Cardiometabolic biomarkers not only share the property of risk prediction for an overlapping cluster of diseases, but they are correlated in the general population. To some extent the correlations will be due to common dependence on a known and measurable characteristic such as BMI, and to some extent on environmental or genetic variation which affects all, most or some of them. The phenotypic correlation matrix from a large Australian dataset82 is shown in Table 2; a similar correlation matrix for some other cardiac biomarkers was published by Drenos.83 At the phenotypic level there is overlap between biomarkers associated with coronary heart disease, Type 2 diabetes, obesity and metabolic syndrome. This could be due to genetic variation with effects on many of these markers, or environmental variation with effects on each. There is evidence for genetic correlation between GGT and other biomarkers or risk factors, particularly for triglycerides and apolipoproteins associated with very-low-density lipoprotein.26 Factor analysis directs attention to a number of groupings containing variables which are correlated and for which we might expect to find common genetic effects. These include the liver markers ALT, AST and GGT, together with ferritin; triglycerides and HDL-C with butyrylcholinesterase, urate and insulin; alkaline phosphatase, CRP and (inversely) bilirubin; and glucose and insulin with (inversely) LDL-C. Either multivariate analysis or GWAS of factor scores may help to identify loci with multiple effects.
Table 2.
Correlation matrix for biomarkers related to lipids, metabolic syndrome, glycaemia and liver function.*
| Urate | 0.01 | ||||||||||||
| Alkaline Phosphatase | 0.11 | −0.11 | |||||||||||
| GGT | 0.20 | 0.29 | |||||||||||
| ALT | 0.23 | 0.22 | 0.51 | ||||||||||
| AST | 0.16 | 0.08 | 0.14 | 0.35 | 0.69 | ||||||||
| Ferritin | 0.16 | 0.06 | 0.06 | 0.24 | 0.27 | 0.17 | |||||||
| CRP | 0.16 | −0.19 | 0.28 | 0.27 | 0.14 | 0.13 | |||||||
| HDL-C | −0.20 | 0.17 | −0.13 | −0.07 | −0.15 | −0.08 | −0.23 | ||||||
| LDL-C | 0.09 | 0.09 | 0.09 | −0.07 | |||||||||
| Triglycerides | 0.23 | −0.28 | 0.20 | 0.26 | 0.30 | 0.13 | 0.17 | 0.21 | −0.49 | 0.15 | |||
| BCHE | 0.23 | −0.10 | 0.20 | 0.24 | 0.26 | 0.13 | 0.18 | 0.14 | −0.17 | 0.21 | 0.34 | ||
| Glucose | 0.03 | 0.10 | 0.13 | 0.11 | 0.07 | 0.13 | −0.10 | 0.12 | 0.06 | ||||
| Insulin | 0.20 | −0.16 | 0.14 | 0.13 | 0.18 | 0.21 | −0.31 | 0.29 | 0.20 | 0.33 | |||
|
|
|||||||||||||
| Urate | Bilirubin | Alkaline Phosphatase | GGT | ALT | AST | Ferritin | CRP | HDL-C | LDL-C | Triglycerides | Butyryl-cholinesterase | Glucose | |
Correlation coefficients are shown for each pair of variables, log-transformed where appropriate and adjusted for sex and age. r ≥ 0.2 are shown in bold and correlations where r < 0.1 are omitted. Data from an Australian adult population-based sample.82
Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase; BCHE, butyrylcholinesterase; CRP, C-reactive protein; GGT, gamma-glutamyl transferase; HDL-C, high-density lipoprotein cholesterol; LDL-C, low-density lipoprotein cholesterol.
Genetic Overlap between Biomarkers
Overlap of published data across biochemical phenotypes is summarised in Table 3. Most of these loci affect multiple lipids such as LDL-C and triglycerides, or else fasting glucose and glycated haemoglobin, which is to be expected as these are to some extent measures of the same phenotype. However, the other loci with multiple effects are less straightforward.
Table 3.
Loci showing effects for multiple biomarker phenotypes.
| Chr | Mbp | Reported Gene(s) | Disease/Trait | Source |
|---|---|---|---|---|
| 1 | 40.0 | MACF1-PABPC4 | HDL-C, CRP | 80, 117 |
| 1 | 63.0 | ANGPTL3-DOCK7 | LDL-C, Triglycerides | 80 |
| 1 | 155.1 | DPM3-EFNA1-PKLR-TRIM46 | GGT, Urate | 118, 119 |
| 1 | 230.3 | GALNT2 | HDL-C, Triglycerides | 80 |
| 2 | 21.2 | APOB | HDL-C, LDL-C, Triglycerides | 80 |
| 2 | 27.7 | GCKR | Albumin, CRP, Glucose, GGT, Platelets, Triglycerides, Urate; (and Crohn’s disease, Kidney disease) | 117,123–127 |
| 2 | 165.5 | COBLL1 | HDL-C, Triglycerides | 80 |
| 2 | 169.8 | G6PC2 | FPG, Glycated haemoglobin | 128, 122 |
| 2 | 227.1 | IRS1 | HDL-C, Triglycerides | 80 |
| 4 | 103.2 | SLC39A8 | BMI, HDL-C | 121, 80 |
| 5 | 156.4 | TIMD4-HAVCR1 | LDL-C, Triglycerides | 80 |
| 6 | 26.1 | HFE | Glycated haemoglobin, LDL-C | 122, 80 |
| 6 | 160.6 | LPA | HDL-C, LDL-C | 80 |
| 7 | 44.2 | GCK | FPG, Glycated haemoglobin | 128, 122 |
| 7 | 3.0 | MLXIPL | CRP, GGT, HDL-C, Triglycerides, Urate | 117, 118, 80, 119 |
| 8 | 9.2 | PPP1R3B | CRP, HDL-C | 117, 80 |
| 8 | 19.8 | LPL | HDL-C, Triglycerides | 80, |
| 8 | 76.5 | HNF4G | BMI, Urate | 121, 119 |
| 8 | 126.5 | TRIB1 | HDL-C, LDL-C, Triglycerides | 80 |
| 11 | 61.6 | FADS1-FADS2-FADS3 | HDL-C, LDL-C, Triglycerides | 80 |
| 11 | 92.7 | MTNR1B | FPG, Glycated haemoglobin | 128, 122 |
| 11 | 116.6 | APOA1-APOC3-APOA4-APOA5 | HDL-C, LDL-C, Triglycerides | 80 |
| 12 | 57.8 | LRP1 | HDL-C, Triglycerides, Urate | 80, 119 |
| 12 | 112.0 | ATXN2-PTPN11-BRAP | LDL-C, Urate | 80, 119 |
| 12 | 121.4 | HNF1A | CRP, GGT, LDL-C, Urate | 117, 118, 80, 119 |
| 12 | 124.5 | CCDC92-ZNF664 | HDL-C, Triglycerides | 80 |
| 15 | 58.7 | LIPC | HDL-C, Triglycerides | 80 |
| 15 | 60.9 | RORA | CRP, GGT | 117, 118 |
| 16 | 57.0 | CETP | HDL-C, LDL-C, Triglycerides | 80 |
| 18 | 57.8 | MC4R | BMI, HDL-C | 121, 80 |
| 19 | 11.2 | LDLR-DOCK6-LOC55908 | LDL-C, HDL-C | 80 |
| 19 | 19.4 | CSPG3-CILP2-PBX4 | LDL-C, Triglycerides | 80 |
| 19 | 45.4 | APOE-APOC1-APOC2 | CRP, HDL-C, LDL-C, Triglycerides | 117, 80 |
| 20 | 43.0 | HNF4A | CRP, HDL-C | 117, 80 |
| 20 | 44.6 | PLTP | HDL-C, Triglycerides | 80 |
Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase; BCHE, butyrylcholinesterase; BMI, body mass index; CRP, C-reactive protein; FPG, fasting plasma glucose; GGT, gamma-glutamyl transferase; HDL-C, high-density lipoprotein cholesterol; LDL-C, low-density lipoprotein cholesterol.
APOE and the nearby APOC genes are well-known for effects on lipid metabolism and (for APOE) Alzheimer’s disease risk, although the two SNPs in APOE which determine the ε2/ε3/ε4 haplotype have differential effects on LDL-C and Alzheimer risk.84 The expected effect found at this locus is for lipids but there is also an effect on CRP, which is paradoxically in the opposite direction (alleles at this locus which increase LDL-C decrease CRP, contrary to the positive association found in the general population and their common status as risk factors).85
GCKR, which has been associated with albumin, CRP, GGT, glucose and insulin, platelet count, triglycerides and other lipids, urate, and also Crohn’s disease and kidney disease, codes for a protein which acts as a regulator of glucokinase (hexokinase) activity in the liver and regulates storage of glucose.86 This places it at an important crossroads of carbohydrate metabolism and it has been reported that SNPs within GCKR have opposing effects on triglycerides and glucose.87 Variation in GCK, which codes for the enzyme regulated by the GCKR protein, is associated with fasting plasma glucose and glycated haemoglobin in GWAS and with MODY Type 2.
