

Abstract
Amphetamine (AMPH) and methamphetamine (METH) alter dopamine (DA) transporter (DAT) function. In vitro heterologous cell line and synaptosome studies demonstrate AMPH-induced DAT internalization, implicating relocalization in reduced DAT uptake following drug exposure. However, few studies have evaluated DAT localization following in vivo AMPH/METH administration. To determine DAT subcellular localization following drug administration, a centrifugation technique was developed to isolate striatal synaptosomal membrane and vesicle fractions. DAT was distributed between the synaptosomal membrane (60%) and endosomal vesicles (40%), and in vitro application of the protein kinase C activator phorbol 12-myristate 13-acetate (PMA) to striatal synaptosomes caused DAT internalization into the vesicle fractions. In contrast, neither single nor repeated in vivo AMPH and/or METH administrations altered DAT localization 5, 15, 30 or 60 min post-treatment, despite reduced DAT uptake. Importantly, repeated METH injections uniformly decreased total DAT immunoreactivity within all fractions 7 d post-treatment. These findings suggest factors other than internalization can contribute to the observed acute and persistent DAT dysfunction and dopaminergic deficits following in vivo AMPH or METH administration.
Keywords: Dopamine Transporter, Amphetamine, Methamphetamine, Striatum, Internalization, Relocalization
Introduction
The dopamine (DA) transporter (DAT) is responsible for moving extracellular DA into presynaptic terminals following neurotransmitter release, and disruption of DAT function profoundly alters intracellular and extracellular DA concentration. A single, high-dose administration of methamphetamine (METH) or amphetamine (AMPH) reversibly reduces DAT function within the striatum (Fleckenstein et al. 1997b) – an effect not due to residual drug from in vivo treatment within the synaptosomal preparation (Fleckenstein et al. 1997b, Kokoshka et al. 1998). In contrast, repeated high-dose METH administrations lead to long-term (greater than 7 d) striatal dopaminergic deficits characterized by loss of DAT immunoreactivity, decreased DA uptake, reduced DA content, loss of tyrosine hydroxlyase activity, and the formation of DAT oligomeric complexes (for review see (Fleckenstein et al. 2007).
AMPH and its analog METH disrupt dopaminergic terminals through several simultaneous mechanisms of action. At low concentrations, amphetamines function as DAT substrates, driving DA efflux through a shift in substrate concentration within the synaptic cleft (Kahlig et al. 2005, Connor & Kuczenski 1986). Higher concentrations of amphetamines diffuse through the plasma membrane and prevent vesicular sequestration of cytoplasmic DA (Fleckenstein et al. 2007, Mack & Bonisch 1979, Kahlig et al. 2005, Sulzer & Rayport 1990), which is rapidly transported into the synaptic cleft through DAT in channel-like fashion (Kahlig et al. 2005). The DAT transport reversal and reduction in activity caused by amphetamines are both regulated by protein kinase C (PKC) phosphorylation of the transporter (Johnson et al. 2005b). Pharmacological PKC inhibition prior to administration of amphetamines preserves DAT function, while PKC activation reduces synaptosomal DAT function (Sandoval et al. 2001, Vaughan et al. 1997). PKC phosphorylation also drives DAT internalization (Melikian & Buckley 1999), suggesting transporter endocytosis may underlie reduced DAT function following exposure to AMPH. Indeed, administration of AMPH to cell lines expressing DAT results in DAT internalization within minutes (Saunders et al. 2000). Subsequent studies, however, demonstrate DAT trafficking as biphasic (Johnson et al. 2005a). Large amounts of DAT are rapidly trafficked to the cell surface within the first minute of AMPH treatment and then internalized as drug exposure persists.
AMPH-induced DAT internalization has been primarily demonstrated in vitro (Melikian & Buckley 1999, Saunders et al. 2000, Chen et al. 2009, Richards & Zahniser 2009, Johnson et al. 2005a), and few studies have investigated in vivo AMPH or METH treatment upon DAT localization. Consequently, this study evaluated terminal DAT localization following single and multiple in vivo AMPH and METH treatment paradigms. To detect drug-induced changes in DAT distribution, plasma membrane and vesicular fractions were isolated from striatal synaptosomes using differential centrifugation combined with equilibrium ultracentrifugation across linear sucrose gradients. Using these techniques, this study suggests in vivo AMPH and METH treatments can decrease DAT function without altering DAT distribution between the membrane and endosomal compartments. Therefore, mechanisms other than internalization are likely responsible for the acute and persistent dopaminergic deficits caused by amphetamines and reported herein.
Materials & Methods
Reagents and Antibodies
All chemicals, unless otherwise noted, were purchased from Sigma Aldrich (St. Louis, MO). Antibodies against DAT (1:500; sc-1433) and the DAT peptide (DAT P; sc-1433P) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies against Na+/K+ ATPase (1:500; 63958) and Rab11 (1:500; 610656) were purchased from BD Biosciences (San Jose, CA). Antibody against lysosomal-associate membrane protein 2 (LAMP2, 1:500; 51-2200) was purchased from Zymed Laboratories (San Francisco, CA). HRP-conjugated secondary antibody against rabbit (1:10000; 711-035-152) and goat (1:10000; 705-035-147) was purchased from Jackson Immuno Research (West Grove, PA).
Animals and Drug Treatment Paradigms
Male Sprague-Dawley rats (300 – 400 g; Charles River Laboratories, Raleigh, NC) were housed with food and water provided ad libidum in a temperature- (22 °C) and light-controlled (14/10 light/dark cycle) environment. Rats were kept in a warm environment (approximately 25 °C) during drug treatment to permit increases in core body temperature. All experiments were performed in accordance with the National Institute of Health Guidelines for the Care and Use of Laboratory Animals, approved by the University of Utah Animal Care and Use Committee, and care was taken to ensure the minimization of animal pain and distress throughout treatment. d/l-METH HCl was generously donated by the National Institute of Drug Abuse and synthesized by Research Triangle Institute (Research Triangle Park, NC), and d-AMPH sulfate was acquired from Sigma-Aldrich (St. Louis, MO). Dosages of both drugs were calculated as free-base and dissolved to a 1 ml/kg concentration in sterile 0.9% saline solution. Rats were sacrificed by decapitation. Striata were dissected and quickly placed in ice-cold 0.32M sucrose buffer (0.32 M sucrose, 3.75 mM NaH2PO4, 12.7 mM Na2HPO4, 1 mM phenylmethanesulfonyl fluoride (PMSF), 10 μg/ml aprotinin, 1 mM Na3VO4, 1 mM NaF, pH 7.4); an environment in which the endocytic pathway, endosomal sorting, intracellular signaling and the vast majority of enzymatic and phosphatase/protease activity is halted. Two drug treatment paradigms, consisting of either single or multiple drug injections, were used in this study and paired with various assessment time points. The single treatment paradigm involved a single, subcutaneous (s.c.) injection of AMPH (15 mg/kg), METH (15 mg/kg) or 0.9% sterile saline solution (1 ml/kg) and decapitation 5, 15, 30 or 60 min later. The multiple treatment paradigm involved four METH (7.5 mg/kg/injection, s.c.) or 0.9% sterile saline solution (1 ml/kg/injection) injections administered at two-hour intervals with decapitation 1 h or 7 d after the last injection.