Several genes of the hepatocyte nuclear factor (HNF) family occur in the list of loci with diverse effects. Variation in HNF1A affects CRP, GGT, LDL-C and urate, encompassing at least four of the domains (inflammation, liver function, lipids and purine metabolism) associated with obesity and cardiometabolic risk. SNPs in HNF4A affect CRP and HDL-C, and SNPs in HNF4G affect BMI and urate. HNFs regulate transcription of many genes, mainly but not exclusively in the liver, and are associated with the MODY forms of diabetes.88 The genes regulated by these proteins include many for carbohydrate and lipid metabolism, which fits with the diverse reported effects on cardiometabolic biomarkers.
MLXIPL variation also affects many cardiometabolic phenotypes; CRP, GGT, HDL-C, triglycerides and urate. This gene codes for a glucose-responsive transcription factor (ChREBP) which affects lipogenesis, and a recent report89 reinforces its importance in human obesity and associated metabolic abnormalities.
RORA, where variation most strongly affects CRP and GGT, is also a transcription factor and regulates expression of genes of lipid metabolism in including apolipoproteins. Gene knockout has implicated RORA in hepatic steatosis,90 perhaps accounting for its effects on GGT.
Overall, the loci which show effects on multiple cardiometabolic phenotypes are associated with control of carbohydrate and lipid metabolism, consistent with these biomarkers’ clinical and epidemiological associations.
Genotypic Overlap between Diseases
A number of examples are known where diseases thought to be distinct show substantial overlap in their genetic basis. The best examples come from mental health, where diagnosis and classification is generally based on symptoms and behaviour rather than known pathology. One way of demonstrating overlap, and of showing that it is likely to be genetic, is to establish increased risk of a comorbid condition in relatives of patients. Another, which has recently developed, is to show that a genetic risk score for one disease is associated also with risk of another. Examples of the genetic classification diverging from conventional classification of disease are starting to appear, for example between bipolar disorder and schizophrenia, extending to other psychiatric conditions to varying degrees;91 between schizophrenia and coronary heart disease;92 and among autoimmune diseases.93 A more ambitious and complex analysis, covering 161 disorders, has been put forward;94 there may be doubts about its specific findings but it illustrates the potential of data-mining from information gathered for other reasons.
Genotypic Overlap between Biomarkers and Disease
Searching for gene variants that affect known risk factors, as a surrogate for searching for variants which affect disease, has advantages but is susceptible to both false negative and false positive results. False negatives result from the existence of loci which affect disease risk through other mechanisms and do not affect the risk factor. False positive results arise if a variant affects the marker (such as LDL-C or glucose) but not the disease (coronary heart disease or Type 2 diabetes). We would expect that the effect would be transmitted from the genetic variation through the risk factor to the disease, but this is not always the case. Testing whether loci which affect putative risk factors do in fact affect the disease has become a useful way of checking for causative relationships.
The development of ever-larger meta-GWAS for coronary heart disease and Type 2 diabetes has been paralleled by meta-analyses on lipids and glycaemic control, and qualitative comparisons of significant loci have shown substantial overlap. The reported associations for coronary heart disease and LDL-C, summarised in Figure 3, reinforce the epidemiological, pathological and therapeutic evidence that LDL-C is a true risk factor. The overlaps between loci for coronary heart disease and Type 2 diabetes, or between LDL-C and Type 2 diabetes, are minimal.
Figure 3.
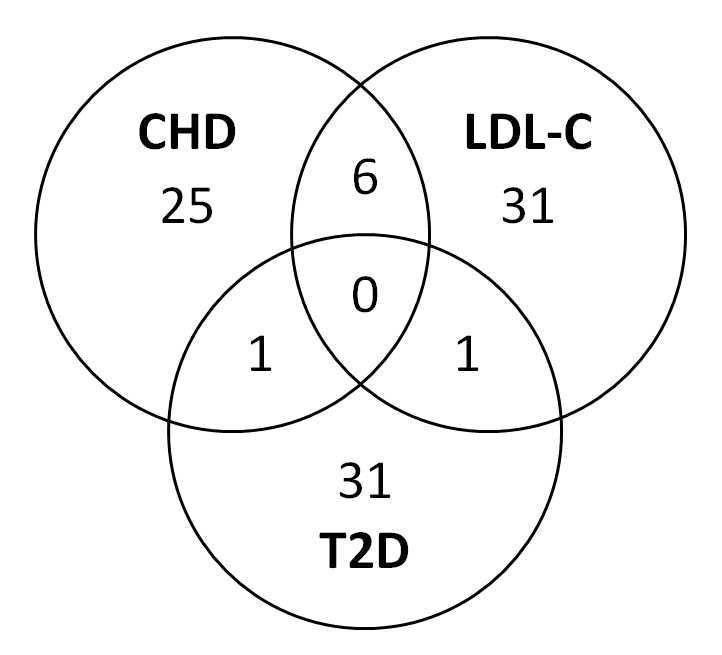
Overlap of genome-wide-significant loci for coronary heart disease (CHD), LDL cholesterol (LDL-C) and type 2 diabetes (T2D). The six significant loci affecting both CHD and LDL-C are ABO, APOA/APOC, CELSR2/SORT1, LDLR, LPA and PCSK9, that for CHD and Type 2 diabetes is CDKN2A/B, and for LDL-C and Type 2 diabetes HNF1A. Data from the National Human Genome Research Institute.48
Studies on risk factors and disease complement each other, and recently a number of such comparisons have led to the conclusion that what was thought to be a primary or causal risk factor is probably only a marker of risk. The important implication of such findings is that drugs or other interventions which change a risk marker (e.g. HDL-C) will not necessarily change the risk of disease. Examples of analyses where SNPs or genetic risk scores have been used in this way are discussed below.
Future Directions for Genetic Association Studies
As information about the human genome has expanded, particularly from haplotype data generated by sequencing in the 1000 Genomes project, it has become possible to infer genotypes at large numbers of SNPs from limited genotyping data. This has allowed refinement of information at known loci, and sometimes identified novel loci where uncommon or ungenotyped variants have significant effects. It was hoped that uncommon variants with minor allele frequencies in the range 0.1% to 5%, or family-specific variants of large effect, would account for some or most of the gap between known SNP effects and estimated heritabilities. Although there are uncommon variants with significant effects, it now seems that the unidentified or ‘missing’ heritability is probably due to variants with effects which are too small to measure accurately with feasible sample sizes. If this is so, then full sequencing of the large number of samples which would be needed to give adequate power will probably not be productive. This has recently been undertaken for HDL-C on almost 1000 people95 and results suggest that common variants (with minor allele frequency >1%) account for almost ten times as much of the variation as rarer ones.
In relation to biomarker investigations, there are a number of additional phenotypes which could usefully be the subject of genome-wide studies. Availability of high-sensitivity assays capable of measuring cardiac troponins in people who have not suffered a clinical event, and of predicting such events,96 may allow detection of further coronary heart disease risk loci. In time, imaging techniques may provide additional phenotypes for genetic association studies but the costs are probably too high to be used in purely research studies; application of genotyping to people who have such investigations for clinical reasons would be more cost-effective. Investigation of pharmacogenetic phenotypes (drug response or non-response, frequency of side-effects) through GWAS may be productive, even with moderate sample sizes. Quite large genetic effects could exist because they would not have been subject to negative selection.
Applications of GWAS Results
Results from GWAS have three main areas of application; the understanding of disease and potential discovery of drug targets; the distinction between causal risk factors and non-causal biomarkers; and clinical prediction. Out of these, improved understanding and clinical prediction of disease were expected but have only partly been realised. The application which has shown unexpected promise has been the use of genomic data to answer questions about cause and effect which have classically been the subject of controlled trials, either when controlled trials are not possible or to supplement their results.
Insight into the Biology of Disease
Genetic studies, and specifically GWAS, have improved our understanding of disease. This is most easily appreciated in relation to the roles of LDL and inflammation in atherosclerosis, and the roles of insulin resistance and beta-cell function in Type 2 diabetes, because these fit with existing knowledge. Other discoveries will require more work before an integrated story is available. It will probably take some time before we can say whether discovery of drug targets has been successful; several known targets have been rediscovered by GWAS, which is encouraging. It is too soon to expect clinical trials of drugs based on GWAS discoveries, although some existing drugs have found new indications or off-label uses as a result of genetic discoveries.
Distinction between Causal Risk Factors and Non-Causal Biomarkers
As mentioned above, SNPs which affect a causal risk factor for disease should also affect the risk of the disease. This has led to the use of genetic information to perform a type of instrumental variable analysis known (rather inaccurately) as Mendelian Randomisation (MR). The basis of this approach is to estimate whether the effect of the gene variant on the disease risk is equal to that expected from the two steps, gene to risk factor and risk factor to disease, where all the necessary relationships can be measured and all the effects of the genetic variation on disease are mediated through the risk factor. The MR approach is advocated as a substitute for randomised trials where these are impractical, because genotype is randomised through the processes of meiosis and fertilisation. The lifelong genetic status of any subject can identify or exclude reverse causation, in which for example CRP is elevated because of pre-disease states rather than elevated CRP causing the disease.