Synaptosomal Isolation, Subcellular Fractionation and Continuous Sucrose Gradients
To isolate striatal synaptosomes, tissues were homogenized in 0.32M sucrose buffer with 25 strokes of a Dounce homogenizer. Figure 1 outlines the subsequent isolation steps employed within this study. Briefly, homogenate was then centrifuged at 800 × g for 12 minutes at 4 °C to pellet cellular debris. The supernatant was transferred to new spin tubes and centrifuged at 22000 × g for 15 minutes at 4 °C. The microsomal supernatant was discarded and the resulting synaptosomal pellet was either resuspended in assay buffer (126 mM NaCl, 4.8 mM KCl, 1.3 mM CaCl2, 1.4 mM MgSO4, 11 mM glucose, 1 mM ascorbic acid, 3.75 mM NaH2PO4, 12.7 mM Na2HPO4, pH 7.4) for [3H]DA uptake assays or lysed in ice-cold ddH20 for further subcellular fractionation.
Figure 1. Illustration of the isolation protocol employed in this study.
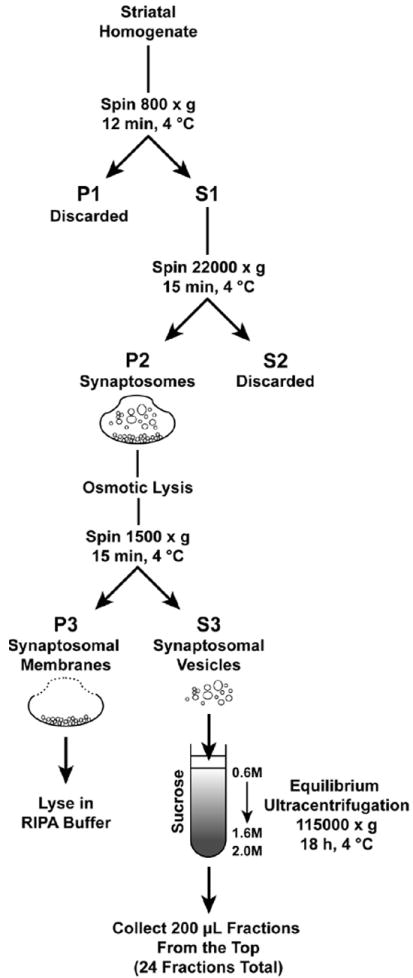
To isolate subcellular structures, synaptosomes were lysed in 450 μl ddH20, triturated 25 times with a glass pipette and disrupted with 5 strokes of a Dounce homogenizer. Samples were then mixed with 50 μl 1M potassium tartrate (KT) and 0.25M HEPES pH 7.4 to yield a 100 mM KT and 25 mM HEPES buffer (final concentration) and halt osmotic lysis. Samples were then centrifuged at 1500 × g for 15 minutes at 4 °C to separate the synaptosomal membrane (pellet, SM) from synaptosomal vesicular structures (supernatant). The SM pellet was resuspended in 1M KT and 0.25M HEPES buffer, the vesicular supernatant was transferred to new spin tubes and all samples were spun again at 1500 × g for 15 minutes at 4 °C to remove contaminants. Two fractions, the SM (pellet) and vesicle (supernatant), were isolated at this stage. The SM pellet was resuspended with 500 μl RIPA lysis buffer (1% Nonidet-P40 (v/v), 24 mM sodium deoxycholate, 3.47 mM sodium dodecyl sulfate, 2 mM ethylenediaminetetraacetic acid, 1X tris-buffered saline (TBS; 20 mM Tris, 137 mM NaCl, pH 8.0), 1 mM PMSF, 10 μg/ml aprotinin, 1 mM Na3VO4, 1 mM NaF) and stored at -80 °C until further use. The vesicle fraction supernatant was either stored at -80 °C until further use or further fractioned across continuous sucrose gradients.
Synaptosomal vesicles were further separated by overlaying the 500 μl of 1500 × g supernatant (vesicle fraction) on 4.0 ml 0.6M – 1.6M continuous sucrose gradients (sucrose mixed in buffer containing 150 mM NaCl, 10 mM HEPES, 1 mM EGTA, 0.1 mM MgCl2, 1 mM PMSF, 10 μg/ml aprotinin, 1 mM Na3VO4, 1 mM NaF) with a 500 μl 2.0M sucrose pad followed by equilibrium centrifugation using a Beckman (Brea, CA) SW55Ti rotor and Optima L-100XP centrifuge to spin at 115000 × g for 18 hours at 4 °C. Twenty-five 200 μl fractions were collected from the top of each sample with a probe (Labconco Auto Densi-flow; Kansas City, MO) and fraction collector (Gilson FC203B; Middleton, WI) then stored at -80 °C until further use.
Synaptosomal [3H]DA Uptake
Uptake of [3H]DA into striatal synaptosomes was performed as previously described (Hadlock et al. 2009). Briefly, samples containing striatal synaptosomes prepared in assay buffer and 1 μM pargyline were incubated at 37 °C for 10 minutes followed by the addition of 0.5 nM [3H]DA for three minutes. Samples with the addition of 50 μM cocaine were included to determine nonspecific uptake. Following incubation samples were passed through 0.05% polyethylenimine-soaked GF/B filter paper (Whatman, Clifton, NJ) and washed three times with ice-cold 0.32M sucrose. Filters were cut and radioactivity trapped within bound synaptosomes was counted using a liquid scintillation counter. Protein concentrations were determined by Bradford assay (Bradford 1976).
SDS-PAGE and Western Blotting
Protein concentration of all samples was determined by bicinchoninic acid protein assay according to manufacturer’s instruction (Pierce Biotechnology). All samples were mixed with Laemmli sample buffer containing 20% β-mercaptoethanol, loaded onto 4-12.5% Tris-HCl Polyacrylamide gels with SeeBlue Plus2 protein ladder (Invitrogen; Carlsbad, CA) and run with constant voltage. Gels were then transferred to polyvinyl difluoride membranes (Whatman) with 6 amps total current, blocked with 5% non-fat dry milk in Tris-buffered saline with Tween-20 (TBS-T; 20 mM Tris, 137 mM NaCl, 0.2% Tween-20, pH 7.6) then probed with primary antibody overnight at 4 °C. Following primary antibody incubation, membranes were washed with TBS-T for 30 minutes, probed with secondary antibody for 1 hour, washed again with TBS-T for 30 minutes, then developed using Super Signal West Pico Chemiluminescent substrate (Thermo Scientific) and imaged with an Alpha Innotech FluorChem HD2 imager.