One practical difficulty is that the effect of any single SNP or locus on the risk factor will usually be small, and the relationship between the risk factor and the disease is usually weak. Therefore a strong allelic effect and a large number of subjects are necessary to provide a valid conclusion about causality. The other common problem is that it can be difficult to be sure that assumptions about paths from gene to disease are met. To improve the power of such analyses, several groups have calculated genetic risk scores incorporating effects of multiple SNPs, each known to affect the proposed risk factor. This too has dangers, particularly if the loci included affect the risk factor through differing pathways of if any of them affect several risk factors in non-uniform ways (as found for some effects on LDL-C and CRP).
A good example of the application of genetic information through MR can be found in the analysis of data on homocysteine and coronary heart disease.97 This calculated the effects of SNPs at 13 loci, accounting for around 6% of variation in homocysteine concentration, on coronary heart disease risk. Although the genetic score was associated with substantial variation in homocysteine concentration it was not associated with disease risk, placing homocysteine in the category of non-causal risk marker. A similar study, focused on the C677T variant in MTHFR, found that the allele which was associated with increased homocysteine was associated with decreased cardiovascular disease risk, emphasising the potential for unexpected outcomes from MR.
A similar application to data on HDL-C98 tested first one SNP (in LIPG), and then a 14-SNP genetic risk score, comparing effects of the SNP or score on HDL-C and the known relationship between HDL-C and risk of myocardial infarction against the effects of SNP or genetic score on the prevalence of myocardial infarction. Results for the single SNP and score were consistent in showing that HDL-C was non-causal, while genetic-score analyses for LDL-C (conducted as a positive control) supported a causal relationship. However, because many loci affect both LDL-C and triglycerides, or triglycerides and HDL-C, selection of loci to include in a genetic score can be difficult.
On the positive side, MR studies using SNPs associated with blood pressure have confirmed a cause-and-effect relationship between higher diastolic and systolic blood pressures and coronary heart disease,99,100 and two recent reports suggest that triglycerides or triglyceride-rich lipoproteins have a causal role in coronary artery disease.101,102 Similarly the causal relationships between variation at the LPA locus, plasma Lp(a) concentration and coronary heart disease have been confirmed through the abolition of the gene-disease association after adjustment for the intermediate phenotype (Lp(a) concentration).59
A wide-ranging analysis of the metabolic and disease consequences of obesity,103 using rs9939609 in FTO, found that obesity was causally related to Type 2 diabetes, metabolic syndrome, blood pressure, glucose and insulin, HDL-C, triglycerides, CRP, GGT and ALT (all of which might have been expected) and also to heart failure. Two studies on urate showed the expected causal relationship with gout but found that the associations with cardiovascular and kidney disease and their biomarkers were not causal.104,105
Future studies of this type are likely to expand the range of SNPs included in calculation of a genetic risk score, including those which do not reach genome-wide significance. This should increase the power of MR analyses but carries the risk that some of the variants included do not meet the assumptions of the method. Nevertheless, MR will help both in understanding the clinical relevance of loci associated with biomarkers and in addressing questions of causality which cannot practically be resolved by experiments or clinical trials. So far the biomarkers studied have been well-established and other types of evidence have been available to support the conclusions, but the search for novel markers through –omic technologies will lead to many situations where genomic MR will help us to understand biomarkers’ characteristics.
Disease Prediction or Risk Stratification
As far as clinical laboratories are concerned, the hope is that testing for a panel of genetic polymorphisms (most simply, of SNPs) will produce useable predictions (better than those available from quantitative risk factors alone). One of the justifications for genetic association studies was the potential to predict common polygenic diseases, but there are several practical limitations. The best possible prediction is limited by heritability, and we know that concordance within pairs of monozygotic or ‘identical’ twins is far from complete. Despite major investments and large studies, the amount of variation explained by known SNP effects is well below this theoretical heritability limit and is likely to remain so.
The sensitivity and specificity of conventional predictive tests is far below that for diagnostic tests because the overlap between those who progress to disease endpoints and those who do not is so great, and comparisons based on receiver operating characteristic (ROC) curves are disappointing. If we aim for risk stratification rather than prediction of outcomes then the picture looks better and from a population treatment perspective (or for identifying high-risk subjects for epidemiological studies) this stratification can be effective.
Many of the published studies have calculated genetic risk scores based on the SNPs which have been shown to have genome-wide-significant effects. The usual approach has been to calculate a score for each person by multiplying the number of risk alleles at each relevant SNP by the beta (effect size) for continuous variables or by the relative risk for binary (affected/unaffected) outcomes, and summing the products across the SNPs. The score is then used as a ‘risk factor’ and tested for its ability to predict either the quantitative variable (such as LDL-C) or the outcome (affected/unaffected) in an independent sample. Because this genetic risk score is a quantitative and quasi-continuous variable, it can be assessed and potentially used for clinical risk assessment in the same way as a measurement of cholesterol or glucose. In general, genetic risk scores for cardiovascular disease or diabetes have not shown better performance than conventional risk factors and they have not added value to the existing approach, but there are some promising aspects.
An early attempt at prediction of cardiovascular disease used risk scores based on SNPs known to affect LDL-C or HDL-C.106 Survival analysis based on genetic risk score categories showed 92% 12-year event-free survival in people in the worst category, and 98% for people in the best category. This validates the choice of SNPs to some extent, though LDL and HDL effects cannot be distinguished. Despite the clear effect of risk score on outcome, ROC curve analysis showed no difference in the predictive value between standard measures and standard measures plus genetic risk score. This is not surprising because the standard risk assessment included LDL-C and HDL-C, and the SNP panel did not include loci affecting cardiovascular disease independent of these risk factors.
A similar design was used to assess genetic prediction of Type 2 diabetes.107 A panel of variants in 11 genes was used to construct the genetic risk score, which was compared against several composites of the known predictors (age, sex, family history, BMI, blood pressure, glucose). Adding the genetic predictor to the clinical model in ROC analysis produced statistically significant but very slight improvement in the area under the curve (0.74 to 0.75). However it appeared that conventional risk prediction was slightly better over shorter periods of follow-up and genetic prediction was slightly better over longer periods. This would be consistent with genetic score being a marker of lifetime risk and the clinical score reflecting metabolic changes leading up to the full expression of the diabetic state.
Since then, many studies of genetic risk scores have been carried out with increasing numbers of SNPs included. Many have focused on testing the relationship between markers and disease, rather than on the predictive value of the score as a potential screening tool. Of those which have assessed predictive performance or the degree of reclassification achieved by adding genetic risk to the predictor, most have shown only minimal effects. This was the case for coronary heart disease108–110 and Type 2 diabetes.111–113 One interesting variation was that a diabetes genetic risk score predicted cardiovascular complications in diabetics, perhaps because of association with poorer diabetic control.114
The frequency distribution of genetic risk scores leads to the conclusion that most people are at about average risk, neither extremely low nor extremely high. This is not surprising, but it means that for people near the middle of the genetic risk distribution, genetic testing makes little difference to their estimated risk (the pre-test and post-test probabilities are similar). However this is a situation we are familiar with from existing risk factors, and they are nevertheless widely used and have contributed to the improvement in cardiovascular mortality seen over the past thirty to forty years.
Prospects for Improved Prediction - More Data?
Given the limitations of current genetic risk scores for prediction and risk assessment for complex disease, how might the situation be improved? Firstly, larger meta-analyses of the current generation of GWAS data could reveal more SNPs to be included in the prediction score. However these will almost certainly have smaller effects than those already discovered and will therefore provide only marginal improvements for risk assessment. Secondly, additional and more comprehensive genotyping of existing cohorts, particularly for less common variants with minor allele frequencies in the range 0.1% to 5% but also for variants not well-tagged by the first generation of genotyping chips, may discover additional variants with substantial effects. Indeed, if effects of uncommon variants are to be detected at all using reasonable numbers of subjects then they must be of comparatively large effect and therefore have risk implications for the people who carry them. This does not solve the problem of the majority of people having average risk, but it would mean that high-risk people would be more differentiated from the bulk of the population.
Thirdly, there may be interactions between independent genetic loci or between genetic variants and environmental effects. Many studies on cardiovascular or metabolic disease have checked for heterogeneity of SNP effects by sex, some with positive results,81 or less frequently by some measure of obesity.115 Interactions with other factors such as smoking, alcohol intake or exercise patterns have not been well-explored and may yet produce results with both mechanistic and predictive value. Interactions between gene loci are certainly possible but a hypothesis-free approach to interactions between approximately 106 independent loci is subject to a massive multiple-testing problem and has not been feasible so far. A more limited approach, testing only known loci for interactions with all other variants, might produce results from existing data. Fourthly, the calculation of a genetic risk score can be extended to cover non-genome-wide-significant SNPs which reach some rather liberal threshold of significance. This will increase the inclusion of false-positive findings, but there is empirical evidence that this does not invalidate the scoring approach. However, the gain in predictive value compared to a more limited set of SNPs is not likely to be great.