Equivalent protein from each sample was loaded onto gels when evaluating the synaptosomal DA uptake (20 μg) experiments and the synaptosomal membrane fractions (20 μg). For continuous sucrose gradient experiments, protein equivalents for all fractions of all samples within a given experiment were normalized to 75 μl volume of each fraction of saline 1. For example, if fraction 3 of saline 1 contained 15 μg protein in 75 μl volume then 15 μg protein from fraction 3 of each subsequent sample was loaded. To normalize across membranes, two samples containing 2.5 μg DAT peptide were loaded onto each gel. For all other experiments, 20 μg protein from each sample was analyzed.
Data Analysis
All data were analyzed with GraphPad Prism 5 (GraphPad Software; La Jolla, CA). DA uptake data were analyzed by one-way ANOVA with Newman-Keuls post-hoc tests. Gradient DAT immunoreactivity data were analyzed by two-way ANOVA with Bonferroni post-hoc tests.
Results
A single, high-dose (15 mg/kg, s.c.) administration of AMPH or METH reduced [3H]DA uptake within striatal synaptosomes prepared 1 h following treatment (Fig. 2; one-way ANOVA Saline vs. AMPH = p < 0.001; Saline vs. METH = p < 0.001). No change in total DAT immunoreactivity, however, was observed among striatal synaptosomes prepared from AMPH-, METH- or saline-treated rats (data not shown).
Figure 2. A single in vivo AMPH or METH treatment reduced synaptosomal DA uptake.
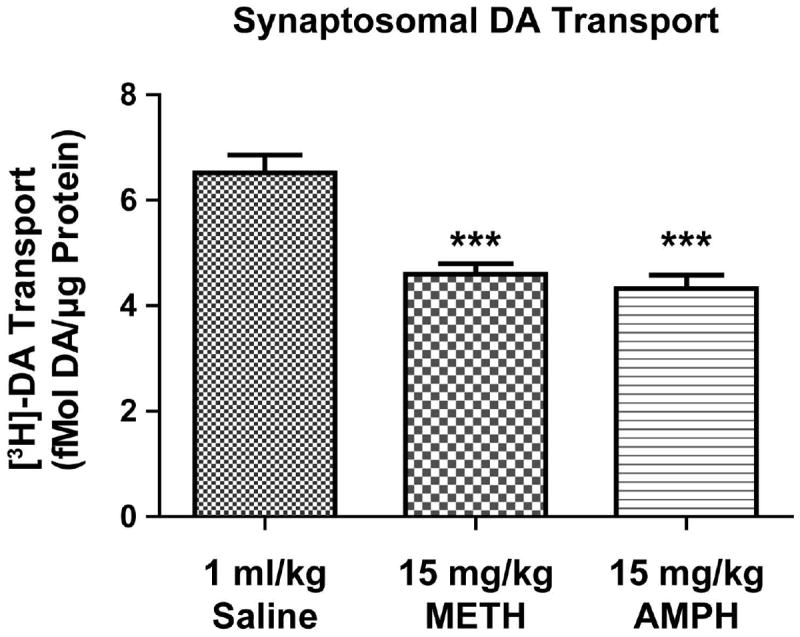
Rats were treated with a single saline (1 ml/kg, s.c.), AMPH (15 mg/kg, s.c.) or METH (15 mg/kg, s.c.) injection and sacrificed 1 h later (n = 8 per treatment group). Striatal synaptosomal DA uptake was calculated as described in Materials and Methods. *** Different than saline (p ≤ 0.001).
To evaluate the effects of AMPH and METH on DAT localization, plasma membrane and intracellular vesicle fractions were isolated from striatal synaptosomes (Figure 1 and Materials & Methods) and DAT distribution within these fractions was determined. Osmotic lysis of untreated synaptosomes followed by differential centrifugation allowed separation of synaptosomal vesicles, a heterogeneous population of synaptic vesicles and endosomal components, from the synaptosomal membrane. These fractions demonstrated 61.1% of the DAT resided at the plasma membrane (i.e., in the 1500 × g pellet; Fig. 3A) and 38.9% of the DAT resided within synaptosomal vesicles (i.e., in the 1500 × g supernatant; Fig. 3A). Equilibrium ultracentrifugation (115000 × g 18 h) of the synaptosomal vesicles fraction (1500 × g supernatant) over 0.6M to 1.6M continuous sucrose gradients provided further discrimination among the vesicle populations. Within the vesicle fraction, DAT immunoreactivity predominantly localized across fractions 3 through 11 with the highest protein concentration found in fraction 5 (Fig. 3B). Endosomal structures were highly enriched within these fractions, as indicated by the recycling endosome marker rab11 within fractions 3 through 7 and the lysosomal-associated membrane protein 2 (LAMP2) marker within fractions 5 through 11 (Fig. 3B). Importantly, the plasma membrane protein Na+/K+-ATPase was highly enriched within the synaptosomal membrane fraction but not within the vesicle fractions, indicating little plasma membrane contamination within the vesicle fractions (Fig. 3B). While nearly all synaptosomal membranes are removed from vesicles, small amounts of vesicular protein (LAMP2, rab11) remain within the synaptosomal membrane fraction.
Figure 3. Distribution of DAT within striatal synaptosomal membrane and vesicle fractions.
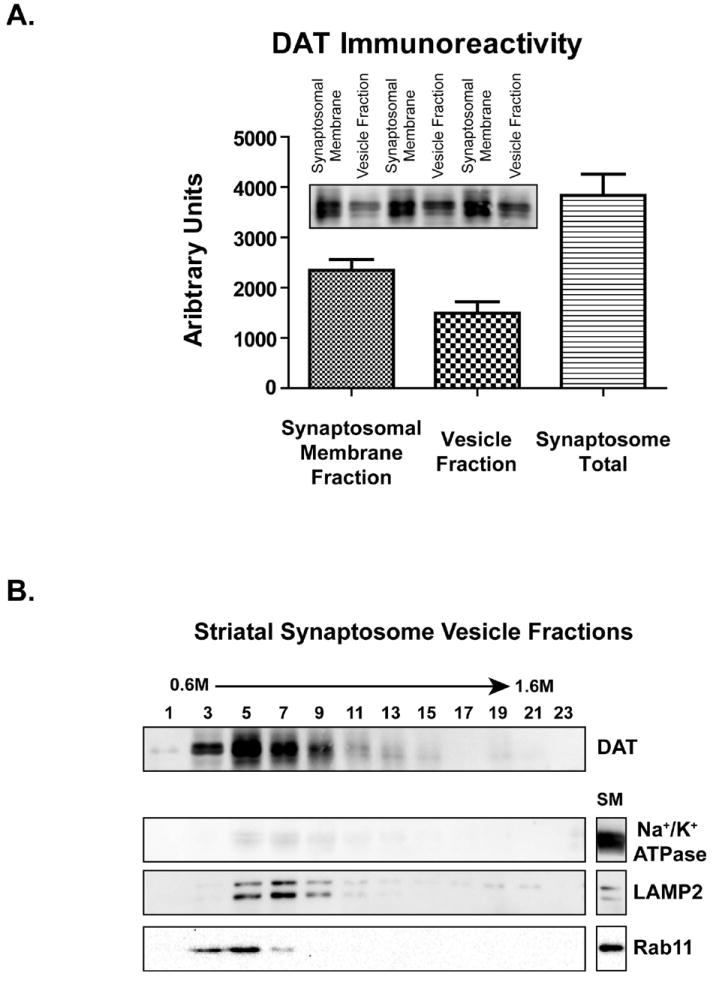
(A) Synaptosomal membrane and total vesicle fractions were isolated from striatal synaptosomes, as described in Materials & Methods, and DAT immunoreactivity was measured by densitometry (n = 3 per group). (B) Distribution of DAT within synaptosomal vesicle fractions isolated across 0.6M – 1.6M continuous sucrose gradients and the synaptosomal membrane fraction. The recycling endosome marker rab11 and lysosome marker LAMP2 localized to DAT within the vesicle fractions, whereas the plasma membrane protein Na+/K+ ATPase did not. ’SM’ indicates synaptosomal membrane fraction.