Prospects for Improved Prediction – Lessons from Existing Prediction Algorithms
We can also attempt to make use of the information we already have, drawing on experience with quantitative risk factors for detection of high-risk people and primary prevention. Just as some people will have an LDL-C result at the upper end of the population distribution and will probably benefit for cholesterol-lowering treatment, some people will have extremely high genetic risk scores and may benefit from intervention. This could form the basis for use of genetic testing for risk stratification for common diseases in which high-risk people would be offered one or more low-risk treatments, but both simulations and trials of such an approach will be needed before we know whether it would be cost-effective and free of unexpected consequences. The limitation is not likely to be the cost of such genetic testing, which is low in comparison with many diagnostic procedures and falling fast, but the predictive validity and the costs and benefits of treatment.
Because the genomic score has inherently high heritability (100%), cascade screening may be useful. A successful and cost-effective cascade testing approach has been implemented for monogenic forms of hypercholesterolaemia, based on the fact that close relatives of patients are at increased risk. This approach has been tried in many centres and guidelines have been published.116 Similarly, relatives of people with a high polygenic risk score will tend to have a high risk score themselves (even though the distribution of scores in first-degree relatives differs from the Mendelian inheritance of familial hypercholesterolaemia). A program which starts with affected patients (most obviously for early-onset coronary heart disease), tests them to produce a genetic risk score, and cascades the testing outwards from anyone who scores in the top decile or quintile of genetic risk, might be able to reproduce the success of testing for familial hypercholesterolaemia (and would probably not depend on sequencing to define a mutation in each family). This is for the future, but the prospects for predictive testing in polygenic or complex diseases are far from hopeless.
Concentrating interventions on the people in the top 10% or 20% of risk may still be productive if the genetic risk score identifies high-risk people who would not be identifiable in existing ways. There is also the theoretical advantage that high-risk people could be identified early, and benefit from change extending over decades; change in results for non-genetic markers over time would provide complementary information about how far the genetic risk had manifested itself. In practice, this would require a method for genotyping a few hundred to a few thousand SNPs at a cost which was comparable to current risk factor measurements, which is a manageable challenge. A genetic risk score would be calculated for each person and this would be applied as an extension to the currently accepted method of basing the decision to treat or not to treat on total risk. It might not be necessary to screen the entire population in this way because genetic risk is greatest in relatives of affected patients. First-degree relatives of patients known to have conditions such as cardiovascular disease or Type 2 diabetes would be tested with genetic as well as existing methods and a proportion would warrant treatment.
Although genetic prediction has not yet reached the stage where trials can be initiated, we should consider the preconditions which would be necessary. Too many tests have been adopted prematurely, or used in ‘off-label’ ways, for us to be sure that inappropriate genetic testing will be avoided. Any trials or even thought experiments will need to consider not only prediction but outcomes, and the major issues of data management, interpretation and communication issues, and health economics which would need to be addressed.
Conclusions
Much time and effort has been invested in genetic association studies on common complex diseases and associated biomarkers. The investment was promoted as a way of discovering more about disease and leading to better treatments, of targeting treatment to individuals’ genetic characteristics, and preventing disease in high-risk people identified through genetic predictors. The improved understanding has occurred for a wide range of diseases. Novel drug targets have been identified, but the lead time for marketable drugs is substantial and although new treatments are appearing it is hard to point to any which are specifically due to GWAS. Genetic prediction for cardiovascular disease and diabetes has not been shown to add to what can be achieved with existing tests or algorithms. An unexpected benefit of GWAS discoveries has been the resolution of questions about causation for several characteristics known to be associated with disease.
Acknowledgments
My work in this area has been supported by grants from the US National Institutes of Health and from the Australian National Health and Medical Research Council. It has been made possible by collaboration with colleagues at Royal Prince Alfred Hospital, Sydney, and more recently at the Queensland Institute of Medical Research (now QIMR Berghofer Medical Research Institute), Brisbane. Working with Rita Middelberg, Beben Benjamin, Grant Montgomery and Nick Martin has made our research on biomarker genetics both possible and enjoyable.
Glossary
Useful definitions related to genetic studies can be found at http://www.snpedia.com/index.php/Glossary or http://ghr.nlm.nih.gov/glossary.
- Allele:
an allele is an alternative within the genome sequence, such as G or A, C or T, for a single nucleotide polymorphism (SNP).
- Complex
(polygenic or multifactorial) diseases and traits are influenced by a combination of many comparatively small environmental and genetic effects, usually acting additively. The disease occurs when the liability from all these sources exceeds some threshold.
- Genotype:
for the autosomal chromosomes in a diploid cell, a SNP will have three possible genotypes because there are copies on each chromosome, for example AA, AG or GG. (But on the X chromosome men are hemizygous so only two genotypes are possible; women may have any of three. Y-chromosome and mitochondrial inheritance are not usually considered for complex traits.)
- GWAS:
a genome-wide association study checks for significant association between SNPs or other polymorphic variation and either case-control status or a quantitative phenotype across the entire genome. In practice, some regions of the genome have been difficult and not all polymorphic variation can be captured by the use of tagging SNPs. Some studies have only reported on variation in autosomal chromosomes.
- Heritability
is the proportion of variance in a phenotype accounted for by all additive genetic effects. (This is the narrow-sense heritability, which is most relevant for complex disease or quantitative traits.) It is usually estimated from resemblance between pairs of relatives such as parents and offspring or twin pairs.
- Linkage disequilibrium
-
(LD) is defined as “co-occurrence of a specific DNA marker and a disease at a higher frequency than would be predicted by random chance” (http://ghr.nlm.nih.gov/glossary). The term is also applied to co-occurrence of alleles in genetic markers such as SNPs. LD is important for GWAS because mutations, or polymorphisms contributing to disease risk or genetic variation in other phenotypes, are assumed to originate on an ancestral chromosome with a haplotype which will be co-inherited across many generations.
Over time, recombination events will break up the haplotype so that only polymorphisms close to the causative variant will remain associated with it. This has the practical advantage that it is not necessary to genotype every SNP in the genome to do a useful GWAS because a sub-set of SNPs can be chosen to tag LD blocks and identify loci for more detailed investigation.
- Mutation:
mutation means change, and in one sense a mutation occurs between one generation and the next. For clarity, a distinction is often made between de-novo mutations (not inherited from either parent) and inherited mutations which are less common than polymorphisms. We can also distinguish between germline mutations, inherited from parents, and somatic mutations which are inherited across cell division.
- Phenotype:
the phenotype is a characteristic which can be observed or measured, such as presence or absence of a disease, hair colour, height, or fasting plasma glucose. In most cases the phenotype will be affected by both genetic and non-genetic (environmental or random) sources of variation; and the phenotype may change over time because of measurement error, biological variation, ageing, or onset of disease.
- Polymorphism:
a polymorphism is a part of a DNA sequence which (as the word implies) can take many forms – but in practice, usually only two. Single-nucleotide polymorphisms or SNPs comprise variation at a single base pair, whereas indels are insertion/deletion polymorphisms which have one base-pair replaced by several. The term polymorphism is usually reserved for variants where the less common (minor) allele has a frequency over 1%. SNPs are identified by rs numbers, for example rs1800562 is the non-synonymous coding variant (cysteine to tyrosine at amino acid 282, C282Y) in the HFE gene, which is associated with the most common form of haemochromatosis.
- Wild-type:
for mutations or for gene knockout in experimental animals, the original, common or ancestral allele is often referred to as wild-type.
Footnotes
Competing Interests: None declared.