To demonstrate that DAT internalization can be detected within the synaptosomal membrane and sucrose gradient fractions described above, striatal synaptosomes were treated with the PKC activator phorbol 12-myristate 13-acetate (PMA). Application of 10 μM PMA to striatal synaptosomes for 30 min (37 °C) increased DAT immunoreactivity within the vesicle fractions as compared to dimethyl sulfoxide (DMSO) vehicle-treated control fractions (Fig. 4A; two-way ANOVA, p = 0.0035 between treatments), indicating movement of DAT into the vesicle fractions (Fraction 3 = 39.8% increase; Fraction 5 = 68.7% increase). This concentration of PMA and time point was selected due to its capacity to phosphorylate striatal synaptosomal PKC within 30 min (Vaughan et al. 1997). Treatment of synaptosomes with concentrations of PMA lower than 10 μM yielded no change in DAT localization (data not shown). PMA treatment did not, however, change synaptosomal membrane DAT immunoreactivity (Fig. 4B), likely due to differences in sensitivity of detection between the vesicle and synaptosomal membrane fractions. Since vesicular proteins are spread across the continuous sucrose gradients, the signal to noise ratio within individual synaptosomal vesicle fractions will be greater than the synaptosomal membrane fraction.
Figure 4. PMA increased DAT immunoreactivity within the synaptosomal vesicle fractions.
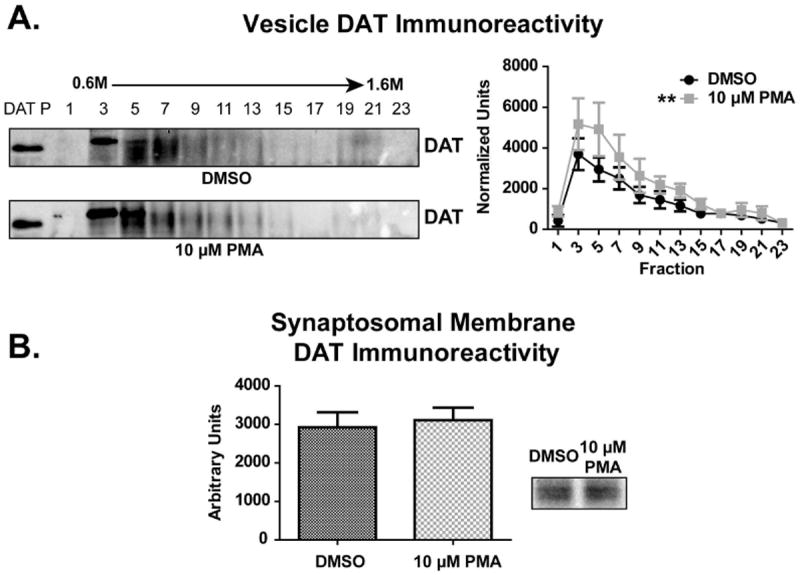
Striatal synaptosomes were treated with 10 μM phorbol 12-myristate 13-acetate (PMA) for 30 min at 37 °C (n = 6 per group). Following treatment, synaptosomal membrane and vesicle fractions were isolated and DAT immunoreactivity was measured by densitometry. (A) DAT immunoreactivity was increased within PMA-treated vesicles as compared to saline-treated vesicles. (B) No difference in DAT immunoreactivity was observed between PMA- and saline-treated synaptosomal membranes. ** Treatment across fractions different than saline-treated fractions (p ≤ 0.01). ‘DAT P’ indicates DAT peptide control lane.
A paradigm previously shown to reduce total DAT immunoreactivity within the striatum (4 × 7.5 mg/kg/injection METH, 2-h intervals, s.c., sacrifice 7 d later) was used to demonstrate the capacity of the synaptosomal membrane and sucrose gradient vesicle fractions to detect loss of DAT (Hadlock et al. 2010). In agreement with previous work (Hadlock et al. 2010), repeated METH administration reduced DAT immunoreactivity within both synaptosomal membrane (Fig. 5B; 55% decrease METH vs. control, t-test p = 0.0030) and vesicle fractions (Fig. 5A; two-way ANOVA, p = 0.0068 between treatments) as compared to saline controls. Importantly, those fractions with the greatest DAT immunoreactivity within saline-treated animals, fractions 3 and 5, had the greatest decrease in DAT immunoreactivity following repeated METH treatment (Fig. 5A; Fraction 3 = 59.0% decrease, p < 0.001; Fraction 5 = 41.4% decrease, p < 0.05). METH-treated rats experienced hyperthermia, as assessed by average core body temperature throughout treatment, as compared to saline controls (METH = 40.0 °C ± 0.2 °C; Saline = 37.4 °C ± 0.1 °C; over the course of repeated METH injections), consistent with previous studies (Metzger et al. 2000).
Figure 5. Repeated METH administrations resulted in persistent loss of striatal DAT immunoreactivity.