References
- 1.Raal FJ, Santos RD. Homozygous familial hypercholesterolemia: Current perspectives on diagnosis and treatment. Atherosclerosis. 2012;223:262–8. doi: 10.1016/j.atherosclerosis.2012.02.019. [DOI] [PubMed] [Google Scholar]
- 2.Faiz F, Hooper AJ, van Bockxmeer FM. Molecular pathology of familial hypercholesterolemia, related dyslipidemias and therapies beyond the statins. Crit Rev Clin Lab Sci. 2012;49:1–17. doi: 10.3109/10408363.2011.646942. [DOI] [PubMed] [Google Scholar]
- 3.Brouwers MC, van Greevenbroek MM, Stehouwer CD, de Graaf J, Stalenhoef AF. The genetics of familial combined hyperlipidaemia. Nat Rev Endocrinol. 2012;8:352–62. doi: 10.1038/nrendo.2012.15. [DOI] [PubMed] [Google Scholar]
- 4.Klupa T, Skupien J, Malecki MT. Monogenic models: what have the single gene disorders taught us? Curr Diab Rep. 2012;12:659–66. doi: 10.1007/s11892-012-0325-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.History of the Framingham Heart Study http://www.framinghamheartstudy.org/about-fhs/history.php (Accessed 10 January 2014)
- 6.Taylor R, Page A, Danquah J. The Australian epidemic of cardiovascular mortality 1935–2005: effects of period and birth cohort. J Epidemiol Community Health. 2012;66:e18. doi: 10.1136/jech.2010.109538. [DOI] [PubMed] [Google Scholar]
- 7.Ford ES, Ajani UA, Croft JB, Critchley JA, Labarthe DR, Kottke TE, et al. Explaining the decrease in U.S. deaths from coronary disease, 1980–2000. N Engl J Med. 2007;356:2388–98. doi: 10.1056/NEJMsa053935. [DOI] [PubMed] [Google Scholar]
- 8.Du G, Song Z, Zhang Q. Gamma-glutamyltransferase is associated with cardiovascular and all-cause mortality: a meta-analysis of prospective cohort studies. Prev Med. 2013;57:31–7. doi: 10.1016/j.ypmed.2013.03.011. [DOI] [PubMed] [Google Scholar]
- 9.Schneider AL, Lazo M, Ndumele CE, Pankow JS, Coresh J, Clark JM, et al. Liver enzymes, race, gender and diabetes risk: the Atherosclerosis Risk in Communities (ARIC) Study. Diabet Med. 2013;30:926–33. doi: 10.1111/dme.12187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schindhelm RK, Dekker JM, Nijpels G, Bouter LM, Stehouwer CD, Heine RJ, et al. Alanine aminotransferase predicts coronary heart disease events: a 10-year follow-up of the Hoorn Study. Atherosclerosis. 2007;191:391–6. doi: 10.1016/j.atherosclerosis.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 11.Monami M, Bardini G, Lamanna C, Pala L, Cresci B, Francesconi P, et al. Liver enzymes and risk of diabetes and cardiovascular disease: results of the Firenze Bagno a Ripoli (FIBAR) study. Metabolism. 2008;57:387–92. doi: 10.1016/j.metabol.2007.10.015. [DOI] [PubMed] [Google Scholar]
- 12.Ruhl CE, Everhart JE. Elevated serum alanine aminotransferase and gamma-glutamyltransferase and mortality in the United States population. Gastroenterology. 2009;136:477–85. e11. doi: 10.1053/j.gastro.2008.10.052. [DOI] [PubMed] [Google Scholar]
- 13.Calderon-Margalit R, Adler B, Abramson JH, Gofin J, Kark JD. Butyrylcholinesterase activity, cardiovascular risk factors, and mortality in middle-aged and elderly men and women in Jerusalem. Clin Chem. 2006;52:845–52. doi: 10.1373/clinchem.2005.059857. [DOI] [PubMed] [Google Scholar]
- 14.Goliasch G, Haschemi A, Marculescu R, Endler G, Maurer G, Wagner O, et al. Butyrylcholinesterase activity predicts long-term survival in patients with coronary artery disease. Clin Chem. 2012;58:1055–8. doi: 10.1373/clinchem.2011.175984. [DOI] [PubMed] [Google Scholar]
- 15.Sato KK, Hayashi T, Maeda I, Koh H, Harita N, Uehara S, et al. Serum butyrylcholinesterase and the risk of future type 2 diabetes: the Kansai Healthcare Study. Clin Endocrinol (Oxf) 2013. 2014;80:362–7. doi: 10.1111/cen.12171. [DOI] [PubMed] [Google Scholar]
- 16.Horsfall LJ, Nazareth I, Petersen I. Cardiovascular events as a function of serum bilirubin levels in a large, statin-treated cohort. Circulation. 2012;126:2556–64. doi: 10.1161/CIRCULATIONAHA.112.114066. [DOI] [PubMed] [Google Scholar]
- 17.Joseph J, Svartberg J, Njolstad I, Schirmer H. Incidence of and risk factors for type-2 diabetes in a general population: the Tromso Study. Scand J Public Health. 2010;38:768–75. doi: 10.1177/1403494810380299. [DOI] [PubMed] [Google Scholar]
- 18.Arsenault BJ, Rana JS, Stroes ES, Despres JP, Shah PK, Kastelein JJ, et al. Beyond low-density lipoprotein cholesterol: respective contributions of non-high-density lipoprotein cholesterol levels, triglycerides, and the total cholesterol/high-density lipoprotein cholesterol ratio to coronary heart disease risk in apparently healthy men and women. J Am Coll Cardiol. 2009;55:35–41. doi: 10.1016/j.jacc.2009.07.057. [DOI] [PubMed] [Google Scholar]
- 19.Sone H, Tanaka S, Iimuro S, Oida K, Yamasaki Y, Oikawa S, et al. Serum level of triglycerides is a potent risk factor comparable to LDL cholesterol for coronary heart disease in Japanese patients with type 2 diabetes: subanalysis of the Japan Diabetes Complications Study (JDCS) J Clin Endocrinol Metab. 2011;96:3448–56. doi: 10.1210/jc.2011-0622. [DOI] [PubMed] [Google Scholar]
- 20.Fang J, Alderman MH. Serum uric acid and cardiovascular mortality the NHANES I epidemiologic follow-up study, 1971–1992. National Health and Nutrition Examination Survey. JAMA. 2000;283:2404–10. doi: 10.1001/jama.283.18.2404. [DOI] [PubMed] [Google Scholar]
- 21.Bhole V, Choi JW, Kim SW, de Vera M, Choi H. Serum uric acid levels and the risk of type 2 diabetes: a prospective study. Am J Med. 2010;123:957–61. doi: 10.1016/j.amjmed.2010.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Humphrey LL, Fu R, Rogers K, Freeman M, Helfand M. Homocysteine level and coronary heart disease incidence: a systematic review and meta-analysis. Mayo Clin Proc. 2008;83:1203–12. doi: 10.4065/83.11.1203. [DOI] [PubMed] [Google Scholar]
- 23.Gwynne P. Diagnostics: The new risk predictors. Nature. 2013;493:S7–S8. doi: 10.1038/493S7a. [DOI] [PubMed] [Google Scholar]
- 24.Wang TJ, Gona P, Larson MG, Tofler GH, Levy D, Newton-Cheh C, et al. Multiple Biomarkers for the Prediction of First Major Cardiovascular Events and Death. New England Journal of Medicine. 2006;355:2631–9. doi: 10.1056/NEJMoa055373. [DOI] [PubMed] [Google Scholar]
- 25.Beekman M, Heijmans BT, Martin NG, Pedersen NL, Whitfield JB, DeFaire U, et al. Heritabilities of apolipoprotein and lipid levels in three countries. Twin Res. 2002;5:87–97. doi: 10.1375/1369052022956. [DOI] [PubMed] [Google Scholar]
- 26.Whitfield JB, Zhu G, Nestler JE, Heath AC, Martin NG. Genetic covariation between serum gamma-glutamyltransferase activity and cardiovascular risk factors. Clin Chem. 2002;48:1426–31. [PubMed] [Google Scholar]
- 27.Loomba R, Rao F, Zhang L, Khandrika S, Ziegler MG, Brenner DA, et al. Genetic covariance between gamma-glutamyl transpeptidase and fatty liver risk factors: role of beta2-adrenergic receptor genetic variation in twins. Gastroenterology. 2010;139:836–45. 45 e1. doi: 10.1053/j.gastro.2010.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fox ER, Benjamin EJ, Sarpong DF, Rotimi CN, Wilson JG, Steffes MW, et al. Epidemiology, heritability, and genetic linkage of C-reactive protein in African Americans (from the Jackson Heart Study) Am J Cardiol. 2008;102:835–41. doi: 10.1016/j.amjcard.2008.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Su S, Snieder H, Miller AH, Ritchie J, Bremner JD, Goldberg J, et al. Genetic and environmental influences on systemic markers of inflammation in middle-aged male twins. Atherosclerosis. 2008;200:213–20. doi: 10.1016/j.atherosclerosis.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schnabel RB, Lunetta KL, Larson MG, Dupuis J, Lipinska I, Rong J, et al. The relation of genetic and environmental factors to systemic inflammatory biomarker concentrations. Circ Cardiovasc Genet. 2009;2:229–37. doi: 10.1161/CIRCGENETICS.108.804245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Whitfield JB, Martin NG. Inheritance and alcohol as factors influencing plasma uric acid levels. Acta GenetMedGemellol(Roma) 1983;32:117–26. doi: 10.1017/s0001566000006401. [DOI] [PubMed] [Google Scholar]
- 32.Bathum L, Petersen I, Christiansen L, Konieczna A, Sorensen TI, Kyvik KO. Genetic and environmental influences on plasma homocysteine: results from a Danish twin study. Clin Chem. 2007;53:971–9. doi: 10.1373/clinchem.2006.082149. [DOI] [PubMed] [Google Scholar]
- 33.Siva A, De Lange M, Clayton D, Monteith S, Spector T, Brown MJ. The heritability of plasma homocysteine, and the influence of genetic variation in the homocysteine methylation pathway. QJM. 2007;100:495–9. doi: 10.1093/qjmed/hcm054. [DOI] [PubMed] [Google Scholar]
- 34.Della-Morte D, Beecham A, Rundek T, Slifer S, Boden-Albala B, McClendon MS, et al. Genetic linkage of serum homocysteine in Dominican families: the Family Study of Stroke Risk and Carotid Atherosclerosis. Stroke. 2010;41:1356–62. doi: 10.1161/STROKEAHA.109.573626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Slack J, Evans KA. The increased risk of death from ischaemic heart disease in first degree relatives of 121 men and 96 women with ischaemic heart disease. J Med Genet. 1966;3:239–57. doi: 10.1136/jmg.3.4.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zdravkovic S, Wienke A, Pedersen NL, Marenberg ME, Yashin AI, De Faire U. Heritability of death from coronary heart disease: a 36-year follow-up of 20 966 Swedish twins. J Intern Med. 2002;252:247–54. doi: 10.1046/j.1365-2796.2002.01029.x. [DOI] [PubMed] [Google Scholar]
- 37.Wienke A, Herskind AM, Christensen K, Skytthe A, Yashin AI. The heritability of CHD mortality in Danish twins after controlling for smoking and BMI. Twin Res Hum Genet. 2005;8:53–9. doi: 10.1375/1832427053435328. [DOI] [PubMed] [Google Scholar]
- 38.Chow CK, Islam S, Bautista L, Rumboldt Z, Yusufali A, Xie C, et al. Parental history and myocardial infarction risk across the world: the INTERHEART Study. J Am Coll Cardiol. 2011;57:619–27. doi: 10.1016/j.jacc.2010.07.054. [DOI] [PubMed] [Google Scholar]
- 39.Sundquist K, Winkleby M, Li X, Ji J, Hemminki K, Sundquist J. Familial transmission of coronary heart disease: a cohort study of 80,214 Swedish adoptees linked to their biological and adoptive parents. Am Heart J. 2011;162:317–23. doi: 10.1016/j.ahj.2011.05.013. [DOI] [PubMed] [Google Scholar]
- 40.Medici F, Hawa M, Ianari A, Pyke DA, Leslie RD. Concordance rate for type II diabetes mellitus in monozygotic twins: actuarial analysis. Diabetologia. 1999;42:146–50. doi: 10.1007/s001250051132. [DOI] [PubMed] [Google Scholar]
- 41.Condon J, Shaw JE, Luciano M, Kyvik KO, Martin NG, Duffy DL. A study of diabetes mellitus within a large sample of Australian twins. Twin Res Hum Genet. 2008;11:28–40. doi: 10.1375/twin.11.1.28. [DOI] [PubMed] [Google Scholar]
- 42.Lehtovirta M, Pietilainen KH, Levalahti E, Heikkila K, Groop L, Silventoinen K, et al. Evidence that BMI and type 2 diabetes share only a minor fraction of genetic variance: a follow-up study of 23,585 monozygotic and dizygotic twins from the Finnish Twin Cohort Study. Diabetologia. 2010;53:1314–21. doi: 10.1007/s00125-010-1746-4. [DOI] [PubMed] [Google Scholar]
- 43.Manolio TA. Genomewide association studies and assessment of the risk of disease. N Engl J Med. 2010;363:166–76. doi: 10.1056/NEJMra0905980. [DOI] [PubMed] [Google Scholar]
- 44.Schork AJ, Thompson WK, Pham P, Torkamani A, Roddey JC, Sullivan PF, et al. All SNPs are not created equal: genome-wide association studies reveal a consistent pattern of enrichment among functionally annotated SNPs. PLoS Genet. 2013;9:e1003449. doi: 10.1371/journal.pgen.1003449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Benyamin B, Middelberg RP, Lind PA, Valle AM, Gordon S, Nyholt DR, et al. GWAS of butyrylcholinesterase activity identifies four novel loci, independent effects within BCHE and secondary associations with metabolic risk factors. Hum Mol Genet. 2011;20:4504–14. doi: 10.1093/hmg/ddr375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Benyamin B, McRae AF, Zhu G, Gordon S, Henders AK, Palotie A, et al. Variants in TF and HFE explain approximately 40% of genetic variation in serum-transferrin levels. Am J Hum Genet. 2009;84:60–5. doi: 10.1016/j.ajhg.2008.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Johnson AD, Kavousi M, Smith AV, Chen MH, Dehghan A, Aspelund T, et al. Genome-wide association meta-analysis for total serum bilirubin levels. Hum Mol Genet. 2009;18:2700–10. doi: 10.1093/hmg/ddp202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.A Catalog of Published Genome-Wide Association Studies http://www.genome.gov/gwastudies/ (Accessed 10 September 2013.
- 49.GWAS Diagram Browser http://www.ebi.ac.uk/fgpt/gwas/ (Accessed 10 September 2013.
- 50.WTCCC Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–78. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Harismendy O, Notani D, Song X, Rahim NG, Tanasa B, Heintzman N, et al. 9p21 DNA variants associated with coronary artery disease impair interferon-gamma signalling response. Nature. 2011;470:264–8. doi: 10.1038/nature09753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cunnington MS, Santibanez Koref M, Mayosi BM, Burn J, Keavney B. Chromosome 9p21 SNPs Associated with Multiple Disease Phenotypes Correlate with ANRIL Expression. PLoS Genet. 2010;6:e1000899. doi: 10.1371/journal.pgen.1000899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Johnson AD, Hwang SJ, Voorman A, Morrison A, Peloso GM, Hsu YH, et al. Resequencing and clinical associations of the 9p21.3 region: a comprehensive investigation in the Framingham heart study. Circulation. 2013;127:799–810. doi: 10.1161/CIRCULATIONAHA.112.111559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Musunuru K. Enduring mystery of the chromosome 9p21.3 locus. Circ Cardiovasc Genet. 2013;6:224–5. doi: 10.1161/CIRCGENETICS.113.000132. [DOI] [PubMed] [Google Scholar]
- 55.Schunkert H, Konig IR, Kathiresan S, Reilly MP, Assimes TL, Holm H, et al. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat Genet. 2011;43:333–8. doi: 10.1038/ng.784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Deloukas P, Kanoni S, Willenborg C, Farrall M, Assimes TL, Thompson JR, et al. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet. 2012;45:25–33. doi: 10.1038/ng.2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tregouet DA, Konig IR, Erdmann J, Munteanu A, Braund PS, Hall AS, et al. Genome-wide haplotype association study identifies the SLC22A3-LPAL2-LPA gene cluster as a risk locus for coronary artery disease. Nat Genet. 2009;41:283–5. doi: 10.1038/ng.314. [DOI] [PubMed] [Google Scholar]
- 58.Kamstrup PR, Tybjaerg-Hansen A, Steffensen R, Nordestgaard BG. Genetically elevated lipoprotein(a) and increased risk of myocardial infarction. JAMA. 2009;301:2331–9. doi: 10.1001/jama.2009.801. [DOI] [PubMed] [Google Scholar]
- 59.Clarke R, Peden JF, Hopewell JC, Kyriakou T, Goel A, Heath SC, et al. Genetic variants associated with Lp(a) lipoprotein level and coronary disease. N Engl J Med. 2009;361:2518–28. doi: 10.1056/NEJMoa0902604. [DOI] [PubMed] [Google Scholar]
- 60.Holliday EG, Maguire JM, Evans TJ, Koblar SA, Jannes J, Sturm JW, et al. Common variants at 6p21.1 are associated with large artery atherosclerotic stroke. Nat Genet. 2012;44:1147–51. doi: 10.1038/ng.2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Traylor M, Farrall M, Holliday EG, Sudlow C, Hopewell JC, Cheng YC, et al. Genetic risk factors for ischaemic stroke and its subtypes (the METASTROKE collaboration): a meta-analysis of genome-wide association studies. Lancet Neurol. 2012;11:951–62. doi: 10.1016/S1474-4422(12)70234-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ehret GB, Munroe PB, Rice KM, Bochud M, Johnson AD, Chasman DI, et al. Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature. 2011;478:103–9. doi: 10.1038/nature10405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wain LV, Verwoert GC, O’Reilly PF, Shi G, Johnson T, Johnson AD, et al. Genome-wide association study identifies six new loci influencing pulse pressure and mean arterial pressure. Nat Genet. 2011;43:1005–11. doi: 10.1038/ng.922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Smith NL, Felix JF, Morrison AC, Demissie S, Glazer NL, Loehr LR, et al. Association of genome-wide variation with the risk of incident heart failure in adults of European and African ancestry: a prospective meta-analysis from the cohorts for heart and aging research in genomic epidemiology (CHARGE) consortium. Circ Cardiovasc Genet. 2010;3:256–66. doi: 10.1161/CIRCGENETICS.109.895763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Helgadottir A, Thorleifsson G, Magnusson KP, Gretarsdottir S, Steinthorsdottir V, Manolescu A, et al. The same sequence variant on 9p21 associates with myocardial infarction, abdominal aortic aneurysm and intracranial aneurysm. Nat Genet. 2008;40:217–24. doi: 10.1038/ng.72. [DOI] [PubMed] [Google Scholar]