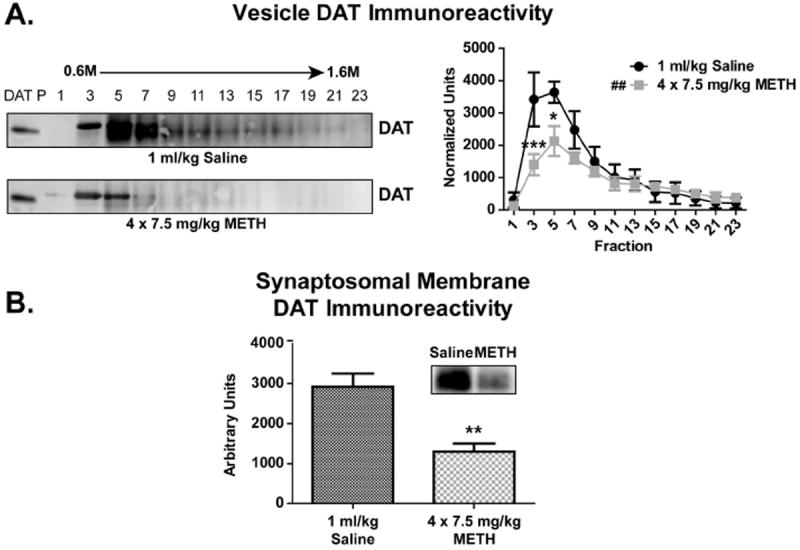
Rats were treated with repeated saline (4 × 1 ml/kg/injection, 2-h intervals, s.c.) or METH (4 × 7.5 mg/kg/injection, 2-h intervals, s.c.) injections and DAT immunoreactivity within synaptosomal membrane and vesicle fractions was determined 7 d later (n = 6 per group). (A) DAT immunoreactivity was reduced within METH-treated fractions as compared to saline-treated control fractions. (B) DAT immunoreactivity was reduced within METH-treated synaptosomal membranes as compared to saline-treated controls. ## Treatment across fractions different than saline-treated fractions (p ≤ 0.01). * Fraction different than comparable saline-treated fraction (p ≤ 0.05). ** Fraction different than comparable saline-treated fraction (p ≤ 0.01). *** Fraction different than comparable saline-treated fraction (p ≤ 0.001). ‘DAT P’ indicates DAT peptide control lane.
A single AMPH or METH injection (15 mg/kg, s.c.; sacrifice 60 min later), regimens that decreased DAT function (Fig. 2), did not alter DAT localization (Figs. 6A, 6C, 8A, 8B). To determine if DAT localization was altered more acutely following AMPH exposure, DAT localization was evaluated in rats given a single AMPH (15 mg/kg, s.c.) injection and sacrificed 5 min, 15 min, or 30 min later (Fig. 6B). No difference in DAT localization was observed between AMPH-treated and saline-treated control rats within the vesicle fractions at any time point (Figs. 6A, 6B). Accordingly, no difference in DAT localization within the striatal synaptosomal membrane fractions was observed between AMPH or saline-treated rats evaluated 5 min, 15 min, 30 min (data not shown), or 60 min (Fig. 6C) following drug exposure.
Figure 6. Single in vivo AMPH treatments did not alter striatal DAT localization 5 - 60 min after drug exposure.
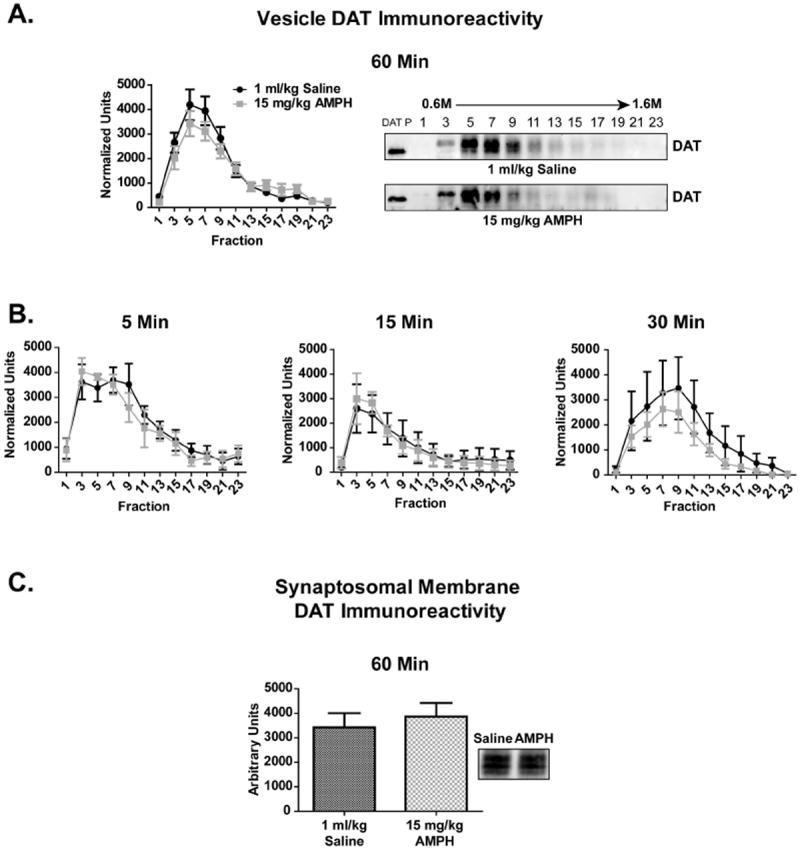
Rats received a single saline (1 ml/kg, s.c.) or AMPH (15 mg/kg, s.c.) injection and were sacrificed 5 min, 15 min, 30 min or 60 min following treatment (n = 6 per group). No difference in DAT immunoreactivity within the vesicle (A, B) or synaptosomal membrane (C) fractions was observed. ‘DAT P’ indicates DAT peptide control lane.
Previous studies have demonstrated that repeated METH administrations (4 × 7.5 mg/kg/injection, 2-h intervals, s.c.) decreased DAT activity by approximately 75% 1 h following treatment (Chu et al. 2008), and that these acute deficits may lead to the persistent dopaminergic deficits caused by METH (e.g., Fig. 5). This treatment paradigm (4 × 7.5 mg/kg/injection, 2-h intervals, s.c., sacrifice 1 h after last injection) did not, however, alter DAT localization or total quantity, within the striatal synaptosomal membrane or vesicle fractions when compared to saline-treated controls (Fig. 8C, 8D). METH-treated rats experienced hyperthermia with an average core temperature of 40.2 °C ± 0.3 °C as compared to 37.4 °C ± 0.1 °C within saline-treated controls.
Discussion
Extensive in vitro work suggests amphetamines cause biphasic DAT relocalization that coincides with reduced extracellular DA uptake (Robertson et al. 2009). When cell lines stably expressing DAT were treated with physiologically relevant concentrations of AMPH (e.g., 2 μM; (Clausing et al. 1995)), DAT moved from endosomal stores to the membrane within the first 2 min and then internalized within 15 min following treatment (Saunders et al. 2000, Johnson et al. 2005a). Similar DAT relocalization occured in striatal synaptosomes treated with AMPH in vitro, but over a longer time period. Treatment of mouse striatal synaptosomes with AMPH increased DAT expression on the plasma membrane within the first 2 min of treatment, and then DAT internalization occurred 1 h following treatment (Chen et al. 2009, Johnson et al. 2005a). Similarly, rat striatal synaptosomal DAT internalization was not observed until 1 h following a large (20 μM) AMPH application (Richards & Zahniser 2009).