- 66.Bown MJ, Jones GT, Harrison SC, Wright BJ, Bumpstead S, Baas AF, et al. Abdominal aortic aneurysm is associated with a variant in low-density lipoprotein receptor-related protein 1. Am J Hum Genet. 2011;89:619–27. doi: 10.1016/j.ajhg.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lemaire SA, McDonald ML, Guo DC, Russell L, Miller CC, 3rd, Johnson RJ, et al. Genome-wide association study identifies a susceptibility locus for thoracic aortic aneurysms and aortic dissections spanning FBN1 at 15q21.1. Nat Genet. 2011;43:996–1000. doi: 10.1038/ng.934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gretarsdottir S, Baas AF, Thorleifsson G, Holm H, den Heijer M, de Vries JP, et al. Genome-wide association study identifies a sequence variant within the DAB2IP gene conferring susceptibility to abdominal aortic aneurysm. Nat Genet. 2010;42:692–7. doi: 10.1038/ng.622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Morris AP, Voight BF, Teslovich TM, Ferreira T, Segre AV, Steinthorsdottir V, et al. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat Genet. 2012;44:981–90. doi: 10.1038/ng.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Voight BF, Scott LJ, Steinthorsdottir V, Morris AP, Dina C, Welch RP, et al. Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat Genet. 2010;42:579–89. doi: 10.1038/ng.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Scott RA, Lagou V, Welch RP, Wheeler E, Montasser ME, Luan J, et al. Large-scale association analyses identify new loci influencing glycemic traits and provide insight into the underlying biological pathways. Nat Genet. 2012;44:991–1005. doi: 10.1038/ng.2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Raj SM, Howson JM, Walker NM, Cooper JD, Smyth DJ, Field SF, et al. No association of multiple type 2 diabetes loci with type 1 diabetes. Diabetologia. 2009;52:2109–16. doi: 10.1007/s00125-009-1391-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Winkler C, Raab J, Grallert H, Ziegler AG. Lack of association of type 2 diabetes susceptibility genotypes and body weight on the development of islet autoimmunity and type 1 diabetes. PLoS One. 2012;7:e35410. doi: 10.1371/journal.pone.0035410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nogueira TC, Paula FM, Villate O, Colli ML, Moura RF, Cunha DA, et al. GLIS3, a susceptibility gene for type 1 and type 2 diabetes, modulates pancreatic beta cell apoptosis via regulation of a splice variant of the BH3-only protein Bim. PLoS Genet. 2013;9:e1003532. doi: 10.1371/journal.pgen.1003532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Alberti KG, Zimmet P, Shaw J. Metabolic syndrome--a new world-wide definition. A Consensus Statement from the International Diabetes Federation. Diabet Med. 2006;23:469–80. doi: 10.1111/j.1464-5491.2006.01858.x. [DOI] [PubMed] [Google Scholar]
- 76.Kraja AT, Vaidya D, Pankow JS, Goodarzi MO, Assimes TL, Kullo IJ, et al. A bivariate genome-wide approach to metabolic syndrome: STAMPEED consortium. Diabetes. 2011;60:1329–39. doi: 10.2337/db10-1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kristiansson K, Perola M, Tikkanen E, Kettunen J, Surakka I, Havulinna AS, et al. Genome-wide screen for metabolic syndrome susceptibility Loci reveals strong lipid gene contribution but no evidence for common genetic basis for clustering of metabolic syndrome traits. Circ Cardiovasc Genet. 2012;5:242–9. doi: 10.1161/CIRCGENETICS.111.961482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sandholm N, Salem RM, McKnight AJ, Brennan EP, Forsblom C, Isakova T, et al. New susceptibility loci associated with kidney disease in type 1 diabetes. PLoS Genet. 2012;8:e1002921. doi: 10.1371/journal.pgen.1002921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Reilly MP, Li M, He J, Ferguson JF, Stylianou IM, Mehta NN, et al. Identification of ADAMTS7 as a novel locus for coronary atherosclerosis and association of ABO with myocardial infarction in the presence of coronary atherosclerosis: two genome-wide association studies. Lancet. 2011;377:383–92. doi: 10.1016/S0140-6736(10)61996-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Teslovich TM, Musunuru K, Smith AV, Edmondson AC, Stylianou IM, Koseki M, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466:707–13. doi: 10.1038/nature09270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Asselbergs FW, Guo Y, van Iperen EP, Sivapalaratnam S, Tragante V, Lanktree MB, et al. Large-Scale Gene-Centric Meta-analysis across 32 Studies Identifies Multiple Lipid Loci. Am J Hum Genet. 2012;91:823–38. doi: 10.1016/j.ajhg.2012.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Whitfield JB, Heath AC, Madden PA, Pergadia ML, Montgomery GW, Martin NG. Metabolic and Biochemical Effects of Low-to-Moderate Alcohol Consumption. Alcohol Clin Exp Res. 2013;37:575–86. doi: 10.1111/acer.12015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Drenos F, Talmud PJ, Casas JP, Smeeth L, Palmen J, Humphries SE, et al. Integrated associations of genotypes with multiple blood biomarkers linked to coronary heart disease risk. Hum Mol Genet. 2009;18:2305–16. doi: 10.1093/hmg/ddp159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bennet AM, Reynolds CA, Gatz M, Blennow K, Pedersen NL, Prince JA. Pleiotropy in the presence of allelic heterogeneity: alternative genetic models for the influence of APOE on serum LDL, CSF amyloid-beta42, and dementia. J Alzheimers Dis. 2010;22:129–34. doi: 10.3233/JAD-2010-100864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Middelberg RP, Ferreira MA, Henders AK, Heath AC, Madden PA, Montgomery GW, et al. Genetic variants in LPL, OASL and TOMM40/APOE-C1-C2-C4 genes are associated with multiple cardiovascular-related traits. BMC Med Genet. 2011;12:123. doi: 10.1186/1471-2350-12-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Iynedjian PB. Molecular physiology of mammalian glucokinase. Cell Mol Life Sci. 2009;66:27–42. doi: 10.1007/s00018-008-8322-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Horvatovich K, Bokor S, Polgar N, Kisfali P, Hadarits F, Jaromi L, et al. Functional glucokinase regulator gene variants have inverse effects on triglyceride and glucose levels, and decrease the risk of obesity in children. Diabetes Metab. 2011;37:432–9. doi: 10.1016/j.diabet.2011.02.003. [DOI] [PubMed] [Google Scholar]
- 88.Ryffel GU. Mutations in the human genes encoding the transcription factors of the hepatocyte nuclear factor (HNF)1 and HNF4 families: functional and pathological consequences. J Mol Endocrinol. 2001;27:11–29. doi: 10.1677/jme.0.0270011. [DOI] [PubMed] [Google Scholar]
- 89.Eissing L, Scherer T, Todter K, Knippschild U, Greve JW, Buurman WA, et al. De novo lipogenesis in human fat and liver is linked to ChREBP-beta and metabolic health. Nat Commun. 2013;4:1528. doi: 10.1038/ncomms2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jetten AM. Retinoid-related orphan receptors (RORs): critical roles in development, immunity, circadian rhythm, and cellular metabolism. Nucl Recept Signal. 2009;7:e003. doi: 10.1621/nrs.07003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lee SH, Ripke S, Neale BM, Faraone SV, Purcell SM, Perlis RH, et al. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat Genet. 2013 doi: 10.1038/ng.2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Andreassen OA, Djurovic S, Thompson WK, Schork AJ, Kendler KS, O’Donovan MC, et al. Improved detection of common variants associated with schizophrenia by leveraging pleiotropy with cardiovascular-disease risk factors. Am J Hum Genet. 2013;92:197–209. doi: 10.1016/j.ajhg.2013.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.International Multiple Sclerosis Genetics Consortium The expanding genetic overlap between multiple sclerosis and type I diabetes. Genes Immun. 2009;10:11–4. doi: 10.1038/gene.2008.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rzhetsky A, Wajngurt D, Park N, Zheng T. Probing genetic overlap among complex human phenotypes. Proc Natl Acad Sci U S A. 2007;104:11694–9. doi: 10.1073/pnas.0704820104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Morrison AC, Voorman A, Johnson AD, Liu X, Yu J, Li A, et al. Whole-genome sequence-based analysis of high-density lipoprotein cholesterol. Nat Genet 2013. 2013;45:899–901. doi: 10.1038/ng.2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Saunders JT, Nambi V, de Lemos JA, Chambless LE, Virani SS, Boerwinkle E, et al. Cardiac troponin T measured by a highly sensitive assay predicts coronary heart disease, heart failure, and mortality in the Atherosclerosis Risk in Communities Study. Circulation. 2011;123:1367–76. doi: 10.1161/CIRCULATIONAHA.110.005264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.van Meurs JB, Pare G, Schwartz SM, Hazra A, Tanaka T, Vermeulen SH, et al. Common genetic loci influencing plasma homocysteine concentrations and their effect on risk of coronary artery disease. Am J Clin Nutr 2013. 2013;98:668–76. doi: 10.3945/ajcn.112.044545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Voight BF, Peloso GM, Orho-Melander M, Frikke-Schmidt R, Barbalic M, Jensen MK, et al. Plasma HDL cholesterol and risk of myocardial infarction: a mendelian randomisation study. Lancet 2012. 2013;34:1826–33. doi: 10.1016/S0140-6736(12)60312-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lieb W, Jansen H, Loley C, Pencina MJ, Nelson CP, Newton-Cheh C, et al. Genetic predisposition to higher blood pressure increases coronary artery disease risk. Hypertension. 2013;61:995–1001. doi: 10.1161/HYPERTENSIONAHA.111.00275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Havulinna AS, Kettunen J, Ukkola O, Osmond C, Eriksson JG, Kesaniemi YA, et al. A blood pressure genetic risk score is a significant predictor of incident cardiovascular events in 32,669 individuals. Hypertension. 2013;61:987–94. doi: 10.1161/HYPERTENSIONAHA.111.00649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Jorgensen AB, Frikke-Schmidt R, West AS, Grande P, Nordestgaard BG, Tybjaerg-Hansen A. Genetically elevated non-fasting triglycerides and calculated remnant cholesterol as causal risk factors for myocardial infarction. Eur Heart J. 2013;34:1826–33. doi: 10.1093/eurheartj/ehs431. [DOI] [PubMed] [Google Scholar]