This study evaluated the effects of in vivo (rather than in vitro) AMPH and METH upon DAT function and localization, demonstrating in vivo drug treatment acutely decreases DAT function in striatal synaptic terminals without concurrent DAT relocalization (Figs. 2, 6, 7, 8). Importantly, thorough washing of synaptosomes in the process of isolation paired with previous analysis of METH levels in synaptosomal preparations suggests this acute loss of DAT function is not due to residual drug within the synaptosomal preparation (Kokoshka et al. 1998, Fleckenstein et al. 1997b). These findings are consistent previous work by Richards and Zahniser (2009), in which no change in DAT localization was observed within striatal synaptosomes prepared 45 min following AMPH treatment (2 mg/kg). Since no change in DAT localization was observed and the amount of total synaptosomal DAT immunoreactivity remained consistent between AMPH- METH- and saline-treated animals 1 h following treatment, DAT was neither transported out of synaptic terminals nor degraded via proteosomal or lysosomal means. Instead, mechanisms other than internalization and relocalization are likely responsible for reduced DAT function at the plasma membrane. However, the possibility of a brief DAT relocalization on a time frame shorter than evaluated within the present work (less than 5 min) cannot be excluded.
Figure 7. A single or repeated METH administration did not alter striatal DAT localization 60 min following treatment.
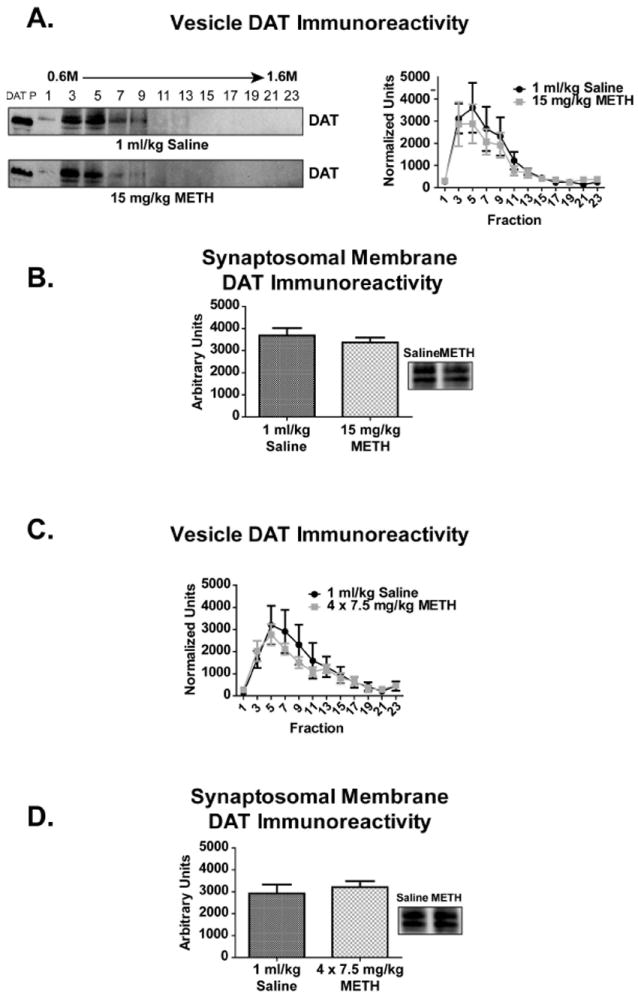
Rats received either single or multiple injections of saline (single = 1 ml/kg, s.c.; multiple = 4 × 1 ml/kg/injection, 2-h intervals, s.c.) or METH (single = 15 mg/kg, s.c.; multiple = 4 × 7.5 mg/kg/injection, 2-h intervals, s.c.) and sacrificed 60 min following treatment (n = 6 per group). No changes in DAT immunoreactivity within the synaptosomal membrane (A, C) or vesicle (B, D) fractions were observed with either treatment paradigm. ‘DAT P’ indicates DAT peptide control lane.
To detect drug-induced changes in synaptosomal DAT localization, a technique was developed to separate the synaptosomal membrane from discrete subcellular fractions using differential centrifugation, with further separation of vesicles using equilibrium ultracentrifugation across continuous sucrose gradients (Fig. 1). Isolation of synaptosomal membrane (P2) from synaptosomal vesicles (S2) revealed approximately 60% of DAT was located at the synaptosomal (plasma) membrane (P2) and 40% of DAT was located within vesicular structures (S2; Fig. 2A) of untreated animals. The synaptosomal vesicles were further separated across 0.6M - 1.6M continuous sucrose gradients using equilibrium ultracentrifugation. These synaptosomal vesicle fractions were enriched with endocytic structures (rab11 and LAMP2) and were not contaminated with plasma membrane, indicated by the lack of Na+/K+ ATPase (Fig. 3). Consistent with other studies (Rao et al. 2010, Chen et al. 2009), DAT localized to those fractions enriched with endocytic vesicles (rab11 and LAMP2). The presence of DAT within mobile endocytic stores supports a proposed model of constitutive DAT cycling between recycling endosomes (rab11) and the plasma membrane (Chen et al. 2009).
The sensitivity of the isolation method to detect movement between and change within the synaptosomal membrane and vesicle fractions was demonstrated through two experiments. First, internalization was detected through increased DAT immunoreactivity within the vesicle fractions of PMA-treated striatal synaptosomes (Fig. 3A). PMA has been previously shown to induce clathrin-mediated DAT endocytosis through phosphorylation by PKC (Melikian & Buckley 1999, Foster et al. 2008, Sorkina et al. 2005, Vaughan et al. 1997), though recent work suggests PKC alone cannot drive substantial DAT internalization (Boudanova et al. 2008, Rao et al. 2010). Our data support the latter assertion, since only a small amount of DAT internalization was observed despite treating synaptosomes with a relatively high concentration of PMA (10 μM; Fig. 3A). Second, DAT immunoreactivity was lost from both synaptosomal membrane and vesicle fractions 7 d following repeated METH injections (Fig. 4), demonstrating a net, uniform subcellular loss of DAT from the striatum and the capacity of this isolation method to detect well-characterized, persistent dopaminergic deficits (Hadlock et al. 2010, Fumagalli et al. 1998).
AMPH- and METH-induced long-term dopaminergic deficits have been associated with acute alteration of DAT function (Fumagalli et al. 1998, Hadlock et al. 2010). Since administration of a METH regimen well documented to cause acute and persistent dopaminergic deficits (4 × 7.5 mg/kg/injection, 2-h intervals, s.c.) did not relocalize DAT (Fig. 4, 7), internalization is not likely a causative factor linking acute and persistent stimulant-induced dopaminergic deficits.