- 102.Do R, Willer CJ, Schmidt EM, Sengupta S, Gao C, Peloso GM, et al. Common variants associated with plasma triglycerides and risk for coronary artery disease. Nat Genet. 2013;45:1345–52. doi: 10.1038/ng.2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Fall T, Hagg S, Magi R, Ploner A, Fischer K, Horikoshi M, et al. The role of adiposity in cardiometabolic traits: a mendelian randomization analysis. PLoS Med. 2013;10:e1001474. doi: 10.1371/journal.pmed.1001474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Stark K, Reinhard W, Grassl M, Erdmann J, Schunkert H, Illig T, et al. Common polymorphisms influencing serum uric acid levels contribute to susceptibility to gout, but not to coronary artery disease. PLoS One. 2009;4:e7729. doi: 10.1371/journal.pone.0007729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Yang Q, Kottgen A, Dehghan A, Smith AV, Glazer NL, Chen MH, et al. Multiple genetic loci influence serum urate levels and their relationship with gout and cardiovascular disease risk factors. Circ Cardiovasc Genet. 2010;3:523–30. doi: 10.1161/CIRCGENETICS.109.934455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kathiresan S, Melander O, Anevski D, Guiducci C, Burtt NP, Roos C, et al. Polymorphisms associated with cholesterol and risk of cardiovascular events. N Engl J Med. 2008;358:1240–9. doi: 10.1056/NEJMoa0706728. [DOI] [PubMed] [Google Scholar]
- 107.Lyssenko V, Jonsson A, Almgren P, Pulizzi N, Isomaa B, Tuomi T, et al. Clinical risk factors, DNA variants, and the development of type 2 diabetes. N Engl J Med. 2008;359:2220–32. doi: 10.1056/NEJMoa0801869. [DOI] [PubMed] [Google Scholar]
- 108.Brautbar A, Pompeii LA, Dehghan A, Ngwa JS, Nambi V, Virani SS, et al. A genetic risk score based on direct associations with coronary heart disease improves coronary heart disease risk prediction in the Atherosclerosis Risk in Communities (ARIC), but not in the Rotterdam and Framingham Offspring, Studies. Atherosclerosis. 2012;223:421–6. doi: 10.1016/j.atherosclerosis.2012.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tikkanen E, Havulinna AS, Palotie A, Salomaa V, Ripatti S. Genetic risk prediction and a 2-stage risk screening strategy for coronary heart disease. Arterioscler Thromb Vasc Biol. 2013;33:2261–6. doi: 10.1161/ATVBAHA.112.301120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ganna A, Magnusson PK, Pedersen NL, de Faire U, Reilly M, Arnlov J, et al. Multilocus Genetic Risk Scores for Coronary Heart Disease Prediction. Arterioscler Thromb Vasc Biol. 2013;33:2267–72. doi: 10.1161/ATVBAHA.113.301218. [DOI] [PubMed] [Google Scholar]
- 111.Anand SS, Meyre D, Pare G, Bailey SD, Xie C, Zhang X, et al. Genetic Information and the Prediction of Incident Type 2 Diabetes in a High-Risk Multi-Ethnic Population: The EpiDREAM Genetic Study. Diabetes Care 2013. 2013;36:2836–42. doi: 10.2337/dc12-2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Muhlenbruch K, Jeppesen C, Joost HG, Boeing H, Schulze MB. The value of genetic information for diabetes risk prediction - differences according to sex, age, family history and obesity. PLoS One. 2013;8:e64307. doi: 10.1371/journal.pone.0064307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Imamura M, Shigemizu D, Tsunoda T, Iwata M, Maegawa H, Watada H, et al. Assessing the clinical utility of a genetic risk score constructed using 49 susceptibility alleles for type 2 diabetes in a Japanese population. J Clin Endocrinol Metab 2013. 2013;98:E1667–73. doi: 10.1210/jc.2013-1642. [DOI] [PubMed] [Google Scholar]
- 114.Qi Q, Meigs JB, Rexrode KM, Hu FB, Qi L. Diabetes genetic predisposition score and cardiovascular complications among patients with type 2 diabetes. Diabetes Care. 2013;36:737–9. doi: 10.2337/dc12-0852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Surakka I, Isaacs A, Karssen LC, Laurila PP, Middelberg RP, Tikkanen E, et al. A genome-wide screen for interactions reveals a new locus on 4p15 modifying the effect of waist-to-hip ratio on total cholesterol. PLoS Genet. 2011;7:e1002333. doi: 10.1371/journal.pgen.1002333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Watts GF, Sullivan DR, van Bockxmeer FM, Poplawski N, Hamilton-Craig I, Clifton PM, et al. A model of care for familial hypercholesterolaemia: key role for clinical biochemistry. Clin Biochem Rev. 2012;33:25–31. [PMC free article] [PubMed] [Google Scholar]
- 117.Dehghan A, Dupuis J, Barbalic M, Bis JC, Eiriksdottir G, Lu C, et al. Meta-Analysis of Genome-Wide Association Studies in >80 000 Subjects Identifies Multiple Loci for C-Reactive Protein Levels. Circulation 2011. 2011;123:731–8. doi: 10.1161/CIRCULATIONAHA.110.948570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Chambers JC, Zhang W, Sehmi J, Li X, Wass MN, Van der Harst P, et al. Genome-wide association study identifies loci influencing concentrations of liver enzymes in plasma. Nat Genet. 2011;43:1131–8. doi: 10.1038/ng.970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kottgen A, Albrecht E, Teumer A, Vitart V, Krumsiek J, Hundertmark C, et al. Genome-wide association analyses identify 18 new loci associated with serum urate concentrations. Nat Genet. 2012;45:145–54. doi: 10.1038/ng.2500. 213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Pare G, Chasman DI, Parker AN, Zee RR, Malarstig A, Seedorf U, et al. Novel associations of CPS1, MUT, NOX4, and DPEP1 with plasma homocysteine in a healthy population: a genome-wide evaluation of 13 974 participants in the Women’s Genome Health Study. Circ Cardiovasc Genet. 2009;2:142–50. doi: 10.1161/CIRCGENETICS.108.829804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Speliotes EK, Willer CJ, Berndt SI, Monda KL, Thorleifsson G, Jackson AU, et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet. 2010;42:937–48. doi: 10.1038/ng.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Soranzo N, Sanna S, Wheeler E, Gieger C, Radke D, Dupuis J, et al. Common variants at 10 genomic loci influence hemoglobin A1c levels via glycemic and nonglycemic pathways. Diabetes. 2010;59:3229–39. doi: 10.2337/db10-0502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kottgen A, Pattaro C, Boger CA, Fuchsberger C, Olden M, Glazer NL, et al. New loci associated with kidney function and chronic kidney disease. Nat Genet. 2010;42:376–84. doi: 10.1038/ng.568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Gieger C, Radhakrishnan A, Cvejic A, Tang W, Porcu E, Pistis G, et al. New gene functions in megakaryopoiesis and platelet formation. Nature. 2011;480:201–8. doi: 10.1038/nature10659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Franke A, McGovern DP, Barrett JC, Wang K, Radford-Smith GL, Ahmad T, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat Genet. 2010;42:1118–25. doi: 10.1038/ng.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Manning AK, Hivert MF, Scott RA, Grimsby JL, Bouatia-Naji N, Chen H, et al. A genome-wide approach accounting for body mass index identifies genetic variants influencing fasting glycemic traits and insulin resistance. Nat Genet. 2012;44:659–69. doi: 10.1038/ng.2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Franceschini N, van Rooij FJ, Prins BP, Feitosa MF, Karakas M, Eckfeldt JH, et al. Discovery and fine mapping of serum protein loci through transethnic meta-analysis. Am J Hum Genet. 2012;91:744–53. doi: 10.1016/j.ajhg.2012.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Prokopenko I, Langenberg C, Florez JC, Saxena R, Soranzo N, Thorleifsson G, et al. Variants in MTNR1B influence fasting glucose levels. Nat Genet. 2009;41:77–81. doi: 10.1038/ng.290. [DOI] [PMC free article] [PubMed] [Google Scholar]