Differences in environmental context may explain the disparity in DAT localization observed between in vivo and in vitro tissue culture/synaptosome models following AMPH and METH administration. Proteins involved in DAT regulation and internalization are not shared between cell lines that exogenously express DAT and dopaminergic neurons. For example, EM4 cells, a derivation of HEK293 cells first used to characterize AMPH-induced DAT internalization (Saunders et al. 2000), lack endogenous expression of calcium-calmodulin dependent protein kinase II (CaMKII), a protein kinase known to phosphorylate and alter DAT function following AMPH treatment (Xu et al. 2008). Since heterologous cell lines do not express DA receptors, they cannot respond to a primary environmental consequence of exposure to amphetamines – elevated extracellular DA – in the same manner as neurons (Zhou et al. 1990). Activation of DA receptors is required for well-characterized METH-induced dopaminergic deficits to develop (Granado et al. 2011). Finally, AMPH and METH impact non-dopaminergic neurons outside of the striatum that connect to and directly influence dopaminergic projections within the striatum. High-dose METH administration enhances the activity of striatonigral GABAergic projections, which increases the release of glutamate within the striatum and leads to long-term striatal DA depletion (Mark et al. 2004). Individually or together, these differences may underlie observed discrepencies in DAT localization between in vivo and in vitro models following AMPH and METH treatment.
While internalization is unlikely to account for AMPH- and METH-induced DAT dysfunction observed herein, a large body of evidence suggests DAT may be structurally altered by drug exposure. A number of interacting protein partners are known to cause post-translational modifications of DAT following exposure to amphetamines, which likely occur at the membrane. PKC, syntaxin1A and CaMKII have all been implicated in changing DAT transport rate or initiating DA efflux through interaction with DAT, and are probable candidates responsible for reduced in vivo transport activity (Binda et al. 2008, Johnson et al. 2005b, Fog et al. 2006). DAT may also be oxidatively inactivated (Fleckenstein et al. 1997a, Berman et al. 1996). Oxidation of excessive terminal DA into reactive oxygen species and quinone formations reduce DA uptake through DAT (Berman et al. 1996, Fleckenstein et al. 1997a, LaVoie & Hastings 1999), which may occur at the membrane.
In summary, this study demonstrates that while in vivo AMPH or METH exposure reduce DAT activity within striatal synaptic terminals, no change in DAT localization is observed at the time points assessed. These studies contrast with previous in vitro AMPH and METH studies, presumably due to differences between in vitro and in vivo models. Mechanisms other than internalization likely cause reduced DAT function and persistent dopaminergic deficits following in vivo AMPH and METH exposure. Substantial evidence suggests modification of the DAT protein itself, through post-translational modification or structural alteration, may underlie drug-induced dysfunction. Consequently, further investigation into the post-translational modifications of striatal DAT and the signaling mechanism involved during AMPH and METH treatment is warranted.
Acknowledgments
This work was supported by grants DA00869, DA04222, DA13367, DA11389, DA019447, and DA00378 from the National Institute on Drug Abuse. We thank Dr. James W. Gibb for his critical reading of this manuscript.
Abbreviations
- METH
methamphetamine
- AMPH
amphetamine
- DA
dopamine
- DAT
dopamine transporter
- PMA
phorbol 12-myristate 13-acetate
Footnotes
The authors have no conflicts of interest to declare.
References
- Berman SB, Zigmond MJ, Hastings TG. Modification of dopamine transporter function: effect of reactive oxygen species and dopamine. Journal of neurochemistry. 1996;67:593–600. doi: 10.1046/j.1471-4159.1996.67020593.x. [DOI] [PubMed] [Google Scholar]
- Binda F, Dipace C, Bowton E, et al. Syntaxin 1A interaction with the dopamine transporter promotes amphetamine-induced dopamine efflux. Molecular pharmacology. 2008;74:1101–1108. doi: 10.1124/mol.108.048447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudanova E, Navaroli DM, Melikian HE. Amphetamine-induced decreases in dopamine transporter surface expression are protein kinase C-independent. Neuropharmacology. 2008;54:605–612. doi: 10.1016/j.neuropharm.2007.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical biochemistry. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Chen R, Furman CA, Zhang M, Kim MN, Gereau RWt, Leitges M, Gnegy ME. Protein kinase Cbeta is a critical regulator of dopamine transporter trafficking and regulates the behavioral response to amphetamine in mice. The Journal of pharmacology and experimental therapeutics. 2009;328:912–920. doi: 10.1124/jpet.108.147959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu PW, Seferian KS, Birdsall E, Truong JG, Riordan JA, Metcalf CS, Hanson GR, Fleckenstein AE. Differential regional effects of methamphetamine on dopamine transport. European journal of pharmacology. 2008;590:105–110. doi: 10.1016/j.ejphar.2008.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausing P, Gough B, Holson RR, Slikker W, Jr, Bowyer JF. Amphetamine levels in brain microdialysate, caudate/putamen, substantia nigra and plasma after dosage that produces either behavioral or neurotoxic effects. The Journal of pharmacology and experimental therapeutics. 1995;274:614–621. [PubMed] [Google Scholar]
- Connor CE, Kuczenski R. Evidence that amphetamine and Na+ gradient reversal increase striatal synaptosomal dopamine synthesis through carrier-mediated efflux of dopamine. Biochemical pharmacology. 1986;35:3123–3130. doi: 10.1016/0006-2952(86)90396-5. [DOI] [PubMed] [Google Scholar]
- Fleckenstein AE, Metzger RR, Beyeler ML, Gibb JW, Hanson GR. Oxygen radicals diminish dopamine transporter function in rat striatum. European journal of pharmacology. 1997a;334:111–114. doi: 10.1016/s0014-2999(97)01175-8. [DOI] [PubMed] [Google Scholar]
- Fleckenstein AE, Metzger RR, Wilkins DG, Gibb JW, Hanson GR. Rapid and reversible effects of methamphetamine on dopamine transporters. The Journal of pharmacology and experimental therapeutics. 1997b;282:834–838. [PubMed] [Google Scholar]
- Fleckenstein AE, Volz TJ, Riddle EL, Gibb JW, Hanson GR. New insights into the mechanism of action of amphetamines. Annual review of pharmacology and toxicology. 2007;47:681–698. doi: 10.1146/annurev.pharmtox.47.120505.105140. [DOI] [PubMed] [Google Scholar]
- Fog JU, Khoshbouei H, Holy M, et al. Calmodulin kinase II interacts with the dopamine transporter C terminus to regulate amphetamine-induced reverse transport. Neuron. 2006;51:417–429. doi: 10.1016/j.neuron.2006.06.028. [DOI] [PubMed] [Google Scholar]
- Foster JD, Adkins SD, Lever JR, Vaughan RA. Phorbol ester induced trafficking-independent regulation and enhanced phosphorylation of the dopamine transporter associated with membrane rafts and cholesterol. Journal of neurochemistry. 2008;105:1683–1699. doi: 10.1111/j.1471-4159.2008.05262.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumagalli F, Gainetdinov RR, Valenzano KJ, Caron MG. Role of dopamine transporter in methamphetamine-induced neurotoxicity: evidence from mice lacking the transporter. J Neurosci. 1998;18:4861–4869. doi: 10.1523/JNEUROSCI.18-13-04861.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granado N, Ares-Santos S, Oliva I, OS E, Martin ED, Colado MI, Moratalla R. Dopamine D2-receptor knockout mice are protected against dopaminergic neurotoxicity induced by methamphetamine or MDMA. Neurobiology of disease. 2011 doi: 10.1016/j.nbd.2011.01.033. [DOI] [PubMed] [Google Scholar]
- Hadlock GC, Baucum AJ, 2nd, King JL, Horner KA, Cook GA, Gibb JW, Wilkins DG, Hanson GR, Fleckenstein AE. Mechanisms underlying methamphetamine-induced dopamine transporter complex formation. The Journal of pharmacology and experimental therapeutics. 2009;329:169–174. doi: 10.1124/jpet.108.145631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadlock GC, Chu PW, Walters ET, Hanson GR, Fleckenstein AE. Methamphetamine-induced dopamine transporter complex formation and dopaminergic deficits: the role of D2 receptor activation. The Journal of pharmacology and experimental therapeutics. 2010;335:207–212. doi: 10.1124/jpet.110.166660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson LA, Furman CA, Zhang M, Guptaroy B, Gnegy ME. Rapid delivery of the dopamine transporter to the plasmalemmal membrane upon amphetamine stimulation. Neuropharmacology. 2005a;49:750–758. doi: 10.1016/j.neuropharm.2005.08.018. [DOI] [PubMed] [Google Scholar]
- Johnson LA, Guptaroy B, Lund D, Shamban S, Gnegy ME. Regulation of amphetamine-stimulated dopamine efflux by protein kinase C beta. The Journal of biological chemistry. 2005b;280:10914–10919. doi: 10.1074/jbc.M413887200. [DOI] [PubMed] [Google Scholar]
- Kahlig KM, Binda F, Khoshbouei H, Blakely RD, McMahon DG, Javitch JA, Galli A. Amphetamine induces dopamine efflux through a dopamine transporter channel. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:3495–3500. doi: 10.1073/pnas.0407737102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokoshka JM, Metzger RR, Wilkins DG, Gibb JW, Hanson GR, Fleckenstein AE. Methamphetamine treatment rapidly inhibits serotonin, but not glutamate, transporters in rat brain. Brain research. 1998;799:78–83. doi: 10.1016/s0006-8993(98)00472-7. [DOI] [PubMed] [Google Scholar]
- LaVoie MJ, Hastings TG. Dopamine quinone formation and protein modification associated with the striatal neurotoxicity of methamphetamine: evidence against a role for extracellular dopamine. J Neurosci. 1999;19:1484–1491. doi: 10.1523/JNEUROSCI.19-04-01484.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack F, Bonisch H. Dissociation constants and lipophilicity of catecholamines and related compounds. Naunyn-Schmiedeberg’s archives of pharmacology. 1979;310:1–9. doi: 10.1007/BF00499868. [DOI] [PubMed] [Google Scholar]
- Mark KA, Soghomonian JJ, Yamamoto BK. High-dose methamphetamine acutely activates the striatonigral pathway to increase striatal glutamate and mediate long-term dopamine toxicity. J Neurosci. 2004;24:11449–11456. doi: 10.1523/JNEUROSCI.3597-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melikian HE, Buckley KM. Membrane trafficking regulates the activity of the human dopamine transporter. J Neurosci. 1999;19:7699–7710. doi: 10.1523/JNEUROSCI.19-18-07699.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger RR, Haughey HM, Wilkins DG, Gibb JW, Hanson GR, Fleckenstein AE. Methamphetamine-induced rapid decrease in dopamine transporter function: role of dopamine and hyperthermia. The Journal of pharmacology and experimental therapeutics. 2000;295:1077–1085. [PubMed] [Google Scholar]
- Rao A, Simmons D, Sorkin A. Differential subcellular distribution of endosomal compartments and the dopamine transporter in dopaminergic neurons. Molecular and cellular neurosciences. 2010;46:148–158. doi: 10.1016/j.mcn.2010.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards TL, Zahniser NR. Rapid substrate-induced down-regulation in function and surface localization of dopamine transporters: rat dorsal striatum versus nucleus accumbens. Journal of neurochemistry. 2009;108:1575–1584. doi: 10.1111/j.1471-4159.2009.05910.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson SD, Matthies HJ, Galli A. A closer look at amphetamine-induced reverse transport and trafficking of the dopamine and norepinephrine transporters. Molecular neurobiology. 2009;39:73–80. doi: 10.1007/s12035-009-8053-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandoval V, Riddle EL, Ugarte YV, Hanson GR, Fleckenstein AE. Methamphetamine-induced rapid and reversible changes in dopamine transporter function: an in vitro model. J Neurosci. 2001;21:1413–1419. doi: 10.1523/JNEUROSCI.21-04-01413.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders C, Ferrer JV, Shi L, et al. Amphetamine-induced loss of human dopamine transporter activity: an internalization-dependent and cocaine-sensitive mechanism. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:6850–6855. doi: 10.1073/pnas.110035297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorkina T, Hoover BR, Zahniser NR, Sorkin A. Constitutive and protein kinase C-induced internalization of the dopamine transporter is mediated by a clathrin-dependent mechanism. Traffic (Copenhagen, Denmark) 2005;6:157–170. doi: 10.1111/j.1600-0854.2005.00259.x. [DOI] [PubMed] [Google Scholar]
- Sulzer D, Rayport S. Amphetamine and other psychostimulants reduce pH gradients in midbrain dopaminergic neurons and chromaffin granules: a mechanism of action. Neuron. 1990;5:797–808. doi: 10.1016/0896-6273(90)90339-h. [DOI] [PubMed] [Google Scholar]
- Vaughan RA, Huff RA, Uhl GR, Kuhar MJ. Protein kinase C-mediated phosphorylation and functional regulation of dopamine transporters in striatal synaptosomes. The Journal of biological chemistry. 1997;272:15541–15546. doi: 10.1074/jbc.272.24.15541. [DOI] [PubMed] [Google Scholar]
- Xu M, Chandler LJ, Woodward JJ. Ethanol inhibition of recombinant NMDA receptors is not altered by coexpression of CaMKII-alpha or CaMKII-beta. Alcohol (Fayetteville, N Y) 2008;42:425–432. doi: 10.1016/j.alcohol.2008.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou QY, Grandy DK, Thambi L, et al. Cloning and expression of human and rat D1 dopamine receptors. Nature. 1990;347:76–80. doi: 10.1038/347076a0. [DOI] [PubMed] [Google Scholar]